
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
Naloxegol
Movantik; NKTR-118; NKTR118; UNII-44T7335BKE; NKTR 118
854601-70-0 cas
1354744-91-4 (Naloxegol Oxalate)
(4R,4aS,7S,7aR,12bS)-7-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]-3-prop-2-enyl-1,2,4,5,6,7,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-4a,9-diol
| MF | C34H53NO11 |
|---|---|
| MW | 651.78472 g/mol |
Naloxegol oxalate (MovantikTM, Moventig)
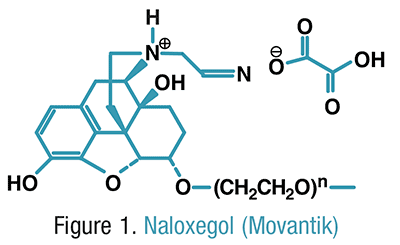
Naloxegol (INN; PEGylated naloxol;[1] trade names Movantik and Moventig) is a peripherally–selective opioid antagonistdeveloped by AstraZeneca, licensed from Nektar Therapeutics, for the treatment of opioid-induced constipation.[2] It was approved in 2014 in adult patients with chronic, non-cancer pain.[3] Doses of 25 mg were found safe and well tolerated for 52 weeks.[4] When given concomitantly with opioid analgesics, naloxegol reduced constipation-related side effects, while maintaining comparable levels of analgesia.[5]

Naloxegol Oxalate was approved by the U.S. Food and Drug Administration (FDA) on Sept 16, 2014, then approved by European Medicine Agency (EMA) on Dec 8, 2014. It was developed and marketed as Movantik®(in the US)/Moventig®(in EU) by AstraZeneca.
Naloxegol oxalate is an antagonist of opioid binding at the mu-opioid receptor. It is indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain.
Movantik® is available as tablets for oral use, containing 12.5 mg or 25 mg of free Naloxegol. The recommended dose is 25 mg once daily (reduce to 12.5 mg if not tolerated).
Chemically, naloxegol is a pegylated (polyethylene glycol-modified) derivative of α-naloxol. Specifically, the 5-α-hydroxyl group of α-naloxol is connected via an ether linkage to the free hydroxyl group of a monomethoxy-terminated n=7 oligomer of PEG, shown extending at the lower left of the molecule image at right. The “n=7” defines the number of two-carbon ethylenes, and so the chain length, of the attached PEG chain, and the “monomethoxy” indicates that the terminal hydroxyl group of the PEG is “capped” with amethyl group.[6] The pegylation of the 5-α-hydroxyl side chain of naloxol prevents the drug from crossing the blood-brain barrier(BBB).[5] As such, it can be considered the antithesis of the peripherally-acting opiate loperamide which is utilized as an opiate-targeting anti-diarrheal agent that does not cause traditional opiate side-effects due to its inability to accumulate in the central nervous system in normal subjects.
Naloxegol was previously a Schedule II drug in the United States because of its chemical similarity to opium alkaloids, but was recently reclassified as a prescription drug after the FDA concluded that the impermeability of the blood-brain barrier to this compound made it non-habit-forming, and so without the potential for abuse — specifically, naloxegol was officially decontrolled on 23. January 2015. [7]

As an opiate antagonist, it is not expected to be capable of inducing the euphoria and anxiolytic effects which are generally cited as the desirable effects of commonly abused opiates (all of which are opiate agonists) if it were to cross the BBB; it would in fact reverse the effects of opiate drugs of abuse if it entered the central nervous system.
Naloxegol is an oral polyethylene glycol (PEG) derivative of naloxone, a peripherally acting µ-opioid receptor antagonist (PAMORA) with limited potential for interfering with centrally mediated opioid analgesia. The incorporation of a polyethylene glycol moiety aims at inhibiting naloxone’s capacity to cross the blood-brain barrier, while preserving the affinity for the µ-opioid receptor [1].

Opioid-induced bowel dysfunction (OIBD) represents a broad spectrum of symptoms that result from the actions of opioids on the CNS as well as the gastrointestinal tract. The majority of gastrointestinal effects seem to be mediated by the high number of µ-receptors that are expressed in the enteric nervous system. Naloxegol was more effective than placebo in increasing the number of spontaneous bowel movements in patients with opioid-induced constipation, including those with an inadequate response to laxatives.
Recognition of Naloxegol as a useful option in the treatment of opioid-induced constipation resulted in its approval by US-FDA for adult patients with chronic, non-cancer pain in 2014.
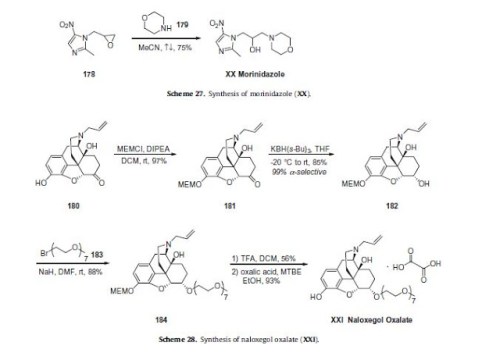
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there havebeen reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is
described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.
152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.
158. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm4183
Naloxegol Synthesis
CREDIT
https://ayurajan.blogspot.in/2016/08/naloxegol.html
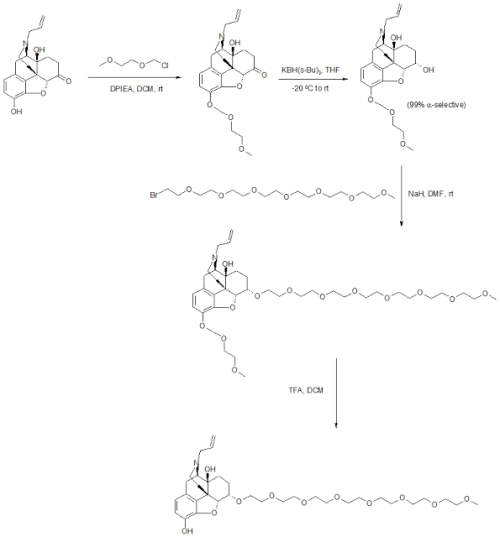
US20050136031A1: The patent reports detailed synthetic procedures to manufacture gram quantities of Naloxegol. The synthesis starts with Naloxone which was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone. Reduction of the ketone with potassium tri-sec-butylborohydride exclusively provided the α-alcohol in 85% yield. Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br provided the pegylated Naloxone in 88% yield.
Identifications:

| 1H NMR (Estimated) for Naloxegol |



PATENT

PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
EXAMPLE 4 SYNTHESIS OF PEG 3-NALθxoL [0211] The structure of the naloxol, an exemplary small molecule drug, is shown below.

Naloxol [0212] This molecule was prepared (having a protected hydroxyl group) as part of a larger synthetic scheme as described in Example 5.
EXAMPLE 5
[0213] α,β-PEGι-naloxol was prepared. The overview of the synthesis is provided below.

(3)

(4)
5.A. Synthesis of 3-MEM-naloxone
[0214] Diisopropylethylamine (390 mg, 3.0 mmole) was added to a solution of naloxone ■ HCl • 2H2O (200 mg, 0.50 mmole) in CH2C12 (10 mL) with stining. Methoxyethyl chloride (“MEMCl,” 250 mg, 2.0 mmole) was then added dropwise to the above solution. The solution was stined at room temperature under N2 overnight.
[0215] The crude product was analyzed by HPLC, which indicated that 3-
MEM-O-naloxone (1) was formed in 97% yield. Solvents were removed by rotary evaporation to yield a sticky oil.
5.B. Synthesis of α and β epimer mixture of 3-MEM-naloxoI (2)
[0216] 3 mL of 0.2 N NaOH was added to a solution of 3-MEM-naloxone
(1) (obtained from 5.A. above, and used without further purification) in 5mL of ethanol. To this was added a solution of NaBHLt (76 mg, 2.0 mmole) in water (1 mL) dropwise. The resulting solution was stined at room temperature for 5 hours. The ethanol was removed by rotary evaporation followed by addition of a solution of 0.1 N HCl solution to destroy excess NaBKj and adjust the pH to a value of 1. The solution was washed with CHC13 to remove excess methoxyethyl chloride and its derivatives (3 x 50 mL), followed by addition of K2OO3 to raise the pH of the solution to 8.0. The product was then extracted with CHC13 (3 x 50 mL) and dried over Na2SO4. The solvent was removed by evaporation to yield a colorless sticky solid (192 mg, 0.46 mmole, 92% isolated yield based on naloxone • HCl • 2H2O).
[0217] HPLC indicated that the product was an α and β epimer mixture of
3-MEM-naloxol (2).
5.C. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-O-3-MEM- naloxol (3a).
[0218] NaH (60% in mineral oil, 55 mg, 1.38 mmole) was added into a solution of 6-hydroxyl-3-MEM-naloxol (2) (192 mg, 0.46 mmole) in dimethylformamide (“DMF,” 6 mL). The mixture was stined at room temperature under N2 for 15 minutes, followed by addition of 2-bromoethyl methyl ether (320 mg, 2.30 mmole) in DMF (1 mL). The solution was then stirred at room temperature under N2 for 3 hours.
[0219] HPLC analysis revealed formation of a mixture of α- and β-6-CH3-OCH2CH2-0-3-MEM-naloxol (3) in about 88% yield. DMF was removed by a rotary evaporation to yield a sticky white solid. The product was used for subsequent transformation without further purification.
5.D. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-naloxoI (4)
[0220] Crude α- and β-6-CH3-OCH2CH2-O-3-MEM-naloxol (3) was dissolved in 5 mL of CH2C12 to form a cloudy solution, to which was added 5 mL of trifluoroacetic acid (“TFA”). The resultant solution was stined at room temperature for 4 hours. The reaction was determined to be complete based upon HPLC assay. CH2C12 was removed by a rotary evaporator, followed by addition of 10 mL of water. To this solution was added sufficient K2OO3 to destroy excess TFA and to adjust the pH to 8. The solution was then extracted with CHC13 (3 x 50 mL), and the extracts were combined and further extracted with 0.1 N HCl solution (3 x 50 mL). The pH of the recovered water phase was adjusted to a pH of 8 by addition of K2CO3>followed by further extraction with CHC13 (3 x 50 mL). The combined organic layer was then dried with Na2SO4. The solvents were removed to yield a colorless sticky solid.
[0221] The solid was purified by passage two times through a silica gel column (2 cm x 30 cm) using CHCl3/CH3OH (30:1) as the eluent to yield a sticky solid. The purified product was determined by 1H NMR to be a mixture of α- and β epimers of 6-CH3-OCH2CH2-naloxol (4) containing ca. 30% α epimer and ca. 70% β epimer [100 mg, 0.26 mmole, 56% isolated yield based on 6-hydroxyl-3-MEM- naloxol (2)].
[0222] 1H NMR (δ, ppm, CDC13): 6.50-6.73 (2 H, multiplet, aromatic proton of naloxol), 5.78 (1 H, multiplet, olefinic proton of naloxone), 5.17 (2 H, multiplet, olefinic protons of naloxol), 4.73 (1 H, doublet, C5 proton of α naloxol), 4.57 (1 H, doublet, C5 proton of β naloxol), 3.91 (1H, multiplet, C6 proton of naloxol), 3.51-3.75 (4 H, multiplet, PEG), 3.39 (3 H, singlet, methoxy protons of PEG, α epimer), 3.36 (3 H, singlet, methoxy protons of PEG, β epimer), 3.23 (1 H, multiplet, C6 proton of β naloxol), 1.46-3.22 (14 H, multiplet, protons of naloxol).
SYN 1

PATENT
https://www.google.com/patents/WO2012044243A1?cl=en
Naloxol-polyethylene glycol conjugates are provided herein in solid phosphate and oxalate salt forms. Methods of preparing the salt forms, and pharmaceutical compositions comprising the salt forms are also provided herein. ACKGROUND
Effective pain management therapy often calls for an opioid analgesic. In addition to the desired analgesic effect, however, certain undesirable side effects, such as bowel dysfunction, nausea, constipation, among others, can accompany the use of an opioid analgesic. Such side effects may be due to opioid receptors being present outside of the central nervous system, principally in the gastrointestinal tract. Clinical and preclinical studies support the use of mPEG7-0-naloxol, a conjugate of the opioid antagonist naloxol and polyethylene glycol, to counteract undesirable side effects associated with use of opioid analgesics. When administered orally to a patient mPEG7-0-naloxol largely does not cross the blood brain barrier into the central nervous system, and has minimal impact on opioid- induced analgesia. See, e.g., WO 2005/058367; WO 2008/057579; Webster et al., “NKTR-118 Significantly Reverses Opioid-Induced Constipation,” Poster 39, 20th AAPM Annual Clinical Meeting (Phoenix, AZ), October 10, 2009.
To move a drug candidate such as mPEG7-O-naloxol to a viable pharmaceutical product, it is important to understand whether the drug candidate has polymorphic forms, as well as the relative stability and interconversions of these forms under conditions likely to be encountered upon large-scale production, transportation, storage and pre-usage preparation. Solid forms of a drug substance are often desired for their convenience in formulating a drug product. No solid form of mPEG7-O-naloxol drug substance has been made available to date, which is currently manufactured and isolated as an oil in a free base form. Exactly how to accomplish this is often not obvious. For example the number of pharmaceutical products that are oxalate salts is limited. The free base form of mPEG7-0-naloxol has not been observed to form a crystalline phase even when cooled to -60 °C and has been observed to exist as a glass with a transition temperature of
approximately -45 °C. Furthermore, mPEG7-0-naloxol in its free base form can undergo oxidative degradation upon exposure to air. Care can be taken in handling the free base, for example, storing it under inert gas, to avoid its degradation. However, a solid form of mPEG7-0-naloxol, preferably one that is stable when kept exposed to air, is desired
a naloxol-polyethlyene glycol conjugate oxalate salt, the salt comprising ionic species of mPEG7-0-naloxol and oxalic acid. The formulas of mPEG7-0-naloxol and oxalic acid are as follows:

In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in ethanol; adding methyl t-butyl ether to the dissolved mPEG?–
O-naloxol solution; adding oxalic acid in methyl t-butyl ether to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in acetonitrile; adding water to the dissolved mPEG7-0-naloxol solution; adding oxalic acid in ethyl acetate to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In some embodiments, the solid salt form of mPEG7-0-naloxol is a crystalline form.
In certain embodiments a solid crystalline salt provided herein is substantially pure, having a purity of at least about 80%, at least about 85%, at least about 90%, at least about 92%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least about 99%.
In certain embodiments, the solid salt form of mPEG7-0-naloxol is a phosphate salt.
In other embodiments, the solid mPEG7-0-naloxol salt form is an oxalate salt. For instance, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form A, as described herein. As another example, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form B, as described herein. In yet other embodiments, an oxalate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In yet other embodiments, an dihydrogenphosphate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In certain embodiments of a solid mPEG7-0-naloxol oxalate salt Form B provided herein, the salt form exhibits a single endothermic peak on differential scanning calorimetry between room temperature and about 150 °C. The single endothermic peak can occur, for instance, between about 91 °C to about 94 °C. For example, in some embodiments the endothermic peak is at about 92 °C, about 92.5 °C, or about93 °C.
PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
PATENT
PATENT
https://www.google.com/patents/CN102174049A?cl=en
References and notes
- Roland Seifert; Thomas Wieland; Raimund Mannhold; Hugo Kubinyi; Gerd Folkers (17 July 2006). G Protein-Coupled Receptors as Drug Targets: Analysis of Activation and Constitutive Activity. John Wiley & Sons. p. 227. ISBN 978-3-527-60695-5. Retrieved 14 May 2012.
- “Nektar | R&D Pipeline | Products in Development | CNS/Pain | Oral Naloxegol (NKTR-118) and Oral NKTR-119”. Retrieved2012-05-14.
- “FDA approves MOVANTIK™ (naloxegol) Tablets C-II for the treatment of opioid-induced constipation in adult patients with chronic non-cancer pain”. 16 September 2014.
- “Randomised clinical trial: the long-term safety and tolerability of naloxegol in patients with pain and opioid-induced constipation.”. Aliment Pharmacol Ther. 40: 771–9. Oct 2014.doi:10.1111/apt.12899. PMID 25112584.
- ^ Jump up to:a b Garnock-Jones KP (2015). “Naloxegol: a review of its use in patients with opioid-induced constipation”. Drugs. 75 (4): 419–425. doi:10.1007/s40265-015-0357-2.
- Technically, the molecule that is attached via the ether link is O-methyl-heptaethylene glycol [that is, methoxyheptaethylene glycol, CH3OCH2CH2O(CH2CH2O)5CH2CH2OH], molecular weight 340.4, CAS number 4437-01-8. See Pubchem Staff (2016). “Compound Summary for CID 526555, Pubchem Compound 4437-01”. PubChem Compound Database. Bethesda, MD, USA: NCBI, U.S. NLM. Retrieved 28 January2016.
- ^http://www.deadiversion.usdoj.gov/fed_regs/rules/2015/fr0123_3.htm
1. WO2012044243A / US12015038524A1.
2. WO2005058367A2 / US7786133B2.
3. US20060182692A1 / US8067431B2.
4. CN101033228A.
5. Fudan Univ. J. Med. Sci. 2007, 34, 888-890.
| WO2008057579A2 * | Nov 7, 2007 | May 15, 2008 | Nektar Therapeutics Al, Corporation | Dosage forms and co-administration of an opioid agonist and an opioid antagonist |
| WO2009137086A1 * | May 7, 2009 | Nov 12, 2009 | Nektar Therapeutics | Oral administration of peripherally-acting opioid antagonists |
| US20050136031 * | Dec 16, 2004 | Jun 23, 2005 | Bentley Michael D. | Chemically modified small molecules |
Patents
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 6 | |
|---|---|
| Patent | 7056500 |
| Expiration | Jun 29, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 2 of 6 | |
|---|---|
| Patent | 7662365 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 3 of 6 | |
|---|---|
| Patent | 8617530 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 4 of 6 | |
|---|---|
| Patent | 9012469 |
| Expiration | Apr 2, 2032 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 5 of 6 | |
|---|---|
| Patent | 7786133 |
| Expiration | Dec 19, 2027 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 6 of 6 | |
|---|---|
| Patent | 8067431 |
| Expiration | Dec 16, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(5α,6α)-4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-propen-1-yl)morphinan-3,14-diol
|
|
| Clinical data | |
| Trade names | Movantik, Moventig |
| AHFS/Drugs.com | movantik |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | ~4.2% |
| Metabolism | Hepatic (CYP3A) |
| Biological half-life | 6–11 h |
| Excretion | Feces (68%), urine (16%) |
| Identifiers | |
| CAS Number | 854601-70-0 |
| ATC code | A06AH03 (WHO) |
| PubChem | CID 56959087 |
| ChemSpider | 28651656 |
| ChEBI | CHEBI:82975 |
| Synonyms | NKTR-118 |
| Chemical data | |
| Formula | C34H53NO11 |
| Molar mass | 651.785 g/mol |
//////////////Naloxegol, Movantik, NKTR-118, NKTR118, UNII-44T7335BKE, NKTR 118, 854601-70-0, EU 2014, FDA 2014
COCCOCCOCCOCCOCCOCCOCCO[C@H]1CC[C@]2([C@H]3Cc4ccc(c5c4[C@]2([C@H]1O5)CCN3CC=C)O)O
Naloxegol oxalate (MovantikTM, Moventig)
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there have been reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.

152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.

