
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

DETOMIDINE1H-Imidazole, 4-[(2,3-dimethylphenyl)methyl]-
4-(2,3-Dimethylbenzyl)-1H-imidazole
507876631-46-4[RN]7N8K34P2XH
- Molecular FormulaC12H14N2
- Average mass186.253 Da
UNII-7N8K34P2XHдетомидинديتوميدين地托咪定
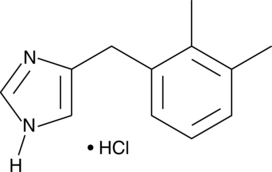
Formal Name5-[(2,3-dimethylphenyl)methyl]-1H-imidazole, monohydrochlorideCAS Number90038-01-0Synonyms
- Domosedan
- MPV 253AII
Molecular FormulaC12H14N2 • HClFormula Weight222.7DetomidineCAS Registry Number: 76631-46-4CAS Name: 4-[(2,3-Dimethylphenyl)methyl]-1H-imidazoleAdditional Names: 4-(2¢,3¢-dimethylbenzyl)imidazoleMolecular Formula: C12H14N2Molecular Weight: 186.25Percent Composition: C 77.38%, H 7.58%, N 15.04%Literature References: a2-Adrenoceptor agonist with sedative and analgesic activity. Prepn: A. J. Karjalayne, K. O. A. Kurkela, EP24829; eidem,US4443466 (1981, 1984 both to Farmos). Physical studies: E. Laine et al.,Acta Pharm. Suec.20, 451 (1983). Crystal structure: L. H. J. Lajunen et al.,ibid.21, 163 (1984). Pharmacology: R. Virtanen, L. Nyman, Eur. J. Pharmacol.108, 163 (1985); R. Virtanen, E. MacDonald, ibid.115, 277 (1985). Mechanism of action: eidem,J. Vet. Pharmacol. Ther.8, 30 (1985).Properties: Crystals from acetone, mp 114-116°. LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela).Melting point: mp 114-116°Toxicity data: LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela) Derivative Type: HydrochlorideTrademarks: Domosedan (Farmos)Molecular Formula: C12H14N2.HClMolecular Weight: 222.71Percent Composition: C 64.72%, H 6.79%, N 12.58%, Cl 15.92%Properties: Crystals, mp 160°. Converts reversibly to monohydrate at room temp, 80% humidity.Melting point: mp 160° Therap-Cat-Vet: Sedative.
Detomidine is an imidazole derivative and α2-adrenergic agonist,used as a large animal sedative, primarily used in horses. It is usually available as the salt detomidine hydrochloride. It is a prescription medication available to veterinarians sold under the trade name Dormosedan.
Currently, detomidine is only licensed for use in horses in the US but it is also licensed for use in cattle in Europe and Australasia.[1]
Properties
Detomidine is a sedative with analgesic properties.[2] α2-adrenergic agonists produce dose-dependent sedative and analgesic effects, mediated by activation of α2 catecholamine receptors, thus inducing a negative feedback response, reducing production of excitatory neurotransmitters. Due to inhibition of the sympathetic nervous system, detomidine also has cardiac and respiratory effects and an antidiuretic action.[3]
Effects
UsesA profound lethargy and characteristic lowering of the head with reduced sensitivity to environmental stimuli (sound, pain, etc.) are seen with detomidine. A short period of reduced coordination is characteristically followed by immobility and a firm stance with front legs spread. Following administration there is an initial increase in blood pressure, followed by bradycardia and second degree atrioventricular block (this is not pathologic in horses). The horse commonly sweats to excess, especially on the flanks and neck. Other side effects reported include pilo erection (hair standing erect), ataxia, salivation, slight muscle tremors, and (rarely) penile prolapse.
Sedation and anaesthetic premedication in horses and other large animals, commonly combined with butorphanol for increased analgesia and depth of sedation. In conjunction with ketamine it may also be used for intravenous anaesthesia of short duration.
The drug is normally administered by the intravenous route, and is fastest and most efficient when given intravenously . However, in recalcitrant animals, detomidine may be administered by the intramuscular or sublingual routes. The dose range advised by the manufacturers is 20–40 µg/kg intravenous for moderate sedation, but this dose may need to be higher if given intramuscularly.
When given intravenously, detomidine usually takes effect in 2–5 minutes, and recovery is full within 30–60 minutes. However, this is highly dependent upon the dosage, environment, and the individual animal; some horses are highly resistant to sedation.
Detomidine is a poor premedication when using ketamine as an anesthetic in horses.As detomidine is an arrhythmogenic agent, extreme care should be exercised in horses with cardiac disease, and in the concurrent administration of other arrhythmogenics. The concurrent use of potentiated sulfonamide antibiotics is considered particularly dangerous.
Anesthetic recoveries in horses that have received ketamine following a detomidine premedication are often violent with the horse having multiple failures to stand resulting in trauma to itself. Xylazine is a superior premedication with ketamine resulting in safer recoveries.
PATENT
EP-03782989
Novel crystalline forms of detomidine hydrochloride monohydrate, processes for their preparation and compositions comprising them are claimed. Also claimed is their use as alpha2-adrenoreceptor agonists.
Detomidine hydrochloride (1H imidazole,4-[(2,3-dimethylphenyl)methyl]-hydrochloride (CAS Number: 90038-01-0) is a synthetic alpha 2-adrenoreceptor agonist with sedative and analgesic properties widely used for sedation of large animals like horses and cattle. This substance displays various other pharmacologic effects related to the cardiovascular and respiratory system as well as on muscles. Detomidine hydrochloride is available as a parenteral solution with 10 mg/ml as active ingredient which is indicated for use as a sedative and analgesic to facilitate minor surgical and diagnostic procedures in mature horses and yearlings (e.g. DORMOSEDAN®). Furthermore, detomidine hydrochloride is supplied as an oromucosal (i.e. sublingual) gel (e.g. DORMOSEDAN GEL®) with 7.6 mg/ml as active ingredient which is indicated for sedation and restraint in horses.
Further details regarding the clinical pharmacology and side effects as well as contraindications for this drug substance (i.e. active pharmaceutical ingredient) can be found in: Veterinary Psychopharmacology; Sharon L. et al., 2nd edition (2019), Wiley & Sons (pages 161 – 162). According to these authors detomidine has not been used in humans to date.
Detomidini hydrochloridum ad usum veterinarium is included in the EUROPEAN PHARMACOPOEIA (Ph. Eur. 9.0) but currently not included in the United States Pharmacopoeia (USP). It has to be noted that in the absence of a statement regarding a specific hydrate form, like a degree of hydration or mono-, di-, etc., in the title of the monograph – as is the case for detomidine hydrochloride – the anhydrous form is indicated for this substance.
According to a prior version of the respective monograph, namely Ph. Eur. 8.0, the substance exists as a white or almost white, hygroscopic, crystalline powder. The substance is soluble in water, freely soluble in ethanol (96 %), very slightly soluble in methylene chloride and practically insoluble in acetone. The molecular weight (M r) amounts to 222.7. The melting point (mp) is specified at about 160 °C. In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 percent (anhydrous substance).
[0003] In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 % (anhydrous substance).
The current monograph includes the three following known impurities:
Impurity A: (RS)-(2,3-dimethylphenyl) (1H-imidazol-4-yl)-methanol
Impurity B: (RS)-(1-benzyl-1H-imidazol-5-yl)(2,3-dimethylphenyl)-methanol
Impurity C: 4-[(2,3-dimethylcylohexyl)methyl]-1H-imidazole
The related substances are specified at ≤ 0.20 % for any unspecified impurities and ≤ 0.5 % for total impurities with a reporting threshold of 0.10 %.
The water content of detomidine hydrochloride as determined by Karl Fischer (KF) titration is limited to ≤ 2.0 % for release as well as shelf-life testing. As detomidine hydrochloride is hygroscopic, the compound has to be stored in airtight containers.
[0004] A synthesis method for detomidine was disclosed in US 4,584,383.
Specific details on the last two steps of a synthesis method for detomidine hydrochloride (including a reaction scheme) were published in Drugs Future 10, 17 (1985).
[0005] Detomidine hydrochloride is known to exist in two crystalline forms, namely the anhydrous form, as described above, and the monohydrate form B (M r: 240.7, CAS Number: 90038-00-9) which can easily interconvert, depending on ambient temperature and air humidity ( Veldre, K. et al., Eur. Journ. Pharm. 44, 273-280 (2011)). At 80 % air humidity and room temperature the monohydrate is reversibly formed. The theoretical water content of detomidine hydrochloride monohydrate amounts to 7.48 %.
[0006] To date, all commercially available (i.e. veterinary) drug products (i.e. parenteral solutions and oromucosal gels) only contain the anhydrous form. In general, hygroscopic substances like detomidine hydrochloride tend to absorb moisture so that they have to be protected from a humid environment during production and storage of the drug substance and corresponding drug product to avoid an inacceptable uptake of water. It has to be noted that such uptake during storage will reduce the content of the drug substance so that this would have to be taken into consideration during production of the corresponding drug product, like pharmaceutical preparation.
[0007] The problem to be solved is to provide a pure and stable active pharmaceutical ingredient (API), namely detomidine hydrochloride monohydrate, that can advantageously be used for the production of pharmaceutical compositions comprising the active pharmaceutical ingredient detomidine hydrochloride.
Example 1
Preparation of detomidine hydrochloride monohydrate (DHM)
[0053] Detomidine hydrochloride was synthesized starting from 1-benzyl-imidazole-4-carboxyaldehyde and 2,3-dimethylphenlymagnesiumbromide according to the two-step synthesis described in Drugs Future 10, 17 (1985).
[0054] For the second step of this synthesis (RS)-(3-Benzyl-3 H-imidazol-4-yl)-(2,3-dimethyl-phenyl)-methanol (HCl) was suspended in a mixture of water and hydrochloric acid. The catalyst (i. e. palladium on activated carbon) suspended in demineralized water was added. Hydrogenation (i.e. removal of the benzyl group and reduction of the hydroxyl group with hydrogen (H 2/Pd-C in HCl)) was performed at elevated temperature (50 – 80 °C) and the obtained suspension was filtered after the hydrogenation was finished. Subsequently ethyl acetate and a solution of ammonium hydroxide were added under continuous stirring. After discontinuation of stirring, phase separation occured after which the aqueous phase was repeatedly extracted with ethyl acetate. The combined organic phases were washed with demineralized water and filtered.
[0055] After addition of 5 – 6 N hydrogen chloride in 2-propanol and cooling precipitation of detomidine hydrochloride occured. After filtration the filtercake (i.e. raw product) was washed with ethyl acetate and dried.
[0056] A fraction of the resulting raw product (i.e. 5 g batch RSO E-190604 RP) was recrystallized from 5 g demineralized water by heating (until complete dissolution was obtained) and subsequent cooling on an ice bath. The resulting crystals were separated by filtration and the resulting filter cake washed with 2-propanol. Subsequently, the washed product was dried under vacuum (10 mbar) at 23 °C. The obtained yield for the white crystalline substance amounted to 66.0 % of the theory.
[0057] The resulting drug substance showed a water content (KF) of 7.49 %. The corresponding DSC curve was in line with the expectation (see for example Figure 1) and showed the two typical peaks routinely observed for DHM. Other than 2-propanol used for final washing none of the other solvents employed during the overall synthesis of this compound were found above the respective LOQ by GC-FID.
Example 2
Impurities after preparation of detomidine hydrochloride monohydrate (DHM)
[0058] A larger batch of detomidine hydrochloride (i.e. 50 g NK E-190709-I A K1) was synthesized in line with Example 1. However, the final crystals obtained after recrystallization from 50 ml demineralized water were washed with 25 ml demineralized water instead of 2-propanol. Drying was performed at 21 °C and 10 mbar until constant weight. The obtained yield for the white crystalline substance amounted to 87.2 % of the theory which was markedly higher than the yield obtained in Example 1. The water content of this substance was determined at 7.54 % (KF) and the corresponding DSC curve showed two peaks with an onset at 95.7 °C and 159.3 °C.
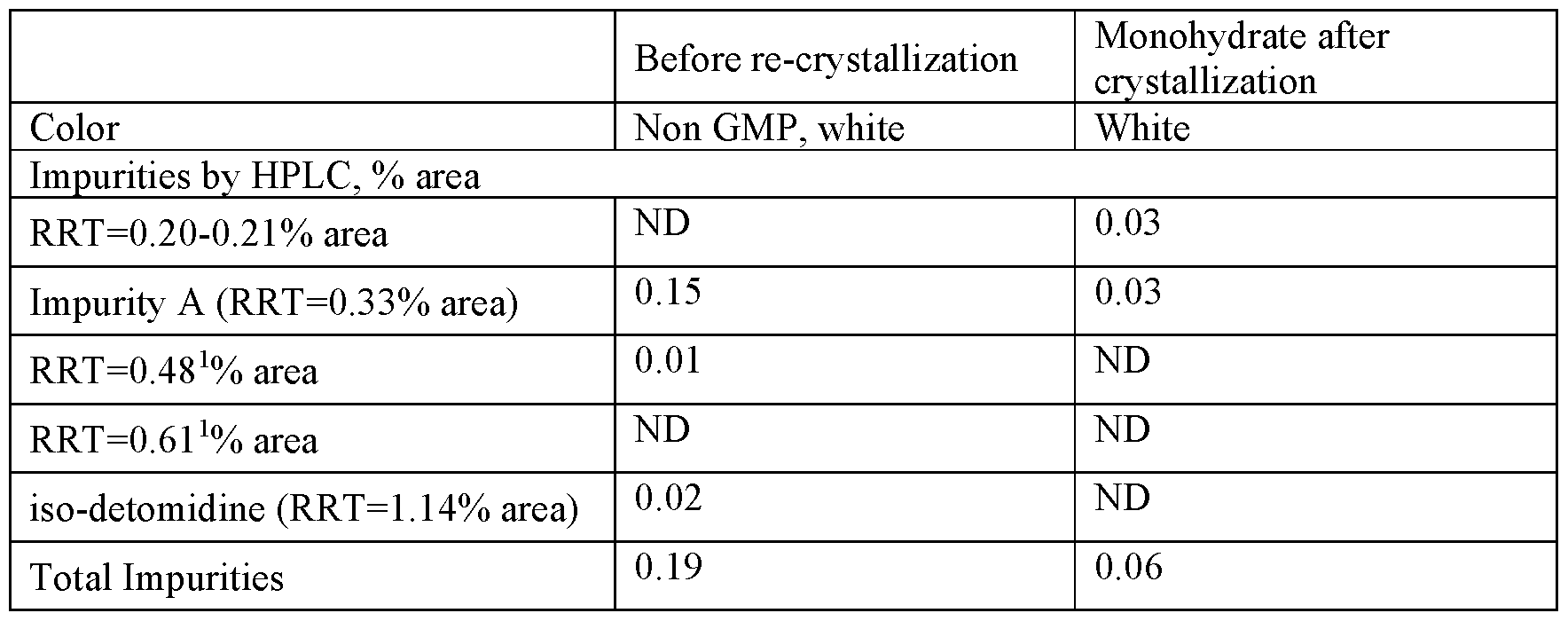
[0059] As shown below, recrystallization of the initial raw product from water (incl. washing) resulted in significant removal/reduction of impurities eluting before the detomidine peak (i.e. more polar compounds, e.g. Impurity A) as well as impurities eluting behind the detomidine peak (i.e. less polar compounds, e.g. Impurity C).
| Sample | Relevant compounds as detected by HPLC [area%]* | ||||
| Impurity A | Impurity RRT 0.84 | Detomidine | Impurity RRT 1.75 | Impurity C | |
| Raw product | 0.11 | 0.33 | 99.40 | 0.04 | 0.04 |
| Final crystallizate (K1) | 0.06 | 0.06 | 99.80 | 0.01 | 0.02 |
| *Table includes all compounds found at or above 0.04 area% in the initial raw product in the order in which they eluted from the HPLC column |
[0060] The final substance showed a very high HPLC purity of 99.80 area% (Ph. Eur. test method) and only a limited number of unknown impurities in addition to those
PATENT
https://patents.google.com/patent/WO2006108910A1/en
Example 1. Preparation of 4-[(2,3-dimethylbenzyl)]imidazole hydrochloride
(detomidine HCl)
l-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 1), 30 % HCl (20 1), ethanol (5 1) and palladium on charcoal 10 % (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75 ± 5 °C for 24 hours. The reaction mixture is filtered at 45 ± 3 0C and the filter cake is washed with water (30 1). 170 1 of water is distilled off under reduced pressure and 30 % HCl (8 1) is added. The solution is cooled to 3 ± 3 0C during 2 h. The solution is seeded with crystals of detomidine HCl at 40 ± 5 °C, 30 ± 5 0C, 20 ± 5 °C and at 10 ± 5 0C, until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 1) are charged. The solution is heated to about 50 °C and stirred for 1 hour. The solution is cooled to 10 °C during 1.5 hour. The solution is filtered and 180 1 of water is distilled off under vacuum. 30 % HCl (20 1) is added and the solution is warmed to 60 0C, and then cooled to 3 ± 3 °C during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifogation and washed with toluene. The product is dried under vacuum at 39 ± 5 °C for 20 hours, at 61 ± 5 °C for 6 hours and at 85 ± 5 °C for 16 hours. The yield is 10.5 kg (78 %).
PATENT
https://patents.google.com/patent/US20080287685A1/en
- Detomidine which is 4-[(2,3-dimethylbenzyl)]imidazole of formula I
- is a well known pharmaceutical agent currently used as its hydrochloride salt in animal sedation.
- [0003]The synthesis of detomidine is described in U.S. Pat. Nos. 4,443,466 and 4,584,383. The preparation of detomidine hydrochloride salt is described in U.S. Pat. No. 4,584,383, wherein detomidine obtained from the hydrogenation step is first recovered from alkaline solution as a free base after which the crystalline product is converted into its hydrochloride salt by treatment with HCl-isopropanol in ethyl acetate.
- [0020]1-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 l), 30% HCl (20 l), ethanol (5 l) and palladium on charcoal 10% (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75±5° C. for 24 hours. The reaction mixture is filtered at 45±3° C. and the filter cake is washed with water (30 l). 170 l of water is distilled off under reduced pressure and 30% HCl (8 l) is added. The solution is cooled to 3±3° C. during 2 h. The solution is seeded with crystals of detomidine HCl at 40±5° C., 30±5° C., 20±5° C. and at 10±5° C., until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 l) are charged. The solution is heated to about 50° C. and stirred for 1 hour. The solution is cooled to 10° C. during 1.5 hour. The solution is filtered and 180 l of water is distilled off under vacuum. 30% HCl (20 l) is added and the solution is warmed to 60° C., and then cooled to 3±3° C. during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The product is dried under vacuum at 39±5° C. for 20 hours, at 61±5° C. for 6 hours and at 85±5° C. for 16 hours. The yield is 10.5 kg (78%).
PATENT
https://patents.google.com/patent/WO2020016827A1/en
Detomidine
Detomidine, 4-[(2,3-dimethylphenyl)methyl]-lH-Imidazole, is an a-2-andregenic agonist available under the brand name Equimidine® and Dormosedan® for use as a veterinary sedative. Detomidine is not currently approved for human use.
Detomidine and related compounds, including its 3,4 dimethyl isomer, iso-detomidine (4-(3,4- Dimethylbenzyl)-lH-imidazole) were first described in US4,443,466. Both the‘466 patent and the later US4, 584,383 describe the synthetic method of manufacturing detomidine as being based on coupling of an imidazole moiety with l-Bromo-2, 3-dimethyl benzene using a Grignard reaction. RU2448095 describes an alternative route of synthesis of the molecule based on coupling in presence of a Titanium catalyst. According to both the‘383 and‘095 patents, detomidine is purified by crystallization of its hydrochloride salt from water. The chemical structures of detomidine HC1 and iso-detomidine are shown below:

Detomidine HC1 Iso-detomidine
Two solid state forms of detomidine HC1 are known, the anhydrous and monohydrate forms.
Synthesis of the anhydrous form by crystallization of the monohydrate and further decomposition at elevated temperatures is described in US7,728,l47. Synthesis of the anhydrous form via decomposition of the monohydrate in reduced pressure is described in Laine et al (1983). According to Veldre et al (2011), the anhydrous and monohydrate forms of detomidine HC1 can easily interconvert depending on temperature and humidity.
The European Pharmacopeia 9.0 monograph (January 2014) describes detomidine HC1 for veterinary use. The monograph lists the established HPLC method for identification of detomidine and its impurities as using a Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of Ammonium phosphate buffer pH 7.9 – 65% and Acetonitrile – 35% at a flow rate of 1.0 mL/min and UV detection at 220 nm. That procedure is listed as recording three distinct impurities of detomidine:
Impurity A: (RS)-(2, 3 -dimethylphenyl)(l/f-imidazol-4-yl)m ethanol
– l/f-imidazol-5-yl)(2,3-dimethylphenyl)m ethanol

Impurity C: 4-| (2.3 -dimcthy ley clohcxyl)m ethyl |- 1 /7-im ida/olc

PCT/US18/012579 discloses topical formulations of detomidine and their uses in treating pain.
Purified detomidine for use in human pharmaceutical formulations is not known in the art.
EXAMPLE 5: Purification of organic impurities from detomidine HC1 monohvdrate
Two potential procedures for purification of organic impurities from sourced monohydrate were compared. The first attempted procedure was by direct re-crystallization of detomidine HC1 from 2.88 volumes of water, while the second included carbon treatment and precipitation of detomidine free base followed by the free base being reacted with HC1 and crystallized as monohydrate. Both procedures used the same non-GMP, off white anhydrous detomidine HC1 starting material which had previously been shown in Table 7 to contain 0.21% of iso-detomidine and 0.07% of Impurity A. All the re-crystallized materials were found to have practically the same purity level. The direct re-crystallization procedure was found to provide a product with a high yield and purity and at the same time provides a practical and scalable crystallization process which could be controlled by process parameters such as seeding and cooling rate.
Example 5 a: Direct recrvstallization
Anhydrous detomidine HC1 (4.5g) was introduced to a round-bottom flask with a magnetic stirrer and thermometer. Deionized water (l3ml) was then added and the mixture stirred and heated in a water bath. At 39°C, the complete dissolution of solids was observed, providing a clear yellow solution with a pH = 4.
The batch was gradually cooled by stirring. At 3 l°C, intensive crystallization was observed. The resulting slurry was cooled in an ice-water bath for 20 min and filtered. Flask and cake were then washed with 2 ml of cold deionized water and 3.97g of a white to cream colored solid was collected. 2.03g of the material was dried in a vacuum desiccator at ambient temperature and 20 mbar to a constant weight over 23 hrs producing a dry monohydrate – l .96g off-white crystalline solid (sample 1).
An additional l .9 lg of the material was dried in a vacuum oven at 90°C under house vacuum to a constant weight over about 24.5 hrs producing a dry anhydrate , l .68g off-white solid (sample 2)
The two samples were subjected to physical characterization and purity analysis by HPLC. The XRPD spectra and DSC and TGA thermograms of sample 1 are presented in Figures 8 -10 and of sample 2 are presented in Figures 11-13, respectively.
As shown in Table 11, direct re-crystallization resulted in the effective purification from all organic impurities, but was not effective for color. The content of iso-detomidine and of Impurity A was reduced to a level below the QL, but the off white color remained after re-crystallization.
Table 11 : properties following direct recrystallization (sample 1)

1 – below the QL
2 – system peak
Example 5b(i): Carbon treatment and detomidine free base isolation
Anhydrous detomidine HC1 (70.3g) and deionized water (220ml) were introduced to a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, thermocoupler and a circulating oil bath for heating and cooling.
The mixture was heated while stirring. At 40°C, complete dissolution was observed. Active carbon (CXV type, 5.2g) was added to the clear yellow solution and the batch stirred at 45°C for 50 minutes. Following this, the batch was filtered on through paper filter on Buchner funnel, reactor and filter washed with deionized water (20ml).
The slightly yellowish clear filtrate was reintroduced to the 0.5 liter reactor, stirred and 40% NaOH solution was added at 40°C. After 10ml NaOH solution was added, a pH of 7 was reached and precipitation began. An additional 13ml of NaOH was added over 1 hour at 42 – 52°C and intensive stirring (400 – 450 rpm) performed. The mixture at the end of the addition of NaOH had a pH of 13.
The batch was stirred at 33 – 35°C overnight then cooled to l6°C over 4 hours and stirred at this temperature for an additional hour. The resultant solid was filtered on Buchner filter, reactor and cake washed with two portions of deionized water (2><200ml). The wet solid (86g) was dried in a vacuum oven at 45°C to constant weight to produce a dry product (53.2g, Yield 90.7%) – white powder, m.p.=l 18.6 – 119.2
The dry detomidine base was analyzed for purity by HPLC, the results presented in Table 12. Table 12: Properties of detomidine base (intermediate in sample 2)

1 – system peak
Example 5b(nT Monohvdrate crystallization from detomidine base
The dry detomidine free base (53.0g) from Example 5b(i) was introduced together with 37% HC1 (29.7g) and deionized water (159g) into a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 45°C, at 37°C complete dissolution of solid was observed. The clear solution had a pH of 1. The solution was cooled gradually to 37°C and seeded with detomidine HC1 monohydrate and cooled gradually to 3°C over 4 hours, and then the batch was stirred for 45 minutes at this temperature. The solid was filtered on Buchner filter, reactor and cake washed with cold deionized water (80ml). The wet solid (61.9g) was dried in vacuum oven for 16 hours at 45°C to produce a dry product (57.8g, Yield 84.3%) – white crystalline powder (sample 2)
The dry detomidine HC1 monohydrate was analyzed for water by CKF (¾0 = 7.46%) and for purity by HPLC with the results presented in Table 13. Microscopic observation for particle morphology (regular prisms) was performed and the microscopic photograph is shown in Figure
14.
Table 13 : Properties of detomidine HC1 (sample 2)

1 – system peak
Example 5c: Re-crvstallization of detomidine HC1 to monohvdrate. bench scale experiment Anhydrous detomidine HC1 (754.6g) 37% HC1 (116. Og) and deionized water (2008g) were introduced to a 3 liter glass jacketed reactor equipped with a mechanical stirrer, two baffles, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 52°C, at 47°C complete dissolution was observed and the clear solution was found to have a pH of 0-0.5.
The solution was cooled gradually and at 45°C seeded with detomidine HC1 monohydrate (0.5g). Crystallization initiation was observed at 43°C and the batch was then cooled to 1.5°C during 5 hours and stirred for 12 hours at this temperature. The solid was filtered on Buchner filter and conditioned on the filter with vacuum for 40 minutes. The wet product (817g) was dried in vacuum oven to constant weight. For the first 13 hours, the material was dried at 30°C and 35-27 mbar, then for an additional 7 hours at 40°C and 30-18 mbar to produce a dry product (771.2g, Yield 94.6%) – white crystalline powder (Batch“90” in Tables 8-9; sample 3)
Dry detomidine HC1 monohydrate was analyzed for water by CKF (FhO = 7.37%) and for purity by HPLC, the results presented in Table 14. The physical characterization results are shown in Table 10 above.
The material was subjected to physical characterization and microscopic observation for particle morphology (regular prisms) microscopic photograph presented in Figure 7.
Table 14: Properties of detomidine HC1 (sample 3)
1 – system peak
EXAMPLE 6: Synthesis of iso -detomidine
Scheme 1 outlines a process for the synthesis of iso-detomidine was developed to produce a solid iso-detomidine HC1 in high yield and substantially free of impurities.

Scheme 1 : Route of synthesis of iso-detomidine
Example 6a: Sandmever Reaction
3,4 dimethyl aniline (150g, 1.24M) was mixed with acetonitrile (0.6 liter) in a 5 liter flask, chilled to lO°C and water (1.2 liter) added dropwise over 5 minutes. The mixture was cooled to 5°C with ice-ethanol bath and concentrated H2SO4 (98% wt, 363g 3.71M) was added dropwise over 30 min at 5-l0°C. Sodium nitrite (NaNC ) aqueous solution (89.7g in 300 ml water, 1.30M) was then added dropwise over 30 min at 0-5°C to give a brown solution. The resulting solution of diazonium salt was stirred at 0-5°C for an additional 30 min.
In another 5 liter flask KI (225g, 1.36M) was dissolved in water (0.8 liter) during stirring and cooled. The diazonium salt solution was added dropwise to the KI solution at 7-l3°C during 35 min, the batch stirred at 7-l3°C for 1.25 hr to give a black solution. MTBE (2.0 liter) was then added to the reaction mixture and Na2SC>4 (23.4g) was introduced in small portions during 5 min.
The mixture was settled and the organic phase separated and washed with two portions of brine (2 500ml). The organic solution was concentrated under vacuum to a volume of about 250ml.
The product was purified by vacuum distillation at ca. 40Pa, BP = 52 – 60°C to give 246g of intermediate 1 as a brown oil with a product yield of 86%.
Example 6b: TRT protection reaction
lH-Imidazole-4-carbaldehyde (45.2g, 0.47M) and acetonitrile (0.8 liter) are introduced into a 2 liter flack and cooled to 8°C, then TRT-C1 (131. Og, 0.47M) was added at 8°C and TEA (57. lg, 0.56M) was added dropwise during 20 min. The reaction mixture was stirred at 8 to l8°C for 2 hrs.
The reaction mixture was poured into a stirring mixture of water (0.72 liter) and MTBE (0.72 liter) and stirred for 10 minutes. The resulting solid was isolated by filtration on Buchner funnel and dissolved with THF (3 liter). The solution was dried over Na2SC>4 and concentrated to remove most of the solvent.
MTBE (400 ml) and PE (200ml) was added to the residue, the mixture stirred at 8°C for 16 hrs. The precipitated solid was isolated by filtration on Buchner filter and dried in air for 16 hrs at room temperature. Then the filter cake is dried by azeotropic drying with 2-Me-THF (2×500 ml) to give l29g of intermediate 2 as white solid with a yield of 66.5%.
Example 6c: Grignard reaction
A 2M solution of i-PrMgCl in THF (0.275 liter, 0.55M) and THF (1.0 liter) was introduced to a 2 liter flask at l2°C. Intermediate 1 (121.8g, 0.525M) was added dropwise during 20 min. The mixture was stirred at l2-l5°C for 3 hrs.
Intermediate 2 (84.6g, 0.25M) was added in small portions without cooling during 30 min, with a temperature rise to 25°C, to give a light brown solution. The solution was stirred for 2.5hrs at l5°C and added to aqueous solution of NH4CI (117g in 0.7 liter water) during 10 min at 5°C. PE (1.6 liter) was added during 5 min and the mixture stirred for extra 25 min.
Precipitated solid filtered on Buchner funnel and then re-slurred with mixture of MTBE (400 ml), water (600 ml) and PE (200 ml). Then the solid was filtered on Buchner funnel and re-slurred with MeOH (700 ml) at 60°C for 10 min, cooled to 20°C with cold water bath and filtered again on Buchner funnel. The solid product was dried in an air oven at 45 °C for 2 hrs to give 112 g of intermediate 3 as a white solid with a yield of 89.9%.
Example 6d: Reductive dehvdroxylation and de-protection
Intermediate 3 (l07g, 0.240M) and DCM (1.10 liter) were introduced to a 2 liter flask at 1 l°C, TFA (214 ml) was added dropwise over 5 mins with a temperature rise to l4°C.
The mixture was stirred for about 5 mins and EhSiH (94.4g, 0.794M) added dropwise during 5 mins. After stirring at 25-30°C for 16 hrs the mixture was concentrated by rotary evaporation at 40°C to a residue.
The residue of evaporation was dissolved in DCM (600 ml) and washed with 1.5M aq. HC1 (0.241iter). Organic phase was separated and washed with aq. NaOH (11.5g in 200ml water), pH of aqueous phase 13. Two phases were separated and the organic phase washed with brine (200 ml) dried over Na2S04 and filtered. The resulting solution was concentrated by rotary evaporation.
The evaporation residue was dissolved in mixture of EtOAc (500 ml) and EtOH (30 ml) and then 4M HC1 solution in dioxane (40 ml) was added dropwise in 5 minutes, pH = 1 – 2 adjusted and a white solid precipitated out.
The solid product was filtered on Buchner funnel, the cake dried in air for 16 hrs to give 36g of white solid.
The solid product was re-crystallized from iPrOH / Acetone. The dry cake (36g) and iPrOH were introduced into a 1 liter flask and heated to dissolution. Acetone (360 ml) was added to the resulting colorless solution at reflux during 10 mins. The mixture was cooled to 8°C and stirred at this temperature for additional 4.5 hrs. The solid product was filtered on Buchner funnel and dried in air for 36 hrs. 29.2g of iso-detomidine as a white solid was obtained with a yield of 54.4%. The 1H-NMR spectra of iso-detomidine is shown in Figure 15. EXAMPLE 7 : Re-crvstallization of detomidine HC1 spiked with 2% iso-detomidine
Detomidine HC1 monohydrate (26. Og), iso-detomidine HC1 (0.52g) and deionized water (68.7g) were introduced to a 100 ml glass jacketed reactor equipped with a mechanical stirrer, a thermocouple and a circulating oil bath for heating and cooling. The batch was stirred and heated to 51°C, at 47°C complete dissolution was observed.
The solution was cooled gradually and at 42°C seeded with detomidine HC1 monohydrate. Crystallization initiation was observed at 39°C and then the batch was cooled to 3°C for 5 hours, filtered on Buchner filter and conditioned on the filter with vacuum. The wet product (20.7 g) was dried in vacuum oven to constant weight to produce a dry product (20.13g, Yield 75.9%) – white crystalline powder
Dry detomidine HC1 monohydrate was analyzed for PSD and morphology, the results are presented in Table 8 (Sample. No. 91). The purity of re-crystallized material was analyzed using the optimized HPLC process disclosed herein, and the results are presented in Table 15.
Table 15 : Properties of detomidine HC1 following recrystallization from iso-detomidine spiked material

a area %
b Spiked amount, calculated
References
- ^ Clarke, Kathy W.; Hall, Leslie W.; Trim, Cynthia M., eds. (2014). “Principles of sedation, anticholinergic agents, and principles of premedication”. Veterinary Anaesthesia. pp. 79–100. doi:10.1016/B978-0-7020-2793-2.00004-9. ISBN 978-0-7020-2793-2.
- ^ England GC, Clarke KW (November 1996). “Alpha 2 adrenoceptor agonists in the horse–a review”. The British Veterinary Journal. 152 (6): 641–57. doi:10.1016/S0007-1935(96)80118-7. PMID 8979422.
- ^ Fornai F, Blandizzi C, del Tacca M (1990). “Central alpha-2 adrenoceptors regulate central and peripheral functions”. Pharmacological Research. 22 (5): 541–54. doi:10.1016/S1043-6618(05)80046-5. PMID 2177556.
External links
- “Medication Protocols for Horses”. The Ontario Association of Equine Practitioners. 2005. Archived from the original on 2007-04-17.
- Compendium of data sheets for animal medicines. National Office of Animal Health. 2005. ISBN 978-0-9548037-0-4.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATCvet code | QN05CM90 (WHO) |
| Legal status | |
| Legal status | Veterinary use only |
| Pharmacokinetic data | |
| Elimination half-life | 30 min |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76631-46-4 |
| PubChem CID | 56032 |
| ChemSpider | 50586 |
| UNII | 7N8K34P2XH |
| KEGG | D07795 |
| ChEMBL | ChEMBL2110829 |
| CompTox Dashboard (EPA) | DTXSID00227457 |
| Chemical and physical data | |
| Formula | C12H14N2 |
| Molar mass | 186.258 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCc2cccc(Cc1cnc[nH]1)c2C | |
| hideInChIInChI=1S/C12H14N2/c1-9-4-3-5-11(10(9)2)6-12-7-13-8-14-12/h3-5,7-8H,6H2,1-2H3,(H,13,14) Key:RHDJRPPFURBGLQ-UHFFFAOYSA-N |
////////////// DETOMIDINE, UNII-7N8K34P2XH , детомидин ,ديتوميدين, 地托咪定 , Domosedan, Farmos, SEDATIVE
#DETOMIDINE, #UNII-7N8K34P2XH , #детомидин ,#ديتوميدين, #地托咪定 , #Domosedan, #Farmos, #SEDATIVE
https://patents.google.com/patent/WO2020016827A1/en
EXAMPLES
EXAMPLE 1 : Elemental analysis of impurities found in commercially available anhydrous detomidine HC1
Example la: Anhydrous detomidine HC1 was sourced from two commercial API suppliers. Properties of the commercial batches, GMP1, GMP2 and GMP3, are presented below.
Elemental impurity analysis was performed by inductively coupled plasma mass spectrometry (ICP-MS) on four different batches of sourced anhydrate. The results of the analysis are found in Table 1.
Table 1 : Elemental impurities in anhydrous detomidine HC1
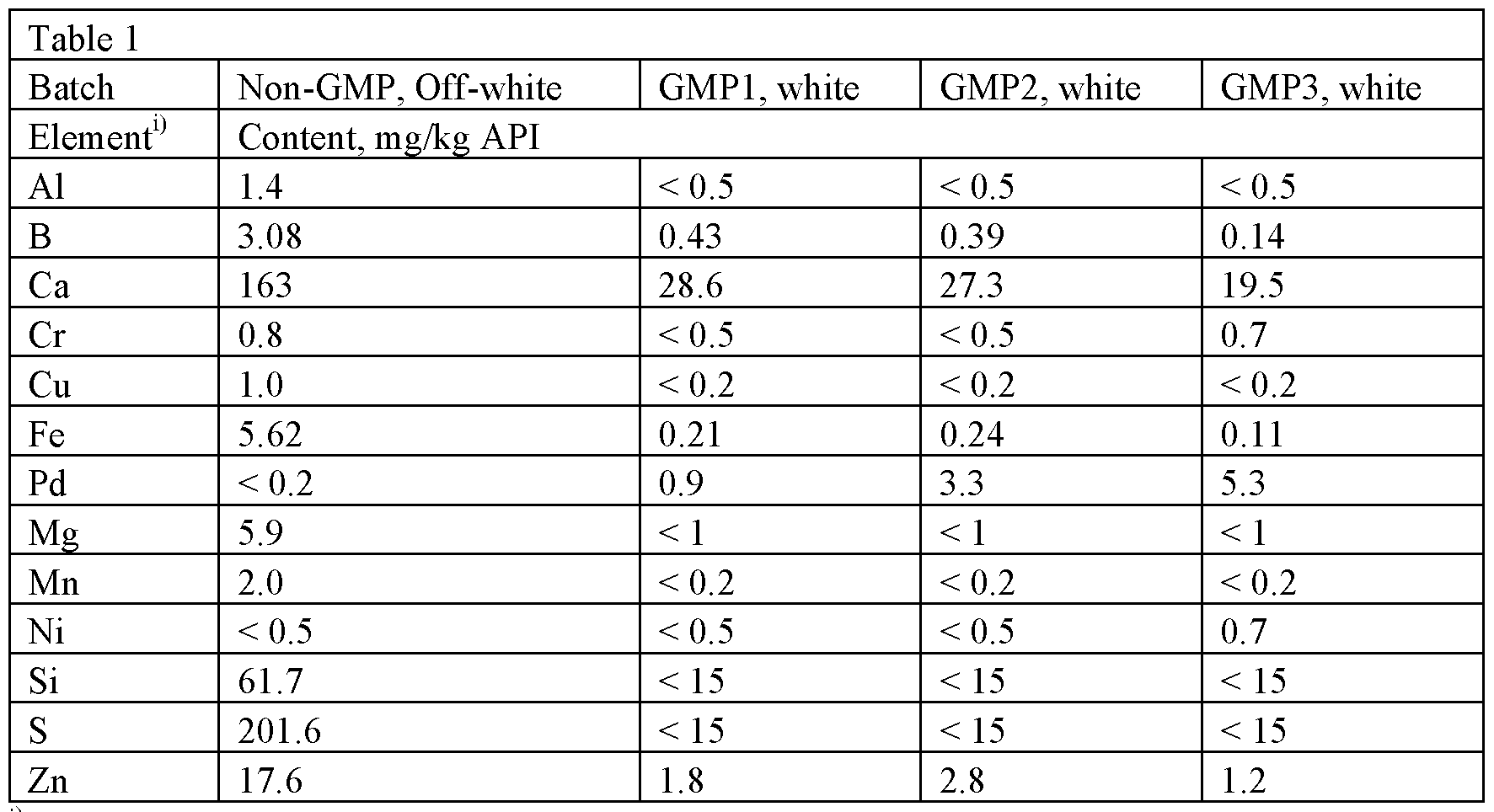
11 Elements having levels L.T. 0.5 mg/kg (Ti, As, Hg, Pb, Mo, Pt, etc) are not presented in the table
The screening of elemental impurities shows that the GMP products contained significant levels of Pd (0.9 – 5.3 mg/kg). Pd is understood to be a catalyst used in the synthesis of detomidine (e.g., in reduction/hydrogenation methods).
Example lb: Characterization of commercially sourced material
Samples of the anhydrous detomidine products described in Table 1 were analyzed for water content and characterized by microscope, XRPD and thermal analyses. The results are summarized in Table 2.
Table 2: Characterization of commercial anhydrous detomidine HC1

a Anhydrous + mono hydrate The values presented in T able 2 demonstrate that the commercial samples of detomidine HC1 labeled as anhydrous contain some amount of monohydrate and this amount varied depending on storage conditions and packaging.
EXAMPLE 2: Stability assessment of anhvdrate and monohvdrate forms of detomidine base and detomidine HC1
Pure forms of crystalline free base, and HC1 salt (both monohydrate and anhydrate) were prepared from commercially sourced anhydrous detomidine HC1 as outlined in Table 3, and characterized using XRPD and thermal analysis. The solids were crystallized from aqueous solutions and then dried under different conditions. The crystallization and drying conditions are summarized in Table 3.
Table 3: Preparation of detomidine HC1 crystalline forms

The properties of the solids crystallized according to Table 3 are described in Table 4.
Table 4: Properties of Detomidine HC1 crystalline forms


These results demonstrate that crystallization from 2.8 – 2.9 volumes of water is effective for isolation and purification of the detomidine HC1 monohydrate drug substance. Drying of the monohydrate under mild conditions (20-40 mbar and temperatures from at least ambient to about 45 °C) provided pure monohydrate without traces of the anhydrous form.
The same monohydrate dried at elevated temperature (30-40 mbar 90°C) converted completely into the anhydrous form. The vacuum dried, hermetically closed anhydrate did not absorb water from the atmosphere and did not convert into the monohydrate. After exposure to atmospheric air, however, the anhydrate absorbed water and converted to a mixture of anhydrate and
monohydrate.
Melting points (m.p.) of the intermediate detomidine free base and hydrochloride of Sample 5 measured in open capillary corresponded with the published literature and the DSC data and are presented in Table 5. In order to evaluate effect of humidity on different forms of detomidine, a hydration study was performed. Samples of detomidine free base and hydrochloride salt were subjected to DVS analysis. These observations are in accordance with the DVS results shown in Figures 5 and 6, for detomidine free base and detomidine HC1, respectively.
Table 5: Composition and properties of known solid forms of detomidine



a -literature data
The free base was found to be crystalline and insoluble in water but it reacted readily with aqueous HC1 giving soluble detomidine hydrochloride.
Crystallization from water provided effective purification of the detomidine HC1 and formation of large regular crystals. Anhydrous detomidine hydrochloride appeared as small irregular particles whereas the possibility to control particle size distribution by crystallization parameters existed for the monohydrate.
The detomidine free base was found to be non-hygroscopic, but also able to absorb more than 1% of water at relative humidity (RH) >50%. An increase of humidity from RH 70% to RH >90% did not lead to absorption of additional water to monohydrate. During the dehydration cycle, the monohydrate began to lose water at RH -10% and converted into the anhydrate at RH =0%. Anhydrate did not absorb water at RH <30% and transformed completely to into the monohydrate at RH between 30% and 50%.
Four cycles of hydration-dehydration demonstrated good reproducibility of anhydrate- monohydrate interconversion.
An anhydrous detomidine HC1 of Sample 2 was shown to absorb water to a level of cKF 7.7% which corresponds well to the theoretical amount of water in the monohydrate form (Table 5).
The hydration profile of detomidine hydrochloride showed that the monohydrate is stable in a wide range of humidity between 10% and >90% RH. At the same time, the anhydrous form is not stable in atmospheric air and absorbs water at RH = 30 – 50%.
This data demonstrates that the anhydrous form is challenging in the aspects of water content and solid form stability and that detomidine HC1 monohydrate is more suitable for pharmaceutical development.
Example 3 : Impurity analysis of commercially sourced detomidine HC1
Using the established Pharmacopeia HPLC protocol (Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of 65% Ammonium phosphate buffer pH 7.9 and 35% Acetonitrile at a flow rate of 1.0 mL/min and UV detection at 220 nm), sourced samples of detomidine HC1 were assayed for impurities. As shown in Figure 1, a previously unreported peak was identified, which partially overlapped with that of detomidine. By LC-MS/MS analysis, this impurity was shown to have the same molecular weight as detomidine.
The established Pharmacopeia HPLC protocol did not separate the detomidine from the impurity. Therefore, for further identification of the elusive impurity, new HPLC protocols for assaying detomidine HC1 were developed. One protocol (“HPLC Protocol A”) comprised using a SunFire C8 column, IOqA, 3.5 pm, 4.6 x l50mm column with an initial mobile phase of 70% Ammonium Phosphate buffer solution, pH 7.9 and 30% Acetonitrile, at a flow rate of 1.0 mL/min and UV detection at 220 nm. To remove late eluting peaks, the flush gradient shown in Table 6 was applied after each run. This HPLC protocol allowed for a resolution factor of 3.9 between detomidine and the unidentified impurity. The quantitation level (QL) for impurities and degradation products is 0.025%. The detection level (DL) for impurities and degradation products is 0.01%.
Table 6: Flush gradient for HPLC protocol

Given its molecular weight, it was hypothesized that the impurity was iso-detomidine.
A solution of 100 pg/ml detomidine HC1 and about 1 pg/mL (about 1% of the working concentration) of detomidine impurity A and iso-detomidine were prepared and assayed using the new HPLC protocol (HPLC Protocol A), disclosed hereinabove. Figure 2 is a chromatogram showing that the previously unreported peak is confirmed as being iso-detomidine.
The analysis of commercially sourced detomidine HC1 revealed a significant additional impurity. Table 7 provides levels of the various detomidine impurities in different commercial batches. In all batches, total impurities were observed at levels of > 0.1% area.
Table 7: Impurity levels (% area) in commercial batches of detomidine.


provided by commercial supplier after undergoing the reciystallization process of Example 5, provided by inventors.
Further analysis of the peak at RRT=0.38 showed that it actually consisted of 2 separate, overlapping peaks. As shown in Figure 3, LC-MS/MS analysis confirmed one of these peaks as iso-impurity A. Further analysis, as shown in Figure 4, identified the second peak as (2,3- dimcthylphcnylX 1 //-imidazol-4-yl) methanone.
EXAMPLE 4: Optimization of the crystallization method of detomidine HC1 monohvdrate from commercial batches of anhydrous detomidine HC1
Crystallization experiments on 25, 65, and 770 gram scale were performed in 100 ml, 500 ml and 3 liter jacketed glass reactors, respectively, equipped with CBT (curved blade turbine) mechanical stirrers, circulating oil bath, thermocouples, and condensers. Stirrer speed in all experiments was between 300 – 600 rpm. Variable process parameters were: amounts of HC1, solvent ratio, cooling time/rate, seeding and cake wash. The parameters and the variation ranges were chosen according to production conditions. The crystallization parameters are summarized in Table 8.
Table 8: Crystallization parameters

a Seeding with detomidine HC1 monohydrate
b Time 24 hrs
c Seeding with anhydrous detomidine HC1
d 5.5 hrs cooling and overnight stirring at 1-3° C
e Spiked with 2% iso -detomidine
The drying parameters and solid properties of batches shown in Table 8 are described in Table 9. Table 9: Drying parameters and solid properties of detomidine monohydrate crystals

microscopic observation: Rods – aspect ratio > 2; prisms – aspect ratio < 2
u)M = mono hydrate
The data presented in Tables 8 and 9 demonstrate that crystallization from water and drying under technical vacuum gives pure detomidine HC1 monohydrate without traces of the detomidine HC1 anhydrous form. Variations of HC1 excess from 0 to 0.5 mole/mole base, cooling time from 1.5 to 24 hours and drying time from 15 to 33 hours appear to have no effect on the obtained properties of the solid form. All crystallization products appeared as pure detomidine HC1 monohydrate.
The crystallization initiation method also had no effect on crystalline form. The batches seeded with anhydrous material gave the same monohydrate as batches seeded with monohydrate and batches which crystallized spontaneously.
Contact with water for 24 hrs completely converted the anhydrous form into the monohydrate, even without complete dissolution (re-slurry).
Crystallization of the monohydrate from water gave large clear crystalline particles with a mean crystal size 0.3 – 0.7 mm, with some crystals larger than 2 mm in size. The shape of the crystals was rod-like or prism-like, if the aspect ratio of the crystals was < 2 the crystals were reported in Table 8 as prisms. A ratio of HC1 to base within the range 1.0 – 1.5 mole : mole and water to solid ratio within the range 2.1 – 2.8 V/wt were found to have no significant effect on the particle size distribution (PSD). However, a ratio of HC1 to base of about 1.5 were found to increase yields of highly pure detomidine HC1 monohydrate from under 90% (60.8%-86.4%) to over 90% (9l .4%-95.9%). Seeding also appeared to have no significant effect on PSD.
The cooling rate was found to have a weak effect on PSD. There was no effect observed for cooling over a time range between 1.5 and 5.5 hrs (mean cooling rate 0.10-0.3 l°C/min).
Slurry -to-slurry recrystallization of anhydrous material resulted in a strong reduction in particle size with the d(0.5) decreasing from 300-500m to 87m. These crystals were found irregular with no signs of prism-like or rod-like habit. In contrast, the re-slurry procedure applied to a mixture of anhydrate and monohydrate (15:85) gave a mixture of rod and prism-like crystals with d(0.5)=4l5p.
Batch size was found to have no significant effect on crystal size and shape. After scaling up from a 26g batch in 100 ml reactor to 770g in a 3 liter reactor, the PSD was very similar to that of small scale batches.
Prolonged cooling resulted in a “rounded” form of crystals. This effect was observed in two experiments, as seen in the microscopic photograph in Figure 7. In the first experiment the crystallizing suspension was cooled for 8 hrs, and in the second one it was stirred at low temperature for 12 hrs (batches 83 and 90 in Tables 8 and 9).
Under the conditions described, cooling had a strong effect on the process yield. Two re-slurry experiments were performed at the same water volume ratio as most of experiments (2.80 V/wt) but these two batches were not cooled and filtered at 24°C. In these experiments the yield dropped from 86% to 60-65% (batches 84, 85 in Tables 8 and 9).
Acceptable yields were obtained in cooled batches within the solvent volume ratio range 2.1 – 2.8 V/wt with the cooling temperature between about l.5°C – 4°C
An increase of HC1 to base molar ratio from 1 to 1.5 was found to raise the yield from 86% to 95%. Cake wash reduced the yield by 2 – 3%. Re-crystallization in presence of 2% iso- detomidine reduced the yield from 84 – 85% to 76%. The purity of the samples prepared according to methods disclosed in Tables 8 and 9, determined using the optimized HPLC method, are presented in Table 10. Table 10

E


{kind=link}
{kind=link}
#DETOMIDINE, #UNII-7N8K34P2XH , #детомидин ,#ديتوميدين, #地托咪定 , #Domosedan, #Farmos, #SEDATIVE
LikeLike