
Home » ANTIBODIES (Page 2)
Category Archives: ANTIBODIES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Daratumumab
![]()
Daratumumab
(Darzalex®)Approved
An anti-CD38 monoclonal antibody used to treat multiple myeloma.
![]()
Research Code HuMax-CD-38; HuMaxCD-38
CAS No.

Daratumumab (HuMax®-CD38)
Daratumumab (Darzalex) is an anti-cancer drug. It binds to CD38.[1] Daratumumab was originally developed by Genmab, but it is now being jointly developed by Genmab along with the Johnson & Johnson subsidiary Janssen Biotech, which acquired worldwide commercialization rights to the drug from Genmab.[2]
Clinical trials
Encouraging preliminary results were reported in June 2012 from a Phase 1/2 clinical trial in relapsed multiple myeloma patients.[3]Updated trial results presented in December 2012 indicate daratumumab is continuing to show promising single-agent anti-myeloma activity.[4] A 2015 study compared monotherapy 8 and 16mg/kg at monthly to weekly intervals.[5]
In November 2015, the U.S. Food and Drug Administration approved daratumumab for treatement of multiple myeloma.[6]

Interference with blood compatibility testing
Daratumumab can also bind to CD38 present on red blood cells and interfere with antibody testing. Patients will show a panreactive antibody panel, including a positive auto-control. Treatment of the antibody panel cells with dithiothreitol (DTT) and repeating testing will effectively negate the binding of daratumumab to CD38 on the RBC surface; however, DTT also inactivates/destroys many antigens on the RBC surface by disrupting disulfide bonds. Fortunately, the only antigen system affected that is associated with common, clinically significant antibodies is Kell, making K-negative RBCs a reasonable alternative when urgent transfusion is indicated.[7]
Daratumumab is a human IgG1k monoclonal antibody (mAb) that binds with high affinity to the CD38 molecule, which is highly expressed on the surface of multiple myeloma cells. It is believed to induce rapid tumor cell death through programmed cell death, or apoptosis, and multiple immune-mediated mechanisms, including complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis and antibody-dependent cellular cytotoxicity.
Daratumumab is approved in the United States for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent.

In May 2013, daratumumab received Fast Track Designation and Breakthrough Therapy Designation from the US FDA for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy including a PI and an immunomodulatory agent or who are double refractory to a PI and an immunomodulatory agent. Breakthrough Therapy Designation is a program intended to expedite the development and review of drugs to treat serious or life-threatening diseases in cases where preliminary clinical evidence shows that the drug may provide substantial improvements over available therapy. Daratumumab has also received Orphan Drug Designation from the US FDA and the EMA for the treatment of multiple myeloma.
Five Phase III clinical studies with daratumumab in relapsed and frontline settings are currently ongoing. Additional studies are ongoing or planned to assess its potential in other malignant and pre-malignant diseases on which CD38 is expressed, such as smoldering myeloma and non-Hodgkin’s lymphoma.
Genmab announced a global license and development agreement for daratumumab with Janssen Biotech, Inc. in August 2012. The agreement became effective in September 2012.
DARZALEX® (daratumumab) Approved by U.S. FDA: First Human Anti-CD38 Monoclonal Antibody Available for the Treatment of Multiple Myeloma
First-in-class immunotherapy approved for multiple myeloma patients who have received three or more prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent or who are double refractory to a PI and immunomodulatory agent
HORSHAM, PA, November 16, 2015 – Janssen Biotech, Inc., a Janssen Pharmaceutical Company of Johnson & Johnson, announced today the U.S. Food and Drug Administration (FDA) has approved DARZALEX® (daratumumab) injection for intravenous infusion for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent.1 This indication is approved under accelerated approval based on response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Multiple myeloma is an incurable blood cancer that occurs when malignant plasma cells grow uncontrollably in the bone marrow.2,3 Refractory cancer occurs when a patient’s disease is resistant to treatment or in the case of multiple myeloma, the disease progresses within 60 days of their last therapy.4,5 Relapsed cancer means the disease has returned after a period of initial, partial or complete remission.6
DARZALEX is the first human anti-CD38 monoclonal antibody (mAb) approved anywhere in the world. CD38 is a surface protein that is expressed by most, if not all, multiple myeloma cells.7 DARZALEX is believed to induce tumor cell death through multiple immune-mediated mechanisms of action,8,9 in addition to apoptosis, in which a series of molecular steps in a cell lead to its death.10 Its approval comes just two months after the Biologics License Application (BLA) was accepted for Priority Review by the FDA in September 2015.11 DARZALEX received Breakthrough Therapy Designation from the FDA for this indication in May 2013.12
“Multiple myeloma is a highly complex disease and remains incurable, with almost all patients relapsing or becoming resistant to therapy,” said DARZALEX clinical trial investigator Paul G. Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center, Dana-Farber Cancer Institute. “With DARZALEX, we have a promising new immunotherapy, which has shown pronounced efficacy as a single agent with an acceptable adverse event profile. This is especially important for treating these heavily pre-treated patients in whom all of the major classes of currently available medicines have failed.”
The pivotal open-label Phase 2 MMY2002 (SIRIUS) study showed treatment with single-agent DARZALEX resulted in an overall response rate (ORR) of 29.2 percent (95% CI; 20.8, 38.9) in patients who received a median of five prior lines of therapy, including a PI and an immunomodulatory agent.1
Stringent complete response (sCR) was reported in 2.8 percent of patients, very good partial response (VGPR) was reported in 9.4 percent of patients, and partial response (PR) was reported in 17 percent of patients.1 These efficacy results were based on ORR as determined by the Independent Review Committee assessment using IMWG (International Myeloma Working Group) criteria and the range for median duration of response.
For responders, the median duration of response was 7.4 months (range 1.2-13.1+ months).1 At baseline, 97 percent of patients were refractory to their last line of therapy, 95 percent were refractory to both a PI and an immunomodulatory agent, and 77 percent were refractory to alkylating agents.1 Additional efficacy data from the Phase 1/2 GEN501 monotherapy study – published in The New England Journal of Medicine in August 2015 – also support this approval.1
“The responses we saw in clinical trials that led to today’s approval were striking, especially considering that these patients received a median of five prior lines of therapy,” said MMY2002 investigator Sagar Lonial, M.D., Chief Medical Officer, Winship Cancer Institute of Emory University and Professor and Executive Vice Chair, Department of Hematology and Medical Oncology, Emory University School of Medicine. “It appears the mechanism of action for daratumumab (DARZALEX) may play an important role in its single-agent activity among this group of advanced-stage multiple myeloma patients.”
“Living with multiple myeloma is challenging, both physically and emotionally, especially as the disease progresses and treatment options become more limited,” said Debby Graff, a patient enrolled in a clinical trial at Dana-Farber Cancer Institute. “I am encouraged by emerging treatments for multiple myeloma, and I have a new outlook on my path forward.”
“While there have been considerable improvements over the past decade in the treatment of people living with multiple myeloma, these patients face a long, hard road – especially those whose disease has relapsed or is no longer responding to current therapies,” said Walter M. Capone, President and Chief Executive Officer of the Multiple Myeloma Research Foundation (MMRF). “With the approval of daratumumab, a new antibody option targeting CD38, along with ongoing work to advance the development of novel classes of therapies by both Janssen and MMRF, we are ushering in a new era of myeloma therapy focused on individualized treatment approaches for patients with significant unmet needs.”
“Our focus is developing transformational medicines for people living with hard-to-treat cancers, such as multiple myeloma,” said Peter F. Lebowitz, M.D., Ph.D., Global Oncology Head, Janssen. “The rapid development and approval of DARZALEX – the first human anti-CD38 monoclonal antibody – is a great example of this commitment and our ongoing work in developing immunotherapies. We will continue to study this compound as both a mono- and a combination therapy to understand its full clinical benefit for patients across the treatment continuum in multiple myeloma and other tumor types.”
The warnings and precautions for DARZALEX include infusion reactions, interference with serological testing and interference with determination of complete response (see Important Safety Information).1 The most frequently reported adverse reactions (incidence ≥20%) were: fatigue, nausea, back pain, pyrexia, cough and upper respiratory tract infection.1
In data from three pooled clinical studies including a total of 156 patients, four percent of patients discontinued treatment due to adverse reactions.1 Infusion reactions were reported in approximately half of all patients treated with DARZALEX.1 Common (≥5 percent) symptoms of infusion reactions included nasal congestion, chills, cough, allergic rhinitis, throat irritation, dyspnea (shortness of breath) and nausea.1 Severe infusion reactions, including bronchospasm, dyspnea, hypoxia and hypertension (<2 percent each).1
The recommended dose of DARZALEX is 16 mg/kg body weight administered as an intravenous infusion.1 The dosing schedule begins with weekly administration (weeks 1-8) and reduces in frequency over time to every two weeks (weeks 9-24) and ultimately every four weeks (week 25 onwards until disease progression).1
In August 2012, Janssen Biotech, Inc. and Genmab A/S entered a worldwide agreement, which granted Janssen an exclusive license to develop, manufacture and commercialize DARZALEX.13 Janssen is currently the global sponsor of all but one clinical study. DARZALEX will be commercialized in the U.S. by Janssen Biotech, Inc.
About Multiple Myeloma
Multiple myeloma is an incurable blood cancer that occurs when malignant plasma cells grow uncontrollably in the bone marrow.2,3 Multiple myeloma is the third most common blood cancer in the U.S., following only leukemia and lymphoma.14 Approximately 26,850 new patients will be diagnosed with multiple myeloma, and approximately 11,240 people will die from the disease in the U.S. in 2015.15 Globally, it is estimated that 124,225 people will be diagnosed, and 87,084 will die from the disease in 2015.16,17 While some patients with multiple myeloma have no symptoms at all, most patients are diagnosed due to symptoms which can include bone problems, low blood counts, calcium elevation, kidney problems or infections.18 Patients who relapse after treatment with standard therapies (including PIs or immunomodulatory agents) typically have poor prognoses and few remaining options.3
Access to DARZALEX® (daratumumab) Injection, for Intravenous Infusion
DARZALEX (daratumumab) injection for intravenous infusion will be available for distribution in the U.S. within two weeks following FDA approval. Janssen Biotech offers comprehensive access and support information, resources and services to assist U.S. patients in gaining access to DARZALEX through the Janssen CarePath Program. For more information, health care providers or patients can contact: 1-844-55DARZA (1-844-553-2792). Information will also be available at www.DARZALEX.com. Dedicated case coordinators are available to work with both healthcare providers and patients.
Patients with private or commercial insurance may be eligible for the Janssen CarePath Savings Program for DARZALEX. Information on the enrollment process will be available online at www.darzalex.com/access-and-cost-support#affordability.
About DARZALEX® (daratumumab) Injection, for Intravenous Infusion
DARZALEX® (daratumumab) injection for intravenous infusion is indicated for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent.1 This indication is approved under accelerated approval based on response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. DARZALEX is the first human anti-CD38 monoclonal antibody (mAb) to receive U.S. Food and Drug Administration (FDA) approval to treat multiple myeloma. DARZALEX is believed to induce tumor cell death through apoptosis, in which a series of molecular steps in a cell lead to its death1,10 and multiple immune-mediated mechanisms of action, including complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP).1,8 More information will be available atwww.DARZALEX.com.

References
- World Health Organization (2009). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 101” (PDF). WHO Drug Information 23 (2).
- “‘Janssen Biotech Announces Global License and Development Agreement for Investigational Anti-Cancer Agent Daratumumab'”. Janssen Biotech. Retrieved 2013-01-31.
- “ASCO: Drug Shows Promise in Myeloma”. MedPage Today.
- “‘Daratumumab Continues To Show Promise For Relapsed/Refractory Myeloma Patients (ASH 2012)'”. The Myeloma Beacon. Retrieved 2013-01-31.
- Lokhorst, Henk M.; Plesner, Torben; Laubach, Jacob P.; Nahi, Hareth; Gimsing, Peter; Hansson, Markus; Minnema, Monique C.; Lassen, Ulrik; Krejcik, Jakub (2015-09-24). “Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma”. The New England Journal of Medicine 373 (13): 1207–1219. doi:10.1056/NEJMoa1506348. ISSN 1533-4406. PMID 26308596.
- http://www.medscape.com/viewarticle/854548?nlid=91686_3663&src=wnl_edit_newsal&uac=78316PX&impID=890536&faf=1
- Chapuy, CI; Nicholson, RT; Aguad, MD; Chapuy, B; Laubach, JP; Richardson, PG; Doshi, P; Kaufman, RM (June 2015). “Resolving the daratumumab interference with blood compatibility testing.”. Transfusion 55 (6 Pt 2): 1545–54. PMID 25764134.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | CD38 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 945721-28-8 |
| ATC code | none |
| ChemSpider | none |
| UNII | 4Z63YK6E0E |
| Chemical data | |
| Formula | C6466H9996N1724O2010S42 |
| Molar mass | 145,391.67 g·mol−1 |
////Daratumumab
Reslizumab


Reslizumab
(Cinqair®) Approved Active, FDA 2016-03-23
An interleukin-5 (IL-5) antagonist used to treat severe asthma.
CAS 241473-69-8
![]()
Research Code CDP-835; CEP-38072; CTx-55700; SCH-5570; SCH-55700; TRFK-5,

Anti-interleukin-5 monoclonal antibody – Celltech/Schering-Plough
Reslizumab was approved by the U.S. Food and Drug Administration (FDA) on March 23, 2016. It was developed and marketed as Cinqair® by Teva.
Reslizumab is an interleukin-5 antagonist, which binds to human IL-5 and prevents it from binding to the IL-5 receptor, thereby reducing eosinophilic inflammation. It is indicated for the maintenance treatment of patients with severe asthma in patients aged 18 years and older.
Cinqair® is available as injection for intravenous infusion, containing 100 mg of reslizumab in 10 mL solution in single-use vials. The recommended dose is 3 mg/kg once every four weeks.
- Originator Celltech R&D; Schering-Plough
- Developer Celltech R&D; Teva Pharmaceutical Industries
- Class Antiasthmatics; Monoclonal antibodies
- Mechanism of Action Interleukin 5 receptor antagonists
- Orphan Drug Status Yes – Oesophagitis
- 23 Mar 2016 Registered for Asthma in USA (IV) – First global approval
- 04 Mar 2016 Pooled efficacy data from two phase III trials in Asthma presented at the 2016 Annual Meeting of the American Academy of Allergy, Asthma and Immunology (AAAAI-2016)
- 10 Dec 2015 Preregistration for Asthma in Canada (IV)
Reslizumab (trade name Cinqair) is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] The FDA approved reslizumab for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older on March 23, 2016. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.[2]

Teva Announces FDA Acceptance of the Biologics License Application for Reslizumab
Investigational Biologic for the Treatment of Inadequately Controlled Asthma in Patients with Elevated Blood Eosinophils Accepted for Review
JERUSALEM–(BUSINESS WIRE)–Jun. 15, 2015– Teva Pharmaceutical Industries Ltd., (NYSE: TEVA) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the Biologics License Application (BLA) for reslizumab, the company’s investigational humanized monoclonal antibody (mAb) which targets interleukin-5 (IL-5), for the treatment of inadequately controlled asthma in adult and adolescent patients with elevated blood eosinophils, despite an inhaled corticosteroid (ICS)-based regimen.
“Despite currently available medicines, uncontrolled asthma remains a serious problem for patients, physicians and healthcare systems, highlighting the need for targeted new treatment options,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The reslizumab BLA filing acceptance represents a significant milestone for Teva as we work toward serving a specific asthma patient population that is defined by elevated blood eosinophil levels and inadequately controlled symptoms despite standard of care therapy. In clinical trials, patients treated with reslizumab showed significant reductions in the rate of asthma exacerbations and significant improvement in lung function. If approved, we believe reslizumab will serve as an important new targeted treatment option to achieve better asthma control for patients with eosinophil-mediated disease.”
The BLA for reslizumab includes data from Teva’s Phase III BREATH clinical trial program. The program consisted of four separate placebo-controlled Phase III trials involving more than 1,700 adult and adolescent asthma patients with elevated blood eosinophils, whose symptoms were inadequately controlled with inhaled corticosteroid-based therapies. Results from these studies demonstrated that reslizumab, in comparison to placebo, reduced asthma exacerbation rates by at least half and provided significant improvement in lung function and other secondary measures of asthma control when added to an existing ICS-based therapy. Common adverse events in the reslizumab treatment group were comparable to placebo and included worsening of asthma, nasopharyngitis, upper respiratory infections, sinusitis, influenza and headache. Two anaphylactic reactions were reported and resolved following medical treatment at the study site.
Results from the reslizumab BREATH program were recently presented at the American Thoracic Society 2015 Annual Meeting and the American Academy of Allergy, Asthma and Immunology 2015 Annual Meeting, in addition to being published in The Lancet Respiratory Medicine. The BLA for reslizumab has been accepted for filing by the FDA for standard review, with FDA Regulatory Action expected in March 2016.
About Reslizumab
Reslizumab is an investigational humanized monoclonal antibody which targets interleukin-5 (IL-5). IL-5 is a key cytokine involved in the maturation, recruitment, and activation of eosinophils, which are inflammatory white blood cells implicated in a number of diseases, such as asthma. Elevated levels of blood eosinophils are a risk factor for future asthma exacerbations. Reslizumab binds circulating IL-5 thereby preventing IL-5 from binding to its receptor.
About Asthma
Asthma is a chronic (long term) disease usually characterized by airway inflammation and narrowing of the airways, which can vary over time. Asthma may cause recurring periods of wheezing (a whistling sound when you breathe), chest tightness, shortness of breath and coughing that often occurs at night or early in the morning. Without appropriate treatment, asthma symptoms may become more severe and result in an asthma attack, which can lead to hospitalization and even death.
About Eosinophils
Eosinophils are a type of white blood cell that are present at elevated levels in the lungs and blood of many asthmatics. Evidence shows that eosinophils play an active role in the pathogenesis of the disease. IL-5 has been shown to play a crucial role in maturation, growth and activation of eosinophils. Increased levels of eosinophils in the sputum and blood have been shown to correlate with severity and frequency of asthma exacerbations.
About Teva
Teva Pharmaceutical Industries Ltd. (NYSE and TASE: TEVA) is a leading global pharmaceutical company that delivers high-quality, patient-centric healthcare solutions to millions of patients every day. Headquartered in Israel, Teva is the world’s largest generic medicines producer, leveraging its portfolio of more than 1,000 molecules to produce a wide range of generic products in nearly every therapeutic area. In specialty medicines, Teva has a world-leading position in innovative treatments for disorders of the central nervous system, including pain, as well as a strong portfolio of respiratory products. Teva integrates its generics and specialty capabilities in its global research and development division to create new ways of addressing unmet patient needs by combining drug development capabilities with devices, services and technologies. Teva’s net revenues in 2014 amounted to $20.3 billion. For more information, visit www.tevapharm.com.
The U.S. Food and Drug Administration today approved Cinqair (reslizumab) for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.
Asthma is a chronic disease that causes inflammation in the airways of the lungs. During an asthma attack, airways become narrow making it hard to breathe. Severe asthma attacks can lead to asthma-related hospitalizations because these attacks can be serious and even life-threatening. According to the Centers for Disease Control and Prevention, as of 2013, more than 22 million people in the U.S. have asthma, and there are more than 400,000 asthma-related hospitalizations each year.
“Health care providers and their patients with severe asthma now have another treatment option to consider when the disease is not well controlled by their current asthma therapies,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Cinqair is administered once every four weeks via intravenous infusion by a health care professional in a clinical setting prepared to manage anaphylaxis. Cinqair is a humanized interleukin-5 antagonist monoclonal antibody produced by recombinant DNA technology in murine myeloma non-secreting 0 (NS0) cells. Cinqair reduces severe asthma attacks by reducing the levels of blood eosinophils, a type of white blood cell that contributes to the development of asthma.
The safety and efficacy of Cinqair were established in four double-blind, randomized, placebo‑controlled trials in patients with severe asthma on currently available therapies. Cinqair or a placebo was administered to patients every four weeks as an add-on asthma treatment. Compared with placebo, patients with severe asthma receiving Cinqair had fewer asthma attacks, and a longer time to the first attack. In addition, treatment with Cinqair resulted in a significant improvement in lung function, as measured by the volume of air exhaled by patients in one second.
Cinqair can cause serious side effects including allergic (hypersensitivity) reactions. These reactions can be life-threatening. The most common side effects in clinical trials for Cinqair included anaphylaxis, cancer, and muscle pain.
Cinqair is made by Teva Pharmaceuticals in Frazer, Pennsylvania.


References
- 1Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics 11 (3): 329–36. PMID 19479666.
- 2http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from rat) |
| Target | IL-5 |
| Clinical data | |
| Trade names | Cinquil |
| Identifiers | |
| ATC code | R03DX08 (WHO) |
| ChemSpider | none |
/////////CDP-835, CEP-38072, CTx-55700, SCH-5570, SCH-55700, TRFK-5, Reslizumab, Cinqair®, teva, interleukin-5 (IL-5) antagonist, severe asthma, FDA 2016, Orphan Drug StatuS
Blinatumomab
Blinatumomab, AMG-103, MEDI-538, MT-103,
(Blincyto®) Approved
A bispecific CD19-directed CD3 T-cell engager used to treat philadelphia chromosome-negative relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
Other Names
1: PN: WO2005052004 SEQID: 1 claimed protein
cas 853426-35-4
![]()
![]() Blinatumomab (trade name Blincyto, previously known as AMG103) is a biopharmaceutical drug used as a second-line treatmentfor Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia. It belongs to a class of constructedmonoclonal antibodies, bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1] In December 2014 it was approved by the US Food and Drug Administration under the accelerated approval program; marketing authorization depended on the outcome of clinical trials that were ongoing at the time of approval.[2][3] When it launched, blinatumomab was priced at $178,000 per year in the United States; only about 1,000 people were eligible to take the drug, based on its label.[4]
Blinatumomab (trade name Blincyto, previously known as AMG103) is a biopharmaceutical drug used as a second-line treatmentfor Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia. It belongs to a class of constructedmonoclonal antibodies, bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1] In December 2014 it was approved by the US Food and Drug Administration under the accelerated approval program; marketing authorization depended on the outcome of clinical trials that were ongoing at the time of approval.[2][3] When it launched, blinatumomab was priced at $178,000 per year in the United States; only about 1,000 people were eligible to take the drug, based on its label.[4]

Medical use
Blinatumomab is used as a second-line treatment for Philadelphia chromosome-negative relapsed or refractory Bcell precursor acute lymphoblastic leukemia.[2]
Mechanism of action

Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: aCD3 site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types and activating the T cell to exert cytotoxic activity on the target cell.[5] CD3 and CD19 are expressed in both pediatric and adult patients, making blinatumomab a potential therapeutic option for both pediatric and adult populations.[6]


History
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchased byAmgen, which has furthered the drug’s clinical trials. In July 2014, the FDA granted breakthrough therapy status to blinatumomab for the treatment of acute lymphoblastic leukemia (ALL).[7] In October 2014, Amgen’s Biologics License Application for blinatumomab was granted priority review designation by the FDA, thus establishing a deadline of May 19, 2015 for completion of the FDA review process.[8]
On December 3, 2014, the drug was approved for use in the United States to treat Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia under the FDA‘s accelerated approval program; marketing authorization depended on the outcome of clinical trials that were ongoing at the time of approval.[2][9]

Cost
When blinatumomab was approved, Amgen announced that the price for the drug would be $178,000 per year, which made it the most expensive cancer drug on the market. Merck’s pembrolizumab was priced at $150,000 per year when it launched; unlike that drug and others, only about 1,000 people can be given the drug, based on its label.[4]
Peter Bach, director of the Center for Health Policy and Outcomes at Memorial Sloan-Kettering Cancer Center, has calculated that according to “value-based pricing,” assuming that the value of a year of life is $120,000 with a 15% “toxicity discount,” the market price of blinaumomab should be $12,612 a month, compared to the market price of $64,260 a month. A representative of Amgen said, “The price of Blincyto reflects the significant clinical, economic and humanistic value of the product to patients and the health-care system. The price also reflects the complexity of developing, manufacturing and reliably supplying innovative biologic medicines.”[10]


Patent
WO 2010052013
http://www.google.co.in/patents/WO2010052013A1?cl=en
Examples:
1. CD19xCD3 bispecific single chain antibody
The generation, expression and cytotoxic activity of the CD19xCD3 bispecific single chain antibody has been described in WO 99/54440. The corresponding amino and nucleic acid sequences of the CD19xCD3 bispecific single chain antibody are shown in SEQ ID NOs. 1 and 2, respectively. The VH and VL regions of the CD3 binding domain of the CD19xCD3 bispecific single chain antibody are shown in SEQ ID NOs. 7 to 10, respectively, whereas the VH and VL regions of the CD19 binding domain of the CD19xCD3 bispecific single chain antibody are shown in SEQ ID NOs 3 to 6, respectively.
PATENT
http://www.google.com.ar/patents/WO2010052014A1?cl=en
PATENT
WO 2015006749
http://www.google.com/patents/WO2015006749A2?cl=un
PATENT
CN 104861067
http://www.google.com/patents/CN104861067A?cl=zh
| WO1998008875A1 * | 18 Aug 1997 | 5 Mar 1998 | Viva Diagnostika Diagnostische Produkte Gmbh | Novel combination preparations and their use in immunodiagnosis and immunotherapy |
| WO1999054440A1 | 21 Apr 1999 | 28 Oct 1999 | Micromet Gesellschaft Für Biomedizinische Forschung Mbh | CD19xCD3 SPECIFIC POLYPEPTIDES AND USES THEREOF |
| WO2004106381A1 | 26 May 2004 | 9 Dec 2004 | Micromet Ag | Pharmaceutical compositions comprising bispecific anti-cd3, anti-cd19 antibody constructs for the treatment of b-cell related disorders |
| WO2007068354A1 | 29 Nov 2006 | 21 Jun 2007 | Micromet Ag | Means and methods for the treatment of tumorous diseases |
References
- “blinatumomab” (PDF). United States Adopted Names Council » Adopted Names.American Medical Association. 2008. N08/16.(registration required)
- Blinatumomab label Updated 12/2014
- Food and Drug Administration December 3, 2014 FDA Press release: Blinatumomab
- Tracy Staton for FiercePharmaMarketing. December 18, 2014 Amgen slaps record-breaking $178K price on rare leukemia drug Blincyto
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”.Molecular Immunology 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032.PMID 17083975.

- Amgen (30 October 2012). Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting 04 December 2012 (PDF) (PDF). Food and Drug Administration. Blinatumomab (AMG 103).
- “Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE® Antibody Blinatumomab In Acute Lymphoblastic Leukemia” (Press release). Amgen. 1 July 2014.
- “Amgen’s BiTE® Immunotherapy Blinatumomab Receives FDA Priority Review Designation In Acute Lymphoblastic Leukemia” (Press release). Amgen. 9 October 2014.
- “Business: Antibody advance”. Seven Days. Nature (paper) 516 (7530): 149. 11 December 2014. doi:10.1038/516148a.

- Peter Loftus (June 18, 2015). “How Much Should Cancer Drugs Cost? Memorial Sloan Kettering doctors create pricing calculator that weighs factors such as side effects, extra years of life”. The Wall Street Journal. Retrieved 22 June 2015.
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Trade names | Blincyto |
| Pregnancy category |
|
| Routes of administration |
intravenous |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Metabolism | degradation into small peptides and amino acids |
| Biological half-life | 2.11 hours |
| Excretion | urine (negligible) |
| Identifiers | |
| CAS Number | 853426-35-4 |
| ATC code | L01XC19 (WHO) |
| ChemSpider | none |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Molar mass | 54.1 kDa |
///////
Fresolimumab

Fresolimumab
GC 1008, GC1008
UNII-375142VBIA
cas 948564-73-6
Structure
- immunoglobulin G4, anti-(human transforming growth factors beta-1, beta-2 (G-TSF or cetermin) and beta-3), human monoclonal GC-1008 γ4 heavy chain (134-215′)-disulfide with human monoclonal GC-1008 κ light chain, dimer (226-226”:229-229”)-bisdisulfide
- immunoglobulin G4, anti-(transforming growth factor β) (human monoclonal GC-1008 heavy chain), disulfide with human monoclonal GC-1008 light chain, dimer
For Idiopathic Pulmonary Fibrosis, Focal Segmental Glomerulosclerosis,and Cancer
An anti-TGF-beta antibody in phase I clinical trials (2011) for treatment-resistant primary focal segmental glomerulosclerosis.
A pan-specific, recombinant, fully human monoclonal antibody directed against human transforming growth factor (TGF) -beta 1, 2 and 3 with potential antineoplastic activity. Fresolimumab binds to and inhibits the activity of all isoforms of TGF-beta, which may result in the inhibition of tumor cell growth, angiogenesis, and migration. TGF-beta, a cytokine often over-expressed in various malignancies, may play an important role in promoting the growth, progression, and migration of tumor cells.

Fresolimumab (GC1008) is a human monoclonal antibody[1] and an immunomodulator. It is intended for the treatment of idiopathic pulmonary fibrosis (IPF), focal segmental glomerulosclerosis, and cancer[2][3] (kidney cancer and melanoma).
It binds to and inhibits all isoforms of the protein transforming growth factor beta (TGF-β).[2]
History
Fresolimumab was discovered by Cambridge Antibody Technology (CAT) scientists[4] and was one of a pair of candidate drugs that were identified for the treatment of the fatal condition scleroderma. CAT chose to co-develop the two drugs metelimumab (CAT-192) and fresolimumab with Genzyme. During early development, around 2004, CAT decided to drop development of metelimumab in favour of fresolimumab.[5]
In February 2011 Sanofi-Aventis agreed to buy Genzyme for US$ 20.1 billion.[6]
As of June 2011 the drug was being tested in humans (clinical trials) against IPF, renal disease, and cancer.[7][8] On 13 August 2012, Genzyme applied to begin a Phase 2 clinical trial in primary focal segmental glomerulosclerosis[9] comparing fresolimumab versus placebo.
As of July 2014, Sanofi-Aventis continue to list fresolimumab in their research and development portfolio under Phase II development.[10]

References
2 National Cancer Institute: Fresolimumab
- 3 Statement On A Nonproprietary Name Adopted By The USAN Council – Fresolimumab
- 4 Grütter, Christian; Wilkinson, Trevor; Turner, Richard; Podichetty, Sadhana; Finch, Donna; McCourt, Matthew; Loning, Scott; Jermutus, Lutz; Grütter, Markus G. (2008-12-23). “A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions”. Proceedings of the National Academy of Sciences 105 (51): 20251–20256. doi:10.1073/pnas.0807200106. ISSN 0027-8424. PMC 2600578. PMID 19073914.
- 5 http://www.independent.co.uk/news/business/news/cat-may-abandon-skin-drug-after-trial-results-disappoint-569445.html
- 6 http://www.bbc.co.uk/news/business-12477750
- 7 http://www.genengnews.com/gen-news-highlights/scientists-trigger-white-fat-to-become-brown-fat-like-to-treat-obesty-and-type-2-diabetes/81245389/
- 8 Clinicaltrials.gov for Fresolimumab
- 9 http://clinicaltrials.gov/show/NCT01665391
- 10 http://en.sanofi.com/rd/rd_portfolio/rd_portfolio.aspx
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | TGF beta 1, 2 and 3 |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 948564-73-6 |
| ATC code | None |
| ChemSpider | none |
| KEGG | D09620 |
| Chemical data | |
| Formula | C6392H9926N1698O2026S44 |
| Molar mass | 144.4 kDa |
////////////
Elotuzumab
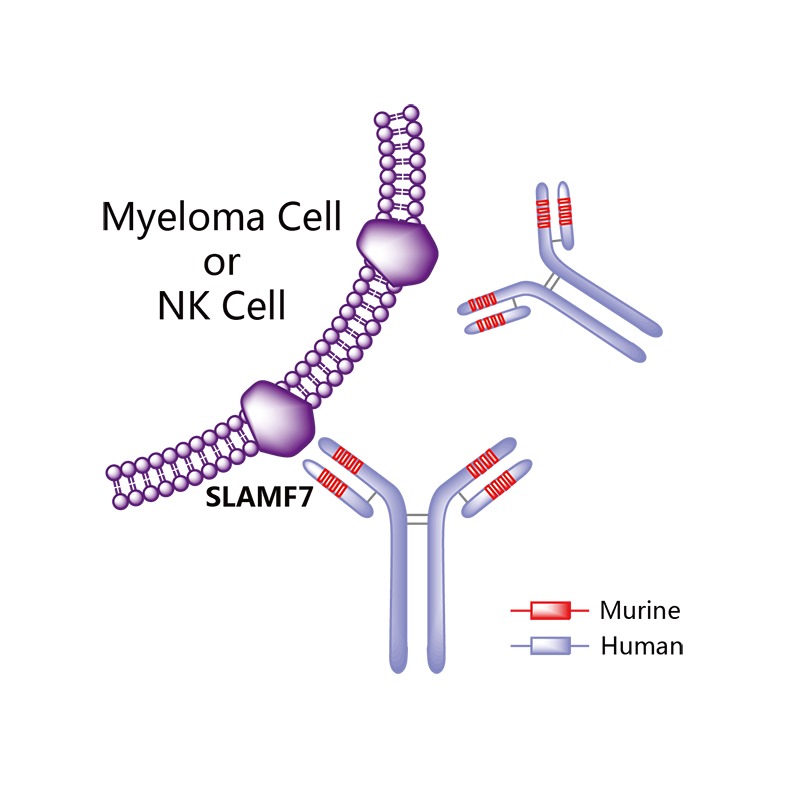
Elotuzumab
Approved nov 30 2012
A SLAMF7-directed immunostimulatory antibody used to treat multiple myeloma.
(Empliciti®)
HuLuc-63;BMS-901608
cas 915296-00-3
![]()


Elotuzumab (brand name Empliciti, previously known as HuLuc63) is a humanized monoclonal antibody used in relapsed multiple myeloma.[1] The package insert denotes its mechanism as a SLAMF7-directed (also known as CD 319) immunostimulatory antibody.[2]
Approvals and indications
In May 2014, it was granted “Breakthrough Therapy” designation by the FDA. [3] On November 30, 2015, FDA approved elotuzumab as a treatment for patients with multiple myeloma who have received one to three prior medications.[1] Elotuzumab was labeled for use with lenalidomide and dexamethasone. Each intravenous injection of elotuzumab should be premedicated with dexamethasone, diphenhydramine, ranitidine and acetaminophen.[2]
Elotuzumab is APPROVED for safety and efficacy in combination with lenalidomide and dexamethasone.
Monoclonal antibody therapy for multiple myeloma, a malignancy of plasma cells, was not very clinically efficacious until the development of cell surface glycoprotein CS1 targeting humanized immunoglobulin G1 monoclonal antibody – Elotuzumab. Elotuzumab is currently APPROVED in relapsed multiple myeloma.
Elotuzumab (HuLuc63) binds to CS1 antigens, highly expressed by multiple myeloma cells but minimally present on normal cells. The binding of elotuzumab to CS1 triggers antibody dependent cellular cytotoxicity in tumor cells expressing CS1. CS1 is a cell surface glycoprotein that belongs to the CD2 subset of immunoglobulin superfamily (IgSF). Preclinical studies showed that elotuzumab initiates cell lysis at high rates. The action of elotuzumab was found to be enhanced when multiple myeloma cells were pretreated with sub-therapeutic doses of lenalidomide and bortezomib. The impressive preclinical findings prompted investigation and analysis of elotuzumab in phase I and phase II studies in combination with lenalidomide and bortezomib.
Elotuzumab As Part of Combination Therapy: Clinical Trial Results
Elotuzumab showed manageable side effect profile and was well tolerated in a population of relapsed/refractory multiple myeloma patients, when treated with intravenous elotuzumab as single agent therapy. Lets’ take a look at how elotuzumab fared in combination therapy trials,
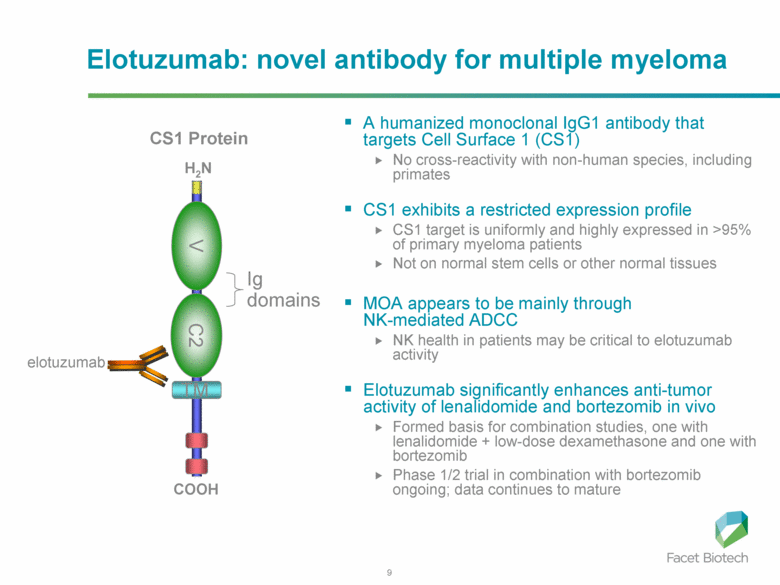
In phase I trial of elotuzumab in combination with Velcade/bortezomib in patients with relapsed/refractory myeloma, the overall response rate was 48% and activity was observed in patients whose disease had stopped responding to Velcade previously. The trial results found that elotuzumab enhanced Velcade activity.
A phase I/II trial in combination with lenalidomide and dexamethasone in refractory/relapsed multiple myeloma patients showed that 82% of patients responded to treatment with a partial response or better and 12% of patients showed complete response. Patients who had received only one prior therapy showed 91% response rate with elotuzumab in combination with lenalidomide and dexamethasone.
Phase I/II trials of the antibody drug has been very impressive and the drug is currently into Phase III trials. Two phase III trials are investigating whether addition of elotuzumab with Revlimid and low dose dexamethasone would increase the time to disease progression. Another phase III trial (ELOQUENT 2) is investigating and comparing safety and efficacy of lenalidomide plus low dose dexamethasone with or without 10mg/kg of elotuzumab in patients with relapsed/refractory multiple myeloma.
Elotuzumab is being investigated in many other trials too. It is being evaluated in combination with Revlimid and low-dose dexamethasone in multiple myeloma patients with various levels of kidney functions, while another phase II study is investigating elotuzumab’s efficacy in patients with high-risk smoldering myeloma.
The main target of multiple myeloma drug development is to satisfy the unmet need for drugs that would improve survival rates. Elotuzumab is an example that mandates much interest in this area and should be followed with diligence.

Empliciti’s Cost
Empliciti will be sold in the U.S. in two vials sizes: A smaller vial that contains 300 mg of the drug, and a larger vial that contains 400 mg.
Bristol-Myers Squibb has informed The Beacon that the wholesale price per vial of Empliciti will be $1,776 for the 300 mg vial and $2,368 for the 400 mg vial.
Using these prices and an assumed patient weight of between 154 and 176 pounds, Empliciti will cost $18,944 per four-week cycle for each of the first two cycles of treatment, and $9,472 per cycle thereafter. This means, in turn, that Empliciti’s cost per year will be $142,080 in the first year and $123,136 in subsequent years.
In comparison, Velcade costs between $4,800 and $8,500 per four-week cycle, depending on how often it is dosed. Ninlaro costs $8,670 per four-week cycle. And Kyprolis costs $10,500 per four-week cycle at the standard (20 – 27 mg/m2) dose.
Additional details about the FDA approval of Empliciti can be found in this press release from the FDA, a related press release from Bristol-Myers Squibb and AbbVie, and the full Empliciti prescribing information.
The results of the ELOQUENT-2 trial were published in Lonial, S. et al., “Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, June 2, 2015 (abstract). Slides from the ASCO presentation summarizing the ELOQUENT-2 results can be viewed here (PDF, courtesy of Dr. Lonial). This Beacon news article provides an in-depth look at the trial results.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | SLAMF7 (CD319) |
| Clinical data | |
| Trade names | Empliciti |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Identifiers | |
| CAS Number | 915296-00-3 |
| ATC code | None |
| IUPHAR/BPS | 8361 |
| UNII | 1351PE5UGS |
| Chemical data | |
| Formula | C6476H9982N1714O2016S42 |
| Molecular mass | 145.5 kDa |
References
1 “Press Announcement—FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma”. U.S. Food and Drug Administration. Retrieved 3 December 2015.
2“Empliciti (elotuzumab) for Injection, for Intravenous Use. Full Prescribing Information” (PDF). Empliciti (elotuzumab) for US Healthcare Professionals. Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.
3 “Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma” (Press release). Princeton, NJ & North Chicago, IL: Bristol-Myers Squibb. 2014-05-19. Retrieved 2015-02-05.
///////
Japanese filing for Amgen’s PCSK9 inhibitor Repatha
![]()
Amgen has filed its closely watched PCSK9 inhibitor Repatha (evolocumab) in Japan for the treatment of high cholesterol.
Repatha is an investigational fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein that reduces the liver’s ability to remove low-density lipoprotein cholesterol (LDL-C), or ‘bad’ cholesterol, from the blood.


| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | PCSK9 |
| Clinical data | |
|
|
| Subcutaneous injection | |
| Identifiers | |
| 1256937-27-5 | |
| C10AX13 | |
| Chemical data | |
| Formula | C6242H9648N1668O1996S56 |
| 141.8 kDa | |

Evolocumab[1] (also known as compound number AMG-145 or AMG145)[2] is a monoclonal antibody designed for the treatment of hyperlipidemia.[3] Evolocumab is a fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9).
PCSK9 is a protein that targets LDL receptors for degradation and thereby reduces the liver’s ability to remove LDL-C, or “bad” cholesterol, from the blood.
Evolocumab, being developed by Amgen scientists, is designed to bind to PCSK9 and inhibit PCSK9 from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from the blood.
Clinical trials
Two trials have been in progress as at mid-2014:
On 23 January 2014 Amgen announced that the Phase 3 GAUSS-2 (Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) trial evaluating evolocumab in patients with high cholesterol who cannot tolerate statins met its co-primary endpoints: the percent reduction from baseline in low-density lipoprotein cholesterol (LDL-C) at week 12 and the mean percent reduction from baseline in LDL-C at weeks 10 and 12. The mean percent reductions in LDL-C, or “bad” cholesterol, compared to ezetimibe were consistent with results observed in the Phase 2 GAUSS study.[4][5]
The GAUSS-2 trial evaluated safety, tolerability and efficacy of evolocumab in 307 patients with high cholesterol who could not tolerate effective doses of at least two different statins due to muscle-related side effects. Patients were randomly assigned to one of four treatment groups: subcutaneous evolocumab 140 mg every two weeks and oral placebo daily; subcutaneous evolocumab 420 mg monthly and oral placebo daily; subcutaneous placebo every two weeks and oral ezetimibe 10 mg daily; or subcutaneous placebo monthly and oral ezetimibe 10 mg daily.
Safety was generally balanced across treatment groups. The most common adverse events (> 5 percent in evolocumab combined group) were headache (7.8 percent evolocumab; 8.8 percent ezetimibe), myalgia (7.8 percent evolocumab; 17.6 percent ezetimibe), pain in extremity (6.8 percent evolocumab; 1.0 percent ezetimibe), and muscle spasms (6.3 percent evolocumab; 3.9 percent ezetimibe).
Cholesterol-lowering treatment with a statin as part of follow-up care can help reduce a patient’s risk after myocardial infarction, ischaemic stroke or TIA.
The FOURIER Phase 3 clinical study http://www.fourierstudy.com/ seeks to find out whether lowering cholesterol by an additional 50% might reduce this risk even further. Several sites in the UK are part of this very large clinical study, lasting up to five years, and it is hoped that the study will help guide future clinical practice.
Evolocumab (also formerly known as AMG145, from Amgen) binds to PCSK9, a natural protein produced by the liver. By binding to PCSK9, evolocumab allows the LDL receptor (a protein present in the liver) to move LDL-cholesterol out of the bloodstream more efficiently. This study is designed to see whether treatment of dyslipidemia with evolocumab in people who have experienced a prior myocardial infarction, ischaemic stroke or TIA, and who are taking a highly effective dose of a statin, reduces the risk of recurring or additional cardiovascular events. Participants in this study have clinically evident cardiovascular disease.
READ AT
MY EARLIER ARTICLE


References
- World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108” (PDF). WHO Drug Information 26 (4).
- 2
- Sheridan, C (2013). “Phase 3 data for PCSK9 inhibitor wows”. Nature Biotechnology 31 (12): 1057–8. doi:10.1038/nbt1213-1057. PMID 24316621.
- 3
- Statement On A Nonproprietary Name Adopted By The USAN Council – Evolocumab
- 4
- Estel Grace Masangkay, “Amgen Phase III GAUSS-2 Trial of Evolocumab (AMG 145) Meets Co-Primary Endpoints Of LDL Cholesterol Reduction”, Bioresearch Online (January 24 2014)
- 5
Pierson, Ransdell (17 March 2014). “Amgen drug meets goal for those with high genetic cholesterol”. Associated Press. Retrieved 19 March 2014.
FDA expands approved use of Opdivo to treat lung cancer
March 4, 2015
Release
The U.S. Food and Drug Administration today expanded the approved use of Opdivo (nivolumab) to treat patients with advanced (metastatic) squamous non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy.
Lung cancer is the leading cause of cancer death in the United States, with an estimated 224,210 new diagnoses and 159,260 deaths in 2014. The most common type of lung cancer, NSCLC affects seven out of eight lung cancer patients, occurring when cancer forms in the cells of the lung.
Opdivo works by inhibiting the cellular pathway known as PD-1 protein on cells that blocks the body’s immune system from attacking cancerous cells. Opdivo is intended for patients who have previously been treated with platinum-based chemotherapy.
“The FDA worked proactively with the company to facilitate the early submission and review of this important clinical trial when results first became available in late December 2014,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide patients and health care providers knowledge of the survival advantage associated with Opdivo and will help guide patient care and future lung cancer trials.”
Opdivo’s efficacy to treat squamous NSCLC was established in a randomized trial of 272 participants, of whom 135 received Opdivo and 137 received docetaxel. The trial was designed to measure the amount of time participants lived after starting treatment (overall survival). On average, participants who received Opdivo lived 3.2 months longer than those participants who received docetaxel.
The safety and efficacy of Opdivo to treat squamous NSCLC was supported by a single-arm trial of 117 participants who had progressed after receiving a platinum-based therapy and at least one additional systemic regimen. The study was designed to measure objective response rate (ORR), or the percentage of participants who experienced partial shrinkage or complete disappearance of the tumor. Results showed 15 percent of participants experienced ORR, of whom 59 percent had response durations of six months or longer.
The most common side effects of Opdivo are fatigue, shortness of breath, musculoskeletal pain, decreased appetite, cough, nausea and constipation. The most serious side effects are severe immune-mediated side effects involving healthy organs, including the lung, colon, liver, kidneys and hormone-producing glands.
Opdivo for squamous NSCLC was reviewed under the FDA’s priority review program, which provides for an expedited review of drugs that treat serious conditions and, if approved, would provide significant improvement in safety or effectiveness in the treatment of a serious condition. Opdivo is being approved more than three months ahead of the prescription drug user fee goal date of June 22, 2015, the date when the agency was scheduled to complete its review of the application.
The FDA previously approved Opdivo to treat patients with unresectable (cannot be removed by surgery) or metastatic melanoma who no longer respond to other drugs.
Opdivo is marketed by Princeton, New Jersey-based Bristol-Myers Squibb.
Shirdi – Wikipedia, the free encyclopedia
pronunciation (help·info) (Marathi: शिर्डी) is a town and falls under the jurisdiction of municipal council popularly known as Shirdi Nagar Panchayat, located …
.




Shraddha Inn,Shirdi

SHIRDI PRASADALAYA BOJAN

Solar Kitchen Feeds Many at Shirdi, India Shrine


Rajdhani Restaurant: Rajdhani at Shirdi

The well equipped kitchen provides food two times a day, daily. Around 27, 000 of people are distributed food at cheap rate. The food comprises of dal,

/////////
Novartis obtains European approval for Cosentyx to treat psoriasis

Novartis obtains European approval for Cosentyx to treat psoriasis
Swiss drug-maker Novartis has received approval from the European Commission (EC) for its Cosentyx (secukinumab, formerly known as AIN457) to treat moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy.SEE
 PSORIAIS
PSORIAIS
secukinumab

Secukinumab is a human monoclonal antibody designed for the treatments of uveitis, rheumatoid arthritis, ankylosing spondylitis, and psoriasis. It targets member A from the cytokine family of interleukin 17.[1][2] At present, Novartis Pharma AG, the drug’s developer, plans to market it under the trade name “Cosentyx.” [3] It is highly specific to the human immunoglobulin G1k (IgG1k) subclass.[2]
In July 2014 secukinumab established superiority to placebo and to etanercept for the treatment of chronic plaque psoriasis in Phase III clinical trials.[4] In October 2014, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee unanimously voted to recommend the drug for FDA approval, although this vote in and of itself does not constitute an approval. However, the FDA typically follows recommendations from these committees.[5] In October 2014, Novartis announced that the drug had achieved a primary clinical endpoint in two phase III clinical trials for ankylosing spondylitis.[6] As of 28 October, the relevant FDA committee had not yet responded to these results. In early November 2014, Novartis also released the results of a Phase 3 study on Psoriatic Arthritis that yielded very promising results.[7]

Although the drug was originally intended to treat rheumatoid arthritis, phase II clinical trials for this condition yielded disappointing results.[8] Similarly, while patients in a phase II clinical trial for [psoriatic arthritis] did show improvement over placebo, the improvement did not meet adequate endpoints and Novartis is considering whether to do more research for this condition.[9] Novartis has said that it is targeting approval and release in early 2015 for plaque psoriasis and ankyloding spondylitis indications.
It is also in a phase II clinical trial for Multiple Sclerosis [10] as it has exhibited efficacy in treating experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833.
- http://www.medscape.com/viewarticle/835331
- Langley RG, Elewski BE, Mark Lebwohl M, et al., for the ERASURE and FIXTURE Study Groups (July 24, 2014). “Secukinumab in Plaque Psoriasis — Results of Two Phase 3 Trials”. N Engl J Med 371: 326–338. doi:10.1056/NEJMoa1314258.
- committees.http://www.familypracticenews.com/index.php?id=2934&type=98&tx_ttnews=306073[dead link]
- http://inpublic.globenewswire.com/2014/10/23/Novartis+AIN457+secukinumab+meets+primary+endpoint+in+two+Phase+III+studies+in+ankylosing+spondylitis+a+debilitating+joint+condition+of+the+spine+HUG1864939.html
- http://www.medpagetoday.com/MeetingCoverage/ACR/48743
- http://www.medscape.com/viewarticle/806510_6
- http://www.ncbi.nlm.nih.gov/pubmed/23361084
- http://clinicaltrials.gov/show/NCT01874340
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL17A |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | |
| ATC code | L04AC10 |
| DrugBank | DB09029 |
| Synonyms | AIN457 |
| Chemical data | |
| Formula | C6584H10134N1754O2042S44 |
| Molecular mass | 147.94 kDa |
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
 Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab linking a T cell to a malignant B cell.
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
December 3, 2014 — The U.S. Food and Drug Administration today
approved Blincyto (blinatumomab) to treat patients with Philadelphia
chromosome-negative precursor B-cell acute lymphoblastic leukemia
(B-cell ALL), an uncommon form of ALL.

Blinatumomab (AMG103) is a drug that has anti-cancer properties. It belongs to a new class of constructed monoclonal antibodies,bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1]
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchases by Amgen, which has furthered the drug’s clinical trials. In July 2014, the FDA granted breakthrough therapy status to blinatumomab for the treatment of acute lymphoblastic leukemia (ALL).[2] In October 2014, Amgen’s Biologics License Application for blinatumomab was granted priority review designation by the FDA, thus establishing a deadline of May 19, 2015 for completion of the FDA review process.[3]
Structure and mechanism of action
Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: a CD3site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types andactivating the T cell to exert cytotoxic activity on the target cell.[4] CD3 and CD19 are expressed in both pediatric and adult patients, making blinatumomab a potential therapeutic option for both pediatric and adult populations.[5]
Therapeutic use
Clinical trials
In a phase 1 clinical study with blinatumomab, patients with non-Hodgkin’s lymphoma showed tumor regression, and in some cases complete remission.[6] There are ongoing phase 1 and phase 2 clinical trials of blinatumomab in patients with acute lymphoblastic leukemia (ALL).[7] One phase II trial for ALL reported good results in 2010 and another is starting.[8]
Adverse effects
Common side effects observed in Phase 2 trials are listed below; they were temporary and typically occurred during the first treatment cycle:[5]
- Flu-like symptoms (i.e. fever, headache, and fatigue)
- Tremor
- Weight increase
- Hypokalemia
- Decrease of blood immunoglobulin
CNS effects were also observed during clinical trials and were treated via a lower dose of blinatumomab, administration of dexamethasone, or treatment discontinuation. Because the side effects were reversible, early monitoring for the CNS symptoms listed below is important:[5]
- Seizure
- Encephalopathy
- Tremor
- Apraxia
- Speech disorders
- Disorientation
Less common side effects include cytokine release syndrome and immunogenicity.[5]
References
- Statement on a Nonproprietary Name adopted by the USAN Council: Blinatumomab
- Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE® Antibody Blinatumomab In Acute Lymphoblastic Leukemia
- Amgen’s BiTE® Immunotherapy Blinatumomab Receives FDA Priority Review Designation In Acute Lymphoblastic Leukemia
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”. Mol Immunol 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032. PMID 17083975.
- Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting 04 December 2012
- Bargou, R; et al. (2008). “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody”. Science 321 (5891): 974–977. doi:10.1126/science.1158545.PMID 18703743.
- ClinicalTrials.gov NCT00560794 Phase II Study of the BiTE Blinatumomab (MT103) in Patients With Minimal Residual Disease of B-precursor Acute ALL
- “Micromet initiates MT103 phase 2 trial in adult ALL patients”. 20 Sep 2010.
External links
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 853426-35-4 |
| ATC code | None |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Mol. mass | 54.1 kDa |
Glenmark’s Enrollment Begins of First Patient in Phase II Vatelizumab (GBR 500) Trial in Relapsing Remitting Multiple Sclerosis

Enrollment Begins of First Patient in Phase II Vatelizumab Trial in Relapsing Remitting Multiple Sclerosis
Glenmark outlicensed Vatelizumab (GBR 500) to Sanofi for all indications in 2011
Mumbai – India, November 4, 2014: Glenmark announced today enrollment of the first patient in a multicenter Phase II clinical trial to evaluate Genzyme’s investigational infusion therapy vatelizumab in patients with relapsing remitting multiple sclerosis (RRMS). The trial, called EMPIRE, is designed to assess the efficacy of vatelizumab vs. placebo in RRMS patients. The safety, tolerability and pharmacokinetics of vatelizumab will also be assessed.
read at
The mechanism of action of vatelizumab, which is developed in a collaboration between Glenmark Pharmaceuticals and Genzyme, is not yet fully understood. However, the researchers believe that it will be able to block VLA-2 on activated immune cells, which may enable the interference with collagen-binding in areas of inflammation, as well as leading to the reduction of inflammatory cascade associated with MS.

“We are excited about the commencement of this trial and are pleased with the continued progress of our partnership with Sanofi/Genzyme,” said the President of Biologics and Chief Scientific Officer of Glenmark Pharmaceuticals Ltd., Michael Buschle. EMPIRE, which will be conducted for 12 weeks, is a global phase 2a/2b double-blind, randomized, placebo-controlled study that will study the efficacy, safety, and dose-response of vatelizumab in 168 patients with active RRMS at55 sites in ten different countries.
Vatelizumab is an immunomodulator. It binds to integrin alpha 2.[1]
| Company | Glenmark Pharmaceuticals Ltd. |
| Description | mAb against integrin alpha(2) (VLA-2; CD49B) |
| Molecular Target | Integrin alpha(2) (VLA-2) (CD49B) |
| Mechanism of Action | Antibody |
| Therapeutic Modality | Biologic: Antibody |
| Latest Stage of Development | Phase I/II |
| Standard Indication | Inflammatory bowel disease (IBD) |
| Indication Details | Treat inflammatory bowel disease (IBD); Treat ulcerative colitis (UC) |
| Regulatory Designation | |
| Partner |
References
- World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 105”(PDF). WHO Drug Information 25 (2).

{kind=link}
{kind=link}