
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Afoxolaner
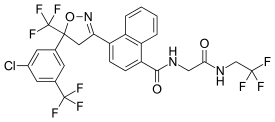
Afoxolaner
- Molecular FormulaC26H17ClF9N3O3
- Average mass625.870 Da
- A1443
- AH252723
1093861-60-9[RN]1-Naphthalenecarboxamide, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,2-trifluoroethyl)amino]ethyl]-4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide
Afoxolaner Merial
On 9 September 2021, the Committee for Medicinal Products for Veterinary Use (CVMP) adopted a positive opinion1, recommending the granting of a variation to the terms of the marketing authorisation for the veterinary medicinal product Frontpro. The marketing authorisation holder for this veterinary medicinal product is Boehringer Ingelheim Vetmedica GmbH. ,,,, https://www.ema.europa.eu/en/medicines/veterinary/summaries-opinion/frontpro-previously-known-afoxolaner-merial
Frontpro is currently authorised as chewable tablets for use in dogs. The variation concerns the change of legal status from prescription-only to non-prescription veterinary medicine. Additionally, the applicant is adding the list of local representatives to the package leaflet.
Detailed conditions for the use of this product are described in the summary of product characteristics (SPC), for which an updated version reflecting the changes will be published in the revised European public assessment report (EPAR) and will be available in all official European Union languages after the variation to the marketing authorisation has been granted by the European Commission.
| Name | Frontpro (previously known as Afoxolaner Merial) |
| Agency product number | EMEA/V/C/005126 |
| International non-proprietary name (INN) or common name | afoxolaner |
| Species | Dogs |
| Active substance | afoxolaner |
| Date opinion adopted | 09/09/2021 |
| Company name | Boehringer Ingelheim Vetmedica GmbH |
| Status | Positive |
| Application type | Post-authorisation |
| Medicine | Frontpro (previously known as Afoxolaner Merial) |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2019-05-20 |
European Medicines Agency (EMA)
| Medicine | Nexgard Spectra |
|---|---|
| Active Substance | afoxolaner, milbemycin oxime |
| INN/Common name | afoxolaner, milbemycin oxime |
| Pharmacotherapeutic Classes | Endectocides, Antiparasitic products, insecticides and repellents, milbemycin oxime, combinations |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2015-01-15 |
| Medicine | NexGard |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Isoxazolines, Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2014-02-11 |
European Medicines Agency (EMA)
SYN WO2009126668,

SYN
IP .COM

PATENT
PATENT
https://patents.google.com/patent/WO2009126668A2/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017176948
A particularly active isoxazoline compound, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,24rifluoroethyl)amino]ethyl]-l-naphthalenecarboxamide, is known by the nonproprietary name afoxolaner. Afoxolaner has the following chemical structure:
Afoxolaner
Other isoxazoline compounds that have been found to be highly active against parasitic insects and arachnids are known by the nonproprietary names fluralaner (see US 7,662,972, which is incorporated herein by reference), sarolaner (see US 8,466, 15, incorporated herein by reference) and lotilaner (see, for example US 8,383,659, incorporated herein by reference). The structures of these compounds are shown below:
In addition, published patent application nos. US 2010/0254960 Al, WO 2007/070606
A2, WO 2007/123855 A2, WO 2010/003923 Al, US7951828 & US7662972, US 2010/0137372 Al, US 2010/0179194 A2, US 2011/0086886 A2, US 2011/0059988 Al, US 2010/0179195 Al and WO 2007/075459 A2 and U.S. Patent No. 7,951,828 (all incorporated herein by reference) describe various other parasiticidal isoxazoline compounds.
It is known in the field that isoxazoline compounds having a chiral quaternary carbon atom such as the carbon atom adjacent to the oxygen on the isoxazoline ring of the compounds described above have at least two optical isomer (enantiomers) that are mirror images of each other. Furthermore, it is sometimes the case with biologically active compounds that one of the enantiomers is more active than the other enantiomer. In addition, it is sometimes the case that one enantiomer of a biologically active compound is less toxic than the other enantiomer.
Therefore, with optically active compounds it is desirable to utilize the enantiomer that is most active and less toxic (eutomer). However, isolating the most active enantiomer from a mixture can be costly and result in waste of up to half of the racemic mixture prepared.
Processes to prepare certain isoxazoline compounds enriched in an enantiomer using some cinchona alkaloid-derived phase transfer catalysts have been described. For example, US 2014/0206633 Al, US 2014/0350261 Al, WO 2013/116236 Al and WO 2014/081800 Al (incorporated herein by reference) describe the synthesis of certain isoxazoline active agents enriched in an enantiomer using cinchona alkaloid-based chiral phase transfer catalysts. Further, Matoba et al., Angew. Chem. 2010, 122, 5898-5902 describes the chiral synthesis of certain pesticidal isoxazoline active agents. However, these documents do not describe the processes and certain catalysts described herein.
Scheme 3
Example 7: Preparation of (S)-afoxolaner using chiral phase transfer catalyst (Ilia- 13-1):
(ΠΑ-1) (^-afoxolaner
1) Starting material (IIA-1) (200g, 1.Oeq, 94.0%) and DCM (6 L, 30 volumes) were placed into a 10 L reactor, the solid was dissolved completely.
2) The mixture was cooled to 0°C, and some starting material precipitated out.
3) The catalyst (Ilia- 13-1) (7.56g, 3% mol, 95.0%) was added to the mixture and the resulting mixture cooled further to -10° C.
4) Hydroxylamine (64.9 g, 3.0 eq, 50% solution in water) was added to a solution of NaOH (52.5g, 4. Oeq, in 5v water) in a separate reactor and stirred for 30 minutes.
5) The resulting hydroxylamine/NaOH solution was then added dropwise to the 10 L reactor containing (IIA-1) over about 4 hours.
6) The resulting mixture was stirred for 12 hours at -10°C and monitored for the extent of reaction until the amount of starting material was < 1.0% by HPLC.
7) The mixture was then warmed to 10°C, 1 liter of water was added and the mixture was stirred for 10 minutes.
8) The mixture was allowed to settle to separate the two phases, and the organic layer was collected.
9) The organic layer was then washed with 2 liters of water, the layers were allowed to separate again and the organic layer was collected.
10) The organic layer was washed with 1 liter of brine, the layers allowed to separate and the organic layer was collected and dried over Na2S04 (200 g).
11) The dried organic layer was concentrated under vacuum to about 2 volumes.
12) Toluene (2 L, 10 volumes) was charged to the concentrated mixture and concentration under vacuum was continued to about 5 volumes. Solvent exchange was repeated twice again.
13) The resulting solution was placed into a 2.0 L reactor and heated to 55-60°C.
14) Cyclohexane (300 ml, 1.5 volumes) was added at 55-60°C.
15) The mixture was then cooled to 40 °C over 1.5 hours and then stirred at 40°C for 3 hours.
16) The mixture was then cooled to 25 °C over 2 hours and stirred at 25°C for a further 3 hours.
17) The resulting mixture was cooled to 0-5 °C over 1 hour and stirred at 5 °C for 12 hours, at which time the mixture was filtered to isolate the product.
18) The filter cake was washed with cold toluene/ Cyclohexane (3 : 1, 1000 ml, 5 volumes).
19) The product was obtained as a white solid. (171.5g, chiral purity > 99.0% by area using the chiral HPLC method described in Example 3, chemical purity > 99.0% by area (HPLC), yield: 83.6%, assay purity: 92%). The 1H NMR and LCMS spectra are consistent with the structure of (^-afoxolaner as the toluene solvate. Figure 3 shows the 1H NMR spectra of (S)-afoxolaner in DMSO-d6 and Figure 4 shows the 1H NMR spectra of afoxolaner (racemic) for comparison. The chiral purity of the product was determined using the chiral HPLC method described in Example 3. Figure 5 shows the chiral HPLC chromatogram of afoxolaner (racemic) and Figure 6 shows the chiral HPLC chromatogram of the product (^-afoxolaner showing one enantiomer.
Example 8: Alternate Process to prepare (^-afoxolaner
An alternate process for the preparation of (S)-afoxolaner was conducted. Some of the key variations in the alternate process are noted below.
1. 1 kilogram of compound (IIA-1) (1 eq.) and 9 liters of DCM are charged to a reactor and stirred to dissolve the compound.
2. The mixture is cooled to about 0° C and 50 grams (5 mole %) of the chiral phase transfer catalyst (Ilia- 13-1) and 1 liter of DCM are charged and the resulting mixture is cooled to about -13° C.
3. A solution of 19% (w/w) hydroxylamine sulfate (294 g, 1.1 eq.) (made with 294 grams of ( H2OH)H2S04 and 141 grams of NaCl in 1112 mL of water) and 4.4 equivalents of NaOH as a 17.6% (w/w) solution (286 grams NaOH and 158 grams of NaCl in 1180 mL water) are charged to the reaction mixture simultaneously.
4. The resulting reaction mixture was aged about 20 hours at about -13° C and then checked for reaction conversion by HPLC (target < 0.5% by area);
5. After completion of the reaction, water (3 vol.) was added at about 0° C. Then, a solution of 709 g of KH2P04 in 4.2 liters of water are added to the mixture to adjust the pH (target 7-8) and the resulting mixture is stirred at about 20° C for 30 minutes.
6. The layers are allowed to settle, the aqueous layer is removed and the organic layer is washed with 3 liters of water twice.
Crystallization of Toluene Solvate
1. After the extraction/washing step, the dichloromethane is removed by distillation under vacuum to about 1-2 volumes and toluene (about 5-10 volumes) is added.
2. The volume is adjusted by further distillation under vacuum and/or addition of more toluene to about 5-6 volumes. The mixture is distilled further while maintaining the volume to completely remove the dichloromethane reaction solvent.
3. The mixture is then cooled to about 10° C and seeded with afoxolaner (racemic compound) and stirred at the same temperature for at least 2 hours;
4. The mixture is heated to about 55-65° C, aged for at least 17 hours and then the solid is filtered off. The filtered solid is washed with toluene;
5. The combined filtrate and wash is adjusted to a volume of about 5-6 volumes by
distillation under vacuum and/or toluene addition;
6. The resulting mixture is cooled to about 10° C and aged for at least 5 hours then filtered.
The cake is washed with toluene.
7. The cake is dried at 50° C under vacuum to obtain a toluene solvate of (S)-afoxolaner containing between about 6% and 8% toluene.
Re-crystallization from cyclohexane/ethanol
The toluene solvate of (S)-afoxolaner was subsequently re-crystallized from a mixture of cyclohexane and ethanol to remove the associated toluene and to further purify the product.
1. 591 grams of the (S)-afoxolaner toluene solvate were charged to a vessel along with 709 mL of ethanol (1.2 vol.) and 1773 mL of cyclohexane (3 vol.) and the mixture heated to about 60° C.
2. To the resulting mixture was added an additional 6383 mL of cyclohexane with stirring.
3. The resulting mixture was cooled to about 30° C and then heated again to 60° C. This process was repeated once.
4. The mixture was slowly cooled to 10° C and stirred for at least 5 hours.
5. The resulting slurry was filtered and the cake washed with cyclohexane.
6. The cake was dried at 50° C under vacuum to provide 453.7 grams of (S)-afoxolaner
Example 9: Comparative selectivity of benzyloxy-substituted chiral phase transfer catalyst (Illa-13) with other cinchona alkaloid-based chiral phase transfer catalysts.
The selectivity of the formation of (S)-afoxolaner from compound IIA-1 as shown above was studied with sixteen chiral phase transfer catalysts (PTC) of different structures. The reaction was conducted using conditions similar to those of example 7. The ratio of (^-afoxolaner and (R)-afoxolaner in the reaction mixture was determined by chiral HPLC using the method described in Example 3. The results of the study are provided in Table 2 below.
Table 2
No. Chiral PTC Ratio of (S)- to (R)-afoxolaner
16 50% : 50%
As shown in the table, the catalyst in which the group R in the structure of formula (Ilia) is 3,4,5-tribenzyloxy phenyl results in a surprising improved selectivity for the (S)-enantiomer compared with other quinine-based phase transfer catalysts in which the group corresponding to R in formula (Ilia) is another group.
Example 10: Improvement of Chiral Purity of (<S)-afoxolaner by Crystallization from Toluene
A sample of reaction mixture containing a ratio (HPLC area) of 92.1 :7.9, (^-afoxolaner to (R)-afoxolaner, was concentrated to dryness and the residue was crystallized from toluene and from ethanol/cyclohexane using a process similar to that described in Example 8. The isolated crystalline solid was analyzed by chiral HPLC to determine the relative amounts of (S)-afoxolaner and (R)-afoxolaner (HPLC method: column – Chiralpak AD-3 150 mm x 4.6 mm x 3.0 μηι, injection volume – 10 μΐ., temperature – 35° C, flow – 0.8 mL/minute, mobile phase -89% hexane/10% isopropanol/1% methanol, detection – 312 nm). The ratio of (^-afoxolaner to (R)-afoxolaner in the solid isolated from the toluene crystallization was found to be 99.0 : 1.0 while the ratio of (S)-afoxolaner to (R)-afoxolaner in the solid crystallized from ethanol/cyclohexane was found to be 95.0 : 5.0.
The example shows that the crystallization (^-afoxolaner from an aromatic solvent such as toluene results in a significant improvement of chiral purity of the product. This is very unexpected and surprising.
Example 1 1 : Comparative selectivity of benzyl oxy vs. alkoxy-substituted chiral phase transfer catalyst of Formula (Ilia- 13)
Three chiral phase transfer catalysts of Formula (IIIa-13), wherein the phenyl ring is substituted with three alkoxy groups and three benzyloxy groups (R = methyl, ethyl and benzyl); R’=OMe, W=vinyl and X=chloro were evaluated in the process to prepare of (,S)-IA from compound IIA-1
as shown below.
The amount of solvents and reagents and the reaction and isolation conditions were as described in Example 7 above. The same procedure was used for each catalyst tested. It was found that the selectivity of the tri-benzyloxy catalyst was surprisingly significantly better than the two alkoxy-substituted catalysts, as shown by the chiral purity of the product. Furthermore, it was found that using the tri-benzyloxy substituted phase transfer catalyst the resulting chemical purity was also much better. The superior selectivity of the benzyloxy-substituted catalyst is significant and surprising and cannot be predicted. Chiral phase transfer catalysts containing a phenyl substituted with benzyloxy and alkoxy groups were found to be superior to catalysts substituted with other groups such as electron-withdrawing groups and alkyl groups. The chiral purity and chemical purity of the product produced from the respective phase-transfer catalysts is shown in the Table 3 below:
Table 3
PATENT
WO 2009002809
WO 2009025983
WO 2009126668
WO 2017176948
WO 2018117034
CN 109879826
JP 2020023442
WO 2020158889
WO 2020171129
WO 2021013825
CN 112457267
CN 112679338
PAPER
IP.com Journal (2009), 9(9B), 35.
Afoxolaner (INN)[2] is an insecticide and acaricide that belongs to the isoxazoline chemical compound group.
It acts as an antagonist at ligand-gated chloride channels, in particular those gated by the neurotransmitter gamma-aminobutyric acid (GABA-receptors). Isoxazolines, among the chloride channel modulators, bind to a distinct and unique target site within the insect GABA-gated chloride channels, thereby blocking pre-and post-synaptic transfer of chloride ions across cell membranes. Prolonged afoxolaner-induced hyperexcitation results in uncontrolled activity of the central nervous system and death of insects and acarines.[3]
Marketing
Afoxolaner is the active principle of the veterinary medicinal products NexGard (alone) and Nexgard Spectra (in combination with milbemycin oxime).[4][5][6] They are indicated for the treatment and prevention of flea infestations, and the treatment and control of tick infestations in dogs and puppies (8 weeks of age and older, weighing 4 pounds (~1.8 kilograms) of body weight or greater) for one month.[7] These products are administered orally and poisons fleas once they start feeding.
The marketing authorization was granted by the European Medicines Agency in February 2014, for NexGard and January 2015, for Nexgard Spectra, after only 14[8] and 12[9] months of quality, safety and efficacy assessment performed by the Committee for Medicinal Products for Veterinary Use (CVMP).[10] Therefore, long-term effects are not known.
List of excipients
In NexGard[11] and NexGard Spectra:[3]
- Maize starch
- Soy protein fines
- Beef braised flavouring
- Povidone (E1201)
- Macrogol 400 (reputed laxatives)
- Macrogol 4000 (reputed laxatives)
- Macrogol 15 hydroxystearate (reputed laxatives)
- Glycerol (E422)
- Triglycerides, medium-chain
Additionally in NexGard Spectra:
Safety
Dosage
Afoxolaner is recommended to be administered at a dose of 2.7–7 mg/kg dog’s body weight.[11]
Toxicity for mammals
According to clinical studies performed prior marketing:
- The oral toxicity profile of afoxolaner consists of a diuretic effect (rats only), effects secondary to a reduction in food consumption (rats and rabbits only) and occasional vomiting and/or diarrhoea (dogs, 120 and 200 mg/kg bodyweight (bw)) following high oral doses. No treatment-related effects on vomiting or diarrhoea were noted following oral doses of up to 31.5 mg/kg bw in the pivotal target animal safety study, nor in the EU field trial.[9]
- mild gastrointestinal effects (vomiting, diarrhoea), pruritus, lethargy, anorexia, and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports, most reported adverse reactions being self-limiting and of short duration,[11]
- (in combination with milbemycin oxime): vomiting, diarrhoea, lethargy, anorexia, and pruritus were observed in 0.2 to 1% of 10,000 animals treated and were generally self-limiting and of short duration,[3]
- In vitro studies reported that afoxolaner can bind to dopamine and norepinephrine cellular transport receptor systems and the CB1 receptor; inhibition of these catecholaminergic systems and certain types of competitive binding at CB1 receptors may mediate pharmacodynamic effects of diuresis, decreased food consumption, and decreased body weight in animals.[9]
According to post-marketing safety experience:
- (in combination with milbemycin oxime): erythema and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports,[3]
- The US FDA reports[12] that some drugs in this class (isoxazolines), including afoxalaner, can have adverse neurologic effects on some dogs, such as muscle tremors, ataxia, and seizures.
- Extralabel use of afoxolaner in a pet pig has been described without any adverse effects.[13] Experimental use in commercial pigs also did not result in any adverse effects.[14]
Selectivity in insects over mammalians
In vivo studies (repeat-dose toxicology in laboratory animals, target animal safety, field studies) provided by MERIAL, the company that produces afoxolaner-derivative medicines, did not show evidence of neurological or behavioural effects suggestive of GABA-mediated perturbations in mammals. The Committee for Medicinal Products for Veterinary Use (CVMP) therefore concluded that binding to dog, rat or human GABA receptors is expected to be low for afoxolaner.[9]
Selectivity for insect over mammalian GABA-receptors has been demonstrated for other isoxazolines.[15] The selectivity might be explained by the number of pharmacological differences that exist between GABA-gated chloride channels of insects and vertebrates.[16]
GEN REF
- Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, Lahm GP, Long JK, Xu M, Wagerle T, Jones GS, Dietrich RF, Cordova D, Schroeder ME, Rhoades DF, Benner EA, Confalone PN: Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs. Vet Parasitol. 2014 Apr 2;201(3-4):179-89. doi: 10.1016/j.vetpar.2014.02.020. Epub 2014 Mar 14. [Article]
References
- ^ Jump up to:a b c “Frontline NexGard (afoxolaner) for the Treatment and Prophylaxis of Ectoparasitic Diseases in Dogs. Full Prescribing Information” (PDF) (in Russian). Sanofi Russia. Retrieved 14 November 2016.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 70” (PDF). World Health Organization. pp. 276–7. Retrieved 14 November 2016.
- ^ Jump up to:a b c d “NexGard Spectra product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Agency. Retrieved 13 November 2019.
- ^ Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, et al. (April 2014). “Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs”. Veterinary Parasitology. 201 (3–4): 179–89. doi:10.1016/j.vetpar.2014.02.020. PMID 24631502.
- ^ Beugnet F, deVos C, Liebenberg J, Halos L, Fourie J (25 August 2014). “Afoxolaner against fleas: immediate efficacy and resultant mortality after short exposure on dogs”. Parasite. 21: 42. doi:10.1051/parasite/2014045. PMC 4141545. PMID 25148564.
- ^ Beugnet F, Crafford D, de Vos C, Kok D, Larsen D, Fourie J (August 2016). “Evaluation of the efficacy of monthly oral administration of afoxolaner plus milbemycin oxime (NexGard Spectra, Merial) in the prevention of adult Spirocerca lupi establishment in experimentally infected dogs”. Veterinary Parasitology. 226: 150–61. doi:10.1016/j.vetpar.2016.07.002. PMID 27514901.
- ^ “Boehringer-Ingelheim companion-animals-product NexGard (afoxolaner)”. Boehringer Ingelheim International GmbH. Retrieved 13 November 2019.
- ^ “CVMP Assessment Report for NEXGARD SPECTRA(EMEA/V/C/003842/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c d “CVMP assessment report for NexGard (EMEA/V/C/002729/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ “Committee for Medicinal Products for Veterinary Use (CVMP) – Section “Role of the CVMP””. European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c “NexGard product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Angency. Retrieved 14 November 2019.
- ^ Medicine, Center for Veterinary. “CVM Updates – Animal Drug Safety Communication: FDA Alerts Pet Owners and Veterinarians About Potential for Neurologic Adverse Events Associated with Certain Flea and Tick Products”. http://www.fda.gov. Retrieved 2018-09-22.
- ^ Smith, Joe S.; Berger, Darren J.; Hoff, Sarah E.; Jesudoss Chelladurai, Jeba R. J.; Martin, Katy A.; Brewer, Matthew T. (2020). “Afoxolaner as a Treatment for a Novel Sarcoptes scabiei Infestation in a Juvenile Potbelly Pig”. Frontiers in Veterinary Science. 7: 473. doi:10.3389/fvets.2020.00473. PMC 7505946. PMID 33102538.
- ^ Bernigaud, C.; Fang, F.; Fischer, K.; Lespine, A.; Aho, L. S.; Mullins, A. J.; Tecle, B.; Kelly, A.; Sutra, J. F.; Moreau, F.; Lilin, T.; Beugnet, F.; Botterel, F.; Chosidow, O.; Guillot, J. (2018). “Efficacy and Pharmacokinetics Evaluation of a Single Oral Dose of Afoxolaner against Sarcoptes scabiei in the Porcine Scabies Model for Human Infestation”. Antimicrobial Agents and Chemotherapy. 62 (9). doi:10.1128/AAC.02334-17. PMC 6125498. PMID 29914951.
- ^ Casida JE (April 2015). “Golden age of RyR and GABA-R diamide and isoxazoline insecticides: common genesis, serendipity, surprises, selectivity, and safety”. Chemical Research in Toxicology. 28 (4): 560–6. doi:10.1021/tx500520w. PMID 25688713.
- ^ Hosie AM, Aronstein K, Sattelle DB, ffrench-Constant RH (December 1997). “Molecular biology of insect neuronal GABA receptors”. Trends in Neurosciences. 20 (12): 578–83. doi:10.1016/S0166-2236(97)01127-2. PMID 9416671. S2CID 5028039.
| Clinical data | |
|---|---|
| Pronunciation | /eɪˌfɒksoʊˈlænər/ ay-FOK-soh-LAN-ər |
| Trade names | NexGard, Frontpro |
| Other names | 4-[(5RS)-5-(5-Chloro-α,α,α-trifluoro-m-tolyl)-4,5-dihydro-5-(trifluoromethyl)-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide |
| License data | US DailyMed: Afoxolaner |
| Routes of administration | By mouth (chewables) |
| ATCvet code | QP53BE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyOTC (RU)[1] |
| Pharmacokinetic data | |
| Bioavailability | 74% (Tmax = 2–4 hours)[1] |
| Elimination half-life | 14 hours[1] |
| Excretion | Bile duct (major route) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1093861-60-9 |
| PubChem CID | 25154249 |
| DrugBank | DB11369 |
| ChemSpider | 28651525 |
| UNII | 02L07H6D0U |
| KEGG | D10361 |
| ChEMBL | ChEMBL2219412 |
| CompTox Dashboard (EPA) | DTXSID50148921 |
| Chemical and physical data | |
| Formula | C26H17ClF9N3O3 |
| Molar mass | 625.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI |
///////////// afoxolaner, A1443, AH252723
FC(F)(F)CNC(=O)CNC(=O)C1=C2C=CC=CC2=C(C=C1)C1=NOC(C1)(C1=CC(=CC(Cl)=C1)C(F)(F)F)C(F)(F)F


NEW DRUG APPROVALS
ONE TIME
$10.00
MAX 40279
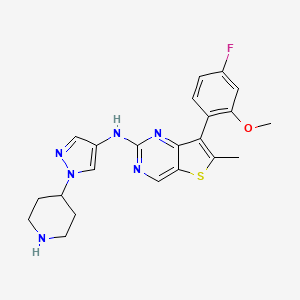
MAX 40279, EX-A4057
Max 4; MAX-40279; MAX-40279-001; MAX-40279-01
UNII-DL772G3NN7
2070931-57-4
C22H23FN6OS, 438.5
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-ylpyrazol-4-yl)thieno[3,2-d]pyrimidin-2-amine
Thieno[3,2-d]pyrimidin-2-amine, 7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-

7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE SEMI-FUMARATE CAS 2388506-43-0
- 7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
- Originator Maxinovel Pharmaceuticals
- ClassAntineoplastics
- Mechanism of ActionFibroblast growth factor receptor antagonists; Fms-like tyrosine kinase 3 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia
- Phase IAcute myeloid leukaemia; Solid tumours
Most Recent Events
- 28 Nov 2019Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) in China (PO) (NCT04183764)
- 16 Apr 2019Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in China (PO) (NCT04187495)
- 23 Jan 2019Guangzhou Maxinovel Pharmaceuticals plans a phase I trial in China (ChiCTR1900020971)
- MaxiNovel Pharmaceuticals, Inc. Announces FDA Orphan Drug Designation for MAX-40279 for the Treatment of Acute Myeloid Leukemia (AML)
March 29, 2018 11:24 AM Eastern Daylight Timehttps://www.businesswire.com/news/home/20180329005826/en/MaxiNovel-Pharmaceuticals-Inc.-Announces-FDA-Orphan-Drug-Designation-for-MAX-40279-for-the-Treatment-of-Acute-Myeloid-Leukemia-AML
GUANGZHOU, China–(BUSINESS WIRE)–MaxiNovel Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (“FDA”) has granted MaxiNovel Orphan Drug Designation for MAX-40279 in the treatment of Acute Myeloid Leukemia (AML).
AML is the most common acute leukemia which accounts for approximately 25% of all adult leukemias worldwide. Approximately one-third of AML patients have a FLT3 gene mutation. Such mutation can result in faster disease progression, higher relapse rates and lower rates of survival than other forms of AML. Inhibition of FLT3 mutation is of high importance in combating AML.
In the preclinical testing, MAX-40279 demonstrated potent inhibition of both FLT3 and FGFR with excellent drug concentration in the bone marrow. It is designed to overcome the observed drug resistance of the current FLT3 inhibitors due to the bone marrow FGF/FGFR pathway activation.
“We are very pleased to receive the ODD,” commented MaxiNovel’s Vice President Dr. Elizabeth Ashraf. “Our objective is to bring the best in class medicine to the patients worldwide.”
The FDA Office of Orphan Products Development grants orphan drug designation to novel drugs and biologics that are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. The designation allows manufacturers to qualify for various incentives including federal grants, tax credits for qualified clinical trials, a waiver of PDUFA filing fees and 7 years of market exclusivity upon regulatory approval.
About MaxiNovel Pharmaceuticals, Inc:
Maxinovel Pharmaceuticals, Inc. is a biotech company focusing on the discovery and development of Immuno-oncology therapy and targeted therapy. It will use its orally active Immuno-oncology product platform to bring effective combo product of multi-components in a single oral pill to the patients worldwide. For more info: www.maxinovel.com
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) signal pathway is a signal transduction pathway stimulated by cytokines discovered in recent years, and it participates in many important biology such as cell proliferation, differentiation, apoptosis and immune regulation. Process (Aaronson, D Set al. Science 2002, 296, 1653-1655; O’Shea, J Jet al. Nat. Rev. Drug Discovery 2004, 3, 555-564). Compared with other signal pathways, the transmission process of this signal pathway is relatively simple. It mainly consists of three components, namely tyrosine kinase-related receptor, tyrosine kinase JAK and transcription factor STAT. JAK (Janus Kinase), a type of molecule in the cell, is rapidly recruited and activated on the receptor after receiving the signal from the upstream receptor molecule. The activated JAK catalyzes the receptor tyrosine phosphorylation, and the phosphorylation of tyrosine on the receptor molecule Amino acid is the recognition and binding site of a kind of signal molecule STAT SH2. Tyrosine phosphorylation occurs after STAT binds to the receptor. Tyrosine phosphorylated STAT forms a dimer and enters the nucleus. As an active transcription factor, dimeric STAT molecules directly affect the expression of related genes, thereby changing the proliferation or differentiation status of target cells.
The JAK-STAT pathway is widely present in various tissues and cells in the body, and has an important role in the differentiation, proliferation, and anti-infection of lymphocytes, and participates in the interaction of various inflammatory factors and signal transduction (Kiesseleva T. et al. . J. Gene, 2002, 285, 1-24). The abnormal activation of this pathway is closely related to a variety of diseases. Finding and screening JAK inhibitors can help in-depth study of the regulatory mechanism of JAK-STAT, thereby providing new drugs and methods for the prevention and treatment of related diseases
The occurrence, growth, invasion and metastasis of tumors are related to the JAK-STAT signal transduction pathway. In normal signal transduction, the activation of STATs is rapid and transient. The continuous activation of STATs is closely related to the process of malignant transformation of cells (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 is the focus of multiple oncogenic tyrosine kinase signal channels such as EGFR, IL-6/JAK, Src, etc. It is activated in a variety of tumor cells and tissues, such as breast cancer, ovarian cancer, and head and neck squamous cells. Like cell carcinoma, prostate cancer, malignant melanoma, multiple myeloma, lymphoma, brain tumor, non-small cell lung cancer and various leukemias, etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008 ). JAK-STAT pathway inhibitors belong to PTK inhibitors, and this enzyme is a member of the oncogene protein and proto-oncoprotein family, and plays an important role in the normal and abnormal cell proliferation. The occurrence and growth of tumors are inseparable from PTK. Therefore, JAK-STAT pathway inhibitors inhibit tumor growth by antagonizing PTK, and have obvious anti-tumor effects (Mora LBet al.J.Cancer Res.2002,62(22) , 6659-6666).
In addition, the latest research shows that: organ transplant rejection, psoriasis, tissue and organ fibrosis, bronchial asthma, ischemic cardiomyopathy, heart failure, myocardial infarction, blood system diseases, and immune system diseases are all related to JAK-STAT signaling. The pathway is closely related. This signaling pathway is not only important for maintaining the normal physiological functions of cells, but also has an important regulatory role for the occurrence and development of diseases.
The Fibroblast Growth Factor Receptor family belongs to a new type of receptor kinase family, which includes four receptor subtypes (FGFR-1,2,3) encoded by four closely related genes. And 4) and some heterogeneous molecules, which form a ternary complex with fibroblast growth factor (FGF) and heparan sulfate, and then trigger a series of signal transduction pathways to participate in the regulation of physiological processes in the organism. FGFR has a wide range of physiological and pathological effects in the body: (1) Embryo development. Studies have shown that during embryonic development, FGFR signal transduction is essential for most organ development and the formation of embryonic patterns. (2) Cell division, migration and differentiation. FGFR stimulates cell proliferation and participates in the regulation of cell transformation in the pathological process. There are many parallel pathways to achieve FGFR-mediated cell division signal transduction, which has been confirmed by many studies (JKWang et al., Oncogene 1997, 14, 1767 -1778.). (3) Bone diseases. The growth and differentiation of bones are also regulated by the FGF family, and mutations in FGFR can cause bone deformities (R. Shang et al., Cell 1994, 78, 335-342.). (4) The occurrence of tumors. FGFR can promote the migration, proliferation and differentiation of endothelial cells, and plays an important role in the regulation of angiogenesis and angiogenesis. Uncontrolled angiogenesis can lead to the occurrence of tumors and the growth of metastases (J.Folkman.Nat.Med.1995) ,1,27-31.).
FMS-like tyrosine kinase 3 (FMS-like tyrosine kinase 3, FLT3) belongs to the type III receptor tyrosine kinase (receptor tyrosine kinase III, RTK III) family member, it is composed of extracellular domain, intracellular domain and The transmembrane region is composed of 3 parts, which are first expressed in human hematopoietic stem cells. FLT3 interacts with its ligand FL to stimulate or act on stem cells, which is of great significance to the growth and differentiation of stem cells. FLT3 kinase has wild-type FLT3-WT and its main activation mutant FLT3-ITD and FLT3-D835Y. FLT3 is mainly expressed in the precursors of normal myeloid cells, but its abnormal expression is also found in a large part of acute myeloid leukemia (AML) cells.
In recent years, many large-scale studies have confirmed that activating mutations of FLT3 play a very important pathological role in the occurrence and progression of acute myeloid leukemia. FLT3 has become an important target for the treatment of acute myeloid leukemia.
rc family kinase (SFK) is a family of non-receptor tyrosine kinases, including c-Src, LYN, FYN, LCK, HCK, FGR, BLK, YES and YRK, among which LYN kinase has LYNα and LYNβ Both subtypes, LYN kinase and its two subtypes can cause similar intracellular tyrosine phosphorylation. According to the amino acid sequence, SFK can be divided into two sub-families: one family is c-Src, FYN, YES and FGR, which are widely expressed in different tissues; the other family is LCK, BLK, LYN and HCK, which are closely related to hematopoietic cells. SFK is connected to multiple signal transduction pathways in the body, and can be activated by growth factors, cytokines and immune cell receptors, G protein-coupled receptors, integrins and other cell adhesion molecules, and then activate the corresponding signal transduction pathways , Causing a variety of physiological effects of cells. The activity of SFK mainly includes the regulation of cell morphology, cell movement, cell proliferation and survival. The abnormal activation and expression of these kinases leads to the occurrence and development of a wide range of diseases, such as a large number of solid tumors, various hematological malignancies and some neuronal pathologies. Therefore, looking for SFK inhibitors is a promising research topic in the field of medicinal chemistry.

NEW DRUG APPROVALS
ONE TIME
$10.00
Patent
CN106366093A
PATENT
WO 2017012559
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559Example 31
N-[7-(4-Fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)- 1H-pyrazole-4-amine (Compound 31)
Synthesis of compound 31-e
2,4-Dichloro-6-methylthiophene [3,2-d] pyrimidine (10g, 45.6mmol) was dissolved in tetrahydrofuran (100mL) and ethanol (100mL), and the reaction solution was cooled to 0°C and divided Sodium borohydride (12.5 g, 198 mmol) was added in batches. The reaction solution was raised to room temperature and continued to stir for 16 hours, diluted with water (500 mL), and then adjusted to pH=7 with 1N aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate (150 mL×3). The organic phase was washed sequentially with water (100mL×3) and saturated brine (100mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-e (7.5g, yield: 88%). The product does not require further purification. LC-MS(ESI): m/z=187[M+H] + .[0492]Synthesis of compound 31-d[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0°C, active manganese dioxide (35 g, 400 mmol) was added, the reaction solution was raised to room temperature and stirring was continued for 16 hours. The reaction solution was filtered through Celite, and the filter cake was washed with chloroform (100 mL×3). The combined filtrates were concentrated under reduced pressure to obtain white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS(ESI): m/z=185[M+H]+.[0494]Synthesis of compound 31-c[0495]Compound 31-d (3.1g, 16.8mmol) was dissolved in trifluoroacetic acid (30mL) at 0℃, N-iodosuccinimide (5.7g, 25.3mmol) was added in batches, and the reaction solution was raised to Keep stirring at room temperature for 1 hour. Water (50 mL) was added to the reaction solution to quench the reaction, and it was extracted with dichloromethane (50 mL×3). The organic phase was washed successively with water (50mL×3) and saturated brine (50mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-c (4.9g, yield: 94%). The product does not require further purification. LC-MS(ESI): m/z=311[M+H] + .[0496]Synthesis of compound 31-b[0497]Compound 31-c (615mg, 1.98mmol), 2-methoxy-4-fluorophenylboronic acid (405mg, 2.38mmol) and sodium carbonate (630mg, 5.94mmol) were suspended in dioxane (5mL) water (5mL) ), add [1,1′-bis(diphenylphosphorus)ferrocene]dichloropalladium dichloromethane complex (163mg, 0.2mmol). Replace with nitrogen 3 times, and heat to 80°C to react for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was partitioned with dichloromethane (50mL) and water (50mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum Ether: dichloromethane=1:1) to obtain a white solid 31-b (240 mg, yield: 39%). LC-MS(ESI): m/z=309[M+H] + .[0498]Synthesis of compound 31-a[0499]Compound 31-b (240mg, 0.78mmol) and compound 32-c (208mg, 0.78mmol) were dissolved in N,N-dimethylformamide (3mL), potassium carbonate (323mg, 2.34mmol) was added, 2- Dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol) and tris(dibenzylideneacetone) dipalladium (134 mg, 0.24 mmol). Under the protection of nitrogen, heat to 110°C to react for 16 hours. After cooling to room temperature, the reaction solution was partitioned with dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin layer chromatography preparation plate (petroleum Ether: ethyl acetate = 1:1) to obtain a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z=539[M+H] + .[0500]Synthesis of compound 31[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), trifluoroacetic acid (3 mL) was added, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. The residue was layered with ethyl acetate (50mL) and 1N aqueous hydrochloric acid (50mL). The aqueous phase was adjusted to pH=10 with saturated aqueous potassium carbonate solution. 3) Washing and vacuum drying the solid to obtain a light yellow solid 31 (22 mg, yield: 14%). LC-MS(ESI): m/z=439[M+H] + .[0502]1 H-NMR (400MHz, MeOD) δ: 8.78 (d, J = 5Hz, 1H), 7.87 (s, 1H), 7.48 (s, 1H), 7.35 (m, 1H), 7.05 (dd, J = 11Hz) ,J = 2Hz, 1H), 6.91 (m, 1H), 4.10 (m, 1H), 3.79 (s, 3H), 3.22 (m, 2H), 2.77 (m, 2H), 2.47 (s, 3H), 2.03(m,2H),1.73(m,2H)ppm
PATENT
WO 2019228171
Example 1 Preparation of fumarate of fused ring pyrimidine compound as shown in formula 2
Weigh the compound N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidine-4- Base)-1H-pyrazol-4-amine (synthesized according to Example 31 of patent CN106366093A) 100mg (0.228mmol, 1eq) into the vial, add 10mL 88% acetone-water solution, add the vial at about 50°C and stir until dissolved clear. 1.1 mL of fumaric acid with a concentration of 0.25 mol/L in ethanol (0.275 mmol, 1.2 eq) was slowly added dropwise to the free base solution of fused ring pyrimidine compounds, and stirred at 50 ℃ for 1 hour, and then the solution was The rate of 5°C/h was slowly reduced to room temperature, and the solid was collected and dried under vacuum at 40°C overnight.
1 H-NMR (400MHz, DMSO-d 6 ) δ: 9.45 (s, 1H), 8.94 (s, 1H), 7.75 (s, 1H), 7.78-7.33 (m, 2H), 7.15 (d, J = 6.4Hz, 1H), 6.99 (dd, J = 7.6 Hz, J = 7.2 Hz, 1H), 6.42 (s, 1H), 4.10 (m, 1H), 3.73 (s, 3H), 3.17 (d, J = 12.4 Hz, 2H), 2.77 (dd, J = 12.4 Hz, J = 11.6 Hz, 2H), 2.40 (s, 3H), 1.94 (d, J = 11.6 Hz, 2H), 1.73 (m, 2H) ppm.
PATENT
7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-yl)-1hydro-pyrazol-4-yl)thieno[3,2 -D]pyrimidine-2-amino is a strong JAK, FGFR, FLT3 kinase inhibitor, and has a good application prospect in the treatment of tumors, immune system diseases, allergic diseases and cardiovascular diseases. This compound is described in patent CN106366093A and has the following chemical structure:
CN106366093A discloses the preparation method of the compound:
In the above synthetic route, NaBH 4 is sodium borohydride, MnO 2 is manganese dioxide, NIS is N-iodosuccinimide, TFA is trifluoroacetic acid, and Pd(dppf)Cl 2 is [1,1′- Bis(diphenylphosphino)ferrocene]palladium dichloride, DIAD is diisopropyl azodicarboxylate, PPh 3 is triphenylphosphine, Pd/C is palladium on carbon, Pd 2 (dba) 3 is Tris(dibenzylideneacetone)dipalladium, RuPhos is 2-bicyclohexylphosphine-2′,6′-diisopropoxybiphenyl.
However, the above method has the problems of a large number of reaction steps, low yield, and requires column chromatography for separation and purification, and is not suitable for industrial scale-up production. Therefore, it is necessary to improve its preparation method.
The present invention provides a method for preparing a compound represented by formula B, which comprises the following steps: under a protective gas atmosphere, in a solvent, in the presence of a catalyst and a base, a compound represented by formula C is combined with a compound represented by formula K The compound can be subjected to the coupling reaction shown below; the catalyst includes a palladium compound and a phosphine ligand;
The preparation method of the compound represented by formula B may further include the following steps: in an organic solvent, in the presence of a base, the compound represented by formula E and the compound represented by formula D are subjected to the substitution reaction shown below, To obtain the compound represented by formula C;
The present invention provides a method for preparing a compound represented by formula C, which comprises the following steps: in an organic solvent, in the presence of a base, a compound represented by formula E and a compound represented by formula D are subjected to the following steps: Substitution reaction is enough;
Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 500L reactor, add 10% palladium on carbon (4.6Kg), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (24.2Kg, 109.5mol), and tetrahydrofuran (150Kg) in sequence And N,N-diisopropylethylamine (17.0Kg, 131.5mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.5 MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 120 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (58Kg) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (60Kg) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 360Kg of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was separated out, centrifuged, and the filter cake was vacuum dried to obtain the product 2-chloro-6-methylthieno[3,2-D]pyrimidine 18.94Kg, yield: 93.2%. LC-MS(ESI): m/z=185.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.30 (s, 1H), 7.34 (s, 1H), 2.73 (s, 3H).
Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
To a 100mL reaction flask, add 10% palladium on carbon (0.17g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), tetrahydrofuran (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 20 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 2.4 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 82%. The LC-MS and 1 H NMR are the same as in Example 1.
Example 3: 7-Bromo 2-chloro-6-methylthieno[3,2-D]pyrimidine (Compound E)
Add trifluoroacetic acid (150Kg) and 2-chloro-6-methylthieno[3,2-D]pyrimidine (18.90Kg, 102.4mol) into a 500L enamel reactor. Add N-bromosuccinimide (18.33Kg, 103.0mol) under temperature control at 15±5℃. After the addition, the temperature is controlled at 25±5℃ to react for 2 hours. Sampling to monitor the reaction, there is still a small amount of raw materials remaining. Additional N-bromosuccinimide (1.0 Kg, 5.6 mol) was added, stirring was continued for 1 hour, sampling and monitoring showed that the reaction was complete. Control the temperature at 10±5°C, and add 274Kg of water dropwise. After the addition, stir at 10±5°C for 2 hours. After centrifugation, the solid was vacuum-dried to obtain the product, 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine, 24.68Kg, yield: 91.4%. LC-MS(ESI): m/z=265.0[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.33 (s, 1H), 2.64 (s, 3H).
Example 4: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Add pyridine (176Kg) and N-BOC-4-hydroxypiperidine (36.00Kg, 178.9mol) to a 500L enamel reactor. Add p-toluenesulfonyl chloride (50.5Kg, 264.9mol) in batches under temperature control at 10±10°C. After the addition, the temperature is controlled at 25±5°C to react for 18 hours. The reaction solution was transferred to a 1000L reactor, the temperature was controlled at 15±5°C, and 710Kg of water was added dropwise. After the addition, stir at 15±5°C for 2 hours. After filtration, the solid was washed with water and dried in vacuum to obtain the product 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester, 59.3Kg, yield: 93.3%. LC-MS(ESI): m/z=378.0[M+Na] + .
Example 5: 4-(4-Nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound F)
Add N,N-dimethylformamide (252Kg), 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester (59.3Kg, 166.8mol), 4-nitro to the reaction kettle Pyrazole (21.5Kg, 190.1mol), and anhydrous potassium carbonate (34.3Kg, 248.2mol). The temperature was controlled at 80±5°C and the reaction was stirred for 18 hours. Cool down to 15±5°C, add 900Kg of water dropwise, control the dropping rate, and keep the temperature at 15±5°C. After the addition, stir at 5±5°C for 2 hours. After filtering, the solid was washed twice with water and dried in vacuum to obtain the product 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 39.92Kg, yield: 80.8%. LC-MS (ESI): m/z=319.1 [M+Na] + .
1 H NMR (400MHz, d 6 -DMSO): δ8.96(s,1H), 8.27(s,1H), 4.44-4.51(m,1H), 4.06-4.08(m,2H), 2.75-2.91( m, 2H), 2.04-2.07 (m, 2H), 1.80-1.89 (m, 2H), 1.41 (s, 9H).
Example 6: 4-(4-Amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound D)
Add 10% palladium-carbon (2.00Kg), 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (39.94Kg, 134.09mol) to the reaction kettle, nothing Water ethanol (314Kg) and ammonia (20.0Kg, 134.09mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.2MPa. Turn on the stirring and keep the temperature at 45±5°C to react for 4 hours. Filter, collect the filtrate, and concentrate the filtrate under reduced pressure. Add ethyl acetate (40Kg) and n-heptane (142Kg) to the concentrate, stir at 25±5°C for 1 hour, and then lower the temperature to 5±5°C and stir for 2 hours. After filtration, the solid was vacuum dried to obtain the product 4-(4-amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 31.85Kg, yield: 88.6%. LC-MS(ESI): m/z=267.2[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ7.06 (s, 1H), 6.91 (s, 1H), 4.08-4.15 (m, 1H), 3.98-4.01 (m, 2H), 3.81 (brs, 2H), 2.83-2.87 (m, 2H), 1.88-1.91 (m, 2H), 1.63-1.72 (m, 2H), 1.41 (s, 9H).
Example 7: 4-(4-(7-Bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl)piperidine-1 -Tert-butyl carbonate (compound C)
Add n-butanol (117Kg), N,N-diisopropylethylamine (15.00Kg, 116.06mol), 4-(4-amino-1hydro-pyrazol-1-yl)piperidine to the reaction kettle 1-tert-butyl carbonate (32.02Kg, 120.22mol) and 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine (24.68Kg, 93.65mol). Turn on the stirring and keep the temperature at 100±5°C to react for 42 hours. Concentrate under reduced pressure. Methanol was added to the concentrate to be beaten. The solid was filtered and dried under vacuum to obtain the product 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl ) Piperidine-1-tert-butyl carbonate 37.26Kg, yield: 80.6%. LC-MS(ESI): m/z=493.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.73 (s, 1H), 8.97 (s, 1H), 8.18 (s, 1H), 7.68 (s, 1H), 4.30-4.36 (m, 1H) ,4.01-4.04(m,2H),2.87-2.93(m,2H),2.53(s,3H),2.00-2.03(m,2H),1.70-1.80(m,2H),1.41(s,9H) .
Example 8: 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1 Hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound B)
Add purified water (113Kg), dioxane (390Kg), 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) into the reactor -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (37.26Kg, 93.65mol), 2-methoxy-4-fluorophenylboronic acid pinacol ester (23.05Kg, 120.22mol) , Anhydrous potassium carbonate (20.95Kg, 151.8mol), palladium acetate (0.18Kg, 0.80mol) and 2-dicyclohexylphosphine-2,4,6-triisopropylbiphenyl (0.90Kg, 1.89mol). Under the protection of nitrogen, the temperature is controlled at 70±5℃ to react for 4 hours. Cool down to 40±5°C, add ammonia water (68Kg), and stir for 8 hours. Cool down to 20±5°C and dilute with water (1110Kg). Dichloromethane extraction twice (244Kg, 170Kg). Combine the organic phases, wash sequentially with water and then with saturated brine. Add 3-mercaptopropyl ethyl sulfide-based silica (4.0Kg, used to remove heavy metal palladium) into the organic phase, and stir at 40±5°C for 20 hours. After filtration, the filtrate was concentrated under reduced pressure. The remainder was slurried sequentially with methyl tert-butyl ether and ethanol. Filter and dry in vacuo to obtain 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 34.6Kg, yield: 68.6%. LC-MS(ESI): m/z=539.3[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.46 (s, 1H), 8.94 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.33 to 7.35 (m, 1H) ,7.08-7.11(m,1H),6.91-6.95(m,1H),4.03-4.12(m,3H),3.73(s,3H),2.85-2.89(m,2H),2.39(s,3H) ,1.90-1.93(m,2H),1.55-1.60(m,2H),1.41(s,9H).
Comparative Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 100mL reaction flask, add 10% palladium on carbon (0.1g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), methanol (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 21 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 1.6 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 54%. Methoxy substituted impurities in 20% yield.
Comparative Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
After replacing the solvent tetrahydrofuran in Example 2 with ethyl acetate, the solubility of 2-chloro-6-methylthieno[3,2-D]pyrimidine in ethyl acetate was poor, and only a small amount of product was formed, which was not calculated Specific yield.
Comparative example 3: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Triethylamine (25mL), N-BOC-4-hydroxypiperidine (5g) were added to a 100mL reaction flask. P-toluenesulfonyl chloride (7.1g) was added in batches while controlling the temperature at 10±10°C. After the addition, the temperature is controlled at 25±5℃ to react for 25 hours. Monitoring by LC-MS showed a large amount of unreacted raw materials and the reaction liquid was black and red.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019228171-A1 | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof | 2018-05-31 | |
| AU-2016295594-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| AU-2016295594-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2020-04-16 |
| EP-3354653-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| EP-3354653-B1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-09-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2018520202-A | Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereof | 2015-07-21 | |
| KR-20180028521-A | Condensed ring pyrimidine-based compounds, intermediates, methods for their preparation, compositions and applications | 2015-07-21 | |
| US-10494378-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-12-03 |
| US-2018208604-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| WO-2017012559-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03412292 | MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML) | Phase 1 | Recruiting | 2021-05-21 |
///////////////Orphan Drug, Acute myeloid leukaemia, MAX 40279, EX-A4057, Max 4, MAX-40279, MAX-40279-001, MAX-40279-01, PHASE 1, Maxinovel Pharmaceuticals
CC1=C(C2=NC(=NC=C2S1)NC3=CN(N=C3)C4CCNCC4)C5=C(C=C(C=C5)F)OC
Sept 2021, This blog New Drug Approvals approaching 34 lakh views
NEW DRUG APPROVALS FAST APPROACING 34 LAKH VIEWS in 225 countries 7 continents. 5 lakh viewers in USA alone
#worlddrugtracker#worldpeaceambassador#india#helpingmillions#amcrasto#medchem#blog#education#service#free#divyang#everydifficultyanopportunity#opensuperstar#pharmadon#qbdking#ict#ictian#pharma

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Saquinavir
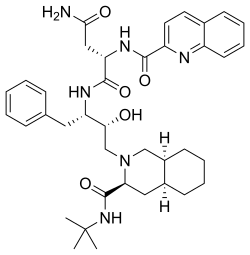
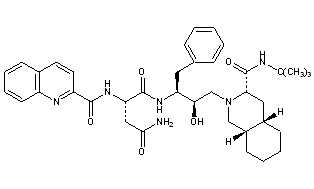
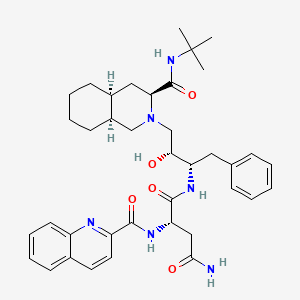
Saquinavir,
Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-decahydroisoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-[(quinolin-2-yl)formamido]butanediamide
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-(quinoline-2-carbonylamino)butanediamide
(-)-cis-N-tert-butyldecahydro-2-{(2R,3S)-2-hydroxy-4-phenyl-3-{[N-(2-quinolylcarbonyl)-L-asparaginyl]amino}butyl}-(3S,4aS,8aS)-isoquinoline-3 carboxamide monomethanesulfonate
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Saquinavir mesylate | UHB9Z3841A | 149845-06-7 | IRHXGOXEBNJUSN-YOXDLBRISA-N |
CAS Registry Number: 127779-20-8
CAS Name: (2S)-N1[(1S,2R)-3-[(3S,4aS,8aS)-3-[[(1,1-Dimethylethyl)amino]carbonyl]octahydro-2(1H)-isoquinolinyl]-2-hydroxy-1-(phenylmethyl)propyl]-2-[(2-quinolinylcarbonyl)amino]butanediamide
Additional Names: (S)-N-[(aS)-a-[(1R)-2-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)octahydro-2(1H)-isoquinolyl]-1-hydroxyethyl]phenethyl]-2-quinaldamido succinamide; N-tert-butyldecahydro-2-[2(R)-hydroxy-4-phenyl-3(S)-[[N-(2-quinolylcarbonyl)-L-asparaginyl]amino]butyl](4aS,8aS)-isoquinoline-3(S)-carboxamide
Manufacturers’ Codes: Ro-31-8959Molecular Formula: C38H50N6O5Molecular Weight: 670.84Percent Composition: C 68.04%, H 7.51%, N 12.53%, O 11.92%
Literature References: Selective HIV protease inhibitor.Prepn: J. A. Martin, S. Redshaw, EP432695; eidem,US5196438 (1991, 1993 both to Hoffmann-LaRoche); K. E. B. Parkes et al.,J. Org. Chem.59, 3656 (1994).In vitro HIV proteinase inhibition: N. A. Roberts et al.,Science248, 358 (1990). Antiviral properties: J. C. Craig et al.,Antiviral Res.16, 295 (1991); S. Galpin et al.,Antiviral Chem. Chemother.5, 43-45 (1994).Clinical evaluation of tolerability and activity: V. S. Kitchen et al.,Lancet345, 952 (1995). Review of pharmacology and clinical experience: S. Kravcik, Expert Opin. Pharmacother.2 303-315 (2001).
Properties: White crystalline solid. [a]D20 -55.9° (c = 0.5 in methanol). Soly (21°): 0.22 g/100 ml water.Optical Rotation: [a]D20 -55.9° (c = 0.5 in methanol)
Derivative Type: Methanesulfonate saltCAS Registry Number: 149845-06-7Additional Names: Saquinavir mesylateManufacturers’ Codes: Ro-31-8959/003Trademarks: Fortovase (Roche); Invirase (Roche)Molecular Formula: C38H50N6O5.CH3SO3HMolecular Weight: 766.95Percent Composition: C 61.08%, H 7.10%, N 10.96%, O 16.69%, S 4.18%
Therap-Cat: Antiviral.Keywords: Antiviral; Peptidomimetics; HIV Protease Inhibitor.
Saquinavir mesylate was first approved by the U.S. Food and Drug Administration (FDA) on Dec 6, 1995, then approved by European Medicine Agency (EMA) on Oct 4, 1996, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Sep 5, 1997. It was developed by Roche, then marketed as Invirase® by Roche in the US and EU and by Chugai in JP.
Saquinavir mesylate is an inhibitor of HIV-1 protease. It is a peptide-like substrate analogue that binds to the protease active site and inhibits the activity of HIV-1 protease that required for the proteolytic cleavage of viral polyprotein precursors into individual functional proteins found in HIV-1 particles. It is indicated for the treatment of HIV-1 infection in combination with ritonavir and other antiretroviral agents in adults (over the age of 16 years).
Invirase® is available as capsule for oral use, containing 200 mg of free Saquinavir. The recommended dose is 1000 mg twice daily in combination with ritonavir 100 mg twice daily for adults.
Human medicines European public assessment report (EPAR): Invirase, saquinavir, HIV Infections, 03/10/1996, 47, Authorised (updated)
EU 08/09/2021
Invirase is an antiviral medicine used to treat adults infected with the human immunodeficiency virus type 1 (HIV 1), a virus that causes acquired immune deficiency syndrome (AIDS). Invirase should only be used in combination with ritonavir (another antiviral medicine) and other antiviral medicines.
Invirase contains the active substance saquinavir.
| Product details | |
|---|---|
| Name | Invirase |
| Agency product number | EMEA/H/C/000113 |
| Active substance | saquinavir |
| International non-proprietary name (INN) or common name | saquinavir |
| Therapeutic area (MeSH) | HIV Infections |
| Anatomical therapeutic chemical (ATC) code | J05AE01 |
| Publication details | |
|---|---|
| Marketing-authorisation holder | Roche Registration GmbH |
| Date of issue of marketing authorisation valid throughout the European Union | 03/10/1996 |
Invirase can only be obtained with a prescription and treatment should be started by a doctor who has experience in the treatment of HIV infection.
Invirase is available as capsules (200 mg) and tablets (500 mg). For patients already taking HIV medicines, the recommended dose of Invirase is 1,000 mg with 100 mg ritonavir twice daily. For patients who are not taking HIV medicines, Invirase is started at 500 mg twice daily with ritonavir 100 mg twice daily for the first 7 days of treatment, given in combination with other HIV medicines. After 7 days, the recommended dose of Invirase is 1,000 mg twice daily with ritonavir 100 mg twice daily in combination with other HIV medicines.
For more information about using Invirase, see the package leaflet or contact a doctor or pharmacist.
The active substance in Invirase, saquinavir, is a ‘protease inhibitor’. It blocks protease, an enzyme involved in the reproduction of HIV. When the enzyme is blocked, the virus does not reproduce normally, slowing down the spread of infection. Ritonavir is another protease inhibitor that is used as a ‘booster’. It slows the breakdown of saquinavir, increasing the levels of saquinavir in the blood. This allows effective treatment while avoiding a higher dose of saquinavir. Invirase, taken in combination with other HIV medicines, reduces the viral load (the amount of HIV in the blood) and keeps it at a low level. Invirase does not cure HIV infection or AIDS, but it may hold off the damage to the immune system and the development of infections and diseases associated with AIDS.
Invirase received a marketing authorisation valid throughout the EU on 4 October 1996.
Drug Name:Saquinavir MesylateResearch Code:Ro-31-8959; Sch-52852Trade Name:Invirase®MOA:HIV-1 protease inhibitorIndication:HIV infectionStatus:ApprovedCompany:Roche (Originator) , ChugaiSales:ATC Code:J05AE01
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2004-12-17 | New dosage form | Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | Priority |
| 1995-12-06 | First approval | Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-10-04 | First approval | Invirase | HIV infection | Capsule | 200 mg | Roche | |
| 1996-10-04 | First approval | Invirase | HIV infection | Tablet, Film coated | 500 mg | Roche |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-09-01 | New dosage form | Invirase | HIV infection | Tablet | 500 mg | Chugai | |
| 1997-09-05 | First approval | Invirase | HIV infection | Capsule | 200 mg | Chugai |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-13 | Marketing approval | 因服雷/Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | |
| 2009-07-01 | Marketing approval | 因服雷/Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche |
Reference:1. US5196438A.Route 2
Reference:1. J. Org. Chem. 1994, 59, 3656-3664.Route 3
Reference:1. WO2006134612A1.
SYN
English: DOI: 10.1021/jo00393a034
DOI: 10.1021/jo00092a026
DOI: 10.1016/S0040-4039(00)77633-7

SYN
In the following, a possible route for the synthesis of Saquinavir is presented. Since Diazomethane is used, the synthesis is not suitable for a scaled up process. Roche has solved this problem with another reaction mechanism. The mechanism for laboratories starts with a ring opening substitution of an epoxid derivative of Phenylalanine with decaisohydroquinoline in dry iso-propanol with nitrogen atmosphere. The intermediate is purified by flash chromatography. In the second step of synthesis, the protection group is removed with gaseous hydrogen and a carbon/palladium catalyst. Furthermore, the new product reacts with N-Benzyloxycarbonyl-Lasparagine(Cbz AsnOH) in the solvents Cbz Asparagine L(Cbz Asn L) and 1- Hydroxybenzotriazolehydrat(HBOT). Afterwards, the protecting group of the former Asparagine is removed with another mixture of gaseous hydrogen and carbon/palladium catalyst. The final intermediate gets stirred in the last step of synthesis together with the solvents Tetrahydrofuran, HBOT and DCC. The mechanism formulated in detail can be found in the Appendix (VIII).7
Kevin E. B. Parkes; David J. Bushnell; et al. Studies toward the Large-Scale Synthesis of the HIV Proteinase Inhibitor Ro 31-8959. J. Org. Chem. 1994, 59, 3656–3664.
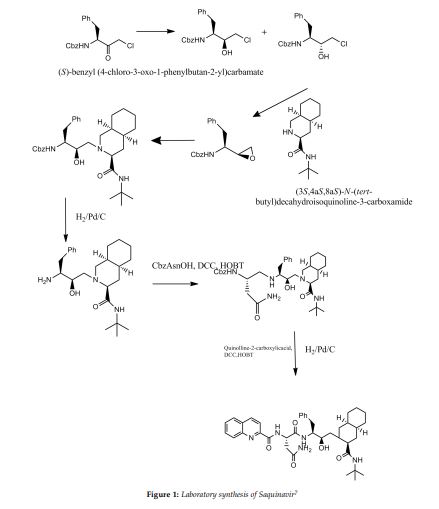
SYN
he synthesis of Ro-31-8959/003 (X) was carried out as follows: Condensation of L-phenylalanine (I) with formaldehyde in concentrated hydrochloric acid gave the tetrahydroisoquinoline (II), which was hydrogenated in 90% acetic acid over rhodium on carbon to yield the decahydroisoquinoline (III) as a mixture of diastereoisomers. Treatment of (III) with benzyl chloroformate in aqueous sodium hydroxide solution gave a mixture of N-protected amino acids which was separated by fractional crystallization of the cyclohexylamine salts to give the (S,S,S)-isomer. Reaction with dicyclohexylcarbodiimide and N-hydroxysuccinimide in dimethoxyethane, followed by treatment of the activated ester with tert-butylamine in dichloromethane and subsequent hydrogenolysis of the benzyloxycarbonyl protecting group gave the decahydroisoquinoline (IV). In the other branch of the synthesis L-phenylalanine was treated with benzyl chloroformate in aqueous sodium hydroxide solution to give the N-protected amino acid. This was converted to the corresponding mixed anhydride with isobutyl chloroformate and N-ethylmorpholine in tetrahydrofuran and immediately reacted with diazomethane in diethyl ether to give the diazomethyl ketone (V). Treatment of (V) with ethereal hydrogen chloride gave the chloromethyl ketone (VI), which on reduction with sodium borohydride in aqueous tetrahydrofuran gave a mixture of diastereoisomeric chlorohydrins. Solvent extraction with boiling n-hexane followed by recrystallization of the less soluble isomer from isopropanol gave pure chlorohydrin (VII), which on treatment with ethanolic potassium hydroxide gave the epoxide (VIII). Condensation of (VIII) with (IV) in ethanol gave the hydroxyethylamine (IX). Hydrogenolysis of (IX) was followed by condensation with N-benzyloxycarbonyl-L-asparagine in tetrahydrofuran in the presence of 1-hydroxybenzotriazole and dicyclohexylcarbodiimide. Hydrogenolysis in ethanol over palladium on charcoal, followed by condensation with quinoline-2-carboxylic acid in tetrahydrofuran in the presence of dicyclohexylcarbodiimide and 1-hydroxybenzotriazole, gave the free base, Ro-31-8959/000. Treatment with methanesulfonic acid in aqueous ethanol then afforded the mesylate salt (X), Ro-31-8959/003.
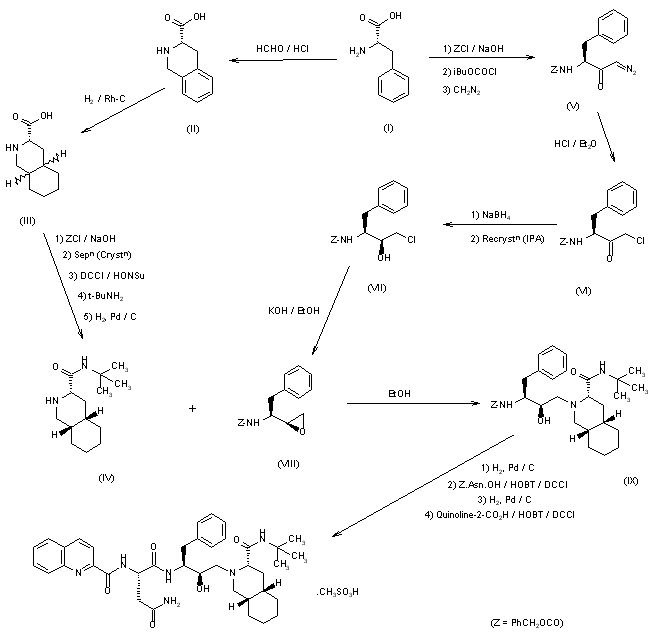
SYN
J Org Chem 1994,59(13),3656
Various new routes for the large-scale synthesis of Ro-31-8959 have been described: 1) The condensation of N-protected-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) gives the keto ester (III), which is enantioselectively reduced with NaBH4 to yield the hydroxy ester (IV). The reaction of (IV) with 2,2-dimethoxypropane (V) by means of p-toluenesulfonic acid affords the oxazolidine (VI), which is hydrolyzed with NaOH in ethanol/water to the corresponding acid (VII). The treatment of (VII) with oxalyl chloride, mercaptopyridine-N-oxide (MPO) and bromotrichloromethane affords the bromomethyloxazolidine (VIII), which, without isolation, is treated with acetic acid to give the N-protected 3(S)-amino-2-bromo-4-phenyl-2(S)-butanol (IX). The reaction of (IX) with KOH in methanol yields the epoxide (X), which is condensed with (3S,4aS,8aS)-N-tert-butyldecahydroisoquinoline-3-carboxamide (XI), yielding the protected condensation product (XII). The deprotection of the amino group of (XII) by hydrogenation with H2 over Pd/C affords the amino derivative (XIII), which is condensed with N-benzyloxycarbonyl-asparagine (XIV) in the usual way, giving the protected peptide (XV). The deprotection of (XV) as before yields compound (XVI), with a free amino group that is finally condensed with quinoline-2-carboxylic acid (XVII) by means of dicyclohexylcarbodiimide and hydroxybenzotriazole.
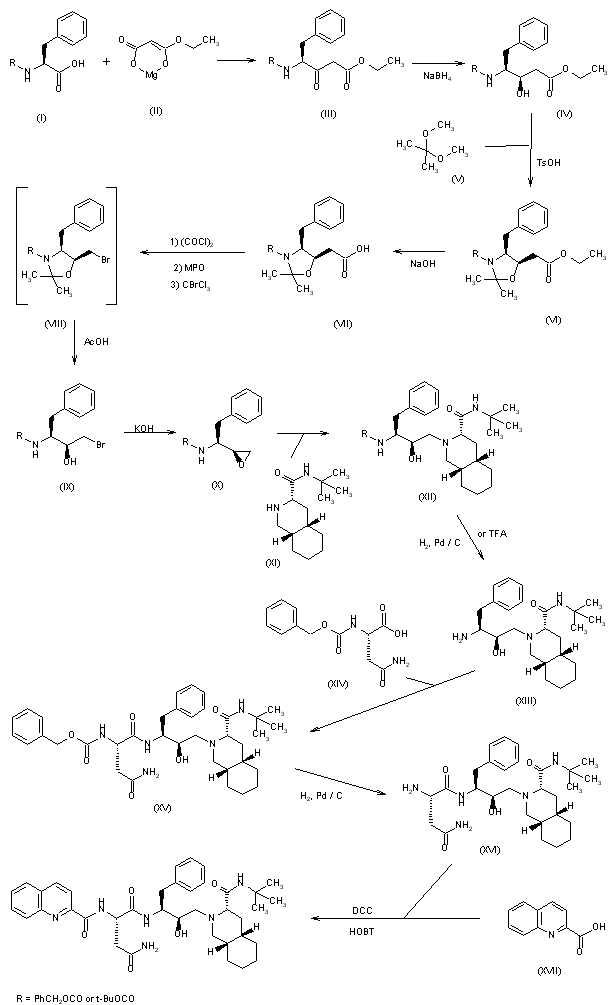
SYN
2) The condensation of N-phthaloyl-L-phenylalaninyl chloride (XVIII) with 1,1,2-tris(trimethylsilyloxy)ethylene (TMS) (XIX) at 90-100 C followed by acidic hydrolysis with HCl gives the acid (XX), which, without isolation, is decarboxylated, yielding 1-hydroxy-3(S)-phthalimido-4-phenyl-2-butanone (XXI). Sequential protection of the OH- group with dihydropyran, reduction of the CO group with NaBH4, mesylation of the resulting OH group with methanesulfonyl chloride and deprotection of the primary OH group gives 2(R)-(methanesulfonyloxy)-4-phenyl-3(S)-phthalimido-1-butanol (XXII). The epoxidation of (XXII) with potassium tert-butoxide yields the epoxide (XXIII), which is condensed with the decahydroisoquinoline (XI) as before, affording the protected condensation product (XXIV). The elimination of the phthalimido group of (XXIV) with methylamine and HCl gives the amino derivative (XIII), already obtained in scheme 16810301a.
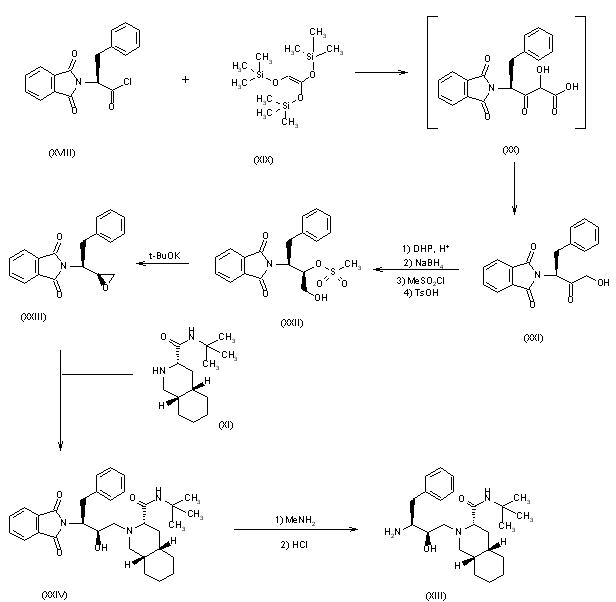
SYN
3) The condensation of N-(tert-butoxycarbonyl)-L-phenylalaninal (XXV) with 2-(trimethylsilyl)thiazole (XXVI) by means of tetrabutylammonium fluoride gives the thiazole derivative (XXVII), which is cleaved by reaction with methyl iodide (formation of the thiazolium derivative) and treated with NaBH4 and HgCl2 to afford the protected 3(S)-amino-2(S)-hydroxy-4-phenylbutanal (XXVIII). Finally, this compound is reductocondensed with isoquinoline (XI) by means of sodium cyanoborohydride to yield the protected condensation product (XII), already obtained in scheme 16810301a.
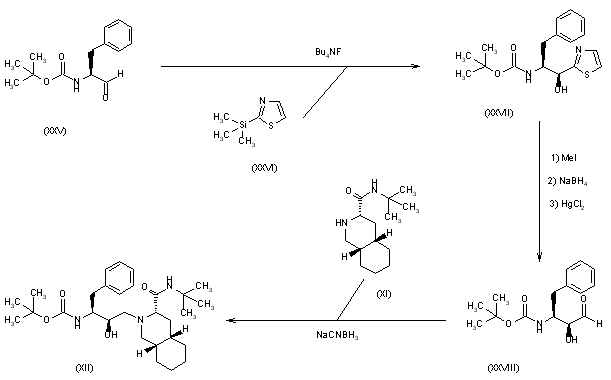
SYN
4) The selective esterification of 3(S)-azido-4-phenylbutane-1,2(S)-diol (XXIX) with 2,4,6-triiosopropylbenzenesulfonyl chloride (XXX) gives the sulfonate ester (XXXI), which by treatment with KOH is converted to the azido epoxide (XXXII). The condensation of (XXXII) with decahydroisoquinoline (XI) affords the azido condensation product (XXXIII), which is finally hydrogenated with H2 over Pd/C to the amino condensation product (XIII), already obtained in scheme 16810301a. 5) The reaction of (XXIX) with SOCl2 and RuCl3 gives the dioxathiole dioxide (XXXIV), which is condensed with decahydroisoquinoline (XI) to afford the azido condensation product (XXXIII), already obtained.
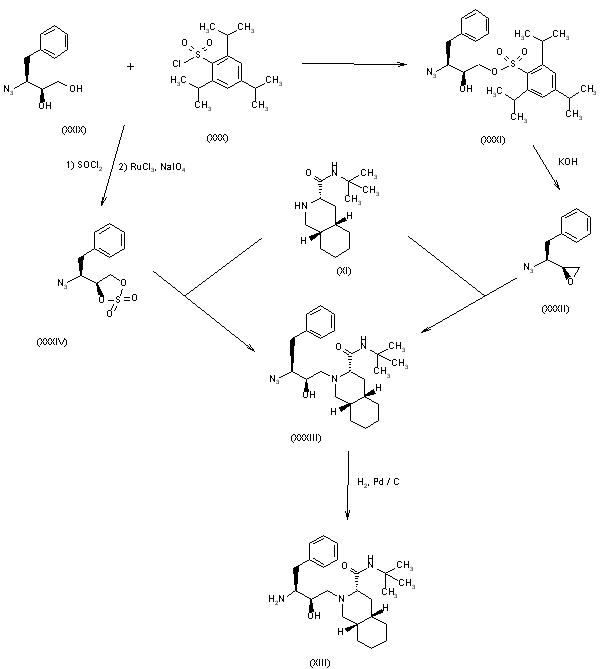
SYN
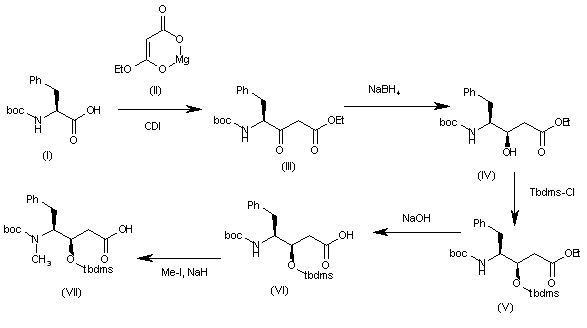
The intermediate (3R,4S)-4-[N-(tert-butoxycarbonyl)-N-methylamino]-5-phenyl-3-(tert-butyldimethylsilyloxy)pentanoic acid (VII) has been obtained as follows: The condensation of N-(tert-butoxycarbonyl)-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) by means of CDI gives the beta-ketoester (III), which is reduced with NaBH4 to yield (3R,4S)-4-(tert-butoxycarbonylamino)-3-hydroxy-5-phenylpentanoic acid ethyl ester (IV). The protection of the OH group of (IV) with Tbdms-Cl and imidazole in DMF affords the silylated ester (V), which is hydrolyzed with NaOH to provide the corresponding carboxylic acid (VI). Finally, this compound is N-methylated by means of Me-I and NaH in THF to obtain the target intermediate (VII).
SYN
J Label Compd Radiopharm 1998,41(12),1103
[14C]-Saquinavir: The cyclization of [ring-14C]-aniline (I) with crotonic aldehyde (II) by means of HCl and acetic anhydride gives labeled 2-methylquinoline (III), which is brominated with Br2 in acetic acid yielding the tribromo derivative (IV). The hydrolysis of (IV) with hot sulfuric acid afforded labeled quinoline-2-carboxylic acid (V), which is finally condensed with Ro-32-0445 (VI) by means of hydroxybenzotriazole (HOBT) and dicyclohexylcarbodiimide (DCC) in THF.
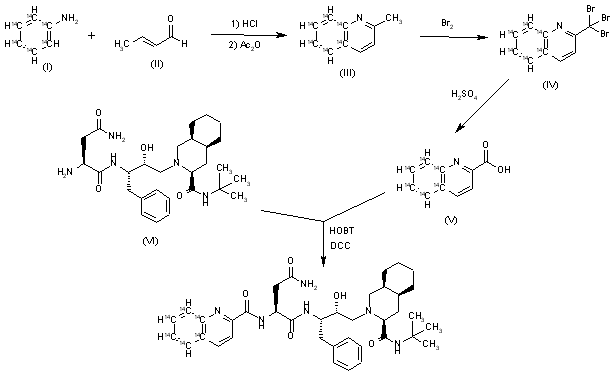
SYN
Pentadeuterated saquinavir: The nitration of hexadeuterobenzene (VII) with HNO3/H2SO4 gives pentadeuteronitrobenzene (VIII), which is hydrogenated with deuterium/Pt in D1-methanol yielding heptadeuteroaniline (IX). The cyclization of (IX) with crotonic aldehyde (II) by means of DCI/D2O and acetic anhydride as before affords hexadeuterated quinoline (X), which is brominated with Br2 as before giving the tribromo derivative (XI). The hydrolysis of (XI) with sulfuric acid as before yields the acid (XII), which is finally condensed with Ro-32-0445 (VI) as before.
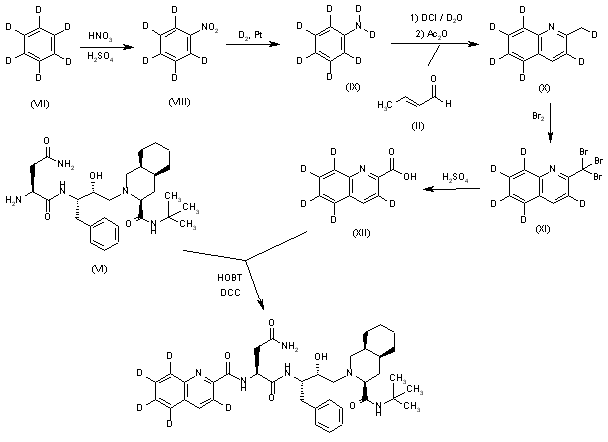
SYN
Tetradeuterated saquinavir: The cyclization of heptadeuteroaniline (IX) with crotonic aldehyde (II) by means of HCl and acetic anhydride as before gives the tetradeuteroquinoline (XIII), which is brominated as described yielding the tribromo derivative (XIV). The hydrolysis of (XIV) with sulfuric acid affords tetradeuterated acid (XV), which is finally condensed with Ro-32-0445 (VI) as indicated.
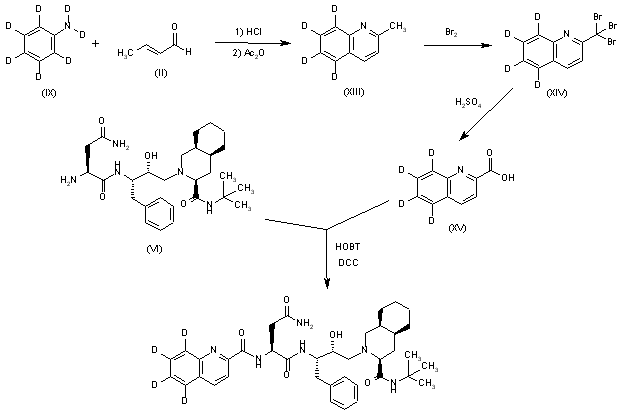
SYN
Tritiated saquinavir: The cyclization of 4-bromoaniline (XVI) with crotonic aldehyde (II) by means of ZnCl2/HCl gives 6-bromo-4-methylquinoline (XVII), which is brominated as before giving tetrabromo derivative (XVIII). The hydrolysis of (XVIII) with sulfuric cid affords 6-bromoquinoline-2-carboxylic acid (XIX), which is condensed with Ro-32-0445 (VI) by means of HOBT and DCC as indicated giving the bromo derivative of saquinavir (XX). Finally, this compound is tritiated with T2 over Pd/C in ethanol.
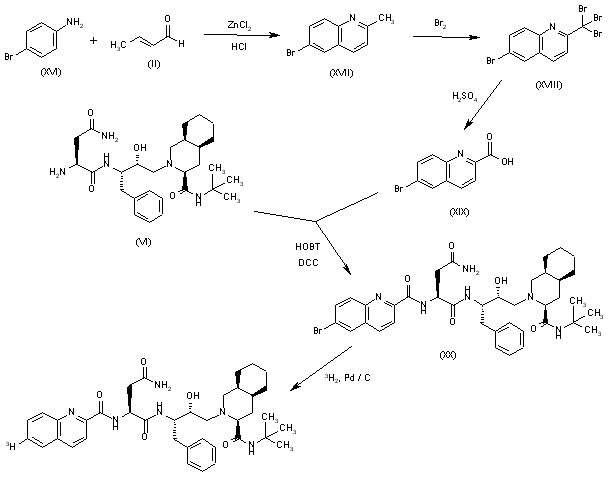
SYN
5)[15N,13C,2H]-Saquinavir: The nitration of [13C6]-benzene (XXI) with [15N]-nitric acid gives the corresponding nitrobenzene (XXII), which is reduced with Sn/HCl to the aniline (XXIII). The cyclization of (XXIII) with crotonic aldehyde (II) by means of ClD/D2O and acetic ahydride yields the tetradeuterated quinoline (XXIV), which is brominated as before givig the tribromo derivative (XXV). The hydrolysis of (XXV) with sulfuric acid as usual affords the [15N,13C6,2H3]-labeled quinoline-2-carboxylic acid (XXVI), which is finally condensed with Ro-32-0445 (VI) by means of HOBT and CDI as indicated.
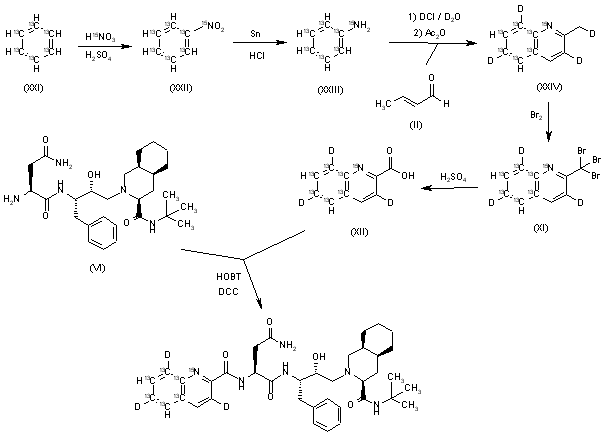
Saquinavir (SQV), sold under the brand names Invirase and Fortovase, is an antiretroviral drug used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3] It is taken by mouth.[3]
Common side effects include nausea, vomiting, diarrhea, and feeling tired.[3] More serious side effects include problems with QT prolongation, heart block, high blood lipids, and liver problems.[3] It appears to be safe in pregnancy.[3] It is in the protease inhibitor class and works by blocking the HIV protease.[3]
Saquinavir was patented in 1988 and first sold in 1995.[4][5]
Medical uses
Saquinavir is used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3]
Side effects
The most frequent adverse events with saquinavir in either formulation are mild gastrointestinal symptoms, including diarrhoea, nausea, loose stools and abdominal discomfort. Invirase is better tolerated than Fortovase.[medical citation needed]
Bioavailability and drug interactions
Saquinavir, in the Invirase formulation, has a low and variable oral bioavailability, when given alone. The Fortovase formulation at the standard dosage delivers approximately eightfold more active drug than Invirase, also at the standard dosage.[6]
In the clinic, it was found that the oral bioavailability of saquinavir in both formulations significantly increases when patients also receive the PI ritonavir. For patients, this has the major benefit that they can take less saquinavir, while maintaining sufficient saquinavir blood plasma levels to efficiently suppress the replication of HIV.[medical citation needed]
The mechanism behind this welcome observation was not directly known, but later it was determined that ritonavir inhibits the cytochrome P450 3A4 isozyme. Normally, this enzyme metabolizes saquinavir to an inactive form, but with the ritonavir inhibiting this enzyme, the saquinavir blood plasma levels increased considerably. Additionally, ritonavir also inhibits multidrug transporters, although to a much lower extent.[medical citation needed]
Unlike other protease inhibitors, the absorption of saquinavir seems to be improved by omeprazole.[7]
Mechanism of action
Saquinavir is a protease inhibitor. Proteases are enzymes that cleave protein molecules into smaller fragments. HIV protease is vital for both viral replication within the cell and release of mature viral particles from an infected cell. Saquinavir binds to the active site of the viral protease and prevents cleavage of viral polyproteins, preventing maturation of the virus. Saquinavir inhibits both HIV-1 and HIV-2 proteases.[8]
History

New HIV infections and deaths, before and after the FDA approval of “highly active antiretroviral therapy”,[9] of which saquinavir, ritonavir and indinavir were key as the first three protease inhibitors.Cully, Megan (28 November 2018). “Protease inhibitors give wings to combination therapy”. nature. Open Publishing. Retrieved 28 October 2020. As a result of the new therapies, HIV deaths in the United States fell dramatically within two years.}}[9]
Saquinavir was developed by the pharmaceutical company Roche.[10] Saquinavir was the sixth antiretroviral and the first protease inhibitor approved by the US Food and Drug Administration (FDA), leading ritonavir and indinavir by a few months.[11] This new class of antiretrovirals played a critical role in the development of highly active antiretroviral therapy (HAART), which helped significantly lower the risk of death from AIDS-related causes, as seen by a reduction of the annual U.S. HIV-associated death rate, from over 50,000 to about 18,000 over a period of two years.[9][12]
Roche requested and received approval of Invirase via the FDA’s “Accelerated Approval” program—a process designed to speed drugs to market for the treatment of serious diseases—a decision that was controversial, as AIDS activists disagreed over the benefits of thorough testing versus early access to new drugs.[13][better source needed] It was approved again on November 7, 1997, as Fortovase,[14] a soft gel capsule reformulated for improved bioavailability. Roche announced in May 2005 that, given reduced demand, Fortovase would cease being marketed early in 2006, in favor of Invirase boosted with ritonavir,[15] owing to the ability of the latter co-formulated drug to inhibit the enzyme that metabolizes the AIDS drugs.[citation needed]
Society and culture
Economics
As of 2015, it is not available as a generic medication.[16]
Formulations
Two formulations have been marketed:
- a hard-gel capsule formulation of the mesylate, with trade name Invirase, which requires combination with ritonavir to increase the saquinavir bioavailability;
- a soft-gel capsule formulation of saquinavir (microemulsion,[17] orally-administered formulation), with trade name Fortovase, which was discontinued worldwide in 2006.[18]
References
- ^ “Saquinavir Use During Pregnancy”. Drugs.com. 20 March 2018. Retrieved 28 January 2020.
- ^ “Invirase- saquinavir mesylate capsule INVIRASE- saquinavir mesylate tablet, film coated”. DailyMed. 26 December 2019. Retrieved 28 January 2020.
- ^ Jump up to:a b c d e f g h i “Saquinavir”. The American Society of Health-System Pharmacists. Archived from the original on 8 September 2015. Retrieved 5 September 2015.
- ^ Minor, Lisa K. (2006). Handbook of Assay Development in Drug Discovery. Hoboken: CRC Press. p. 117. ISBN 9781420015706. Archived from the original on 31 March 2016.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 509. ISBN 9783527607495.
- ^ “Fortovase”. Drugs.com. 22 March 2019. Retrieved 28 January2020.
- ^ Winston A, Back D, Fletcher C, et al. (2006). “Effect of omeprazole on the pharmacokinetics of saquinavir-500 mg formulation with ritonavir in healthy male and female volunteers”. AIDS. 20 (10): 1401–6. doi:10.1097/01.aids.0000233573.41597.8a. PMID 16791014. S2CID 44506039.
- ^ Raphael Dolin, Henry Masur, Michael S. Saag. “AIDS Therapy“, Churchill Livingstone, (1999), p. 129.
- ^ Jump up to:a b c “HIV Surveillance—United States, 1981-2008”. Archivedfrom the original on 9 November 2013. Retrieved 8 November 2013.
- ^ J. Hilts, Philip (8 December 1995). “MF.D.A. Backs A New Drug To Fight AIDS”. New York Times. Retrieved 28 October 2020.
- ^ “Antiretroviral Drug Discovery and Development”. NIH. 26 November 2018. Retrieved 29 October 2020.
- ^ The CDC, in its Morbidity and Mortality Weekly Report, ascribes this to “highly active antiretroviral therapy”, without mention of either of these drugs, see the preceding citation. A further citation is needed to make this accurate connection between this drop and the introduction of the protease inhibitors.
- ^ “Drugs! Drugs! Drugs! An Overview of the Approved Anti-HIV Medications”. The Body. Archived from the original on 9 November 2013. Retrieved 20 February 2013.
- ^ “Drug Approval Package: Fortovase/Saquinavir NDA 20828”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 28 January 2020.
- ^ Withdrawal of Fortovase (PDF) Archived 2006-05-14 at the Wayback Machine
- ^ “Generic Invirase Availability”. Drugs.com. Retrieved 9 July2020.
- ^ Gibaud S, Attivi D (August 2012). “Microemulsions for oral administration and their therapeutic applications” (PDF). Expert Opinion on Drug Delivery. 9 (8): 937–51. doi:10.1517/17425247.2012.694865. PMID 22663249. S2CID 28468973.
- ^ News-Medical.Net. May 18, 2005 Roche to discontinue the sale and distribution of Fortovase (saquinavir) Archived 2015-02-22 at the Wayback Machine
External links
- “Saquinavir”. Drug Information Portal. U.S. National Library of Medicine.
links
| Clinical data | |
|---|---|
| Trade names | Invirase, Fortovase |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696001 |
| License data | EU EMA: by INNUS DailyMed: Saquinavir |
| Pregnancy category | AU: B1[1] |
| ATC code | J05AE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | ~4% (without ritonavir boosting)[2] |
| Protein binding | 98% |
| Metabolism | Liver, mainly by CYP3A4 |
| Elimination half-life | 9–15 hours |
| Excretion | feces (81%) and urine (3%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 127779-20-8 |
| PubChem CID | 441243 |
| IUPHAR/BPS | 4813 |
| DrugBank | DB01232 |
| ChemSpider | 390016 |
| UNII | L3JE09KZ2F |
| KEGG | D00429 |
| ChEMBL | ChEMBL114 |
| NIAID ChemDB | 000640 |
| PDB ligand | ROC (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6044012 |
| Chemical and physical data | |
| Formula | C38H50N6O5 |
| Molar mass | 670.855 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
///////////////saquinavir, Antiviral, Peptidomimetics, HIV Protease Inhibitor, Ro-31-8959, EU 2021, APPROVALS 2021, Invirase, Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
[H][C@@]12CCCC[C@]1([H])CN(C[C@@H](O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CC(N)=O)NC(=O)C1=NC3=C(C=CC=C3)C=C1)[C@@H](C2)C(=O)NC(C)(C)C

NEW DRUG APPROVALS
ONE TIME
$10.00
Benzonatate


Benzonatate
- Molecular FormulaC30H53NO11
- Average mass603.742 Da
104-31-4[RN]2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-yl 4-(butylamino)benzoateбензонататبنزوناتات苯佐那酯ベンゾナテート;KM 652,5,8,11,14,17,20,23,26-nonaoxaoctacosan-28-yl 4-(butylamino)benzoate2-[2-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl 4-(butylamino)benzoate
Benzonatate bulk and Benzonatate capsules 100mg, cdsco india 2021, 15.07.2021
For the treatment of refractory coughCAS Registry Number: 104-31-4CAS Name: 4-(Butylamino)benzoic acid 3,6,9,12,15,18,21,24,27-nonaoxaoctacos-1-yl esterAdditional Names: nonaethyleneglycol monomethyl ether p-n-butylaminobenzoate; p-butylaminobenzoic acid w-O-methylnonaethyleneglycol ester; benzononatineTrademarks: Exangit; Tessalon (Forest)Molecular Formula: C30H53NO11Molecular Weight: 603.74Percent Composition: C 59.68%, H 8.85%, N 2.32%, O 29.15%Literature References: Prepn: Matter, US2714608 (1955 to Ciba).Properties: Colorless to faintly yellow oil. Soluble in most organic solvents except aliphatic hydrocarbons.Therap-Cat: Antitussive.Keywords: Antitussive.
Synthesis Reference
Matter, M.; U.S. Patent 2,714,608; August 2, 1955; assigned to Ciba Pharmaceutical Products, Inc.
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 94-32-6 | C13H19NO2 | ethyl 4-butylaminobenzoate | Benzoic acid, 4-(butylamino)-, ethyl ester |
| 6048-68-6 | C19H40O10 | nonaethylene glycol monomethyl ether | 2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-ol |
Benzonatate, sold under the brand name Tessalon among others, is a medication used to try to help with the symptoms of cough and hiccups.[1][2] It is taken by mouth.[1] Use is not recommended in those under the age of ten.[3] Effects generally begin within 20 minutes and last up to eight hours.[1][4]
Side effects include sleepiness, dizziness, headache, upset stomach, skin rash, hallucinations, and allergic reactions.[1] Excessive doses may cause seizures, irregular heartbeat, and death.[3] Chewing or sucking on the capsule can lead to laryngospasm, bronchospasm, and circulatory collapse.[1] It is unclear if use in pregnancy or breastfeeding is safe.[5] It works by numbing stretch receptors in the lungs and suppressing the cough reflex in the brain.[1]
Benzonatate was approved for medical use in the United States in 1958.[1] It is available as a generic medication.[3] It is not available in many countries.[6] In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Medical uses

100mg generic benzonatate capsules
Cough
Benzonatate is a prescription non-opioid alternative for the symptomatic relief of cough.[1][3] It has been shown to improve cough associated with a variety of respiratory conditions including asthma, bronchitis, pneumonia, tuberculosis, pneumothorax, opiate-resistant cough in lung cancer, and emphysema.[1][9][10]
Benzonatate also reduces the consistency and volume of sputum production associated with cough in those with chronic obstructive pulmonary disorder (COPD).[9]
Compared to codeine, benzonatate has been shown to be more effective in reducing the frequency of induced cough in experiments.[1]
Benzonatate does not treat the underlying cause of the cough.[11]
Hiccups
Benzonatate has been shown to have use in the suppression of hiccups.[2]
Intubation
Benzonatate acts as a local anesthetic and the liquid inside the capsule can be applied in the mouth to numb the oropharynx for awake intubation.[1] However, there can be life-threatening adverse effects when the medication is absorbed by the oral mucosa, including choking, hypersensitivity reactions, and circulatory collapse.[1]
Contraindications
Hypersensitivity to benzonatate or any related compounds is a contraindication to its administration.[4]
Side effects
Benzonatate is generally well-tolerated[vague][specify] if the liquid-capsule is swallowed intact.[1] Potential adverse effects to benzonatate include:
- Constipation, dizziness, fatigue, stuffy nose, nausea, headache are frequently reported.[12]
- Sedation, a feeling of numbness in the chest, sensation of burning in the eyes, a vague “chilly” sensation, itchiness, and rashes are also possible.[1][4]
- Ingestion of a small handful of capsules has caused seizures, cardiac arrhythmia, and death in adults.[13]
Hypersensitivity reactions
Benzonatate is structurally related to anesthetic medications of the para-aminobenzoic acid (PABA) class which includes procaine and tetracaine.[4][13] Procaine and tetracaine, previously used heavily in the fields of dentistry and anesthesiology, have fallen out of favor due to allergies associated with their metabolites.[13] Similarly, severe hypersensitivity reactions to benzonatate have been reported and include symptoms of laryngospasm, bronchospasm, and cardiovascular collapse.[4][14] These reactions are possibly associated with chewing, sucking, or crushing the capsule in the mouth.[4][13]
Improper use
Benzonatate should be swallowed whole.[4] Crushing or sucking on the liquid-filled capsule, or “softgel,” will cause release of benzonatate from the capsule and can produce a temporary local anesthesia of the oral mucosa.[4] Rapid development of numbness of the tongue and choking can occur.[4][13] In severe cases, excessive absorption can lead to laryngospasm, bronchospasm, seizures, and circulatory collapse.[4][13] This may be due to a hypersensitivity reaction to benzonatate or a systemic local anesthetic toxicity, both of which have similar symptoms.[13] There is a potential for these adverse effects to occur at a therapeutic dose, that is, a single capsule, if chewed or sucked on in the mouth.[13]
Psychiatric effects
Isolated cases of bizarre behavior, mental confusion, and visual hallucinations have been reported during concurrent use with other prescribed medications.[4] Central nervous system effects associated with other para-aminobenozic acid (PABA) derivative local anesthetics, for example procaine or tetracaine, could occur with benzonatate and should be considered.[1]
Children
Safety and efficacy in children below the age of 10 have not been established.[4] Accidental ingestion resulting in death has been reported in children below the age of 10.[4] Benzonatate may be attractive to children due to its appearance, a round-shaped liquid-filled gelatin capsule, which looks like candy.[14][15] Chewing or sucking of a single capsule can cause death of a small child.[4][15] Signs and symptoms can occur rapidly after ingestion (within 15–20 minutes) and include restlessness, tremors, convulsions, coma, and cardiac arrest.[15] Death has been reported within one hour of ingestion.[12][15]
Pregnancy and breast feeding
In the U.S., benzonatate is classified by the U.S. Food and Drug Administration (FDA) as pregnancy category C.[5] It is not known if benzonatate can cause fetal harm to a pregnant woman or if it can affect reproduction capacity.[4][5] Animal reproductive studies have not yet been conducted with benzonatate to evaluate its teratogenicity.[4] Benzonatate should only be given to a pregnant woman if it is clearly needed.[4][5]
It is not known whether benzonatate is excreted in human milk.[4][5] It is recommended to exercise caution when benzonatate is given to a nursing woman.[4][5]
Overdose
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology and toxicology.[13]
Benzonatate overdose is characterized by symptoms of restlessness, tremors, seizures, abnormal heart rhythms (cardiac arrhythmia), cerebral edema, absent breathing (apnea), fast heart beat (tachycardia), and in severe cases, coma and death.[1][4][16][11] Symptoms develop rapidly, typically within 1 hour of ingestion.[4][11] Treatment focuses on removal of gastric contents and on managing symptoms of sedation, convulsions, apnea, and cardiac arrhythmia.[4]
Despite a long history of safe and appropriate usage, the safety margin of benzonatate is reportedly narrow.[13] Toxicity above the therapeutic dose is relatively low and ingestion of a small handful of pills can cause symptoms of overdose.[13][11] Children are at an increased risk for toxicity, which have occurred with administration of only one or two capsules.[15][16][11]
Due to increasing usage of benzonatate and rapid onset of symptoms, there are accumulating cases of benzonatate overdose deaths, especially in children.[11]
Pharmacology
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology.[13]
Mechanism of action
Similar to other local anesthetics, benzonatate is a potent voltage-gated sodium channel inhibitor.[13] After absorption and circulation to the respiratory tract, benzonatate acts as a local anesthetic, decreasing the sensitivity of vagal afferent fibers and stretch receptors in the bronchi, alveoli, and pleura in the lower airway and lung.[1][2] This dampens their activity and reduces the cough reflex.[1][4] Benzonatate also has central antitussive activity on the cough center in central nervous system at the level of the medulla.[1][9] However, there is minimal inhibition of the respiratory center at a therapeutic dosage.[4]
Pharmacokinetics
The antitussive effect of benzonatate begins within 15 to 20 minutes after oral administration and typically lasts between 3 and 8 hours.[4][9]
Benzonatate is hydrolyzed by plasma butyrylcholinesterase (BChE) to the metabolite 4-(butylamino)benzoic acid (BABA) as well as polyethylene glycol monomethyl esters.[13] Like many other local anesthetic esters, the hydrolysis of the parent compound is rapid.[13] There are concerns that those with pseudocholinesterase deficiencies may have an increased sensitivity to benzonatate as this hydrolysis is impaired, leading to increased levels of circulating medication.[13]
Chemical structure
Benzonatate is a butylamine, structurally related to other polyglycol ester local anesthetics such as procaine and tetracaine.[13] The molecular weight of benzonatate is 603.7 g/mol.[4] However, the reference standard for benzonatate is a mixture of n-ethoxy compounds, differing in the abundance of 7-9 repeating units, with an average molecular weight of 612.23 g/mol.[13] There is also evidence that the compound is not uniform between manufacturers.[13]
Society and culture
Benzonatate was first made available in the U.S. in 1958 as a prescription medication for the treatment of cough in individuals over the age of 10.[15][16] There are a variety of prescription opioid-based cough relievers, such as hydrocodone and codeine, but have unwanted side effects and potential of abuse and diversion.[13] However, benzonatate is currently the only prescription non-opioid antitussive and its usage has been rapidly increasing.[13][11] The exact reasons of this increase are unclear.[11]
Economics
In the United States between 2004 and 2009, prescriptions increased 50% from 3.1 million to 4.7 million, the market share of benzonatate among antitussives increased from 6.3% to 13%, and the estimated number of children under the age of 10 years receiving benzonatate increased from 10,000 to 19,000.[13][11] Throughout this same period, greater than 90% of prescriptions were given to those 18 or older.[11] The majority of prescriptions were given by general, family, internal, and osteopathic physicians with pediatricians account for about 3% of prescribed benzonatate.[11]
In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Brand names
Tessalon is a brand name version of benzonatate manufactured by Pfizer, Inc.[13][11] It is available as perles or capsules.[17] Zonatuss was a brand name manufactured by Atley Pharmaceuticals, Inc. and Vertical Pharmaceuticals, Inc.[18][19]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s “Benzonatate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 23 March 2019.
- ^ Jump up to:a b c Becker, DE (2010). “Nausea, vomiting, and hiccups: a review of mechanisms and treatment”. Anesthesia Progress. 57 (4): 150–6, quiz 157. doi:10.2344/0003-3006-57.4.150. PMC 3006663. PMID 21174569.
- ^ Jump up to:a b c d “Drugs for cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z “Tessalon – benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Benzonatate Use During Pregnancy”. Drugs.com. 10 October 2019. Retrieved 20 February 2020.
- ^ Walsh, T. Declan; Caraceni, Augusto T.; Fainsinger, Robin; Foley, Kathleen M.; Glare, Paul; Goh, Cynthia; Lloyd-Williams, Mari; Olarte, Juan Nunez; Radbruch, Lukas (2008). Palliative Medicine E-Book. Elsevier Health Sciences. p. 751. ISBN 9781437721942.
- ^ Jump up to:a b “The Top 300 of 2021”. ClinCalc. Retrieved 18 February2021.
- ^ Jump up to:a b “Benzonatate – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b c d Homsi, J.; Walsh, D.; Nelson, K. A. (November 2001). “Important drugs for cough in advanced cancer”. Supportive Care in Cancer. 9 (8): 565–574. doi:10.1007/s005200100252. ISSN 0941-4355. PMID 11762966. S2CID 25881426.
- ^ Estfan, Bassam; LeGrand, Susan (November 2004). “Management of cough in advanced cancer”. The Journal of Supportive Oncology. 2 (6): 523–527. ISSN 1544-6794. PMID 16302303.
- ^ Jump up to:a b c d e f g h i j k l McLawhorn, Melinda W.; Goulding, Margie R.; Gill, Rajdeep K.; Michele, Theresa M. (January 2013). “Analysis of benzonatate overdoses among adults and children from 1969-2010 by the United States Food and Drug Administration”. Pharmacotherapy. 33 (1): 38–43. doi:10.1002/phar.1153. ISSN 1875-9114. PMID 23307543. S2CID 35165660.
- ^ Jump up to:a b “Benzonatate (Professional Patient Advice)”. Drugs.com. 4 March 2020. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w Bishop-Freeman SC, Shonsey EM, Friederich LW, Beuhler MC, Winecker RE (June 2017). “Benzonatate Toxicity: Nothing to Cough At”. J Anal Toxicol. 41 (5): 461–463. doi:10.1093/jat/bkx021. PMID 28334901.
- ^ Jump up to:a b “Drugs for Cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f “FDA Drug Safety Communication: Death resulting from overdose after accidental ingestion of Tessalon (benzonatate) by children under 10 years of age”. U.S. Food and Drug Administration (FDA). 28 June 2019. Retrieved 22 April 2020.
- ^ Jump up to:a b c “In brief: benzonatate warning”. The Medical Letter on Drugs and Therapeutics. 53 (1357): 9. 7 February 2011. ISSN 1523-2859. PMID 21304443.
- ^ “Tessalon- benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 25 April 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 2 June 2010. Retrieved 20 August 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 31 October 2016. Retrieved 20 August 2020.
External links
- “Benzonatate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Tessalon, Zonatuss, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682640 |
| License data | US DailyMed: Benzonatate |
| Routes of administration | By mouth |
| ATC code | R05DB01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Elimination half-life | 3-8 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 32760-16-0 |
| PubChem CID | 7699 |
| IUPHAR/BPS | 7611 |
| DrugBank | DB00868 |
| ChemSpider | 7413 |
| UNII | 5P4DHS6ENR |
| KEGG | D00242 |
| ChEBI | CHEBI:3032 |
| ChEMBL | ChEMBL1374379 |
| CompTox Dashboard (EPA) | DTXSID9022655 |
| ECHA InfoCard | 100.002.904 |
| Chemical and physical data | |
| Formula | C30H53NO11 |
| Molar mass | 603.750 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////Benzonatate, refractory cough , INDIA 2021, APPROVALS 2021, бензонатат , بنزوناتات , 苯佐那酯 , KM 65 , ベンゾナテート, ANTITUSSIVE, IND 2021
CCCCNC1=CC=C(C=C1)C(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOC

NEW DRUG APPROVALS
ONE TIME
$10.00
Plasminogen
Plasminogen
FDA APPROVED 2021, Ryplazim, 2021/6/4
Plasminogen;
Glu-plasminogen;
Plasminogen, human-tvmh;
Ryplazim (TN)
RYPLAZIM (plasminogen, human-tvmh)
Enzyme replacement (plasminogen), Plasminogen deficiency type 1
CAS: 9001-91-6
STN: 125659
Proper Name: plasminogen, human-tvmh
Tradename: RYPLAZIM
Manufacturer: Prometic Biotherapeutics Inc.
Indication:
For the treatment of patients with plasminogen deficiency type 1 (hypoplasminogenemia)
READ https://diapharma.com/plasminogen-plg/
On August 11, 2017 Prometic Biotherapeutics submitted a BLA (STN 125659) for a Drug Product (DP) RYPLAZIM, Plasminogen (Human). This drug product is indicated for replacement therapy in children and adults with plasminogen deficiency.
Plasmin is an important enzyme (EC 3.4.21.7) present in blood that degrades many blood plasma proteins, including fibrin clots. The degradation of fibrin is termed fibrinolysis. In humans, the plasmin protein is encoded by the PLG gene.[5]
Function
Fibrinolysis (simplified). Blue arrows denote stimulation, and red arrows inhibition.
Plasmin is a serine protease that acts to dissolve fibrin blood clots. Apart from fibrinolysis, plasmin proteolyses proteins in various other systems: It activates collagenases, some mediators of the complement system, and weakens the wall of the Graafian follicle, leading to ovulation. Plasmin is also integrally involved in inflammation.[6] It cleaves fibrin, fibronectin, thrombospondin, laminin, and von Willebrand factor. Plasmin, like trypsin, belongs to the family of serine proteases.
Plasmin is released as a zymogen called plasminogen (PLG) from the liver into the systemic circulation. Two major glycoforms of plasminogen are present in humans – type I plasminogen contains two glycosylation moieties (N-linked to N289 and O-linked to T346), whereas type II plasminogen contains only a single O-linked sugar (O-linked to T346). Type II plasminogen is preferentially recruited to the cell surface over the type I glycoform. Conversely, type I plasminogen appears more readily recruited to blood clots.
In circulation, plasminogen adopts a closed, activation-resistant conformation. Upon binding to clots, or to the cell surface, plasminogen adopts an open form that can be converted into active plasmin by a variety of enzymes, including tissue plasminogen activator (tPA), urokinase plasminogen activator (uPA), kallikrein, and factor XII (Hageman factor). Fibrin is a cofactor for plasminogen activation by tissue plasminogen activator. Urokinase plasminogen activator receptor (uPAR) is a cofactor for plasminogen activation by urokinase plasminogen activator. The conversion of plasminogen to plasmin involves the cleavage of the peptide bond between Arg-561 and Val-562.[5][7][8][9]
Plasmin cleavage produces angiostatin.
Mechanism of plasminogen activation
Full length plasminogen comprises seven domains. In addition to a C-terminal chymotrypsin-like serine protease domain, plasminogen contains an N-terminal Pan Apple domain (PAp) together with five Kringle domains (KR1-5). The Pan-Apple domain contains important determinants for maintaining plasminogen in the closed form, and the kringle domains are responsible for binding to lysine residues present in receptors and substrates.
The X-ray crystal structure of closed plasminogen reveals that the PAp and SP domains maintain the closed conformation through interactions made throughout the kringle array .[9] Chloride ions further bridge the PAp / KR4 and SP / KR2 interfaces, explaining the physiological role of serum chloride in stabilizing the closed conformer. The structural studies also reveal that differences in glycosylation alter the position of KR3. These data help explain the functional differences between the type I and type II plasminogen glycoforms.[citation needed]
In closed plasminogen, access to the activation bond (R561/V562) targeted for cleavage by tPA and uPA is blocked through the position of the KR3/KR4 linker sequence and the O-linked sugar on T346. The position of KR3 may also hinder access to the activation loop. The Inter-domain interactions also block all kringle ligand-binding sites apart from that of KR-1, suggesting that the latter domain governs pro-enzyme recruitment to targets. Analysis of an intermediate plasminogen structure suggests that plasminogen conformational change to the open form is initiated through KR-5 transiently peeling away from the PAp domain. These movements expose the KR5 lysine-binding site to potential binding partners, and suggest a requirement for spatially distinct lysine residues in eliciting plasminogen recruitment and conformational change respectively.[9]
Mechanism of plasmin inactivation
Plasmin is inactivated by proteins such as α2-macroglobulin and α2-antiplasmin.[10] The mechanism of plasmin inactivation involves the cleavage of an α2-macroglobulin at the bait region (a segment of the aM that is particularly susceptible to proteolytic cleavage) by plasmin. This initiates a conformational change such that the α2-macroglobulin collapses about the plasmin. In the resulting α2-macroglobulin-plasmin complex, the active site of plasmin is sterically shielded, thus substantially decreasing the plasmin’s access to protein substrates. Two additional events occur as a consequence of bait region cleavage, namely (i) a h-cysteinyl-g-glutamyl thiol ester of the α2-macroglobulin becomes highly reactive and (ii) a major conformational change exposes a conserved COOH-terminal receptor binding domain. The exposure of this receptor binding domain allows the α2-macroglobulin protease complex to bind to clearance receptors and be removed from circulation.
Pathology
Plasmin deficiency may lead to thrombosis, as the clots are not adequately degraded. Plasminogen deficiency in mice leads to defective liver repair,[11] defective wound healing, reproductive abnormalities.[citation needed]
In humans, a rare disorder called plasminogen deficiency type I (Online Mendelian Inheritance in Man (OMIM): 217090) is caused by mutations of the PLG gene and is often manifested by ligneous conjunctivitis.
Interactions
Plasmin has been shown to interact with Thrombospondin 1,[12][13] Alpha 2-antiplasmin[14][15] and IGFBP3.[16] Moreover, plasmin induces the generation of bradykinin in mice and humans through high-molecular-weight kininogen cleavage.[17]
References
- ^ Jump up to:a b c GRCh38: Ensembl release 89: ENSG00000122194 – Ensembl, May 2017
- ^ Jump up to:a b c GRCm38: Ensembl release 89: ENSMUSG00000059481 – Ensembl, May 2017
- ^ “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Jump up to:a b “Entrez Gene: plasminogen”.
- ^ Atsev S, Tomov N (December 2020). “Using antifibrinolytics to tackle neuroinflammation”. Neural Regeneration Research. 15(12): 2203–2206. doi:10.4103/1673-5374.284979. PMC 7749481. PMID 32594031.
- ^ Miyata T, Iwanaga S, Sakata Y, Aoki N (October 1982). “Plasminogen Tochigi: inactive plasmin resulting from replacement of alanine-600 by threonine in the active site”. Proc. Natl. Acad. Sci. U.S.A. 79 (20): 6132–6. Bibcode:1982PNAS…79.6132M. doi:10.1073/pnas.79.20.6132. PMC 347073. PMID 6216475.
- ^ Forsgren M, Råden B, Israelsson M, Larsson K, Hedén LO (March 1987). “Molecular cloning and characterization of a full-length cDNA clone for human plasminogen”. FEBS Lett. 213 (2): 254–60. doi:10.1016/0014-5793(87)81501-6. PMID 3030813. S2CID 9075872.
- ^ Jump up to:a b c Law RH, Caradoc-Davies T, Cowieson N, Horvath AJ, Quek AJ, Encarnacao JA, Steer D, Cowan A, Zhang Q, Lu BG, Pike RN, Smith AI, Coughlin PB, Whisstock JC (2012). “The X-ray crystal structure of full-length human plasminogen”. Cell Rep. 1 (3): 185–90. doi:10.1016/j.celrep.2012.02.012. PMID 22832192.
- ^ Wu, Guojie; Quek, Adam J.; Caradoc-Davies, Tom T.; Ekkel, Sue M.; Mazzitelli, Blake; Whisstock, James C.; Law, Ruby H.P. (2019-03-05). “Structural studies of plasmin inhibition”. Biochemical Society Transactions. 47 (2): 541–557. doi:10.1042/bst20180211. ISSN 0300-5127. PMID 30837322.
- ^ Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, Degen JL (December 21, 1999). “Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver”. Proc. Natl. Acad. Sci. U.S.A. Proceedings of the National Academy of Sciences of the United States of America. 96 (26): 15143–8. Bibcode:1999PNAS…9615143B. doi:10.1073/pnas.96.26.15143. PMC 24787. PMID 10611352.
- ^ Silverstein RL, Leung LL, Harpel PC, Nachman RL (November 1984). “Complex formation of platelet thrombospondin with plasminogen. Modulation of activation by tissue activator”. J. Clin. Invest. 74 (5): 1625–33. doi:10.1172/JCI111578. PMC 425339. PMID 6438154.
- ^ DePoli P, Bacon-Baguley T, Kendra-Franczak S, Cederholm MT, Walz DA (March 1989). “Thrombospondin interaction with plasminogen. Evidence for binding to a specific region of the kringle structure of plasminogen”. Blood. 73 (4): 976–82. doi:10.1182/blood.V73.4.976.976. PMID 2522013.
- ^ Wiman B, Collen D (September 1979). “On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin”. J. Biol. Chem. 254 (18): 9291–7. doi:10.1016/S0021-9258(19)86843-6. PMID 158022.
- ^ Shieh BH, Travis J (May 1987). “The reactive site of human alpha 2-antiplasmin”. J. Biol. Chem. 262 (13): 6055–9. doi:10.1016/S0021-9258(18)45536-6. PMID 2437112.
- ^ Campbell PG, Durham SK, Suwanichkul A, Hayes JD, Powell DR (August 1998). “Plasminogen binds the heparin-binding domain of insulin-like growth factor-binding protein-3”. Am. J. Physiol. 275 (2 Pt 1): E321-31. doi:10.1152/ajpendo.1998.275.2.E321. PMID 9688635.
- ^ Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, Frigout Y, Hommet Y, Lebouvier L, Montaner J, Vivien D, Gauberti M (August 2016). “Hyperfibrinolysis increases blood brain barrier permeability by a plasmin and bradykinin-dependent mechanism”. Blood. 128 (20): 2423–2434. doi:10.1182/blood-2016-03-705384. PMID 27531677.
Further reading
- Shanmukhappa K, Mourya R, Sabla GE, Degen JL, Bezerra JA (July 2005). “Hepatic to pancreatic switch defines a role for hemostatic factors in cellular plasticity in mice”. Proc. Natl. Acad. Sci. U.S.A. 102 (29): 10182–7. Bibcode:2005PNAS..10210182S. doi:10.1073/pnas.0501691102. PMC 1177369. PMID 16006527.
- Anglés-Cano E, Rojas G (2002). “Apolipoprotein(a): structure-function relationship at the lysine-binding site and plasminogen activator cleavage site”. Biol. Chem. 383 (1): 93–9. doi:10.1515/BC.2002.009. PMID 11928826. S2CID 29248198.
- Ranson M, Andronicos NM (2003). “Plasminogen binding and cancer: promises and pitfalls”. Front. Biosci. 8 (6): s294-304. doi:10.2741/1044. PMID 12700073.
External links
- The MEROPS online database for peptidases and their inhibitors: S01.233
- Plasmin at the US National Library of Medicine Medical Subject Headings (MeSH)
| PLG | ||
|---|---|---|
| Available structuresPDBOrtholog search: PDBe RCSBshowList of PDB id codes | ||
| Identifiers | ||
| Aliases | PLG, plasminogen, plasmin, HAE4 | |
| External IDs | OMIM: 173350 MGI: 97620 HomoloGene: 55452 GeneCards: PLG | |
| showGene location (Human) | ||
| showGene location (Mouse) | ||
| showRNA expression pattern | ||
| showGene ontology | ||
| Orthologs | ||
| Species | Human | Mouse |
| Entrez | 5340 | 18815 |
| Ensembl | ENSG00000122194 | ENSMUSG00000059481 |
| UniProt | P00747 | P20918 |
| RefSeq (mRNA) | NM_001168338 NM_000301 | NM_008877 |
| RefSeq (protein) | NP_000292 NP_001161810 | NP_032903 |
| Location (UCSC) | Chr 6: 160.7 – 160.75 Mb | Chr 17: 12.38 – 12.42 Mb |
| PubMed search | [3] | [4] |
| Wikidata | ||
| View/Edit HumanView/Edit Mouse |
///////////Plasminogen, FDA 2021, APPROVALS 2021, Ryplazim

NEW DRUG APPROVALS
ONE TIME
$10.00
ABX 464

ABX-464
- Molecular FormulaC16H10ClF3N2O
- Averrage mass338.712 Da
SPL-4641258453-75-6[RN]26RU378B9V2-Quinolinamine, 8-chloro-N-[4-(trifluoromethoxy)phenyl]-8-Chloro-N-[4-(trifluoromethoxy)phenyl]-2-quinolinamine
EX-A3322, DB14828, SB18690, BS-14770
Abivax is developing ABX464 a lead from HIV-1 splicing inhibitors, which modulates biogenesis of viral RNA, and acts by targeting the Rev protein, for treating HIV infection, rheumatoid arthritis, ulcerative colitis and COVID-19 infection.
In August 2021, ABX464 was reported to be in phase 3 clinical development.
ABX464 is an oral, first-in-class, small molecule that has demonstrated safety and profound anti-inflammatory activity in preclinical trials and in Phase 2a and Phase 2b induction trials to treat ulcerative colitis (UC). Patients who completed the induction studies had the option to roll over into the respective open-label extension studies.
In May 2021, Abivax communicated the top-line results of its randomized, double-blind and placebo-controlled Phase 2b induction trial conducted in 15 European countries, the US and Canada in 254 patients. The primary endpoint (statistically significant reduction of Modified Mayo Score) was met with once-daily ABX464 (25mg, 50mg, 100mg) at week 8.
Further, all key secondary endpoints, including endoscopic improvement, clinical remission, clinical response and the reduction of fecal calprotectin showed significant difference in patients dosed with ABX464 compared to placebo. Importantly, ABX464 also showed rapid efficacy in patients who were previously exposed to biologics and/or JAK inhibitors treatment.
In addition to the top-line induction results, preliminary data from the first 51 patients treated with 50mg ABX464 in the Phase 2b open-label maintenance study showed increased and durable clinical remission and endoscopic improvement after 48 weeks of treatment.
Based on the positive results from the Phase 2a and Phase 2b studies, Abivax plans to advance ABX464 into a Phase 3 clinical program by the end of 2021.
- Originator Splicos
- Developer Abivax
- Class Anti-inflammatories; Antirheumatics; Antivirals; Small molecules
- Mechanism of Action MicroRNA stimulants; Rev gene product inhibitors; RNA cap-binding protein modulators
- Phase II/III COVID 2019 infections
- Phase II Crohn’s disease; Rheumatoid arthritis; Ulcerative colitis
- DiscontinuedHIV infections
- 24 Jun 2021 Discontinued – Phase-II for HIV infections (Adjunctive treatment, Treatment-experienced) in France (PO) (Abivax pipeline, June 2021)
- 24 Jun 2021 Discontinued – Phase-II for HIV infections (Treatment-experienced, Adjunctive treatment) in Belgium (PO) (Abivax pipeline, June 2021)
- 24 Jun 2021
- Discontinued – Phase-II for HIV infections (Treatment-experienced, Adjunctive treatment) in Spain (PO) (Abivax pipeline, June 2021)
Evotec and Abivax in small-molecule pact
by Michael McCoy
September 18, 2017 | A version of this story appeared in Volume 95, Issue 37

The contract research firm Evotec will work with Abivax, a French biotech company, to develop new treatments for viral diseases. Abivax has developed a library of more than 1,000 small molecules designed to inhibit mRNA biogenesis. At its facility in Toulouse, France, Evotec will optimize Abivax’s drug candidates and help develop new drugs for influenza, Dengue, and other viral infections. Abivax’s lead candidate, ABX464, is in Phase II clinical trials as an HIV/AIDS treatment.
PATENT
WO 2010143170
WO 2010143168
WO 2010143169
EP 2974729
WO 2016009065
WO 2017158201
PATENT
WO2016009065
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016009065

Buchwald-Hartwig coupling of 2,8-dichloroquinoline (I) with 4-(trifluoromethoxy)aniline (II) using Pd(OAc)2, Cs2CO3 and xantphos or Pd2dba3, K2CO3 and xphos in t-BuOH
PATENT
https://patents.google.com/patent/US10253020B2/en
US 20170226095
COMPD 90
- (90) 8-chloro-N-[4-(trifluoromethoxy)phenyl]quinolin-2-amine
Example 5: Compound (90) of the Table IAccording to route (A), a mixture of 2,8-dichloroquinoline (984 mg) and 4-(trifluoromethoxy)aniline (743 μL), Pd(OAc)2 (22 mg), XantPhos (58 mg) and Cs2CO3 (4.6 g) in 20 mL of t-BuOH gave compound (90) (1.1 g).1H NMR (300 MHz, CDCl3) δ 7.84 (d, J=9.1, 2H), 7.79 (d, J=8.9, 1H), 7.67 (dd, J=1.2, 7.6, 1H), 7.48 (dd, J=1.1, 8.0, 1H), 7.18 (s, 3H), 6.89 (s, 1H), 6.75 (d, J=8.9, 1H).13C NMR (75 MHz, CDCl3) δ 153.88, 144.30, 143.91, 139.00, 138.25, 131.13, 130.13, 126.55, 125.42, 123.45, 122.50, 122.17, 120.49, 119.10, 113.24.
| 90 | 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 9.1, 2H), 7.79 (d, J = 8.9, 1H), 7.67 (dd, J = 1.2, |
| 7.6, 1H), 7.48 (dd, J = 1.1, 8.0, 1H), 7.18 (s, 3H), 6.89 (s, 1H), 6.75 (d, J = 8.9, | |
| 1H) | |
| 13C NMR (75 MHz, CDCl3) δ 153.88, 144.30, 143.91, 139.00, 138.25, 131.13, | |
| 130.13, 126.55, 125.42, 123.45, 122.50, 122.17, 120.49, 119.10, 113.24. | |
| MS (ESI) [M + H]+ = 339 |
PAPER
Tetrahedron Letters (2018), 59(23), 2277-2280.
https://www.sciencedirect.com/science/article/abs/pii/S0040403918305641
Abstract
A solvent-free Buchwald-Hartwig amination had been developed under high-speed ball-milling conditions, which afforded the desired products with moderate to high yields. The addition of sodium sulfate was found to be crucial for improving both the performance and the reproducibility. Comparative solvent-free stirring experiments implicated the importance of mechanical interaction for the transformation, and the inert gas was proved to be unnecessary for this amination.
Graphical abstract

PATENT
WO2015001518
COMPD 90
PATENT
WO-2021152131
Novel co-crystalline polymorphic forms and salts of ABX464 , useful for treating inflammatory diseases, cancer, and diseases caused by viruses eg HIV, severe acute respiratory syndrome caused by SARS-CoV or SARS-CoV-2 infection including strains responsible for COVID-19 and their mutants.
W02010/143169 application describes the preparation and use of compounds, and in particular quinoline derivatives including certain pharmaceutically acceptable salts useful in the treatment of HIV infection. Said application in particular discloses 8-Chloro-N-(4-(trifluoromethoxy)phenyl)quinolin-2-amine also named (8-chloro-quinoline-2-yl)-(4-trifluoromethoxy-phenyl) -amine which is currently under clinical development. The inventors have stated that ABX464 is naturally highly crystalliferous and thus is spontaneously present under a specific unique stable and crystalline form named “crystalline form I”.
W02017/158201 application deals with certain mineral acid or sulfonic acid salts of ABX464.
ABX464 has a poor solubility in aqueous solutions. The main drawback of said poor solubility is that the active ingredient cannot entirely reach their targets in the body if the drug remains undissolved in the gastrointestinal system.
PATENT
WO2021152129 ,
amorphous solid dispersion (eg tablet) comprising ABX464.
PATENT
WO2020127839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020127839
use of quinoline derivatives (ie ABX464) for treating cancer and dysplasia.
///////////ABX464, ABX 464, phase 3 , SPL 464, EX A3322, DB14828, SB18690, BS 14770

NEWDRUG APPROVALS
ONE TIME
$10.00
THIAMINE, Vitamin B1


THIAMINE
- Molecular FormulaC12H17N4OS
- Average mass265.354 Da
- Thiazolium, 3-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methyl-, chloride, hydrochloride (1:1:1), Thiamine CL hcl, 67-03-8, (Component: 70-16-6) 1;1;1,
- C12 H17 N4 O S . Cl H . Cl
3595616 [Beilstein]
3-[(4-Amino-2-methyl-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methylthiazolium
thiamin hydrochloride
Vitamin B1 hydrochloride
thiamine hydrochloride
aneurin hydrochloride
3-(4-amino-2-methyl-5-pyrimidinyl)methyl-5-(2-hydroxyethyl)-4-methylthiazolium chloride hydrochlorideThiamineCAS Registry Number: 59-43-8CAS Name: 3-[(4-Amino-2-methyl-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methylthiazolium chlorideAdditional Names: vitamin B1; aneurin; thiamine monochloride; thiaminium chlorideMolecular Formula: C12H17ClN4OSMolecular Weight: 300.81Percent Composition: C 47.91%, H 5.70%, Cl 11.79%, N 18.63%, O 5.32%, S 10.66%Literature References: Essential nutrient required for carbohydrate metabolism; also involved in nerve function. Biosynthesized by microorganisms and plants. Dietary sources include whole grains, meat products, vegetables, milk, legumes and fruit. Also present in rice husks and yeast. Converted in vivo to thiamine diphosphate, a coenzyme in the decarboxylation of a-keto acids. Chronic deficiency may lead to neurological impairment, beriberi, Wernicke-Korsakoff syndrome. Isoln from rice bran: B. C. P. Jansen, W. F. Donath, Chem. Weekbl.23, 201 (1926).
Structure: R. R. Williams, J. Am. Chem. Soc.58, 1063 (1936); R. R. Williams, J. K. Cline, ibid. 1504; R. R. Williams et al.,ibid.59, 526 (1937). Review of syntheses: Knobloch in H. Vogel, Chemie und Technik der Vitaminevol. II (Stuttgart, 1953) pp 1-128. Toxicity data: D. Winter et al.,Int. Z. Vitaminforsch.37, 82 (1967). HPLC determn in foods, pharmaceuticals, body tissues: T. Kawaski, Methods Enzymol.122, 15 (1986); in plasma and pharmacokinetics: H. Mascher, C. Kikuta, J. Pharm. Sci.82, 56 (1993).
Review of bioavailability, absorption, and role in nutrition: F. L. Iber et al.,Am. J. Clin. Nutr.36, 1067-1082 (1982). Reviews: “Thiamin: Twenty Years of Progress”, Ann. N.Y. Acad. Sci.378, H. Z. Sable, C. J. Grubier, Eds. (1982) 470 pp; “Thiamin, Vitamin B1, Aneurin” in Vitamins, W. Friedrich, Ed. (de Gruyter, Berlin, 1988) pp 339-401.
Derivative Type: HydrochlorideCAS Registry Number: 67-03-8Additional Names: Thiamine chloride hydrochloride; thiamine dichlorideTrademarks: Benerva (Roche); Betabion (Merck KGaA); Betalin S (Lilly); Betaxin (Sterling Winthrop); Bewon (Wyeth); Metabolin (Takeda); Vitaneurin (Mepha)Molecular Formula: C12H17ClN4OS.HClMolecular Weight: 337.27Percent Composition: C 42.73%, H 5.38%, Cl 21.02%, N 16.61%, O 4.74%, S 9.51%Literature References: Comprehensive description: K. A. M. Al-Rashood et al.,Anal. Profiles Drug Subs.18, 413-458 (1989).Properties: Monoclinic plates in rosette-like clusters. Slight thiazole odor. Bitter taste. dec 248°. One gram dissolves in ~1 ml water, 18 ml glycerol, 100 ml 95% alcohol, 315 ml abs alcohol; more sol in methanol. Sol in propylene glycol. Practically insol in ether, benzene, hexane, chloroform. pH of a 1% w/v soln in water 3.13; pH of a 0.1% w/v soln in water 3.58.
On exposure to air of average humidity, the vitamin absorbs an amount of water corresponding to nearly one mol, forming a hydrate. LD50 in mice (mg/kg): 89.2 i.v.; 8224 orally (Winter).Toxicity data: LD50 in mice (mg/kg): 89.2 i.v.; 8224 orally (Winter)
Derivative Type: MononitrateCAS Registry Number: 532-43-4Molecular Formula: C12H17N5O4SMolecular Weight: 327.36Percent Composition: C 44.03%, H 5.23%, N 21.39%, O 19.55%, S 9.80%Literature References: Prepn: R. J. Turner, G. J. Schmitt, US2844579 (1958 to Am. Cyanamid).Properties: Crystals, mp 196-200° (dec). Practically nonhygroscopic. pKa 4.8. Soly in water (g/100 ml): 2.7 (25°); ~30 (100°). pH of 2% aq soln 6.5 to 7.1. More stable than the hydrochloride; suitable for enrichment of flours and feeds, multivitamin prepns.Melting point: mp 196-200° (dec)pKa: pKa 4.8
Therap-Cat: Vitamin (enzyme cofactor).Therap-Cat-Vet: Vitamin (enzyme cofactor).Keywords: Enzyme Cofactor; Vitamin/Vitamin Source; Vitamin B1.
Vitamin B1 (Thiamine)
Deficiency of this causes beriberi


Some symptoms of ‘dry’ beriberi. There is also a ‘wet’ version of beriberi which mainly affects the heart and circulatory system,
with shortness of breath, swelling of the lower legs, and increased heart rate.
According to the global “Vitamin B1 (Thiamine Mononitrate) Market 2020” research report, the global vitamin B1 market revenue was USD 648.8 million in 2020 and will be projected to reach USD 854.7 million by 2026.Global Vitamin B1 (Thiamine Mononitrate) Sales Market Report 2020, 2020. Fully Continuous Flow Synthesis of 3-Chloro-4-oxopentyl Acetate: An Important Intermediate for Vitamin B1
M Jiang, M Liu, C Yu, D Cheng… – … Process Research & …, 2021 – ACS Publications
… Journal Logo. Fully Continuous Flow Synthesis of 3-Chloro-4-oxopentyl Acetate:
An Important Intermediate for Vitamin B1. Meifen Jiang* Meifen Jiang. Shanghai
Engineering Center of Industrial Asymmetric Catalysis for Chiral …
SPECTROSCOPY

Compound Name:
Thiamin hydrochlorideMolecular Formula: C12H17ClN4OSMolecular Weight: 300.8CAS Registry No.:
67-03-8 MASS

13C NMR D2O


1H NMR : 400 MHz in DMSO-d6


IR


SynCN108239084 – PRODUCTION DEVICE OF MEDICINE THIAMINE HYDROCHLORIDE FOR TREATING NEURITIS

https://patentscope.wipo.int/search/en/detail.jsf?docId=CN223080274&_cid=P12-KT00YC-33991-1
The production device for the treatment drug thiamine hydrochloride for neuritis. The production process is as follows: add acetamidine hydrochloride and α-dimethoxymethyl-β-methoxymethylpropionitrile into the reactor D101, and condense in an alkaline medium Is 3,6-dimethyl 1,2-dihydro-2,4,5,7-tetrazine (Ⅱ), which is then hydrolyzed to obtain the intermediate product (Ⅲ), which is then closed to form 2-methyl in alkaline 4-amino-5-aminomethyl pyrimidine (IV), introduced into D102, continue to react with carbon disulfide and ammonia to obtain (Ⅴ), then condense with acetic acid-γ-chloro-γ propyl acetate, and then in hydrochloric acid After hydrolysis and cyclization, thiothiamine hydrochloride is obtained, which is pumped into D103, neutralized with ammonia water, oxidized by hydrogen peroxide, and then converted into ammonium nitrate thiamine with nitric acid, and finally hydrochloric acid is added to obtain the product. The invention has the advantages of reducing the intermediate links of the reaction, reducing the reaction temperature and the reaction time, and improving the reaction yield.
SYNhttps://pubs.acs.org/doi/abs/10.1021/jo00277a036 Journal of Organic Chemistry, 54(16), 3941-5; 1989

NEW DRUG APPROVALS
ONE TIME
$10.00
SYNhttps://pubs.acs.org/doi/abs/10.1021/acs.oprd.1c00065A fully continuous flow synthesis of 3-chloro-4-oxopentyl acetate (2), an important intermediate for vitamin B1 (1), was developed. This continuous flow manufacturing included two chemical transformations and an inline extraction step without intermediate purification and solvent exchange. In this work, the traditional synthetic route for batch operation was efficiently simplified via a series of separated screening tests in flows under various conditions. We found that the chlorination reaction can be carried out in only 30 s at room temperature by flow. We also simplified the decarboxylation/acylation step by using a cross-mixer, so that acetic anhydride was no longer required in the acylation reaction. A computational fluid dynamics simulation was carried out to study the improved micromixing of liquid–liquid two-phase streams. Finally, 3-chloro-4-oxopentyl acetate (2) was obtained in a 90% isolated yield with a product purity of 96% and a total residence time of approximately 32 min. This fully continuous process was operated smoothly for 12 h, and approximately 19.1 g of the desired product was generated with a production rate of 1.79 g h–1.


Batch operation for the decarboxylation/acylation reaction Procedure: 1) Mix acetic acid (3.2 eq.), water (1.1 eq.), and 35 % hydrochloric acid (0.1eq.); 2) Add 1 eq. of 3-acetyl-3-chlorodihydrofuran-2(3H)-one (3) into the mixture at room temperature; 3) Increase the reaction temperature to 120 ℃ to reflux for about 2 hours; 4) Add 2 eq. of acetic anhydride to the mixture; 5) Keep reluxing for another 3 hours; 6) After reaction (analysed by GC-MS), add saturated sodium bicarbonate solution for neutralization to make the pH to be around 7; 7) Add DCM solvent to extract the product for 3 times; 8) Concentrate the DCM solution and distill under vacuum distillation to collect the highly pure product of 3-chloro-4-oxopentyl acetate (2). Distillation condition: 90 ℃, 3-7 mmHg. After 6 hours reaction,the yield of crude product is obtained as 63 % and the purity is around 92 %. After distillation, the purity increases to 95% with an isolation yield of 60%.The production rate for batch is about 1.47 g/h, which is less than the continuous process(1.79g/h).
syn
CN108239084 – PRODUCTION DEVICE OF MEDICINE THIAMINE HYDROCHLORIDE FOR TREATING NEURITIS
SYN
Bulletin of the Chemical Society of Japan, 45(7), 2010-15; 1972
https://www.journal.csj.jp/doi/10.1246/bcsj.45.2010
The reaction of 2-dimethoxymethyl-3-methoxypropionitrile (1) with acetamidine produces pyrimidopyrimidine (8) via the consecutive process of 1→an intermediate→8. The intermediate was not isolated, but two structures have been proposed for it. We have now succeeded in the isolation of the intermediate and determined it to be 2-methyl-4-amino-5-dimethoxymethyl-5,6-dihydropyrimidine (4). Several key intermediates were also successfully isolated. The novel reaction pathway for the title reaction was concluded to be as follows: the elimination of methanol from 1, followed by the addition of acetamidine affords 3-acetamidinopropionitrile (3), the subsequent quick cyclization of which produces the intermediate, 4; the further elimination of methanol from 4, followed by a replacement reaction with acetamidine, gives an acetamidinomethylene compound (6), which is converted into the final product, 8, via an intermediate (7). Some minor pathways will also be presented.
syn
CN109467553-PURIFICATION METHOD OF FORMYL PYRIMIDINE AND SYNTHETIC METHOD OF VITAMIN B1
![Synthesis of thiamine, method by Williams and Cline [90].](https://www.researchgate.net/profile/Artur-Ratkiewicz/publication/321626762/figure/fig5/AS:661906792648715@1534822285512/Synthesis-of-thiamine-method-by-Williams-and-Cline-90.png)
90 Williams, R.R. and Cline, J.K. (1936) Synthesis of vitamin B1. J. Am. Chem. Soc. 58, 1504–1505, https://doi.org/10.1021/ja01299a505SYN
Thiaminpyrophosphate (11) (Figure 1) is an essential cofactor in all forms of life and it plays a key role in carbohydrate and amino acid metabolism by stabilizing acyl carbanion biosynthons. The mechanistic enzymology of thiamin pyrophosphate-dependent enzymes is described in detail in the chapter by Frank Jordan.1 Here, we will review recent progress on the biosynthesis of thiamin pyrophosphate in bacteria and Saccharomyces cerevisiae with an emphasis on some of the novel organic chemistry that has emerged from these studies. Recent reviews describing the regulation of the pathway,2,3 the identification of biosynthetic precursors,4 and the structural biology of the pathway5–7 have been published.

SYN
Vitamin B1 338 Commercial production involves a six-step synthetic procedure (Williams & Cline, 1936). Beginning with 339 ethyl 3-ethoxypropionate as the feedstock for vitamin B1 production, the synthetic reactions include (1) 340 formylation using ethyl formate, (2) reaction with acetamidine hydrochloride leading to aminopyrimidine 341 ring formation, (3) replacement of aminopyrimidine hydroxyl group with a chlorine atom (chlorination) 342 using phosphorus(V) oxychloride, (4) replacement of the labile chlorine atom with an amino group using 343 alcoholic ammonia, (5) ammonium salt formation using hydrobromic acid, (6) introduction of the thiazole 344 ring using 4-methyl 5-hydroxyethyl thiazole.

A search of the patent literature revealed two methods for vitamin B1 (thiamine) production by 349 fermentative methods. The first patent describes the development of mutants of the genus Saccharomyces 350 Meyen emend Reess (yeast) for synthesizing vitamin B1 from sugars and inorganic salts (Silhankova, 1980). A 351 more recent invention provides a method for producing thiamine products using a microorganism of the 352 genus Bacillus containing a mutation (i.e., gene deletions or other mutations) that causes it to overproduce 353 and release thiamine products into the medium (Goese, 2012).
PATENT
CN109467553 – PURIFICATION METHOD OF FORMYL PYRIMIDINE AND SYNTHETIC METHOD OF VITAMIN B1
The invention relates to the field of vitamin B1 synthesis, and particularly relates to a purification method of formyl pyrimidine and a synthetic method of vitamin B1. The purification method of formyl pyrimidine comprises the following steps: washing formyl pyrimidine with alcohol; washing formyl pyrimidine with water; dissolving formyl pyrimidine with alcohol, and decoloring formyl pyrimidine with activated carbon to obtain a formyl pyrimidine solution; and separating out formyl pyrimidine in the formyl pyrimidine solution and separating the formyl pyrimidine from the solution to obtain purified formyl pyrimidine. According to the purification method of formyl pyrimidine, by washing the formyl pyrimidine with alcohol and water, decoloring the formyl pyrimidine with activated carbon in an alcohol solution and separation the purification method of formyl pyrimidine by water, impurities in the formyl pyrimidine are removed, the content of the formyl pyrimidine reaches 99.5% over, and agood basis is provided for further synthesizing vitamin B1.
| Example 1 |
| A method for purifying formyl pyrimidine, the steps are: |
| a. Wash formyl pyrimidine with methanol to remove impurities dissolved in methanol in formyl pyrimidine. The weight ratio of formyl pyrimidine to methanol is 1:2. |
| b. Add water to wash formyl pyrimidine to remove impurities dissolved in water in formyl pyrimidine. The weight ratio of formyl pyrimidine to water is 1:2. |
| c. Dry the washed formylpyrimidine, add methanol at a weight ratio of 1:1, reflux and heat to 40-50°C to completely dissolve. |
| d. Add activated carbon while hot for decolorization, the weight ratio of formylpyrimidine solution to activated carbon is 1:0.01, quickly stir and decolorize for 15min, and filter out formylpyrimidine solution while hot. |
| e. Cool down to 0-10°C and formyl pyrimidine precipitates out, filter and dry to obtain formyl pyrimidine solid. |
| The obtained formylpyrimidine solid was tested, as shown in Figure 1. |
| The information in Figure 1 is shown in Table 1. |
| Table 1 Detection peak information |
| |
| |
| The formula for calculating the content of formyl pyrimidine in solid formyl pyrimidine is as follows: |
| |
| A—formylpyrimidine content; |
| S 1 —Sample peak area; |
| S 2 —Standard peak area; |
| M 1 —Standard quality; |
| M2—sample quality; |
| W 1 —The concentration of the standard. |
| According to calculation, the content of formyl pyrimidine purified by this method can reach 99.7%, and the content of formyl pyrimidine in the unpurified formyl pyrimidine is 91%. |
| After testing, the yield was 94% based on the mass of the formyl pyrimidine before purification. |
| The formyl pyrimidine obtained by the above purification method is reacted to obtain vitamin B1. Subsequent detection shows that the quality of vitamin B1 is higher, and the content of impurities in the detection data such as related substances and chromatographic purity is lower. The chromatographic purity of the impurity before purification was 0.8, and the chromatographic purity after purification was about 0.1. The content of each impurity in related substances decreased year-on-year. The average compliance rate of the final vitamin B1 is 100%. |
PAPER
HELVETICA CHIMICA ACTA ~ Vol. 73 (1990)


1. 3-Mercapto-4-oxopentyl Acetate (5a). Anh. KSH (7.22 g, 0.1 mol) was suspended in 50 ml of abs. MeOH. The mixture was cooled to 0″ in an ice-bath and 3-chloro-4-oxopentyl acetate (3; 17.9 g, 0.1 mol), previously dissolved in 50 ml of abs. MeOH, was added dropwise in order to maintain the temp. in the mixture between 0 and 5″. After complete addition, stirring was continued at r.t. for 1 h, while a slow stream of N, was passed through the mixture to remove residual H2S. The precipitated KC1 was filtered off and the solvent evaporated under reduced pressure. The residue was taken up in 50 ml of CH,C12 and the insoluble material removed by filtration. Evaporation of the solvent in uamo at 30″ gave 14.9 g of slightly yellow liquid. Bulb-to-bulb distillation of the crude mixture at 120″/0.3 mm yielded 12.95 g (0.07 mol, 73.5%) of 5a as a colourless liquid7). IR (film): 2960w, 2550~. 1740s, 17153, 1370m, 1245s, 1050m. ‘H-NMR (CDCI,): 1.74 (d, J= 12, SH); 1.95-2.25 (m, CH,); 2.05 (s, AcO); 2.35(s,Me);3.42(td,J= 12,5.7,SCH);4.2(t,J=5.7,CH20).EI-MS: 134(2), 116(36),74(21),73(58),43(100). Anal. calc. for C7HI2O,S (176.23): C 47.71, H 6.86, S 18.19; found: C 47.94, H 6.95, S 17.24.
2. 3,4-Dihydro-7-methylpyrimido[4,5-d]pyrimidine (4). From 4-amino-2-methyl-5-(aminomethyl)pyrimidine (Za) and DMF-DMA. In a flask equipped with a Vigreux column and a Liebig condenser, Zag) (69 g, 0.5 mol) was suspended in dimethylformamide dimethyl acetal(59.6 g, 0.5 mol). The stirred suspension was slowly heated to ca. 8&85″, until the temp. at the head of the Vigreux column reached 60°9). The MeOH/Me,NH mixture was then distilled off, until the mixture in the flask became a thick mass. The temp. was increased to 90″ for 30 min, 250 ml of toluene were added, and the obtained suspension was further stirred for 1 h at 90°. It was then allowed to cool to r.t., filtered, and washed twice with 100 ml of hexane. The crude material was dried at SOo under reduced pressure: 69.6 g of a tan solid was obtained, which was then sublimated at 1 SOo (oil-bath temp.) under high vacuum (0.2 mm) togive65.5g(0.44mol,88.5%)of4asawhitesolid. M.p. 173″(dec.).UV:202(4),298(3,7).1R(KBr): 3430m(br.), 2860m, 2840s, 16703, 1620s, 15803, 15303, 1450s. 1210s. ‘H-NMR ((D,)DMSO): 2.4 (s, Me); 4.5 (s, CH,); 7.2 (br. s, vinyl. CH); 8.03 (s, arom. H); 9.9 (br. s, NH). EI-MS: 148 (50, M’), 147 (loo), 106 (12), 53 (17), 42 (20). Anal. calc. for C7H,N, (148.169): C 56.74, H 5.44, N 37.81; found: C 56.79, H 5.44, N 37.75.
From 2a and Triethyl Orthoformate. In a flask equipped with a 20-cm Vigreux column and a Liebig condenser, Zag) (69 g, 0.5 mol), triethyl orthoformate (148.2 g, 1 rnol), and TsOH (2.5 g)”) were introduced. The stirred suspension was slowly heated to ca. 110″ so that the temp. at the head of the Vigreux column reached 80-85″. The EtOH was then distilled off, until the mixture in the flask became a thick mass. The temp. was maintained at 100-1 10″ for 30 min, then 250 ml of toluene were added, and theobtained suspension was further stirred for 1 h at90°. It was cooled to r.t. and placed overnight in the refrigerator. The light-brown precipitate was filtered and washed twice with 50 ml of toluene. The crude material was dried at 50″ under reduced pressure to give 59.3 g of a beige solid which was sublimated at 150″ (oil-bath temp.) under high vacuuni (0.2 mm) to yield 52.5 g (0.35 mol, 71 %) of 4 as a white solid. M.p. 182O (dec.).
3. 3-1 (4-Amino-2-methylpyrimidin-5-yl)methyl]-5-(2-hydroxyethyl)-4-methylthiazolium Chloride Hydrochloride (Thiamine Hydrochloride, la). Compound 4 (7.4 g, 0.05 mol) was dissolved in 100 ml of HCOOH. To this slightly yellow soh, 5a (9.25 g, 0.052 mol) was immediately added at such a rate so that the temp. did not exceed 3540″. The mixture was further stirred for 30 min at r.t. and then 25 ml of a freshly prepared sat. soh. of HCI in abs. EtOH was added dropwise. The temp. rose to 35-36O, and the mixture was further stirred for 30 min at r.t.”), The crude mixture was then poured into a 500-ml flask and evaporated at 50″ under reduced pressure to give 26.07 g of a green-yellow solid residue, which was taken up in 100 ml of ahs. EtOH. Aq. HCI soh. (25%, 30 ml) was then added and the crude mixture heated on a steam-bath, until a clear soln. was obtained. The soln. was cooled to r.t. and placed overnight in the refrigerator. The resulting white crystals were collected and dried in vucuo to yield 14.56 g (86.3%) of la. M.p. 245-246′ (dec.). The mother-liquor was then evaporated at 50O under reduced pressure and the residue taken up in 50 ml of H,O. The aq. phase was then washed twice with 25 ml of CH2C1, and evaporated under reduced pressure to give 3.29 g of a still slightly greenish residue, which was again taken up in 20 ml of abs. EtOH. Aq. HCI soln. (25%, 5 ml) was added and the mixture heated on a steam-bath, until a clear soln. was obtained. It was then cooled to r.t. and kept overnight in the refrigerator. The white crystals were filtered to give 1.42 g (8.4%) of la. M.p. 244-24So(dec.) (combined yieldI2) of la: 94.7% based on 4).
Recrystallization. The two crops of la were combined and dissolved in 100 ml of warm abs. EtOH. Aq. HCI soh (25 %, 40 ml) was added. The soln. was then allowed to cool slowly to r.t. and kept at Oo overnight. The white crystals were filtered and dried in vucuo at 50″ to give 13.6 g (0.04 mol, 80.6 %) of la.
M.p. 243-244″ (dec.). UV: 234 (4.1), 266 (3.9).
IR (KBr): 3500m, 3430m. 3340m. 3240m. 3065s. 2615m. 1660s, 1607m, 1380m.
‘H-NMR (D,O): 2.54(s,Me);2.62(s,Me);3.19(t,J= 5.8,CH2);3.88(t,J= 5.8,CH20);5.56(s,1H,CH2N);8.02(s,1arom.H); proton of thiazole ring is exchanged with deuterium of D,O.
FAB-MS: 265 (100, M+), 181 (18), 144 (30), 123 (65), 122 (65), 91 (78).
Anal. calc. for C,2H18C1,N40S (337.27): C 42.74, H 5.38, N 16.61, S 9.51, CI 21.02; found: C 42.93, H 5.28, N 16.70, S 9.61, C121.17.
////////////////////////////////////////////////////////////////////////////////////////////////////
Thiamine, also known as thiamin or vitamin B1, is a vitamin found in food and manufactured as a dietary supplement and medication.[1][4] Food sources of thiamine include whole grains, legumes, and some meats and fish.[1] Grain processing removes much of the thiamine content, so in many countries cereals and flours are enriched with thiamine.[1][5] Supplements and medications are available to treat and prevent thiamine deficiency and disorders that result from it, including beriberi and Wernicke encephalopathy.[3] Other uses include the treatment of maple syrup urine disease and Leigh syndrome.[3] They are typically taken by mouth, but may also be given by intravenous or intramuscular injection.[3][6]
Thiamine supplements are generally well tolerated.[3][7] Allergic reactions, including anaphylaxis, may occur when repeated doses are given by injection.[3][7] Thiamine is in the B complex family.[3] It is an essential micronutrient, which cannot be made in the body.[8] Thiamine is required for metabolism including that of glucose, amino acids, and lipids.[1]
Thiamine was discovered in 1897, was the first B vitamin to be isolated in 1926, and was first made in 1936.[9] It is on the World Health Organization’s List of Essential Medicines.[10] Thiamine is available as a generic medication, and as an over-the-counter drug.[3]
Medical uses
Thiamine deficiency
Main article: Thiamine deficiency
Thiamine is used to treat thiamine deficiency which when severe can prove fatal.[11] In less severe cases, non-specific signs include malaise, weight loss, irritability and confusion.[12] Well-known disorders caused by thiamine deficiency include beriberi, Wernicke–Korsakoff syndrome, optic neuropathy, Leigh’s disease, African seasonal ataxia (or Nigerian seasonal ataxia), and central pontine myelinolysis.[13]
In Western countries, thiamine deficiency is seen mainly in chronic alcoholism.[14] Thiamine deficiency is often present in alcohol misuse disorder. Also at risk are older adults, persons with HIV/AIDS or diabetes, and persons who have had bariatric surgery.[1] Varying degrees of thiamine deficiency have been associated with the long-term use of high doses of diuretics, particularly furosemide in the treatment of heart failure.[15]
Prenatal supplementation
See also: Prenatal vitamins
Women who are pregnant or lactating require more thiamine. For pregnant and lactating women, the consequences of thiamine deficiency are the same as those of the general population but the risk is greater due to their temporarily increased need for this nutrient. In pregnancy, this is likely due to thiamine being preferentially sent to the fetus and placenta, especially during the third trimester. For lactating women, thiamine is delivered in breast milk even if it results in thiamine deficiency in the mother.[16] Pregnant women with hyperemesis gravidarum are also at an increased risk for thiamine deficiency due to losses when vomiting.[17]
Thiamine is important for not only mitochondrial membrane development, but also synaptosomal membrane function.[18] It has also been suggested that thiamine deficiency plays a role in the poor development of the infant brain that can lead to sudden infant death syndrome (SIDS).[19]
Other uses
Thiamine is a treatment for some types of maple syrup urine disease and Leigh disease.[3]
Adverse effects
Thiamine is generally well tolerated and non-toxic when administered orally.[3] Rarely, adverse side effects have been reported when thiamine is given intravenously including allergic reactions, nausea, lethargy, and impaired coordination.[20][21]
Chemistry
Thiamine is a colorless organosulfur compound with an unpleasant sulfur odor and the chemical formula C12H17N4O S. Its structure consists of an aminopyrimidine and a thiazolium ring linked by a methylene bridge. The thiazole is substituted with methyl and hydroxyethyl side chains. Thiamine is soluble in water, methanol, and glycerol and practically insoluble in less polar organic solvents. As a base it can form salts with acids, such as hydrochloride. It is stable at acidic pH, but is unstable in alkaline solutions.[11][22] Thiamine, which is a persistent carbene, is used by enzymes to catalyze benzoin condensations in vivo.[23] Thiamine is unstable to heat, but stable during frozen storage.[24] It is unstable when exposed to ultraviolet light[22] and gamma irradiation.[25][26] Thiamine reacts strongly in Maillard-type reactions.[11]
Biosynthesis

A 3D representation of the TPP riboswitch with thiamine bound
Complex thiamine biosynthesis occurs in bacteria, some protozoans, plants, and fungi.[27][28] The thiazole and pyrimidine moieties are biosynthesized separately and then combined to form thiamine monophosphate (ThMP) by the action of thiamine-phosphate synthase (EC 2.5.1.3). The biosynthetic pathways may differ among organisms. In E. coli and other enterobacteriaceae, ThMP may be phosphorylated to the cofactor thiamine diphospate (ThDP) by a thiamine-phosphate kinase (ThMP + ATP → ThDP + ADP, EC 2.7.4.16). In most bacteria and in eukaryotes, ThMP is hydrolyzed to thiamine, which may then be pyrophosphorylated to ThDP by thiamine diphosphokinase (thiamine + ATP → ThDP + AMP, EC 2.7.6.2).
The biosynthetic pathways are regulated by riboswitches.[21] If there is sufficient thiamine present in the cell then the thiamine binds to the mRNAs for the enzymes that are required in the pathway and prevents their translation. If there is no thiamine present then there is no inhibition, and the enzymes required for the biosynthesis are produced. The specific riboswitch, the TPP riboswitch (or ThDP), is the only riboswitch identified in both eukaryotic and prokaryotic organisms.[29]
Nutrition
Occurrence in foods
Thiamine is found in a wide variety of processed and whole foods. Whole grains, legumes, pork, fruits, and yeast are rich sources.[30][31]
The salt thiamine mononitrate, rather than thiamine hydrochloride, is used for food fortification, as the mononitrate is more stable, and does not absorb water from natural humidity (is non-hygroscopic), whereas thiamine hydrochloride is hygroscopic.[citation needed] When thiamine mononitrate dissolves in water, it releases nitrate (about 19% of its weight) and is thereafter absorbed as the thiamine cation.
Dietary recommendations
In the U.S. the Estimated Average Requirements (EARs) and Recommended Dietary Allowances (RDAs) for thiamine were updated in 1998, by the Institute of Medicine now known as the National Academy of Medicine (NAM).[32]
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For women (including those pregnant or lactating), men and children the PRI is 0.1 mg thiamine per megajoule (MJ) of energy consumed. As the conversion is 1 MJ = 239 kcal, an adult consuming 2390 kilocalories should be consuming 1.0 mg thiamine. This is slightly lower than the U.S. RDA.[33] The EFSA reviewed the same safety question and also reached the conclusion that there was not sufficient evidence to set a UL for thiamine.[20]
| United States | ||
| Age group | RDA (mg/day) | Tolerable upper intake level[32] |
|---|---|---|
| Infants 0–6 months | 0.2* | ND |
| Infants 6–12 months | 0.3* | |
| 1–3 years | 0.5 | |
| 4–8 years | 0.6 | |
| 9–13 years | 0.9 | |
| Females 14–18 years | 1.0 | |
| Males 14+ years | 1.2 | |
| Females 19+ years | 1.1 | |
| Pregnant/lactating females 14–50 | 1.4 | |
| * Adequate intake for infants, as an RDA has yet to be established[32] | ||
| European Food Safety Authority | ||
| Age group | Adequate Intake (mg/MJ)[20] | Tolerable upper limit[20] |
| All persons 7 months+ | 0.1 | ND |
To aid with adequate micronutrient intake, pregnant women are often advised to take a daily prenatal multivitamin. While micronutrient compositions vary among different vitamins, a typical prenatal vitamin contains around 1.5 mg of thiamine.[34]
For U.S. food and dietary supplement labeling purposes the amount in a serving is expressed as a percentage of Daily Value (%DV). For thiamine labeling purposes 100% of the Daily Value was 1.5 mg, but as of 27 May 2016 it was revised to 1.2 mg to bring it into agreement with the RDA.[35][36] Compliance with the updated labeling regulations was required by 1 January 2020 for manufacturers with US$10 million or more in annual food sales, and by 1 January 2021 for manufacturers with lower volume food sales.[37][38] A table of the old and new adult daily values is provided at Reference Daily Intake.
Antagonists
Thiamine in foods can be degraded in a variety of ways. Sulfites, which are added to foods usually as a preservative,[39] will attack thiamine at the methylene bridge in the structure, cleaving the pyrimidine ring from the thiazole ring.[12] The rate of this reaction is increased under acidic conditions. Thiamine is degraded by thermolabile thiaminases (present in raw fish and shellfish).[11] Some thiaminases are produced by bacteria. Bacterial thiaminases are cell surface enzymes that must dissociate from the membrane before being activated; the dissociation can occur in ruminants under acidotic conditions. Rumen bacteria also reduce sulfate to sulfite, therefore high dietary intakes of sulfate can have thiamine-antagonistic activities.
Plant thiamine antagonists are heat-stable and occur as both the ortho- and para-hydroxyphenols. Some examples of these antagonists are caffeic acid, chlorogenic acid, and tannic acid. These compounds interact with the thiamine to oxidize the thiazole ring, thus rendering it unable to be absorbed. Two flavonoids, quercetin and rutin, have also been implicated as thiamine antagonists.[12]
Food fortification
Main article: Food fortification
Refining grain removes its bran and germ, and thus subtracts its naturally occurring vitamins and minerals. In the United States, B-vitamin deficiencies became common in the first half of the 20th century due to white flour consumption. The American Medical Association successfully lobbied for restoring these vitamins by enrichment of grain, which began in the US in 1939. The UK followed in 1940 and Denmark in 1953. As of 2016, about 85 countries had passed legislation mandating fortification of wheat flour with at least some nutrients, and 28% of industrially milled flour was fortified, often with thiamine and other B vitamins.[40]
Absorption and transport
Absorption
Thiamine is released by the action of phosphatase and pyrophosphatase in the upper small intestine. At low concentrations, the process is carrier-mediated. At higher concentrations, absorption also occurs via passive diffusion. Active transport is greatest in the jejunum and ileum, but it can be inhibited by alcohol consumption or by folate deficiency.[11] Decline in thiamine absorption occurs at intakes above 5 mg/day.[41] On the serosal side of the intestine, discharge of the vitamin by those cells is dependent on Na+-dependent ATPase.[12]
Bound to serum proteins
The majority of thiamine in serum is bound to proteins, mainly albumin. Approximately 90% of total thiamine in blood is in erythrocytes. A specific binding protein called thiamine-binding protein (TBP) has been identified in rat serum and is believed to be a hormone-regulated carrier protein important for tissue distribution of thiamine.[12]
Cellular uptake
Uptake of thiamine by cells of the blood and other tissues occurs via active transport and passive diffusion.[11] About 80% of intracellular thiamine is phosphorylated and most is bound to proteins. Two members of the SLC gene family of transporter proteins, SLC19A2 and SLC19A3, are capable of the thiamine transport.[19] In some tissues, thiamine uptake and secretion appears to be mediated by a soluble thiamine transporter that is dependent on Na+ and a transcellular proton gradient.[12]
Tissue distribution
Human storage of thiamine is about 25 to 30 mg, with the greatest concentrations in skeletal muscle, heart, brain, liver, and kidneys. ThMP and free (unphosphorylated) thiamine is present in plasma, milk, cerebrospinal fluid, and, it is presumed, all extracellular fluid. Unlike the highly phosphorylated forms of thiamine, ThMP and free thiamine are capable of crossing cell membranes. Calcium and magnesium have been shown to affect the distribution of thiamine in the body and magnesium deficiency has been shown to aggravate thiamine deficiency.[19] Thiamine contents in human tissues are less than those of other species.[12][42]
Excretion
Thiamine and its acid metabolites (2-methyl-4-amino-5-pyrimidine carboxylic acid, 4-methyl-thiazole-5-acetic acid, and thiamine acetic acid) are excreted principally in the urine.[22]
Function
Its phosphate derivatives are involved in many cellular processes. The best-characterized form is thiamine pyrophosphate (TPP), a coenzyme in the catabolism of sugars and amino acids. In yeast, TPP is also required in the first step of alcoholic fermentation. All organisms use thiamine, but it is made only in bacteria, fungi, and plants. Animals must obtain it from their diet, and thus, for humans, it is an essential nutrient. Insufficient intake in birds produces a characteristic polyneuritis.
Thiamine is usually considered as the transport form of the vitamin. Five natural thiamine phosphate derivatives are known: thiamine monophosphate (ThMP), thiamine diphosphate (ThDP), also sometimes called thiamine pyrophosphate (TPP), thiamine triphosphate (ThTP), the most recently discovered adenosine thiamine triphosphate (AThTP), and adenosine thiamine diphosphate (AThDP). While the coenzyme role of thiamine diphosphate is well-known and extensively characterized, the non-coenzyme action of thiamine and derivatives may be realized through binding to a number of recently identified proteins which do not use the catalytic action of thiamine diphosphate.[43]
Thiamine diphosphate
No physiological role is known for thiamine monophosphate (ThMP); however, the diphosphate is physiologically relevant. The synthesis of thiamine diphosphate (ThDP), also known as thiamine pyrophosphate (TPP) or cocarboxylase, is catalyzed by an enzyme called thiamine diphosphokinase according to the reaction thiamine + ATP → ThDP + AMP (EC 2.7.6.2). ThDP is a coenzyme for several enzymes that catalyze the transfer of two-carbon units and in particular the dehydrogenation (decarboxylation and subsequent conjugation with coenzyme A) of 2-oxoacids (alpha-keto acids). Examples include:
- Present in most species
- Present in some species:
- pyruvate decarboxylase (in yeast)
- several additional bacterial enzymes
The enzymes transketolase, pyruvate dehydrogenase (PDH), and 2-oxoglutarate dehydrogenase (OGDH) are all important in carbohydrate metabolism. The cytosolic enzyme transketolase is a key player in the pentose phosphate pathway, a major route for the biosynthesis of the pentose sugars deoxyribose and ribose. The mitochondrial PDH and OGDH are part of biochemical pathways that result in the generation of adenosine triphosphate (ATP), which is a major form of energy for the cell. PDH links glycolysis to the citric acid cycle, while the reaction catalyzed by OGDH is a rate-limiting step in the citric acid cycle. In the nervous system, PDH is also involved in the production of acetylcholine, a neurotransmitter, and for myelin synthesis.[44]
Thiamine triphosphate
Thiamine triphosphate (ThTP) was long considered a specific neuroactive form of thiamine, playing a role in chloride channels in the neurons of mammals and other animals, although this is not completely understood.[19] However, recently it was shown that ThTP exists in bacteria, fungi, plants and animals suggesting a much more general cellular role.[45] In particular in E. coli, it seems to play a role in response to amino acid starvation.[46]
Adenosine thiamine triphosphate
Adenosine thiamine triphosphate (AThTP) or thiaminylated adenosine triphosphate has recently been discovered in Escherichia coli, where it accumulates as a result of carbon starvation.[47] In E. coli, AThTP may account for up to 20% of total thiamine. It also exists in lesser amounts in yeast, roots of higher plants and animal tissue.[48]
Adenosine thiamine diphosphate
Adenosine thiamine diphosphate (AThDP) or thiaminylated adenosine diphosphate exists in small amounts in vertebrate liver, but its role remains unknown.[48]
History
Further information: Vitamin § History
Thiamine was the first of the water-soluble vitamins to be described,[11] leading to the discovery of more essential nutrients and to the notion of vitamin.
In 1884, Takaki Kanehiro (1849–1920), a surgeon general in the Japanese navy, rejected the previous germ theory for beriberi and hypothesized that the disease was due to insufficiencies in the diet instead.[49] Switching diets on a navy ship, he discovered that replacing a diet of white rice only with one also containing barley, meat, milk, bread, and vegetables, nearly eliminated beriberi on a nine-month sea voyage. However, Takaki had added many foods to the successful diet and he incorrectly attributed the benefit to increased protein intake, as vitamins were unknown substances at the time. The Navy was not convinced of the need for so expensive a program of dietary improvement, and many men continued to die of beriberi, even during the Russo-Japanese war of 1904–5. Not until 1905, after the anti-beriberi factor had been discovered in rice bran (removed by polishing into white rice) and in barley bran, was Takaki’s experiment rewarded by making him a baron in the Japanese peerage system, after which he was affectionately called “Barley Baron”.
The specific connection to grain was made in 1897 by Christiaan Eijkman (1858–1930), a military doctor in the Dutch Indies, who discovered that fowl fed on a diet of cooked, polished rice developed paralysis, which could be reversed by discontinuing rice polishing.[50] He attributed beriberi to the high levels of starch in rice being toxic. He believed that the toxicity was countered in a compound present in the rice polishings.[51] An associate, Gerrit Grijns (1865–1944), correctly interpreted the connection between excessive consumption of polished rice and beriberi in 1901: He concluded that rice contains an essential nutrient in the outer layers of the grain that is removed by polishing.[52] Eijkman was eventually awarded the Nobel Prize in Physiology and Medicine in 1929, because his observations led to the discovery of vitamins.
In 1910, a Japanese agricultural chemist of Tokyo Imperial University, Umetaro Suzuki (1874-1943), first isolated a water-soluble thiamine compound from rice bran and named it as aberic acid (He renamed it as Orizanin later). He described the compound is not only anti beri-beri factor but also essential nutrition to human in the paper, however, this finding failed to gain publicity outside of Japan, because a claim that the compound is a new finding was omitted in translation from Japanese to German.[53] In 1911 a Polish biochemist Casimir Funk isolated the antineuritic substance from rice bran (the modern thiamine) that he called a “vitamine” (on account of its containing an amino group).[54][55] However, Funk did not completely characterize its chemical structure. Dutch chemists, Barend Coenraad Petrus Jansen (1884–1962) and his closest collaborator Willem Frederik Donath (1889–1957), went on to isolate and crystallize the active agent in 1926,[56] whose structure was determined by Robert Runnels Williams (1886–1965), a US chemist, in 1934. Thiamine was named by the Williams team as “thio” or “sulfur-containing vitamin”, with the term “vitamin” coming indirectly, by way of Funk, from the amine group of thiamine itself (by this time in 1936, vitamins were known to not always be amines, for example, vitamin C). Thiamine was synthesized in 1936 by the Williams group.[57]
Thiamine was first named “aneurin” (for anti-neuritic vitamin).[58] Sir Rudolph Peters, in Oxford, introduced thiamine-deprived pigeons as a model for understanding how thiamine deficiency can lead to the pathological-physiological symptoms of beriberi. Indeed, feeding the pigeons upon polished rice leads to an easily recognizable behavior of head retraction, a condition called opisthotonos. If not treated, the animals died after a few days. Administration of thiamine at the stage of opisthotonos led to a complete cure within 30 minutes. As no morphological modifications were observed in the brain of the pigeons before and after treatment with thiamine, Peters introduced the concept of a biochemical lesion.[59]
When Lohman and Schuster (1937) showed that the diphosphorylated thiamine derivative (thiamine diphosphate, ThDP) was a cofactor required for the oxydative decarboxylation of pyruvate,[60] a reaction now known to be catalyzed by pyruvate dehydrogenase, the mechanism of action of thiamine in the cellular metabolism seemed to be elucidated. At present, this view seems to be oversimplified: pyruvate dehydrogenase is only one of several enzymes requiring thiamine diphosphate as a cofactor; moreover, other thiamine phosphate derivatives have been discovered since then, and they may also contribute to the symptoms observed during thiamine deficiency. Lastly, the mechanism by which the thiamine moiety of ThDP exerts its coenzyme function by proton substitution on position 2 of the thiazole ring was elucidated by Ronald Breslow in 1958.[61]
- Some contributors to the discovery of thiamine
- Takaki Kanehiro
- Christiaan Eijkman
- Gerrit Grijns
- Umetaro Suzuki
- Casimir Funk
- Rudolph Peters
- Ronald Breslow
See also
References
- ^ Jump up to:a b c d e f “Office of Dietary Supplements – Thiamin”. ods.od.nih.gov. 11 February 2016. Archived from the original on 30 December 2016. Retrieved 30 December 2016.
- ^ Royer-Morrot MJ, Zhiri A, Paille F, Royer RJ (1992). “Plasma thiamine concentrations after intramuscular and oral multiple dosage regimens in healthy men”. European Journal of Clinical Pharmacology. 42 (2): 219–22. doi:10.1007/BF00278489. PMID 1618256. S2CID 19924442.
- ^ Jump up to:a b c d e f g h i j American Society of Health-System Pharmacists. “Thiamine Hydrochloride”. Drugsite Trust (Drugs.com). Retrieved 17 April 2018.
- ^ “Thiamine: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 30 April 2018.
- ^ Guidelines on food fortification with micronutrients (PDF). WHO and FAO. 2006. pp. 13–14. ISBN 92-4-159401-2. Retrieved 5 May2018.
- ^ “Thiamine”. drugbank.ca. Retrieved 30 April 2018.
- ^ Jump up to:a b Kliegman RM, Stanton B (2016). Nelson Textbook of Pediatrics. Elsevier Health Sciences. p. 322. ISBN 9781455775668.
There are no cases of adverse effects of excess thiamine… A few isolated cases of puritis…
- ^ Constable PD, Hinchcliff KW, Done SH, Gruenberg W (2017). Diseases of the Nervous System – Veterinary Medicine (Eleventh Edition) – 14. pp. 1155–1370. ISBN 978-0-7020-5246-0.
Thiamine (vitamin B1) is synthesized only in bacteria, fungi, and plants but is an essential nutrient for animals.
- ^ Squires VR (2011). The Role of Food, Agriculture, Forestry and Fisheries in Human Nutrition – Volume IV. EOLSS Publications. p. 121. ISBN 9781848261952. Archived from the original on 30 December 2016.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g Mahan LK, Escott-Stump S, eds. (2000). Krause’s food, nutrition, & diet therapy (10th ed.). Philadelphia: W.B. Saunders Company. ISBN 978-0-7216-7904-4.
- ^ Jump up to:a b c d e f g Combs Jr GF (2008). The Vitamins: Fundamental Aspects in Nutrition and Health (3rd ed.). Ithaca, NY: Elsevier Academic Press. ISBN 978-0-12-183493-7.
- ^ McCandless D (2010). Thiamine Deficiency and Associate Clinical Disorders. New York, NY: Humana Press. pp. 157–159. ISBN 978-1-60761-310-7.
- ^ The Editors of Encyclopaedia Britannica (19 December 2017). “Beriberi”. Encyclopædia Britannica. Retrieved 13 April 2018.
- ^ Katta N, Balla S, Alpert MA (July 2016). “Does Long-Term Furosemide Therapy Cause Thiamine Deficiency in Patients with Heart Failure? A Focused Review”. The American Journal of Medicine. 129 (7): 753.e7–753.e11. doi:10.1016/j.amjmed.2016.01.037. PMID 26899752.
- ^ Butterworth RF (December 2001). “Maternal thiamine deficiency: still a problem in some world communities”. The American Journal of Clinical Nutrition. 74 (6): 712–3. doi:10.1093/ajcn/74.6.712. PMID 11722950.
- ^ Oudman E, Wijnia JW, Oey M, van Dam M, Painter RC, Postma A (May 2019). “Wernicke’s encephalopathy in hyperemesis gravidarum: A systematic review”. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 236: 84–93. doi:10.1016/j.ejogrb.2019.03.006. PMID 30889425.
- ^ Kloss O, Eskin NA, Suh M (April 2018). “Thiamin deficiency on fetal brain development with and without prenatal alcohol exposure”. Biochemistry and Cell Biology. 96 (2): 169–177. doi:10.1139/bcb-2017-0082. hdl:1807/87775. PMID 28915355.
- ^ Jump up to:a b c d Lonsdale D (March 2006). “A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives”. Evidence-Based Complementary and Alternative Medicine. 3 (1): 49–59. doi:10.1093/ecam/nek009. PMC 1375232. PMID 16550223.
- ^ Jump up to:a b c d Tolerable Upper Intake Levels For Vitamins And Minerals (PDF), European Food Safety Authority, 2006, archived (PDF) from the original on 16 March 2016
- ^ Jump up to:a b Bettendorff L (2020). “Thiamine”. In BP Marriott, DF Birt, VA Stallings, AA Yates (eds.). Present Knowledge in Nutrition, Eleventh Edition. London, United Kingdom: Academic Press (Elsevier). pp. 171–88. ISBN 978-0-323-66162-1.
- ^ Jump up to:a b c Tanphaichitr V (1999). “Thiamin”. In Shils ME, Olsen JA, Shike M, et al. (eds.). Modern Nutrition in Health and Disease(9th ed.). Baltimore: Lippincott Williams & Wilkins.
- ^ “Archived copy” (PDF). Archived (PDF) from the original on 14 February 2012. Retrieved 18 March 2011.
- ^ “Vitamin B1 (Thiamine)”. Medicine LibreTexts. 12 May 2017.
- ^ Luczak M (1968). “Changes occurring in milk powder subjected to gamma rays”. Zeszyty Problemowe Postepow Nauk Rolniczych. 80(497–501).Chem Abstr 1969;71,2267g
- ^ Syunyakova ZM, Karpova IN (1966). “The effect of γ-rays and thermal sterilization on the content of thiamine, riboflavine, nicotinic acid, and tocopherol in beef”. Vop Pitan. 25 (2): 52–5. Chem Abstr1966;65,1297b
- ^ Webb ME, Marquet A, Mendel RR, Rébeillé F, Smith AG (October 2007). “Elucidating biosynthetic pathways for vitamins and cofactors”. Natural Product Reports. 24 (5): 988–1008. doi:10.1039/b703105j. PMID 17898894.
- ^ Begley TP, Chatterjee A, Hanes JW, Hazra A, Ealick SE (April 2008). “Cofactor biosynthesis–still yielding fascinating new biological chemistry”. Current Opinion in Chemical Biology. 12(2): 118–25. doi:10.1016/j.cbpa.2008.02.006. PMC 2677635. PMID 18314013.
- ^ Bocobza SE, Aharoni A (October 2008). “Switching the light on plant riboswitches”. Trends in Plant Science. 13 (10): 526–33. doi:10.1016/j.tplants.2008.07.004. PMID 18778966.
- ^ “Thiamin content per 100 grams; select food subset, abridged list by food groups”. United States Department of Agriculture, Agricultural Research Service, USDA Branded Food Products Database v.3.6.4.1. 17 January 2017. Archived from the original on 2 February 2017. Retrieved 27 January 2017.
- ^ “Thiamin, Food sources”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 2013. Archived from the original on 2 February 2017. Retrieved 27 January 2017.
- ^ Jump up to:a b c Institute of Medicine (1998). “Thiamin”. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. pp. 58–86. ISBN 978-0-309-06554-2. Archived from the original on 16 July 2015. Retrieved 29 August2017.
- ^ “Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies” (PDF). 2017. Archived (PDF) from the original on 28 August 2017.
- ^ Kominiarek MA, Rajan P (November 2016). “Nutrition Recommendations in Pregnancy and Lactation”. The Medical Clinics of North America. 100 (6): 1199–1215. doi:10.1016/j.mcna.2016.06.004. PMC 5104202. PMID 27745590.
- ^ “Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels. FR page 33982” (PDF). Archived (PDF) from the original on 8 August 2016.
- ^ “Daily Value Reference of the Dietary Supplement Label Database (DSLD)”. Dietary Supplement Label Database (DSLD). Retrieved 16 May 2020.
- ^ “Changes to the Nutrition Facts Label”. U.S. Food and Drug Administration (FDA). 27 May 2016. Retrieved 16 May 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Industry Resources on the Changes to the Nutrition Facts Label”. U.S. Food and Drug Administration (FDA). 21 December 2018. Retrieved 16 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ McGuire M, Beerman KA (2007). Nutritional Sciences: From Fundamentals to Foods. California: Thomas Wadsworth.
- ^ Annemarie Hoogendoorn, Corey Luthringer, Ibrahim Parvanta and Greg S. Garrett (2016). “Food Fortification Global Mapping Study” (PDF). European Commission. pp. 121–128.
- ^ Hayes KC, Hegsted DM. Toxicity of the Vitamins. In: National Research Council (U.S.). Food Protection Committee. Toxicants Occurring Naturally in Foods. 2nd ed. Washington DCL: National Academy Press; 1973.
- ^ Bettendorff L, Mastrogiacomo F, Kish SJ, Grisar T (January 1996). “Thiamine, thiamine phosphates, and their metabolizing enzymes in human brain”. Journal of Neurochemistry. 66 (1): 250–8. doi:10.1046/j.1471-4159.1996.66010250.x. PMID 8522961. S2CID 7161882.
- ^ Molecular mechanisms of the non-coenzyme action of thiamin in brain: biochemical, structural and pathway analysis : Scientific Reports Archived 31 July 2015 at the Wayback Machine
- ^ Butterworth RF (2006). “Thiamin”. In Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ (eds.). Modern Nutrition in Health and Disease (10th ed.). Baltimore: Lippincott Williams & Wilkins.
- ^ Makarchikov AF, Lakaye B, Gulyai IE, Czerniecki J, Coumans B, Wins P, et al. (July 2003). “Thiamine triphosphate and thiamine triphosphatase activities: from bacteria to mammals”. Cellular and Molecular Life Sciences. 60 (7): 1477–88. doi:10.1007/s00018-003-3098-4. PMID 12943234. S2CID 25400487.
- ^ Lakaye B, Wirtzfeld B, Wins P, Grisar T, Bettendorff L (April 2004). “Thiamine triphosphate, a new signal required for optimal growth of Escherichia coli during amino acid starvation”. The Journal of Biological Chemistry. 279 (17): 17142–7. doi:10.1074/jbc.M313569200. PMID 14769791.
- ^ Bettendorff L, Wirtzfeld B, Makarchikov AF, Mazzucchelli G, Frédérich M, Gigliobianco T, et al. (April 2007). “Discovery of a natural thiamine adenine nucleotide”. Nature Chemical Biology. 3(4): 211–2. doi:10.1038/nchembio867. PMID 17334376.
- ^ Jump up to:a b Frédérich M, Delvaux D, Gigliobianco T, Gangolf M, Dive G, Mazzucchelli G, et al. (June 2009). “Thiaminylated adenine nucleotides. Chemical synthesis, structural characterization and natural occurrence”. The FEBS Journal. 276 (12): 3256–68. doi:10.1111/j.1742-4658.2009.07040.x. PMID 19438713. S2CID 23313946.
- ^ McCollum EV. A History of Nutrition. Cambridge, Massachusetts: Riverside Press, Houghton Mifflin; 1957.
- ^ Eijkman C (1897). “Eine Beriberiähnliche Krankheit der Hühner”[A disease of chickens which is similar to beri-beri]. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 148 (3): 523–532. doi:10.1007/BF01937576. S2CID 38445999.
- ^ “The Nobel Prize and the Discovery of Vitamins”. nobelprize.org.
- ^ Grijns G (1901). “Over polyneuritis gallinarum” [On polyneuritis gallinarum]. Geneeskundig Tijdschrift voor Nederlandsch-Indië (Medical Journal for the Dutch East Indies). 41 (1): 3–110.
- ^ Suzuki U, Shimamura T (1911). “Active constituent of rice grits preventing bird polyneuritis”. Tokyo Kagaku Kaishi. 32: 4–7, 144–146, 335–358. doi:10.1246/nikkashi1880.32.4.
- ^ Funk, Casimir (1911). “On the chemical nature of the substance which cures polyneuritis in birds induced by a diet of polished rice”. The Journal of Physiology. 43 (5): 395–400. doi:10.1113/jphysiol.1911.sp001481. PMC 1512869. PMID 16993097.
- ^ Funk, Casimir (1912). “The etiology of the deficiency diseases. Beri-beri, polyneuritis in birds, epidemic dropsy, scurvy, experimental scurvy in animals, infantile scurvy, ship beri-beri, pellagra”. Journal of State Medicine. 20: 341–368. The word “vitamine” is coined on p. 342: “It is now known that all these diseases, with the exception of pellagra, can be prevented and cured by the addition of certain preventative substances; the deficient substances, which are of the nature of organic bases, we will call “vitamines”; and we will speak of a beri-beri or scurvy vitamine, which means a substance preventing the special disease.”
- ^ Jansen BC, Donath WF (1926). “On the isolation of antiberiberi vitamin”. Proc. Kon. Ned. Akad. Wet. 29: 1390–1400.
- ^ Williams RR, Cline JK (1936). “Synthesis of vitamin B1“. J. Am. Chem. Soc. 58 (8): 1504–1505. doi:10.1021/ja01299a505.
- ^ Carpenter KJ (2000). “Beriberi, white rice, and vitamin B: a disease, a cause, and a cure”. Berkeley, CA: University of California Press.
- ^ Peters RA (1936). “The biochemical lesion in vitamin B1deficiency. Application of modern biochemical analysis in its diagnosis”. Lancet. 230 (5882): 1161–1164. doi:10.1016/S0140-6736(01)28025-8.
- ^ Lohmann K, Schuster P (1937). “Untersuchungen über die Cocarboxylase”. Biochem. Z. 294: 188–214.
- ^ Breslow R (1958). “On the mechanism of thiamine action. IV.1 Evidence from studies on model systems”. J Am Chem Soc. 80(14): 3719–3726. doi:10.1021/ja01547a064.
External links
- “Thiamine”. Drug Information Portal. U.S. National Library of Medicine.
| Skeletal formula and ball-and-stick model of the cation in thiamine | |
| Clinical data | |
|---|---|
| Pronunciation | /ˈθaɪ.əmɪn/ THY-ə-min |
| Other names | Vitamin B1, aneurine, thiamin |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: ThiamineUS FDA: Thiamine |
| Routes of administration | by mouth, IV, IM[1] |
| Drug class | vitamin |
| ATC code | A11DA01 (WHO) |
| Legal status | |
| Legal status | US: OTC |
| Pharmacokinetic data | |
| Bioavailability | 3.7% to 5.3%[medical citation needed] |
| Elimination half-life | 1.8d[2][better source needed] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 59-43-8 59-43-8 HCl: 67-03-8 |
| PubChem CID | 1130 |
| DrugBank | DB00152 |
| ChemSpider | 1098 |
| UNII | X66NSO3N35HCl: M572600E5P |
| KEGG | C00378 |
| ChEBI | CHEBI:18385 |
| ChEMBL | ChEMBL1547 |
| CompTox Dashboard (EPA) | DTXSID50220251 |
| Chemical and physical data | |
| Formula | C12H17N4OS+ |
| Molar mass | 265.35 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Bibliography
- Wikipedia: Beriberi; Christiaan Eijkman; Adolphe_Vorderman; Casimir_Funk; Rice Polishing; White rice; Thiamine; Thiamine_pyrophosphate; Citric Acid Cycle
- A. Bay, “Beriberi in Modern Japan: The Making of a National Disease”, University of Rochester Press (2012).
- K.J. Carpenter. Beriberi, White Rice and Vitamin B. University of California Press, 2000
- http://www.healthline.com/health/beriberi
- M.C. Latham, . “Chapter 16. Beriberi and thiamine deficiency” in Human nutrition in the developing world, 29 [Rome, Food and Agriculture Organization of the United Nations, 1997].
- D.-T. Nguyen-Khoa, Beriberi (Thiamine Deficiency) Treatment & Management
- M. Golden, Mike . “Diagnosing Beriberi in Emergency Situations”. Field Exchange 1 (1997) 18.
- Y. Itokawa, . “Kanehiro Takaki (1849–1920): A Biographical Sketch”. J. Nutrit. 106 (1976) 581–8.
- R. Breslow. “On the mechanism of thiamine action. IV.1 Evidence from studies on model systems”. J. Am. Chem. Soc. 80 (1958) 3719–3726.
- R.R. Williams, J.K. Cline,. “Synthesis of vitamin B1“. J. Am. Chem. Soc. 58 (1936) 1504–1505.
- T.P. Begley, A.Chatterjee, J.W. Hanes, A. Hazra, S.E. Ealick,. “Cofactor biosynthesis—still yielding fascinating new biological chemistry”. Curr. Opin. in Chem. Biol. 12 (2008) 118–125.
- L. Bettendorff, F. Mastrogiacomo, S.J. Kish, T. Grisar, “Thiamine, thiamine phosphates and their metabolizing enzymes in human brain”. J. Neurochem. 66 (1996) 250–258.
- B.C.P. Jansen, W.F. Donath, “On the isolation of antiberiberi vitamin”. Proc. Kon. Ned. Akad. Wet. 29 (1926) 1390–1400.
- C. Nordqvist, “What is Thiamin, or Vitamin B1?“, Medical News Today, (2016)
- Thiamin, NIH Fact Sheet for Health Professionals.
- Thiamine, Oregon State University
//////////THIAMINE, aneurin hydrochloride, vitamin b1
Cc2ncc(C[n+]1csc(CCO)c1C)c(N)n2
Tegoprazan
Tegoprazan
RN: 942195-55-3
UNII: W017G7IF4S
ROTATION (-)
Molecular Formula, C20-H19-F2-N3-O3, Molecular Weight, 387.3841
(S)-4-(5,7-difluorochroman-4-yloxy)-N,N,2-trimethyl-lH-benzo[d]imidazole-6-carboxamide).
- 1H-Benzimidazole-5-carboxamide, 7-(((4S)-5,7-difluoro-3,4-dihydro-2H-1-benzopyran-4-yl)oxy)-N,N,2-trimethyl-
- 7-(((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy)-N,N,2-trimethyl-1H-benzimidazole-5-carboxamide
- (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-carboxamide
HK inno.N/RaQualia Pharma
Alternative Names: CJ-12420; IN-A001; K-CAB; LXI-15028; RQ-00000004; RQ-4Тегопразан [Russian] [INN]تيغوبرازان [Arabic] [INN]替戈拉生 [Chinese] [INN]
- A novel Potassium-competitive acid blocker.
- OriginatorPfizer
- Tegoprazan, a reversible H+/K+-ATPase inhibitor developed by CJ Healthcare (now inno.N), was first approved and launched in South Korea in 2019 for the treatment of gastroesophageal reflux disease (GERD).
- DeveloperCJ Cheiljedang Corp.; HK inno.N; RaQualia Pharma; Shandong Luoxin Pharmaceutical
- ClassAmides; Anti-inflammatories; Antibacterials; Antiulcers; Benzimidazoles; Benzopyrans; Fluorobenzenes; Small molecules
- Mechanism of ActionH(+) K(+)-exchanging ATPase inhibitors; Potassium-competitive acid blockers
- MarketedErosive oesophagitis; Gastro-oesophageal reflux
- Phase IIIGastric ulcer; Helicobacter infections; Peptic ulcer
- 28 Aug 2021No recent reports of development identified for phase-I development in Gastro-oesophageal-reflux in Japan (PO, Tablet)
- 28 Aug 2021No recent reports of development identified for phase-I development in Gastro-oesophageal-reflux in USA (PO, Tablet)
- 18 Aug 2021Shandong Luoxin Pharmaceutical Group plans a phase III trial for Duodenal ulcer in China (PO, Tablet) (NCT05010954)
PATENT
https://patents.google.com/patent/US20070142448A1/en
STEP 1: N-{4-Bromo-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide
- [0371]
To a solution of 4-bromo-2-nitro-6-[(phenylmethyl)oxy]aniline (33.0 g, 102 mmol, WO 2004054984) and acetic anhydride (14.5 mL, 153 mmol) in acetic acid (90 mL) was added concentrated sulfuric acid (2 drops) at 70° C. The mixture was stirred at 70° C. for 20 minutes. After cooling to room temperature, water (800 mL) was added, and the formed precipitate was collected by filtration, and washed with diisopropyl ether to give the title compound as a brown solid (30.9 g, 83%). - [0372]
1H NMR (CDCl3, 270 MHz) δ: 7.69 (d, J=2.0 Hz, 1H), 7.56 (br. s, 1H), 7.47-7.38 (m, 5H), 7.34 (d, J=2.0 Hz, 1H), 5.14 (s, 2H), 2.16 (s, 3H) ppm. - [0373]
MS (ESI) m/z: 365 (M+H)+.
STEP 2: N-{4-Cyano-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide
- [0374]
A mixture of N-{4-bromo-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (6.5 g, 17.8 mmol, STEP 1), zinc cyanide (4.18 g, 35.6 mmol), and tetrakis(triphenylphosphine)palladium (2.06 g, 1.78 mmol) in N,N-dimethylformamide (100 mL) was heated to 170° C. for 20 minutes in the microwave synthesizer (Biotage, Emrys Optimizer). After cooling to room temperature, the suspension was filtered, and washed with ethyl acetate. The organic layers were combined, washed with water, dried over magnesium sulfate, and concentrated in vacuum. The residual solid was purified by column chromatography on silica gel eluting with hexane/ethyl acetate (3:1) to afford the title compound as a white solid (5.5 g, 99%). - [0375]
1H NMR (CDCl3, 300 MHz) δ: 7.92 (s, 1H), 7.83 (s, 1H), 7.53-7.33 (m, 5H), 7.39 (s, 1H), 5.21 (s, 2H), 2.21 (s, 3H) ppm. - [0376]
MS (ESI) m/z: 312 (M+H)+, 310 (M−H)−.
STEP 3: 2-Methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carbonitrile
- [0377]
A mixture of N-{4-cyano-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (5.5 g, 17.7 mmol, STEP 2) and iron powder (2.96 g, 53.0 mmol) in acetic acid (90 mL) was refluxed with stirring for 2 hours. After cooling to room temperature, the mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was poured into water, and the aqueous layer was extracted with ethyl acetate/methanol (20:1). The organic layers were combined, washed with brine, dried over magnesium sulfate, and concentrated in vacuum to afford the title compound as a brown solid (3.82 g, 82%). - [0378]
1H NMR (DMSO-d6, 300 MHz) δ: 7.64 (s, 1H), 7.64-7.27 (m, 6H), 7.19 (s, 1H), 5.34 (s, 2H), 2.50 (s, 3H) ppm. - [0379]
MS (ESI) m/z: 264 (M+H)+, 262 (M−H)−.
STEP 4: 2-Methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxylic Acid
- [0380]
A solution of 2-methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carbonitrile (3.82 g, 14.5 mmol, STEP 3) and potassium hydroxide (85%, 10.2 g, 15.4 mmol) in ethylene glycol (50 mL) was heated to 170° C. for 20 minutes in the microwave synthesizer (Biotage, Emrys Optimizer). After cooling to room temperature, the mixture was acidified with 2M hydrochloric acid aqueous solution (pH=3). The precipitated solid was collected by filtration to afford the title compound as a white solid (3.83 g, 93%). - [0381]
1H NMR (DMSO-d6, 270 MHz) δ: 12.68 (br. s, 1H), 7.74 (s, 1H), 7.64-7.01 (m, 7H), 5.33 (s, 2H), 2.50 (s, 3H) ppm. - [0382]
MS (ESI) m/z: 283 (M+H)+, 281 (M−H)−.
STEP 5: N,N,2-Trimethyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide
- [0383]
A mixture of 2-methyl-4-[(phenylmethyl)oxy-1H-benzimidazole-6-carboxylic acid (5.0 g, 17.7 mmol, STEP 4), dimethylamine hydrochloride (4.33 g, 53.1 mmol), 2-[1H-benzotriazole-1-yl]-1,1,3,3-tetramethyluronium hexafluorophosphate (10.1 g, 26.6 mmol), and triethylamine (10.7 g, 106 mmol) in N,N-dimethylformamide (80 mL) was stirred at room temperature for 1 hour. The mixture was diluted with ethyl acetate/methanol (20:1), and washed with saturated ammonium chloride aqueous solution. The organic layer was dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from ethyl acetate only to ethyl acetate methanol 5:1) to afford the title compound as a white solid (4.90 g, 89%). - [0384]
1H NMR (CDCl3, 270 MHz) δ: 7.47-7.23 (m, 5H), 7.20 (s, 1H), 6.75 (s, 1H), 5.22 (s, 2H), 2.95 (br. s, 6H), 2.54 (s, 3H) ppm (—NH was not observed). - [0385]
MS (ESI) m/z: 310 (M+H)+, 308 (M−H)−.
STEP 6: N,N,2-Trimethyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide
- [0386]
To a suspension of N,N,2-trimethyl-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (928 mg, 3.0 mmol, STEP 5) in N,N-dimethylformamide (20 mL) was added sodium hydride (60% in mineral oil, 180 mg, 4.50 mmol) at 0° C. After stirring at room temperature for 30 minutes, the reaction mixture was cooled to 0° C. To the mixture was added 4-methylbenzenesulfonyl chloride (572 mg, 3.00 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 2 hours. The mixture was poured into water, and the aqueous layer was extracted with ethyl acetate. The organic layers were combined, washed with water, dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from dichloromethane only to ethyl acetate only) to afford the title compound as a white solid (1.00 g, 72%). - [0387]
1H NMR (CDCl3,270 MHz) δ: 7.80 (d, J=8.1 Hz, 2H), 7.70 (s, 1H), 7.45 (d, J=831 Hz, 2H), 7.40-7.22 (m, 5H), 6.86 (s, 1H), 5.32 (s, 2H), 3.11 (br. s, 3H), 2.89 (br s, 3H), 2.81 (s, 3H), 2.40 (s, 3H) ppm. - [0388]
MS (ESI) m/z: 464 (M+H)+.
STEP 7: 4-Hydroxy-N N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide
- [0389]
A mixture of N,N,2-trimethyl-1-(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (350 mg, 0.756 mmol, STEP 6) and 20% palladium hydroxide (1.20 g) in acetic acid (20 mL) was stirred under hydrogen gas (4 atmospheres) for 4 hours. The resulted mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from ethyl acetate only to ethyl acetate:methanol 5:1) to afford the title compound as a white solid (131 mg, 36%). - [0390]
1H NMR (CDCl3, 270 MHz) δ: 7.82 (d, J=8.1 Hz, 2H), 7.63 (s, 1H), 7.31 (d, J=8.1 Hz, 2H), 6.92 (s, 1H), 3.14 (br. s, 3H), 3.01 (br. s, 3H), 2.79 (s, 3H), 2.40 (s, 3H) ppm (—OH was not observed). - [0391]
MS (ESI) m/z: 374 (M+H)+, 372 (M−H)−.
STEP 8: 4-[(5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamideSTEP 8-1: 5,7-Difluoro-3,4-dihydro-2H-chromen-4-ol
- [0392]
To a solution of 5,7-difluoro-2,3-dihydro-4H-chromen-4-one (14.2 g, 77.0 mmol, US 20050038032) in methanol (200 mL) was added sodium borohydride (3.50 g, 92.5 mmol) at 0° C. The reaction mixture was stirred at the same temperature for 1 hour, and evaporated to remove methanol. The residue was quenched with water, and extracted with ethyl acetate. The extract was washed with brine, dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on silica gel (hexane:ethyl acetate=3:1 as an eluent) to afford the title compound as a pale gray solid (9.64 g, 67%). - [0393]
1H NMR (CDCl3, 270 MHz) δ: 6.47-6.36 (m, 2H), 5.05-4.97 (m, 1H), 4.36-4.20 (m, 2H), 2.16-1.92 (m, 3H) ppm.
STEP 8-2: 4-[(5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-methylphenyl)sulfo nyl]-1H-benzimidazole-6-carboxamide
- [0394]
To a stirred mixture of 4-hydroxy-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide (110 mg, 0.294 mmol, STEP 7), 5,7-difluoro-3,4-dihydro-2H-chromen-4-ol (164 mg, 0.884 mmol, STEP 8-1) and triphenylphosphine (232 mg, 0.884 mmol) in toluene (5 mL) was added diisopropyl azodicarboxylate (DIAD) (179 mg, 0.884 mmol) at room temperature. The reaction mixture was stirred at room temperature for 6 hours and concentrated in vacuum. The residue was purified by column chromatography on silica gel (ethyl acetate:hexane gradient elution from 1:20 to 10:1) to afford a mixture of the title compound and triphenylphosphine oxide (280 mg, crude) as white solids, which was used in the next step without further purification. - [0395]
1H NMR (CDCl3, 270 MHz) δ: 7.81 (d, J=8.1 Hz, 2H), 7.51 (s, 1H), 7.31 (d, J=8.1 Hz, 2H), 7.07 (s, 1H), 6.54-6.22 (m, 2H), 5.93 (br. s, 1H), 4.40 (t, J=10.8 Hz, 1H), 4.27 (t, J=10.8 Hz, 1H), 3.15 (br. s, 3H), 3.03 (br. s, 3H), 2.79 (s, 3H), 2.39 (s, 3H), 2.40-2.21 (m, 1H), 2.19-1.73 (m, 1H) ppm. - [0396]
MS (ESI) m/z: 542 (M+H)+, 540 (M−H)−.
STEP 9: 4-[(5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide
- [0397]
To a solution of 4-[(5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy-N,N,2-trimethyl-1-[(4-methylphenyl)-sulfonyl]-1H-benzi midazole-6-carboxamide (280 mg, crude, STEP 8) in tetrahydrofuran (8 mL) and methanol (4 mL) was added sodium hydroxide (165 mg, 4.1 mmol) at room temperature. After stirring at room temperature for 1 hour, the mixture was quenched with saturated sodium dihydrogenphosphate aqueous solution, and extracted with ethyl acetate. The organic layers were combined, dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (gradient elution from dichloromethane only to ethyl acetate:methanol 10:1) to afford the title compound as a white solid (74 mg, 65% for 2 steps). - [0398]
1H NMR (CDCl3, 270 MHz) δ: 7.27 (s, 1H), 6.95 (s, 1H), 6.51-6.33 (m, 2H), 5.87-5.69 (m, 1H), 4.41-4.25 (m, 2H), 3.10 (br. s, 6H), 2.56 (s, 3H), 2.44-2.34 (m, 1H), 2.14-1.98 (m, 1H) ppm (—NH was not observed). - [0399]
MS (ESI) m/z: 388 (M+H)+, 386 (M−H)−.
Example 2(−)-4-[((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1 fl-benzimidazole-6-carb oxamide andExample 3(−)-4-]((4R)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benximidazole-6-carboxamide
- [0400]
- [0401]
The fraction-1 (582 mg) and fraction-2 (562 mg) were prepared from racemic 4-[(5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N, 1,2-trimethyl-1H-benzimidazole-6-carboxamide (1.63 g, STEP 9 in Example 1) by HPLC as follows. - Isolation Condition
- [0402]
Column: CHIRALCEL OJ-H (20 mm×250 mm, DAICEL) - [0403]
Mobile phase: n-Hexane/Ethanol/Diethylamine (95/5/0.1) - [0404]
Flow fate: 18.9 mL/min
(−)-4-[((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide (fraction-1)
- [0405]
1H NMR: spectrum data were identical with those of the racemate - [0406]
optical rotation: [α]D 23=−101.1° (c=1.00, Methanol) - [0407]
retention time: 14 min
(+)-4-[((4R)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide (fraction-2)
- [0408]
1H NMR: spectrum data were identical with those of the racemate - [0409]
optical rotation: [α]D 23=+104.2° (c=1.00, Methanol) - [0410]
retention time: 18 min - The following is the alternative method for synthesizing (−)-4-[((4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide.
STEP 1: 6-Bromo-2-methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole
- [0411]
A mixture of N-{4-bromo-2-nitro-6-[(phenylmethyl)oxy]phenyl}acetamide (120 g, 329 mmol, STEP 1 in Example 1) and iron powder (55.1 g, 986 mmol) in acetic acid (500 mL) was refluxed with stirring for 6 hours. After cooling to room temperature, the mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum. The residue was diluted with ethyl acetate (1.5 L). The resulted precipitates were filtered through a pad of Celite, and washed with ethyl acetate (500 mL). The filterate was concentrated in vacuum, and the residue was diluted with ethyl acetate (200 mL). The brine (800 mL) was added to the organic mixture, the resulted white precipitates were collected by filtration, and washed with water (200 mL) and diethyl ether (200 mL). The white solid was dissolved with dichloromethane/methanol (10:1, 1.0 L), dried over magnesium sulfate, and concentrated. The solid was triturated with diethyl ether (300 mL), collected by filtration, and dried in vacuum to afford the title compound as a white solid (54.7 g, 53%). - [0412]
1H NMR (DMSO-d6, 270 MHz) δ: 7.63-7.28 (m, 7H), 5.38 (s, 2H), 2.69 (s, 3H) ppm. (NH was not observed.) - [0413]
MS (ESI) m/z: 317 (M+H)+, 315 (M−H)−.
STEP 2: 6-Bromo-2-methyl-1-[(4-methylphenyl)sulfonyl]-4-[(Phenylmethyl)oxy]-1H-benzimidazole
- [0414]
To a suspension of 6-bromo-2-methyl-4-[(phenylmethyl)oxy]-1H-benzimidazole (79.2 g, 250 mmol, STEP 1) in N,N-dimethylformamide (500 mL) was added sodium hydride (60% in mineral oil, 12.0 g, 300 mmol) at 0° C. After stirring at room temperature for 20 minutes, the reaction mixture was cooled to 0° C. To the mixture was added 4-methylbenzenesulfonyl chloride (47.6 g, 250 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 30 minutes. The mixture was quenched with water (800 mL), and the white precipitates were collected by filtration, washed with diisopropyl ether (500 mL), and dried in vacuum at 70° C. for 7 hours to afford the title compound as a white solid (116 g, 98%). - [0415]
1H NMR (DMSO-d6, 270 MHz) δ: 7.98 (d, J=8.1 Hz, 2H), 7.64 (d, J=1.9 Hz, 1H), 7.53-7.34 (m, 7H), 7.22 (d, J=1.9 Hz, 1H), 5.28 (s, 2H), 2.74 (s, 3H), 2.38 (s, 3H) ppm. - [0416]
MS (ESI) m/z: 471 (M+H)+, 469 (M−H)−.
STEP 3: N,N,2-Trimethyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide
- [0417]
A mixture of 6-bromo-2-methyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole (53.0 g, 112 mmol, STEP 2) and tetrakis(triphenylphosphine)palladium(0) (25.9 g, 22.4 mmol) in 2M dimethylamine tetrahydrofuran solution (580 mL) was stirred at 65° C. under carbon mono-oxide gas (1 atmosphere) for 32 hours. The mixture was cooled to room temperature, and diluted with ethyl acetate (600 mL). The organic mixture was washed with saturated ammonium chloride aqueous solution (800 mL) and brine (500 mL), dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on silica gel (hexane:ethyl acetate gradient elution from 1:2 to 1:3) to afford the title compound as a white solid (21.8 g, 42%). - [0418]
1H NMR: spectrum data were identical with STEP 6 in Example 1.
STEP 4: 4-Hydroxy-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide
- [0419]
A mixture of N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-4-[(phenylmethyl)oxy]-1H-benzimidazole-6-carboxamide (29.0 g, 62.6 mmol, STEP 3) and 10% palladium on carbon (6.0 g) in tetrahydrofuran (200 mL) was stirred under hydrogen gas (1 atmosphere) at room temperature for 24 hours. Another 4.0 g of 10% palladium on carbon was added, and the mixture was stirred under hydrogen gas (1 atmosphere) at room temperature for additional 6 hours. The resulted mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuum to afford the title compound as a white solid (23.0 g, 98%). - [0420]
1H NMR: spectrum data were identical with STEP 7 in Example 1.
STEP 5: Methyl 3-(3,5-difluorophenoxy)acrylate
- [0421]
A solution of 3,5-difluorophenol (35.5 g, 273 mmol) and methyl propiolate (25.0 mL, 300 mmol) in acetonitrile (109 mL) was added to a solution of tetrabutylammonium fluoride in tetrahydrofuran (1.0 M commercial solution, 109 mL, 109 mmol) at room temperature over a period of 2 hours. After complete addition of the solution, the mixture was stirred for 1 hour. The reaction mixture was diluted with toluene (350 mL) and the organic mixture was washed twice with water (250 mL×2), dried over magnesium sulfate, and concentrated in vacuum. The residue was purified by column chromatography on amino gel (hexane:ethyl acetate=3:2 as an eluent) to afford the title compound as a yellow solid (60.0 g, quant, 1:1 mixture of cis- and trans-isomers). - [0422]
1H NMR (CDCl3, 270 MHz,) δ: 7.72 (d, J=10.8 Hz, 0.5H), 6.83 (d, J=5.4 Hz, 0.5H), 6.74-6.49 (m, 3H), 5.68 (d, J=10.8 Hz, 0.5H), 5.28 (d, J=5.4 Hz, 0.5H), 3.76 (s, 3H) ppm.
STEP 6: Methyl 3-(3,5-difluorophenoxy)propanoate
- [0423]
A mixture of methyl 3-(3,5-difluorophenoxy)acrylate (60.0 g, 280 mmol, STEP 5), and 10% palladium on carbon (1.0 g) in methanol (300 mL) was stirred under hydrogen gas (1 atmosphere) at room temperature for 18 hours. The reaction mixture was filtered through a pad of Celite, and washed with toluene (100 mL). The filtrate was concentrated in vacuum to afford the title compound (61.0 g, quant) as a colorless oil, which was used in the next step without further purification. - [0424]
1H NMR (CDCl3, 270 MHz) δ: 6.56-6.21 (m, 3H), 4.21 (t, J=5.4 Hz, 2H), 3.74 (s, 3H), 2.80 (t, J=5.4 Hz, 2H) ppm.
STEP 7: 5,7-Difluoro-2,3-dihydro-4H-chromen-4-one
- [0425]
A mixture of methyl 3-(3,5-difluorophenoxy)propanoate (11.6 g, 53.7 mmol, STEP 6) and trifluoromethanesulfonic acid (23.2 mL, 2.0 mL/g of substrate) was stirred at 80° C. for 2 hours. After cooling to room temperature, the reaction mixture was diluted with water (120 mL), and extracted with toluene (120 mL). The organic layer was washed successively with aqueous solution of potassium carbonate (50 mL), water (50 mL), and dried over magnesium sulfate. The organic mixture was concentrated in vacuum to afford the title compound (8.75 g, 88%) as a white solid, which was used in the next step without further purification. - [0426]
1H NMR (CDCl3, 270 MHz) δ: 6.51-6.40 (m, 2H), 4.55-4.50 (m, 2H), 2.86-2.75 (m, 2H) ppm.
STEP 8: (+)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-ol
- [0427]
To a mixture of 1 M (S)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c][1,3,2]oxazaborole toluene solution (5.43 mL, 5.43 mmol) and tetrahydrofuran (40 mL) was added 2M borane-methyl sulfide complex tetrahydrofuran solution (29.8 mL, 59.7 mmol) at 0° C. and the mixture was stirred for 20 minutes. To the mixture was added a solution of 5,7-difluoro-2,3-dihydro-4H-chromen-4-one (10.0 g, 54.3 mmol, STEP 7) in tetrahydrofuran (70 mL) at 0° C. over a period of 1 hour, and the mixture was stirred at the same temperature for 1 hour. The reaction mixture was quenched with methanol (50 mL) and stirred for 30 minutes at room temperature. The mixture was concentrated in vacuum and the residue was purified by column chromatography on silica gel (hexane:ethyl acetate=4:1 as an eluent) to afford crude white solids (8.85 g, 86% ee). The solids were recrystallized from hexane (300 mL) to give the title compound as a colorless needle crystal (5.90 g, 58%, >99% ee). - [0428]
1H NMR: spectrum data were identical with those of the racemate (STEP 8-1 in Example 1). - [0429]
optical rotation: [α]D 24=+143.6° (c=1.00, Methanol).
STEP 9: (−)-4-[((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-methylphenyl) sulfonyl]-1H-benzimidazole-6-carboxamide
- [0430]
To a stirred mixture of 4-hydroxy-N,N,2-trimethyl-1-[(4-methylphenyl)sulfonyl]-1H-benzimidazole-6-carboxamide (21.2 g, 56.8 mmol, STEP 4), (+)-5,7-difluoro-3,4-dihydro-2H-chromen-4-ol (15.86 g, 85.1 mmol, STEP 8) and tributylphosphine (22.9 g, 113 mmol) in toluene (840 mL) was added 1,1′-(azodicarbonyl)dipiperidine (ADDP) (19.3 g, 76.5 mmol) at room temperature. After stirring at room temperature for 2 hours, the reaction mixture was filtered through a pad of Celite and washed with ethyl acetate (300 mL). The filtrate was concentrated in vacuum. The residue was purified by column chromatography on silica get (ethyl acetate:hexane gradient elution from 1:20 to 20:1) to afford crude solids (27.0 g). The solids were recrystallized from 2-propanol (130 mL) to give the title compound as a colorless crystal (23.2 g, 75%, >99% ee) - [0431]
1H NMR: spectrum data were identical with those of the racemate (STEP 8-2 in Example 1). - [0432]
optical rotation: [α]D 24=80.4° (c=0.50, Methanol).
STEP 10: (−)-4-[((4S)-5,7-Difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1H-benzimidazole-6-carboxamide
- [0433]
To a solution of (−)-4-[((4S)-5,7-difluoro-3,4-dihydro-2H-chromen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-methylphenyl)-sulfonyl]-1H-benzimidazole-6-carboxamide (24.2 g, 44.7 mmol, STEP 9) in tetrahydrofuran (65 mL) and 2-propanol (220 mL) was added 2M sodium hydroxide aqueous solution (220 mL, 440 mmol) at room temperature. After stirring at room temperature for 4 hours, the mixture was diluted with ethyl acetate (1.20 L) and washed with saturated ammonium chloride aqueous solution (500 mL). The organic solution was dried over magnesium sulfate and concentrated in vacuum. The residue was purified by column chromatography on amino gel (ethyl acetate:methanol gradient elution from 50:1 to 20:1) to afford the title compound as a white solid (15.2 g, 87%, >99% ee). - [0434]
1H NMR: spectrum data were identical with those of the racemate (STEP 9 in Example 1). - [0435]
Optical rotation and retention time were identical with the above.
PATENT
Tegoprazan is the world’s first potassium-competitive acid blocker (P-CAB), has a mechanism similar to that of an acid pump antagonist (APA), and blocks gastric acid secretion by competing with potassium ions for binding to the enzyme H+/K+– ATPase (proton pump) that secretes H+ ions, which are a component of gastric acid, from the gastric parietal cells into the gastric lumen. Since tegoprazan is not a prodrug such as a proton pump inhibitor (PPI), it does not require an activation process, and thus acts not only on an active proton pump but also on an inactive proton pump. Thus, tegoprazan has the advantages of exhibiting its effect rapidly and reaching the maximum effect within one hour.
Meanwhile, in general, in order for a drug to exhibit an expected effect, the blood concentration of the drug needs to be maintained at a certain level or higher. To maintain the blood concentration of the drug, a patient is required to take the prescribed drug repeatedly according to a certain schedule.
In this case, taking the drug frequently decreases the patient’s medication compliance, and as a result, there are many cases where the expected therapeutic effect is not obtained. Thus, in a disease for which a drug needs to be taken for a long period of time or the blood concentration of the drug at a time when the patient cannot take the drug needs to be maintained at a certain level or higher, the frequency and method of taking the drug is also an important factor to be considered for increasing the therapeutic effect of the drug.
Accordingly, there is a need to develop a formulation capable of maintaining a therapeutically effective blood concentration of a drug because there is no problem in the absorption rate of the drug while modifying the release of the drug.
DISCLOSURE
PATENT
Gastric acid-related gastrointestinal diseases, such as gastroesophageal reflux disease, non-erosive reflux disease, gastric ulcers, and ulcers caused by non-steroidal anti-inflammatory drugs are the most common diseases of the gastrointestinal tract. Histamine 2 receptor blockers and proton pump inhibitors (PPIs) are used in the treatment of the above symptoms, showing good curative effects and greatly improving the quality of life of patients. However, the degree of satisfaction with existing drugs for the treatment of gastrointestinal diseases related to gastric acid is still not high. For example, during the process of taking proton pump inhibitors, the symptoms of heartburn and esophageal reflux at night are still difficult to overcome, and the related symptoms cannot be effectively relieved 3 days before taking the medicine.
Potassium ion competitive acid blocker (P-CAB) is a new mechanism of H + -K + -ATPase inhibitor, which is a reversible proton pump inhibitor. Currently on the market are Revaprazan, Vonoprazan and Tegoprazan.
Tegoprazan’s chemical name is (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo [d] Imidazole-6-carboxamide, the structure is shown in formula (1):
| Both WO2007072146 and CN101341149B disclose two synthetic methods of Tegoprazan: |
| Method one (milligram preparation method): |
WO2007072146 and CN101341149B quote the synthesis method of WO2004054984 to prepare A-3 compound, then acetylate under concentrated sulfuric acid/acetic anhydride, introduce cyano group through microwave reaction to obtain A-5 compound, and then undergo reduction, ring closure, hydrolysis, condensation, and Toluenesulfonyl protection, ether hydrogenolysis, Mitsunobu reaction (Mitsunobu reaction) to obtain A-11 compound, after hydrolysis to remove the p-toluenesulfonyl protecting group to obtain A-12 compound, namely Tegorazan racemate, and finally through a chiral column Split to obtain Tegorazan with optical activity.
This synthetic route requires 12 steps of reactions (not including the preparation of 5,7-difluorochroman-4-ol), and the synthesis yield is only 2.0%; zinc cyanide is used in the reaction, which requires special treatment of wastewater; In the reaction, the protecting group (benzyl protection, p-toluenesulfonyl protection) and the removal of the protecting group need to be carried out twice. Suitable for industrial production.
Method two (ten-gram preparation method):
The obtained A-4 compound is reduced and fused under the condition of iron powder/acetic acid to obtain A-13 compound, which is protected by p-toluenesulfonyl, amidation, and debenzyl protection to obtain A-10 compound, and finally combined with a chiral alcohol The Tegorazan precursor is obtained by the Mitsunobu reaction, and then the Tegorazan is obtained by hydrolysis to protect it.
Although method 2 has been shortened compared with method 1, the synthetic route still requires 9-step reaction (excluding the preparation of chiral alcohol), the route is still longer, and the total yield is 6.8%; carbon monoxide gas is used in the reaction to pass through the coupling Co-preparation of amides requires special equipment to carry out the reaction, which poses a safety hazard; two protective groups (benzyl protection, p-toluenesulfonyl protection) and two removal of protective groups are still required in the reaction, and the reaction steps are also added. This results in low synthesis efficiency, which is not conducive to industrial production.
The comparative document CN101341149B discloses the preparation method of compound 5, that is, the tetrahydrofuran solution of 5,7-difluorochroman-4-one is added to the chiral reagent (S)-1-methyl-3,3- Diphenyl-1H,3H-pyrrolo[1,2-c][1,3,2]oxazolborane, borane-dimethyl sulfide complex and tetrahydrofuran in a mixed solution, wait until the reaction is complete After purification by column chromatography, the chiral purity was 86% ee, and then recrystallized with hexane to obtain compound 5, the optical purity of which was >99% ee, and the yield was 58%.
The comparative document CN107849003A discloses the preparation methods of compounds 3 and 5, that is, 5,7-difluorochroman-4-one is used as a raw material for reduction with a chiral ruthenium catalyst, and the yield of compound 3 is 85%. The purity is 100% ee, the yield of the obtained compound 5 is 91%, and the chiral purity is 100% ee. This method involves ruthenium reagents that are difficult to purchase commercially and are expensive.
Patent EP2390254A1 discloses the preparation method of compound 2, which uses 3-fluoro-4nitrobenzoic acid in dichloromethane with oxalyl chloride and N,N-dimethylformamide to obtain acid chloride after concentration, and then the obtained The acid chloride is dissolved in dichloromethane, and then added dropwise to a mixed solution containing dimethylamine hydrochloride and triethylamine for preparation, and the purification method adopts column chromatography for purification.
| Example 1 |
| Preparation of (S)-5,7-difluorochroman-4-ol (3) |
| |
| Take a 2L three-necked flask, add anhydrous THF (400mL) and R-Me-CBS (1mol/L toluene solution, 53mL, 53mmol), protect with argon, and inject borane dimethyl sulfide complex at room temperature (10mol/L, 58.6mL, 586mmol). 5,7-Difluorochroman-4-one (98g, 533mmol) was dissolved in anhydrous tetrahydrofuran (600mL), and slowly dripped into the above system. The entire dripping process lasted 9 hours. After dripping, let it stand overnight. The reaction solution was slowly poured into methanol cooled in an ice-water bath to generate a large number of bubbles, stirred until no obvious bubbles were generated, and concentrated to remove the solvent. Add 350 mL of ethyl acetate to dissolve, wash the organic phase with water (200 mL, 200 mL) and brine (100 mL) successively, dry over anhydrous sodium sulfate, filter, and concentrate to obtain a pale yellow solid. The chiral purity measured by chiral HPLC was 94.18%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1 mL/min, detection wavelength 220nm). |
| The above solid was heated and dissolved in a mixed solvent composed of n-hexane and ethyl acetate (n-hexane/ethyl acetate = 17:1), decolorized with activated carbon and then cooled and crystallized to obtain 77.8 g of off-white solid with a yield of 78.5% . [α] D 23 = -141.4 (c = 1, MeOH). The chiral purity measured by chiral HPLC is >99.9%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1mL/min, detection wavelength 220nm). |
| 1 HNMR(400MHz, CDCl 3 )δ: 6.46-6.34 (m, 2H), 5.00 (t, J=2.8Hz, 1H), 4.36-4.19 (m, 2H), 2.11-1.91 (m, 3H). |
| Example 2 |
| Preparation of (R)-5,7-difluorochroman-4-ol (5) |
| |
| Take a 1L three-necked flask, add anhydrous THF (66mL) and S-Me-CBS (1mol/L toluene solution, 9mL, 9mmol), protect with argon, and inject borane dimethyl sulfide complex at room temperature (10mol/L, 9.9mL, 99mmol). Dissolve 5,7-difluorochroman-4-one (16.6 g, 90 mmol) in anhydrous tetrahydrofuran (166 mL) and slowly drip into the above system. The entire dripping process lasted 5.5 hours. After dripping, let it stand overnight. The reaction solution was slowly poured into methanol cooled in an ice-water bath to generate a large number of bubbles, stirred until no obvious bubbles were generated, and concentrated to remove the solvent. Add 100 mL of ethyl acetate to dissolve, wash the organic phase with water (50 mL, 30 mL) and brine (20 mL) successively, dry over anhydrous sodium sulfate, filter, and concentrate to obtain an oil, which is placed at room temperature as a yellow solid. The chiral purity measured by chiral HPLC was 93.6%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1mL/min, detection wavelength 220nm). |
| The above solid was heated and dissolved in a mixed solvent consisting of n-hexane and ethyl acetate (n-hexane/ethyl acetate=17:1), and 11.1 g of needle crystals were obtained by recrystallization, with a yield of 66.5%. [α] D 20 = +141.9 (c=1, MeOH). The chiral purity measured by chiral HPLC is >99.9%ee (OZ-H column, n-hexane/isopropanol=95/5, flow rate=1 mL/min, detection wavelength 220nm). |
| 1 HNMR(400MHz, CDCl 3 )δ: 6.46-6.34 (m, 2H), 5.00 (t, J=2.8Hz, 1H), 4.36-4.19 (m, 2H), 2.11-1.91 (m, 3H). |
| Example 3 |
| Preparation of 3-fluoro-N,N-dimethyl-4-nitrobenzamide (2) |
| |
| Suspend 3-fluoro-4-nitrobenzoic acid (60g, 324mmol) in dichloromethane (400mL), add DMF (1mL), cool in an ice water bath, add oxalyl chloride (33mL, 389mmol) dropwise, after the addition is complete Incubate and stir for 2.5h. Dimethylamine hydrochloride (26.4g, 324mmol) was added to it, the temperature was lowered to -10°C, and a mixed solution composed of triethylamine (118mL, 842mmol) and dichloromethane (120mL) was added dropwise. After the addition was completed, the temperature was kept and stirred for 20 minute. Wash with 1 mol/L hydrochloric acid (100 mL), water (50 mL, 100 mL, 100 mL), half-saturated sodium bicarbonate solution (100 mL), and brine (100 mL) in sequence. It was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to remove most of the solvent. About 100 mL was left. 300 mL of n-hexane was added to make slurry, filtered, and washed twice with 100 mL of n-hexane, and dried to obtain 62.5 g of light yellow solid with a yield of 91.0%. |
| 1 H NMR(400MHz, CDCl 3 )δ8.10(dd,J=7.2Hz,8.4Hz,1H), 7.38-7.29(m, 2 H), 3.12(s, 3H), 2.97(s, 3H). |
| Example 4 |
| Preparation of 3-hydroxy-N,N-dimethyl-4-nitrobenzamide (4) |
| |
| Put 3-hydroxy-4-nitrobenzoic acid (20.58g, 112mmol), dimethylamine hydrochloride (9.2g, 112mmol), EDCI (23.6g, 123mmol), HOBt (15.1g, 112mmol) in 1L In the reaction flask, acetonitrile (250 mL) was added, followed by triethylamine (31.2 mL, 224 mmol), and the mixture was stirred at room temperature overnight. Concentrate to remove acetonitrile, add water (250mL), extract 8 times with dichloromethane, 150mL each time, combine the organic phases and wash with saturated sodium bicarbonate solution twice, 200mL each time, and then wash once with saturated brine (100mL) , Dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 19.7 g of yellow solid with a yield of 83.9%. |
| 1 HNMR(400MHz, CDCl 3 )δ: 10.63 (brs, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.18 (d, J = 1.6 Hz, 1H), 7.01 (dd, J = 1.6 Hz, 8.4 Hz, 1H), 3.12 (s,3H), 2.97(s,3H). |
| Example 5 |
| Preparation of 3-hydroxy-N,N-dimethyl-4-nitrobenzamide (4) |
| |
| Place 3-hydroxy-4-nitrobenzoic acid (9.15g, 50mmol) in a 500mL reaction flask, add dichloromethane (100mL), and then add 1 drop of DMF. After cooling in an ice water bath, add dropwise oxalyl chloride (5.1mL, 60mmol). Heat to reflux for 1 hour, and concentrate to remove the solvent. Add dichloromethane (100 mL) to dissolve into a solution for later use. Take another reaction flask, add 50 mL of dichloromethane and 20 mL of 33% dimethylamine aqueous solution, and cool in an ice-water bath. Add the dichloromethane solution of acid chloride dropwise to the above system while stirring, and stir for 10 minutes after dropping. The dichloromethane layer was separated, and the aqueous phase was extracted with dichloromethane 6 times, 100 mL each time. The organic phases were combined and washed with saturated brine (60 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to obtain 10 g of yellow solid. The yield was 94.9%. |
| 1 HNMR(400MHz,DMSO-d 6 )δ: 11.29 (brs, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.08 (s, 1H), 6.96 (dd, J = 0.8 Hz, 8.0 Hz, 1H), 2.99 (s, 3H) , 2.98(s, 3H). |
| Example 6 |
| (S)-3-((5,7-Difluorochroman-4-yl)oxy)-N,N-dimethyl-4-nitrobenzamide (6) |
| |
| Compound potassium tert-butoxide (0.44g, 3.9mmol) was dissolved in anhydrous tetrahydrofuran (9mL), protected by argon, cooled in an ice water bath, and compound 3 (0.61g, 3.3mmol) in anhydrous tetrahydrofuran solution (3 mL) was added dropwise ), keep and stir for 10 minutes after the addition is complete, add dropwise an anhydrous tetrahydrofuran solution (3 mL) of compound 2 (636 mg, 3 mmol), and after the addition is complete, keep and keep stirring for 10 minutes. Add 10mL of water, extract twice with ethyl acetate, 20mL each time, combine the organic phases, wash with brine, dry with anhydrous sodium sulfate, filter, and concentrate to obtain a yellow oil, add n-hexane to make a slurry, filter, and dry to obtain 1.0g off-white Solid, the yield is 90.9%. |
| 1 H NMR(400MHz, CDCl 3 )δ7.81(d,J=8.0Hz,1H),7.40(d,J=1.2Hz,1H), 7.11(dd,J=1.2Hz,8.0Hz,1H),6.52-6.33(m,2H) , 5.64 (brs, 1H), 4.48-4.32 (m, 2 H), 3.14 (s, 3H), 2.99 (s, 3H), 2.36-2.24 (m, 1H), 2.14-2.02 (m, 1H). |
| Example 7 |
| (S)-3-((5,7-Difluorochroman-4-yl)oxy)-N,N-dimethyl-4-nitrobenzamide (6) |
| Compound potassium tert-butoxide (41g, 368mmol) was dissolved in anhydrous tetrahydrofuran (500mL), protected by argon, cooled in an ice water bath, compound 3 (57.9g, 311mmol) in anhydrous tetrahydrofuran solution (250 mL) was added dropwise. After the addition was completed, the mixture was kept and stirred for 10 minutes, and an anhydrous tetrahydrofuran solution (250 mL) of compound 2 (60 g, 283 mmol) was added dropwise. After the addition, the mixture was kept and stirred for 10 minutes. Add 200 mL of ice water, concentrate to remove the organic solvent, add 800 mL of water, and extract four times with ethyl acetate, 500 mL each time. Combine the obtained organic phases, wash with half-saturated brine (1L), saturated brine (500mL), dry with anhydrous sodium sulfate, filter, and concentrate to obtain a brown oil. Pour 50mL of isopropanol while hot, and add petroleum ether (500mL). ) Be beaten, filter, wash twice with a mixture of isopropanol/petroleum ether=10/100, 100mL each time, and then wash twice with a mixture of isopropanol/petroleum ether=5/100, 100mL each time Finally, it was washed with petroleum ether (100 mL) once, and left to dry at room temperature to obtain compound 6, 97.3 g of pale yellow solid, with a yield of 91.0%. |
| 1 H NMR(400MHz, CDCl 3 )δ7.81(d,J=8.0Hz,1H),7.40(d,J=1.2Hz,1H), 7.11(dd,J=1.2Hz,8.0Hz,1H),6.52-6.33(m,2H) , 5.64 (brs, 1H), 4.48-4.32 (m, 2 H), 3.14 (s, 3H), 2.99 (s, 3H), 2.36-2.24 (m, 1H), 2.14-2.02 (m, 1H). |
| Example 8 |
| (S)-3-((5,7-Difluorochroman-4-yl)oxy)-N,N-dimethyl-4-nitrobenzamide (6) |
| |
| Dissolve compound 4 (1g, 4.76mmol), compound 5 (0.93g, 5mmol), and triphenylphosphine (1.5g, 5.71mmol) in anhydrous ethyl acetate (25mL), protected by argon, and cooled in an ice water bath. A mixed solution consisting of DIAD (1.1 mL, 5.71 mmol) and anhydrous ethyl acetate (1.5 mL) was added dropwise, and the mixture was stirred for 2 hours after dropping. Anhydrous zinc chloride (0.86 g, 6.3 mmol) was added, and after stirring for 1 hour, the insoluble matter was removed by filtration, and the filter cake was washed twice with 10 mL of ethyl acetate. The filtrate was washed once with a mixed solution of ammonia water (2.5 mL) and water (20 mL), then washed with water (30 mL) once, washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain an oily substance. Add isopropanol (2.4 mL) to dissolve, slowly add n-hexane (24 mL) dropwise, and stir at room temperature for 1 hour, stir and heat to 80 degrees for 30 minutes, cool down and stir overnight. Filtered to obtain 1.86g of white solid (containing hydrazine-1,2-dicarboxylic acid diisopropyl ester), chiral purity>99%ee (OZ-H chiral column, flow rate 1mL/min, detection wavelength 254nm, normal hexane Alkyl-isopropanol=80mL-20mL, temperature 28°C) was used directly in the next step without further purification. |
| A small amount of crude product was purified by silica gel column chromatography (0~2% ethyl acetate in dichloromethane solution), the nuclear magnetic data is: 1 HNMR (400MHz, CDCl 3 )δ7.82(d,J=8.0Hz,1H),7.40(d,J=1.6Hz,1H),7.12 (dd,J=1.6Hz,8.4Hz,1H),6.52-6.29(m,2H) , 5.64(brs,1H), 4.47-4.30(m,2H), 3.13(s,3H), 3.00(s,3H), 2.34-2.26(m,1H), 2.14-2.23(m,1H). |
| Example 9 |
| (S)-3-((5,7-Difluorochroman-4-yl)oxy)-N,N-dimethyl-4-nitrobenzamide (6) |
| Compound 4 (210mg, 1mmol), compound 5 (186mg, 1mmol), triphenylphosphine (314 mg, 1.2mmol) were dissolved in anhydrous THF (5mL), protected by argon, cooled in an ice water bath, and then DIAD ( A mixed solution consisting of 236 μL, 1.2 mmol) and anhydrous THF (0.3 mL) was dripped and stirred for 5 hours. Concentrate and purify by silica gel column chromatography (0-2% ethyl acetate in dichloromethane solution). Compound 6 was obtained, with a total of 339 mg of off-white solid, with a yield of 89.7%. |
| Example 10 |
| Preparation of (S)-4-amino-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylformamide (7) |
| |
| The compound 6 (1.86 g) obtained in Example 8 was dissolved in methanol (60 mL), and dry palladium on carbon (10% palladium on carbon, 194 mg) was added. The mixture was stirred at room temperature under normal pressure for 12 hours in a hydrogen atmosphere, filtered, washed with methanol, and the filtrate was concentrated. A purple solid was obtained, and 25 mL of isopropyl ether was added for beating to obtain 1.3 g of a slightly pink solid. The yield of the two steps was 78.3%. |
| 1 H NMR(400MHz, CDCl 3 )δ7.17 (d, J = 1.6 Hz, 1H), 6.96 (dd, J = 1.6 Hz, 8.0 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.49-6.37 (m, 2H) ,5.51(brs,1H),4.41-4.23(m,2H),4.15-3.77(brs,2H),3.07(s,6H),2.37-2.26(m,1H),2.08-1.93(m,1H) ). |
| Example 11 |
| Preparation of (S)-4-amino-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylformamide (7) |
| The compound 6 (96 g, 254 mmol) obtained in Example 7 was dissolved in a mixed solution (500 mL) composed of methanol/tetrahydrofuran = 1/4, and 50% water content wet palladium on carbon (10% supported on carbon, 19.2 g) was added. Shake hydrogenation at ~25psi pressure. After 3 hours, it was filtered, the filtrate was concentrated to a slurry, 300 mL of isopropyl ether was added to make a slurry, and dried to obtain compound 7, 78 g of an off-white solid, with a yield of 88.6%. |
| 1 H NMR(400MHz, CDCl 3 )δ7.17(d,J=1.6Hz,1H), 6.96(dd,J=1.6,8.0Hz, 1H), 6.69(d,J=8.0Hz,1H), 6.52-6.35(m,2H), 5.51(brs,1H),4.42-4.23(m,2H), 4.21-3.76(brs,2H),3.07(s,6H),2.35-2.27(m,1H),2.08-1.94(m,1H). |
| Example 12 |
| Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8) |
| |
| Compound 7 (174mg, 0.5mmol), potassium phosphate (127mg, 0.6mmol) were suspended in dichloromethane (5mL), and 2,2,2-trichloroethylacetimide hydrochloride (9-1, 135mg , 0.6mmol), stirred at room temperature for 24h. Add 5 mL of saturated potassium carbonate solution and 15 mL of ethyl acetate and stir for 5 minutes, separate the organic phase, and extract the aqueous phase twice with ethyl acetate, each time 10 mL. The organic phases were combined, washed with brine, dried with anhydrous sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (methanol/ammonia/dichloromethane=1/1/100~3/1/100) to obtain compound 8. 60mg pale yellow foamy solid, yield 30.0%. |
| HR-MS: [M+H] + : Measured value 390.1601 |
| Example 13 to Example 21 |
| Compound 7 (174mg, 0.5mmol) was used for feeding, referring to Example 12. The specific compound 9-1, base, solvent (5mL), ratio and yield of compound 8 used are shown in the following table: |
| ExampleCompound 9-1Ratio of compound 9-1 to compound 7/baseSolventYield (%)Example 130.6mmol1.2/ Disodium hydrogen phosphate dodecahydrateDichloromethane43.8Example 140.6mmol1.2/sodium carbonateDichloromethane51.5Example 150.6mmol1.2/sodium acetateDichloromethane69.4Example 160.6mmol1.2/sodium acetateEthyl acetate72.0Example 170.6mmol1.2/sodium acetateChloroform86.0Example 180.6mmol1.2/sodium acetateEthanol30.8Example 190.6mmolNo alkaliDichloromethane51.5Example 200.75mmol1.5/sodium acetateDichloromethane88.6Example 211.0mmol2.0/sodium acetateDichloromethane100.0 |
| Example 22 |
| Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8) |
| Compound 7 (1.2g, 3.4mmol) was suspended in dichloromethane (14mL), sodium acetate (367mg, 4.5mmol) and 2,2,2-trichloroethylacetimide hydrochloride ( 500mg, 2.3mmol), add three batches, and stir for 5 hours after the addition. Extract 4 times with water, 15 mL each time, combine the water phases, and backwash the water phase with isopropyl ether (25 mL) once. The resulting aqueous phase was adjusted to alkaline with potassium carbonate (2g), extracted with ethyl acetate (20mL, 15mL, 10mL), the organic phases were combined and washed once with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain compound 8, 1.3 g White foamy solid, yield 98.5%. |
| 1 H NMR(400MHz, CDCl 3 )δ: 7.24 (s, 1H), 7.11 (d, J = 8.0 Hz, 1H), 6.91 (brs, 1 H), 6.49-6.30 (m, 2H), 5.43 (s, 1H), 4.47-4.24 ( m,3H),3.07(brs,6H),2.26-2.15(m,1H),1.94-1.81(m,1H). |
| Example 23 |
| Preparation of (S)-4-iminoacetamido-3-((5,7-difluorochroman-4-yl)oxy)-N,N-dimethylbenzamide (8) |
| |
| Compound 7 (1.66g, 4.76mmol) was dissolved in dichloromethane (14mL), sodium acetate (390mg, 4.76mmol) and ethylacetimide hydrochloride (9-2, 440mg, 3.57mmol) were added every 1 hour ), add a total of four batches, and stir for 1 hour after the addition. Concentrate to remove dichloromethane, add 35 mL of water, extract 3 times with ethyl acetate, 15 mL each time, and discard. The aqueous phase was adjusted to alkaline with potassium carbonate (1.3g), extracted with ethyl acetate (30 mL, 20 mL, 20 mL, 10 mL), the organic phases were combined and washed with brine once, dried over anhydrous sodium sulfate, filtered, and concentrated to obtain the compound 8. 1.37g white foam, yield 74.0%. |
| Example 24 |
| (S)-4-((5,7-Difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-carboxamide (1 ) Preparation |
| |
| Compound 8 (1.3 g, 3.4 mmol) was dissolved in acetonitrile (13 mL), cooled to 5° C. in an ice-water bath, N-chlorosuccinimide (454 mg, 3.4 mmol) was added in batches, and the mixture was kept warm and stirred for 35 minutes. A solution containing sodium hydroxide (0.68 g, 17 mmol) and water (4 mL) was added, and the mixture was stirred at room temperature for 2 hours. Concentrate to remove acetonitrile, add 25mL of water, adjust the pH to about 3-4 with 1mol/L hydrochloric acid solution (17mL), extract the resulting aqueous solution with ethyl acetate (25mL, 25mL, 20mL), and then further distill the organic solvent from the aqueous phase. Adjust the pH to 8 with saturated sodium bicarbonate solution, and a white solid can be precipitated. After suction filtration, washing with water, and drying, 0.94 g of off-white solid was obtained with a yield of 72.3%. [α] D 24 = -97.8 (c = 1, MeOH). |
| HR-MS: [M+H] + C 20 H 20 F 2 N 3 O 3 The calculated value is 388.1467, and the measured value is 388.1470. |
| 1 H NMR(400MHz, DMSO-d 6 )δ12.57(brs,1H),7.15(s,1H),6.95(s,1H), 6.88-6.78(m,1H),6.74-6.67(m,1H),6.04(s,1H), 4.41 -4.33 (m, 1H), 4.30-4.20 (m, 1H), 2.98 (s, 6H), 2.46 (s, 3H), 2.30-2.19 (m, 1H), 2.14-2.01 (m, 1H). |
| 1 H NMR(400MHz, CDCl 3 )δ: 7.19 (s, 1H), 6.91 (s, 1H), 6.48-6.29 (m, 2H), 5.76 (brs, 1H), 4.40-4.18 (m, 2H), 3.11 & 3.04 (br, 6H) ), 2.47(s,3H),2.36-2.26(m,1H), 2.08-1.94(m,1H). |
| Example 25 |
| (S)-4-((5,7-Difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-carboxamide(1) Preparation |
| Compound 8 (1.5g, 3.9mmol) was dissolved in 2,2,2-trifluoroethanol (19mL), cesium carbonate (1.38g, 4.25mmol) was added, cooled in an ice water bath, and diacetyl iodobenzene (1.37g, 4.25mmol), keep stirring for 40 minutes, add water, extract twice with ethyl acetate, wash with brine, dry with anhydrous sodium sulfate, filter, and concentrate to obtain an oily substance, which is subjected to silica gel column chromatography (3-4% methanol in dichloromethane solution ) 0.6 g of off-white foamy solid was obtained, with a yield of 40.3%. |
| 1 H NMR(400MHz, CDCl 3 )δ: 7.19 (s, 1H), 6.91 (s, 1H), 6.48-6.29 (m, 2H), 5.76 (brs, 1H), 4.40-4.18 (m, 2H), 3.11 & 3.04 (br, 6H) ), 2.47(s,3H), 2.36-2.26(m,1H), 2.08-1.94(m,1H). |
PATENT
| Tegorazan, also known as Tegoprazan, Tegoprazan, CJ-12420, was approved by the Korean Ministry of Food and Drug Safety (MFDS) in July 2018 for the treatment of gastroesophageal reflux disease and erosive esophagitis . |
| Tegoprazan was originally developed by Pfizer. In 2008, it was licensed to RaQualia Pharma (from Pfizer) for cooperative development. In 2014, it was licensed by RaQualia Pharma to CJ Health Care. Finally, CJ Health Care was successfully developed and marketed in Korea. Tegoprazan is a competitive potassium ion acid blocker (P-CAB) and hydrogen ion/potassium ion exchange ATPase (H + /K + ATPase) inhibitor. It has a fast onset and can control the pH of gastric juice for a long time. The drug was first launched in South Korea and is a brand new drug for the treatment of gastroesophageal reflux disease and erosive esophagitis. |
| Gastric proton pump hydrogen ion/potassium ion exchange ATPase is the main pharmacological target for the treatment of gastric acid-related diseases. Potassium Competitive Acid Blocker (P-CAB) can inhibit gastric acid secretion by competitively binding to K + H + /K + -ATPase. Studies have found that Tegoprazan is such a potassium-competitive acid blocker, which is considered to be the most advanced drug for the treatment of gastroesophageal reflux disease, because proton pump inhibitors are the most commonly used drugs for the treatment of gastroesophageal reflux disease, and Tegoprazan It just can overcome the shortcomings of proton pump inhibitors. The effectiveness and safety of Tegoprazan are mainly based on two phase III clinical trials. One of them is a double-blind, actively controlled phase III study (NCT02456935), which was conducted in South Korea, with 280 patients with erosive esophagitis as the research object, and the cumulative healing rate of erosive esophagitis at the 8th week as the primary endpoint. To compare the safety and effectiveness of Tegoprazan and the proton pump inhibitor esomeprazole. Another phase III clinical trial is a double-blind, randomized, placebo-controlled trial (NCT02556021). The trial was conducted in 324 patients in South Korea. The primary endpoint was the percentage of patients whose main symptoms (heartburn and reflux) completely resolved at 4 weeks using the reflux disease questionnaire (RDQ) to evaluate the once-daily Tegoprazan tablet ( 50mg and 100mg) in the safety and effectiveness of patients with non-erosive reflux disease. The approval of the drug on the market provides a new option for the treatment of this type of disease, and to a certain extent makes up for the shortcomings of other drugs, so that this type of disease can be better treated. |
| Tegoprazan chemical name is (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-methan Amide, the chemical structure contains a benzimidazole structure and a chiral 5,7-difluorochroman-4-oxyl structure, the specific chemical structure is as follows: |
| |
| Patent CN101341149B discloses the preparation method of Tegoprazan, specifically 4-hydroxy-N,N,2-trimethyl-1-[(4-tolyl)sulfonyl]-1H-benzo[d]imidazole-6-methan Amide and (S)-5,7-difluoro-3,4-dihydro-2H-chromenen-4-ol undergo condensation reaction under the action of tributylphosphine/ADDP to prepare (-)-4- [((4S)-5,7-Difluoro-3,4-2H-chromogen-4-yl)oxy]-N,N,2-trimethyl-1-[(4-tolyl) Sulfonyl]-1H-benzo[d]imidazole-6-carboxamide intermediate, the latter removes the protective group under the action of a base to complete the preparation of Tegoprazan. The specific synthesis route is as follows: |
| |
| Based on the description of the above patent, the preparation of Tegoprazan mainly involves 4-hydroxy-N,N,2-trimethyl-1-[(4-tolyl)sulfonyl]-1H-benzo[d]imidazole-6- The condensation reaction of formamide and (S)-5,7-difluoro-3,4-dihydro-2H-chromenen-4-ol, this condensation reaction not only involves the use of dangerous reagents tributylphosphine and coupling Nitrogen compounds with low yield and high cost. |
| Therefore, the development of a new synthetic method suitable for industrialization and cost-effective synthesis of Tegoprazan and its analogs can not only reduce the risk of industrial production of Tegoprazan, but also provide more analogs for potential drugs with higher activity. Research. |
| The synthetic route of the present invention is as follows: |
| |
| Example 1: 4-[((4S)-5,7-difluoro-3,4-2H-chromogen-4-yl)oxy]-2-methyl-1-p-toluenesulfonyl-1H -Preparation of benzo[d]imidazole-6-carboxylic acid tert-butyl ester |
| The 4-chloro-2-methyl-1-p-toluenesulfonyl-1H-benzo[d]imidazole-6-carboxylic acid tert-butyl ester (42.10g, 0.10mol), (S)-5,7-two Fluoro-3,4-dihydro-2H-chromenen-4-ol (28.0g, 0.15mol), copper acetate (1.0g, 5.0mmol), potassium tert-butoxide (17.0g, 0.152mol) and N 1 ,N 2 -Bis (naphthalene-1-ylmethyl)oxalamide (3.7g, 10.05mmol) was added to the reaction flask, followed by nitrogen replacement three times, and then anhydrous 1,4-dioxide was added to the reaction flask Six rings (150 mL), the reaction system was replaced with nitrogen again three times. Subsequently, the reaction system was heated to 100°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (500 mL), stirred vigorously for 0.5 hours, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. Add dichloromethane (1.0L) and H to the residue 2 O (400 mL), the system was stirred for 15 minutes, the organic phase was separated, the aqueous phase was extracted 3 times with dichloromethane (3×400 mL), the organic phases were combined, the solvent was removed from the organic phase under reduced pressure, and the residue was added to heptane (500 mL) Stir vigorously overnight and filter. The obtained solid compound is dried and recrystallized from ethyl acetate/heptane to obtain an off-white solid (42.83 g, 75.1%). |
| Example 2: (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,N,2-trimethyl-1H-benzo(d)imidazole-6-methan Preparation of Tegoprazan |
| Add 4-bromo-N,N,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide (2.82g, 10.0mmol), (S)-5,7-bis Fluoro-3,4-dihydro-2H-chromenen-4-ol (2.80g, 15mmol), cuprous iodide (100mg, 0.53mmol), sodium tert-butoxide (1.45g, 15.1mmol) and N 1 ,N 2 -Bis (phenylethyl)oxalamide (150mg, 0.51mmol) was added to the reaction flask, followed by nitrogen replacement three times, then anhydrous DMF (15mL) was added to the reaction flask, and the reaction system was replaced with nitrogen again three times. Subsequently, the reaction system was heated to 85°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (200 mL), stirred vigorously for 0.5 hours, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. The residue was purified by column chromatography (ethyl acetate/heptane) to obtain a white solid (3.32 g, 85.7%). |
| Example 3: (S)-4-((5,7-difluorochroman-4-yl)oxy)-N,1,2-trimethyl-1H-benzo(d)imidazole-6-methan Amide |
| Add 4-iodo-N,1,2-trimethyl-1H-benzo[d]imidazole-6-carboxamide (3.30g, 10.0mmol), (S)-5,7-difluoro to the reaction flask successively -3,4-Dihydro-2H-chromenen-4-ol (2.80g, 15mmol), cuprous iodide (60mg, 0.32mmol), sodium tert-butoxide (1.15g, 11.97mmol) and N 1 , N 2 -bis(benzyl)oxalyl diamide (135 mg, 0.50 mmol) was added to the reaction flask, followed by nitrogen replacement three times, then anhydrous DMF (15 mL) was added to the reaction flask, and the reaction system was again nitrogen replaced three times. Subsequently, the reaction system was heated to 75°C for 24 hours with stirring. After the reaction, the system naturally dropped to room temperature. The reaction system was diluted with ethyl acetate (200 mL), stirred vigorously for 1 hour, and filtered through Celite. The filtrate was desolventized under reduced pressure to remove the organic solvent. The residue was purified by column chromatography (ethyl acetate/heptane) to obtain an off-white solid (2.77 g, 71.5%). |
Patents
CN 112851646
CN 111303131,
US 20070142448
| //////////////////////////////////////////////////////////////////////////////////////////// |
Tegoprazan was approved by the Ministry of Food and Drug Safety (MFDS) for marketing in July 2018 for the treatment of gastroesophageal reflux disease and erosive esophagitis. Tegoprazan was originally developed by Pfizer. In 2008, it was licensed to RaQualiaPharma (separated from Pfizer) for joint development. In 2014, Tegoprazan was licensed to CJHealthCare by RaQualiaPharma. Finally, CJHealthCare was successfully developed and marketed in Korea. Tegoprazan is a competitive potassium ion acid blocker (P-CAB) and hydrogen ion/potassium ion exchange ATPase (H+/K+ATPase) inhibitor. The drug was first marketed in South Korea. Medicines for treating gastroesophageal reflux disease and erosive esophagitis. Proton pump hydrogen ion/potassium ion exchange ATPase is the main pharmacological target for the treatment of gastric acid-related diseases. Potassium-competitive acid blocker (P-CAB) can inhibit gastric acid secretion by competitively binding to K+ with H+/K+-ATPase. Research finds that Tegoprazan is such a potassium-competitive acid blocker and is considered to be the most advanced drug for treating gastroesophageal reflux disease, because proton pump inhibitors are the most commonly used drugs for treating gastroesophageal reflux disease. Tegoprazan The shortcomings of proton pump inhibitors can be just overcome. Tegoprazan’s effectiveness and safety are mainly based on two phase III clinical trials. One of them is a double-blind, active-controlled phase III study. This study was conducted in South Korea. The study used 280 patients with erosive esophagitis as the primary endpoint[1].
Fig 1. Chemical structure formula and three-dimensional structure of Tegoprazan
Tegoprazan, a potassium-competitive acid blocker, is a potent, oral active and highly selective inhibitor of gastric H+/K+-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H+/K+-ATPases in vitro.
Tegoprazan inhibits porcine, canine, and human H+/K+-ATPase activity. Tegoprazan inhibits gastric H+/K+-ATPase in a potassium-competitive and reversible manner. Tegoprazan (3 μM) inhibits 86% of H+/K+-ATPase activity, whereas the inhibition is decreased to 34% after the dilution of Tegoprazan concentration to 0.15 μM[2].
Tegoprazan (1.0 mg/kg, p.o.) potently and completely inhibits histamine-induced gastric acid secretion in dogs. Tegoprazan (1.0-3.0 mg/kg, p.o.) reverses the pentagastrin-induced acidified gastric pH to the neutral range. Tegoprazan (3 mg/kg, p.o.) immediately evokes a gastric phase III contraction of the migrating motor complex in pentagastrin-treated dogs[3].
The invention relates to a method for preparing Tegoprazan chiral alcohols, in particular to the preparation method of (S) 5,7 difluoro 3,4 dihydro 2H chromogenic ene 4 alcohol. Using 5,7-difluoro-4H-benzopyran-4-ketone as starting material, the method realizes the preparation of (S)5,7-difluoro-3,4-dihydro-2H-chromogenic enone-4-alcohol by asymmetric reduction of ketone carbonyl with chiral reagent and subsequent conventional hydrogenation reaction[4].
Tegoprazan, a reversible H+/K+-ATPase inhibitor developed by CJ Healthcare (now inno.N), was first approved and launched in South Korea in 2019 for the treatment of gastroesophageal reflux disease (GERD). In 2020, the product attained supplemental approval for the treatment of gastric ulcers and Helicobacter pylori infection. Additional phase III clinical trials are being conducted by Shandong Luoxin Pharmacy Group, CJ Healthcare’s Chinese licensee. Tegoprazan was originally developed by RaQualia and licensed to CJ CheilJedang (the parent company of CJ Healthcare) in 2010 in Southeastern Asian markets; this agreement was later extended to Europe and North America in 2019. In 2015, a Chinese sublicense was granted to Shandong Luoxin Pharmacy Group. CJ Healthcare was acquired by Kolmar Korea in 2018, and renamed as inno.N in 2020.

NEW DRUG APPROVALS
one time
$10.00
References
[1] Takahashi N, et al. Tegoprazan, a Novel Potassium-Competitive Acid Blocker to Control Gastric Acid Secretion and Motility. J Pharmacol Exp Ther. 2018 Feb;364(2):275-286.
[2] Nobuyuki Takahashi and Yukinori Take.Journal of Pharmacology and Experimental Therapeutics February 2018, 364 (2) 275-286.
[3] Kim HK, Park SH, Cheung DY, Cho YS, Kim JI, Kim SS, Chae HS, Kim JK, and Chung IS (2010) Clinical trial: inhibitory effect of revaprazan on gastric acid secretion in healthy male subjects. J Gastroenterol Hepatol 25:1618–1625.
Mikami T, Ochi Y, Suzuki K, Saito T, Sugie Y, and Sakakibara M (2008) 5-Amino-6-chloro-N-[(1-isobutylpiperidin-4-yl)methyl]-2-methylimidazo[1,2-α]pyridine-8-carboxamide (CJ-033,466), a novel and selective 5-hydroxytryptamine4 receptor partial agonist: pharmacological profile in vitro and gastroprokinetic effect in conscious dogs. J Pharmacol Exp Ther 325:190–199.
/////// tegoprazan, Тегопразан , تيغوبرازان , 替戈拉生 , CJ-12420, IN-A001, K-CAB, LXI-15028, RQ-00000004, RQ-4, CJ 12420, IN A001, K CAB, LXI 15028, RQ 00000004, RQ 4, korea 2019
CN(C)C(=O)c1cc(O[C@H]2CCOc3cc(F)cc(F)c23)c4[nH]c(C)nc4c1
FLUVATINIB

4-(2-Fluoro-3chloro-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
6-Quinolinecarboxamide, 4-[3-chloro-4-[[(cyclopropylamino)carbonyl]amino]-2-fluorophenoxy]-7-methoxy-
N-(4-(6-Aminocarbonyl-7-methoxyquinolin-4-yl)oxy-2-chloro-3-fluorophenyl)-N’-cyclopropylurea
cas 2304405-29-4
C21 H18 Cl F N4 O4
444.84CN109134365 discloses an active compound or medicinal salt with multi-target effects of VEGFR1~3, fibroblast growth factor receptor 1~3, RET, Kit and PDGFR, and its chemical structure formula is as follows: Formula I:
Chemical name: 4-(2-Fluoro-3chloro-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinoline carboxamide, the drug name is fluvatinib. The compound has strong activity and provides a potential new treatment option for patients with tumors such as liver and kidney.
PATENT
CN109134365
PATENT
WO 2020187188
https://patents.google.com/patent/WO2020187188A1/enProcess A

Example 1A

At 20-30°C, 4-chloro-7-methoxyquinoline-6-carboxamide (550.0 g) was added to the reaction kettle. At 20-30°C, DMSO (16.5L) was added to the reactor. At 20-30°C, 2-fluoro-3chloro-4-aminophenol was added to the reactor. At 20-35°C, sodium tert-butoxide (229g) was slowly added to the reaction kettle under stirring for 10-15 minutes. The reaction kettle was heated to 96°C (internal temperature) in 1.5 hours. The reaction was stirred at 96-100°C for 6.5 hours, and no 4-amino-3-chloro-2 fluorophenol remained. The reaction was cooled to 20-30°C. Under stirring, 23.1L of water was slowly added to the reaction solution. During the process, a dark brown solid was precipitated. Keep the internal temperature below 40°C. Stir at 30-40°C for 0.5 hour. Cool to 20-30°C and filter. At 20-30°C, the filter cake and 3.5L of water are added to the reactor. Stir for 0.5 hour at 20-30°C. filter. At 20-30°C, the filter cake and 4.0L of water are added to the reactor. Stir for 0.5 hour at 20-30°C. After filtering, the filter cake was dried in a vacuum dryer at 40°C for 18 hours (phosphorus pentoxide used as a desiccant, and the oil pump was vacuumed). The solid was pulverized to obtain 758 g of off-white solid and dried at 40° C. for 18 hours (phosphorus pentoxide was used as the desiccant, and the oil pump was vacuumed) to obtain Example 1A.LCMS(ESI)m/z:362.0[M+1] +1 H NMR (400MHz, DMSO-d 6 ) δppm 8.68 (br s, 2H), 7.82-7.96 (m, 1H), 7.67-7.82 (m, 1H), 7.46-7.59 (m, 1H), 7.12-7.26 (m, 1H), 6.67-6.80 (m, 1H), 6.43-6.58 (m, 1H), 5.84 (s, 2H), 4.04 (s, 3H).Example 1B

Example 1A (6.05g) was added to a three-necked flask containing NMP (60mL), pyridine (1.32g) and phenyl chloroformate (5.20g) were added to the reaction system, and the reaction system was at room temperature (25-30°C). ) After stirring for 1 hour, the reaction was complete. Cyclopropylamine (2.84g) was also added to the reaction system. The reaction solution was stirred at room temperature (25-30°C) for 0.5 hours. The reaction was completed. Add 20 mL of ethanol to the reaction system and stir. Tap water (500 mL) was added to the reaction system, a solid was precipitated, filtered, and the filter cake was spin-dried under reduced pressure to obtain a crude product (orange solid, 5.26 g); the crude product was passed through a chromatography column (DCM: MeOH = 20/1~10 /1) Purification to obtain the product (orange solid, 3.12 g), the product was added with 4 mL of absolute ethanol and stirred at room temperature for 18 hours, filtered, the filter cake was washed with 1 mL of ethanol, and dried under reduced pressure to obtain Example 1B. This compound is obtained by adding 1 equivalent of hydrochloric acid, sulfuric acid or methanesulfonic acid in acetone or ethanol solution to obtain the corresponding salt.LCMS(ESI)m/z:445.0[M+1] +1 H NMR (400MHz, DMSO-d 6 ) ppm 8.66-8.71 (m, 2H), 8.12-8.20 (m, 2H), 7.72-7.93 (m, 2H), 7.45 (t, J = 9.16 Hz, 1H) ,7.28(d,J=2.76Hz,1H),6.58(d,J=5.02Hz,1H),4.05(s,3H),2.56-2.64(m,1H),0.38-0.77(m,4H)Example 1

Example 1B (1.5g, 3.37mmol) was added to EtOH (45mL), the reaction temperature was raised to 60°C, at this temperature, CH 3 SO 3 H (324.07mg, 3.37mmol, 240.05μL) was added dropwise to the reaction In the solution, after the dripping is completed, the reaction solution is dissolved, and the temperature of the reaction solution is naturally cooled to 15-20°C under stirring, and the reaction solution is stirred at this temperature for 2 hours. A large amount of brown solid precipitated, filtered, and the filter cake was rinsed with absolute ethanol (5 mL), and the obtained filter cake was spin-dried under reduced pressure at 50° C. without purification, and Example 1 was obtained.LCMS(ESI)m/z:445.0[M+1] +1 H NMR(400MHz,DMSO-d 6 )δppm 9.02(d,J=6.53Hz,1H)8.72(s,1H)8.18-8.27(m,2H)7.87-8.03(m,2H)7.65(s,1H )7.53(t,J=9.03Hz,1H)7.32(br s,1H)7.11(d,J=6.27Hz,1H)4.08(s,3H)2.55-2.62(m,1H)2.35(s,3H) 0.34-0.75(m,4H)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021143954&tab=FULLTEXT&_cid=P12-KSZPW4-91508-1Example 1 Preparation of fluvatinib crystal form I
Add the free base of fluvatinib of formula I (50mg, 112.40umol) to EtOH (2mL), stir at 15-20℃ for 12h, filter to obtain a filter cake, add the filter cake to 200mL acetone, stir at 15-20℃ After 12h, filter and spin-dry the filter cake under reduced pressure at 40°C to obtain fluvatinib solid. The result of XRPD detection is shown in Figure 1, named as the crystalline form I of fluvatinib, and the detection results of DSC and TGA are shown in Figure 2. And Figure 3.
Example 2 Preparation of crystal form I of fluvatinib mesylate (also referred to herein as “fluvatinib mesylate”)
The 4-[3-chloro-4-(cyclopropylaminocarbonylamino)-2-fluoro-phenoxy]-7-methoxy-quinoline-6-carboxamide i.e. fluvatinib (0.5g, 1.12mmol) was added to EtOH (10mL) solvent, heated to 55~60℃, and methanesulfonic acid (108.02mg, 1.12mmol, 80.02μL, 1eq) was added to the reaction flask under stirring at this temperature, and the reaction solution was dissolved. , The reaction solution was cooled to 20 ~ 30 ℃, stirred at this temperature for 1 h, a brown solid precipitated out under vacuum filtration, the filter cake was rinsed with ethanol (2mL*2), and the filter cake was spin-dried at 40 ~ 50 ℃ under reduced pressure. The solid product, named as the crystalline form I of fluvatinib mesylate, was tested by XRPD, DSC, and TGA. The XRPD test results are shown in Table 1 and Figure 4 below, and the DSC and TGA test results are shown in Figure 5. Melting point is about 232-237°C.
/////////////
NC(=O)c1cc2c(ccnc2cc1OC)Oc1ccc(NC(=O)NC2CC2)c(Cl)c1F

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}