PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
To treat adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer after receiving a ROS1 kinase inhibitor
FDA 2026, APPROVALS 2026, Jideytro, NVL-520, NUV-520, NU-520, NVL 520, NUV 520, NU 520, MX5KQV5XHC
Zidesamtinib (sold under the brand name Jideytro) is an oral, highly selective, next-generation kinase inhibitor approved by the U.S. Food and Drug Administration (FDA) on July 22, 2026, to treat adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC) who have previously been treated with at least one ROS1 kinase inhibitor. Developed originally by Nuvalent and subsequently acquired by GSK, it represents a major milestone as GSK’s first approved therapeutic targeting lung cancer.
Mechanism of Action
Zidesamtinib functions by targeting and inhibiting the receptor tyrosine kinase c-ros oncogene 1 (ROS1). It is custom-engineered to solve the primary clinical challenges that limit previous therapies:
Overcoming Resistance Mutations: It binds tightly to wild-type ROS1 and remains robustly active against a broad array of treatment-emergent point mutants. This includes G2032R (the most common solvent-front resistance mutation), as well as S1986Y/F, L2026M, and D2033N mutations.
Blood-Brain Barrier Penetration: It features high central nervous system (CNS) penetrance to effectively treat and control brain metastases, which are frequent in aggressive ROS1-positive cancers.
TRK-Sparing Design: Unlike older dual-acting inhibitors, it deliberately avoids inhibiting the structurally similar tropomyosin receptor kinase (TRK) family. This minimizes off-target TRK-related neurological toxicities like severe dizziness and ataxia.
Clinical Trial Outcomes
The FDA approval was heavily supported by data from the ongoing global, single-arm, Phase 1/2 ARROS-1 clinical trial (N=117 heavily pretreated patients):
Overall Response: Delivered an Objective Response Rate (ORR) of 44% in patients who had exhausted alternative TKI options.
Subgroup Efficacy: Achieved a 51% ORR in patients who had received only one prior ROS1 inhibitor, a 54% ORR in those harboring the G2032R mutation, and an intracranial ORR of 48% for patients with active brain metastases.
Durability: Showed prolonged disease control, with a 12-month duration of response (DOR) rate standing at 69%.
Administration and Side Effects
Jideytro is formulated as an oral tablet taken once daily, with or without food. It demonstrates a highly tolerable safety profile, with only a 10% dose reduction rate and a 2% treatment discontinuation rate due to adverse events.
Common Adverse Reactions (≥ 15%): Edema (swelling), peripheral neuropathy, constipation, fatigue, and dyspnea (shortness of breath).
Warnings & Precautions: Includes risks of mild CNS reactions (dizziness, cognitive alterations), QTc interval prolongation, skeletal fractures, pancreatic toxicity, and interstitial lung disease (ILD)/pneumonitis.
Zidesamtinib, sold under the brand name Jideytro, is an anti-cancer medication used for the treatment of previously treated locally advanced or metastatic ROS1+ non-small cell lung cancer.[1][2][3] It is taken by mouth once daily.[1][2][3]
Medical uses
Indication
Zidesamtinib is a prescription medicine used to treat adults with non-small cell lung cancer that has spread within the chest or other parts of the body and is caused by an abnormal ROS1 gene, and who have received a ROS1 kinase inhibitor.[1][2][3]
Mechanism of action
Zidesamtinib is a kinase inhibitor that works by blocking ROS1, an abnormal protein that drives some lung cancers to grow, including forms that have become resistant to earlier ROS1 treatments.[4] Jideytro also works on the related proteins ALK and TRK. In laboratory and animal studies, Zidesamtinib stopped cancer cells with ROS1 changes from growing and slowed tumor growth, including tumors in the brain.[2]
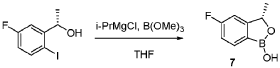
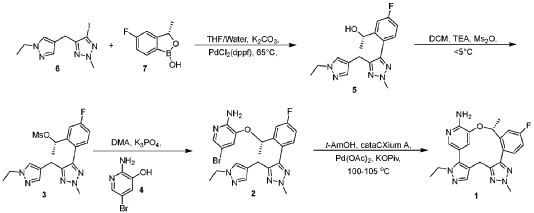
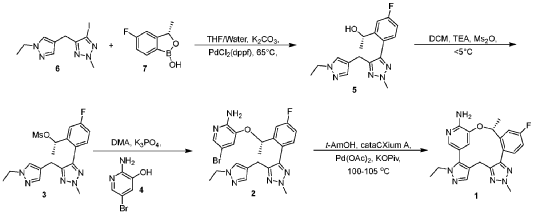
[00548] Synthesis of Compound 5. To a reactor was charged THF (10 vol ), water (1 vol ), followed by Compound 6 (850.0 g, 2.68 mol, 1 equiv.) and Compound 7 (534.0 g, 3.22 mol, 1.2 equiv ) at 20~30°C. The solids were completely dissolved at 20~30°C while stirring for 15 min and K2CO3 (1.11 kg, 3 equiv.) was added in portions over 10-15 min at 20~30°C. The reaction mixture was fully refdled with nitrogen, and was added Pd(dppf)C12 (78.5 g, 0.04 equiv.) in one portion under nitrogen. The reaction mixture was fully refdled with nitrogen again, then heated to 60-65 °C and stirred at 60~65°C for 16 h under nitrogen. The reaction mixture was cooled to 20~30°C, fdtered through a 10 cm celite pad (2X, 2.4 kg celite). The combined fdtrates were washed with EtOAc (10 vol., 21 L) and separated. The organic phase was washed with water (5 vol., 10.5 L) and separated. The organic phase was stirred for 1 h at 40-45°C in 5 w% aqueous L-cysteine (2.0 eq., 1.61 kg in 30.6 kg water) and separated. The organic phase was washed with water (5 vol., 10.5 L) and separated. The resulting organic phase was concentrated at 45-50°C in vacuum to afford crude product as a light brown oil (2.28 kg). To the crude product was charged MTBE (228 mL, 0.1 vol. relative to crude product), heated to 50°C over 15 min, followed by isopropyl ether (2.28 L, 1 vol.) dropwise over 1 h at 45~50°C, then cooled to 10°C over 2 h. A large amount of solids came out and the resulting slurry was stirred for 2 h at 10-15°C. The solids were collected by fdtration, dried in oven at 45°C for 16 h to get crude Compound 5 as a pale-yellow solid (1.67 kg, 96.3% /220 nm, >99.9%/220 nm chiral purity). 1.67 kg of crude Compound 5 was purified by silica gel chromatography (EtOAc/ n-heptane=l: 1, 2.5X silica gel, 100-200 meshes) to get Compound 5 as an off-white solid (1.58 kg, 99.6%/220 nm, >99.9%/220 nm chiral purity, 97.9 w%, 72% yield). H NMR (400 MHz, DMSO) 5 7.44 (dd, J = 10.5, 2.5
[00549] In another example, a similar procedure was run in a 0.5:2 biphasic mixture of toluene and water (2.5 vol.) with a catalystic amount (e.g. 0.002 mol equiv.) Pd(Amphos)C12 (instead of 0.04 mol equiv. of PdidppfhCE) used as the catalyst. Potassium phosphate (K3PO4 3 H2O) substituted potassium carbonate (K2CO3) 3.0 mol equiv. as the base, and the amount of Compound 7 employed was 1.02 mol equiv. The improved process was conducted at 50 °C. At the end of the reaction, the organic layer was fdtered and treated with activated carbon and concentrated, and the final material was crystallized from toluene/heptane/water to give Compound 5 in 92% yield and 99.9% purity.
[00550] Synthesis of Compound 3. To a 50 L reactor was charged dichloromethane (11.25 L), Compound 5 (750 g, >99.9%/220 nm chiral purity) and triethylamine (920.0 g) at r t. (20~30°C). The resulting mixture was refilled with nitrogen and cooled to 0°C. To it was added a solution of MS2O (793.0 g) in dichloromethane (3.75 L) drop-wise over 45 min while keeping the temperature at 0~5°C. The reaction mixture was stirred at 0~5°C for 1 h under nitrogen. The reaction mixture was quenched with cooled water (7.5 L) at 5~15°C and separated. The organic phase was washed with cooled water (3.75 L) and separated. The organic phase was dried over anhydrous Na2SC>4, filtered and concentrated at 25~30°C in vacuum to around 2 vol., then switched to n-heptane (2.25 L) and concentrated at 25~30°C in vacuum to around 2 vol. of Compound 3 in n-heptane. n-heptane /EtOAc (3.0 L, lOv/lv) was added to the above mixture and the mixture was slurried for 1 h at 0~10°C under nitrogen and filtered. The filter cake was washed with n-heptane (1.5 L), dried in vacuum at 25~30°C for 5 h to afford Compound 3 as an off-white solid (845 g, 98.9 w%, 99.98%/220 nm chiral purity, 91% yield). H NMR (400 MHz, CDC13) 5 7.35 (dd, J = 9.6, 2.5 Hz, 1H), 7.24 – 7.18 (m, 2H), 7.12 (s, 1H), 7.08 (td, J = 8.3, 2.6 Hz, 1H), 5.78 (d, J = 6.4 Hz, 1H), 4.21 (s, 3H), 4.05 (q, J = 7.3 Hz, 2H), 3.90 – 3.76 (m, 2H), 2.78 (s, 3H), 1.58 (d, J = 6.5 Hz, 3H), 1.40 (t, J = 7.3 Hz, 3H). MS (ESI, m/z): 408.20 (M + H)+.
[00551] In another example, triethylamine base (1.3 mol equiv.), MS2O (1.2 mol equiv.), and dichloromethane solvent (10 vol) were used. The reaction mixture was quenched with aqueous sodium bicarbonate to remove excess MS2O, and crystallization from dichloromenthane/hexane results in 98% yield with 100% purity of Compound 3.
[00552] Synthesis of Compound 2. A 20 L reactor was refilled with nitrogen, then charged with DMA (12.6 L) at r.t. (20~25°C) To the reactor was charged Compound 4 (390.0 g) and Compound 3 (840.0 g, 99.98%/220 nm chiral purity) in one portion at 20~25°C through a dry nitrogen flow. The reaction mixture was heated to 35°C over 15 min and stirred for 5-10 min at 35~40°C to get a clear solution. To the reaction mixture was charged powder K3PO4 (875.0 g) in one portion at 35~45°C. After complete addition, the resulting mixture was heated to 60°C over 20 min and stirred at 58~63°C for 1.5 h through a dry nitrogen flow. The reaction mixture was cooled to 25~30°C, filtered through a celite pad (5 cm, 1.5 kg) and rinsed the filter cake with EtOAc (2 L, 2-3 vol.). The filtrate was poured into water (16.8 L, 20 vol.) at 0-10°C, extracted with EtOAc (10 L, 12 vol.) and separated. The aqueous phase was extracted with EtOAc (5 L, 6 vol.). The combined organic phases were washed with water (5 L*3, 6 vol. *3), concentrated at 50°C in vacuum to afford crude product as a gray solid (956 g). The crude product was dissolved in EtOAc (950 mL, 1 vol. relative to crude product) at 35~40°C, then was added dropwise n-heptane (950 mL, 1 vol. relative to crude product) at 30~40°C over 20 min. The resulting mixture was cooled to 20~25°C over 30 min and stirred for 1 h at 30-40°C. Some solids came out slowly and n-heptane (1.9 L, 2 vol. relative to crude product) was added dropwise to the slurry mixture at 20~25°C over 30 min. The precipitates were stirred at 15~20°C for 3 h and filtered. The filter cake was washed with n-heptane (1.5 L) and dried in oven at 45-50°C for 16 h to afford Compound 2 as a pale-yellow solid (743 g, 98.6%/220 nm, 96.9 w%, 99.98%/220 nm chiral purity, 0.48%KF, 72% yield). H NMR (400 MHz, DMSO) 5 7.54 (dd, J = 10.2, 2.7 Hz, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.42 (s, 1H), 7.31 (dd, J = 8.5, 5.8 Hz, 1H), 7.22 (td, J = 8.4, 2.7 Hz, 1H), 7.17 (s, 1H), 6.92 (d, J = 1.8 Hz, 1H), 6.14 (s, 2H), 5.47 (q, J = 6.0 Hz, 1H), 4.22 (s, 3H), 4.02 (q, J = 7.3 Hz, 2H), 3.78 (q, J = 16.1 Hz, 2H), 1.40 (d, J = 6.3 Hz, 3H), 1.29 (t, J = 7.3 Hz, 3H). MS (ESI, m/z): 500.30 (M + H)+.
[00553] In another example, a process was developed where Compound 4 (1.1 mol equiv. to Compound 3) was used. Potassium phosphate base (K2PO4, 4. 1 mol equiv.) and DMA (16 vol.) were substituted with cesium carbonate (CS2CO3, 2.2 mol equiv.) and NMP (5.6 vol.). The reaction was carried out at 20~30°C. Following completion of the reaction, the crude product was precipitated with water. The material was then dissolved in ethyl acetate, washed with water, and treated with activated carbon. The product is subsequently crystallized from toluene/ethyl acetate/heptane to give Compound 2 in 80% yield and 99.9% purity.
[00554] Synthesis of Compound 1. To a reactor was charged t-AmOH (20 vol.), Compound 2 (700.0 g, 99.99% chiral purity) and potassium pivalate (588.0 g). The reaction mixture was fully refilled with nitrogen. To the reaction mixture was added cataCXium A (120.4 g) and Pd(OAc)2 (37.8 g) at r.t. under nitrogen. The resulting mixture was heated to 100°C and stirred for 18 h under nitrogen. The reaction mixture was cooled to 30°C , filtered through a celite pad and washed the filter cake with EtOAc (3 vol.). The filtrate was washed with water (5 vol. *2) and separated. The upper organic phase was concentrated in vacuum to afford a brown oil. The oil was dissolved in EtOAc (27 L) then added 5w% aqueous L-cysteine (0.98 kg in 18.6 kg water), stirred for 1 h at 40~45°C and separated. The organic phase was washed with water (6.75 L) and separated. 5w% aqueous L-cysteine (0.98 kg in 18.6 kg water) was charged to the above organic phase, stirred for 1 h at 40~45°C and separated. The organic phase was washed with water (6.75 L.) and separated. The organic phase was concentrated in vacuum at 45~50°C to afford a brown solid (1.12 kg). The crude solid (1.12 kg) was further purified by silica gel chromatography eluted with EtOAc/DCM (dry loading, 3X, 100-200 meshes, EtOAc:DCM=l : 1) to afford a pale-yellow solid ( 1.02 kg). The solid was dissolved in EtOAc (600 mL, 2 vol.) at 50~60°C, then was added n-heptane (1.8 L, 6 vol.) dropwise over 50 min at 50~60°C. A large of solids came out during addition. The resulting slurry was cooled to 15~20°C over 50 min and stirred for 30 min at 15~20°C. The slurry was concentrated in vacuum at 45~50°C to 2-3 vol. mixture, n-heptane (1.2 L, 4 vol.) was added to the
above mixture (2-3 vol.), concentrated in vacuum at 45~50°C to 2-3 vol. mixture. The mixture was cooled to 10~15°C over 2 h, stirred at 10~15°C for 1 h and filtered. The filtered cake was rinsed with n-heptane (600 mb) and dried in vacuum at 50°C for 20 h to afford Form 1 of Compound 1 as an off-white solid (280 g, 99.0%). H NMR (400 MHz, DMSO) 57.79 (dd, J = 10.3, 2.2 Hz, 1H), 7.58 (s, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.24 – 7.16 (m, 2H), 6.13 (s, 2H), 6.08 (d, J = 1.7 Hz, 1H), 5.31 – 5.23 (m, 1H), 4.16 (s, 3H), 4.05 – 3.94 (m, 2H), 3.78 (d, J = 15.6 Hz, 1H), 2.98 (d, J = 15.5 Hz, 1H), 1.71 (d, J = 6.2 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H). MS (ESI, m/z): 420.30 (M + H)+. XRPD (FIG. 1), TG/DTA (FIG. 2), DSC (FIG. 3), DVS (FIG. 4), and FT-IR (FIG. 5) results for a sample of Form 1 were obtained.
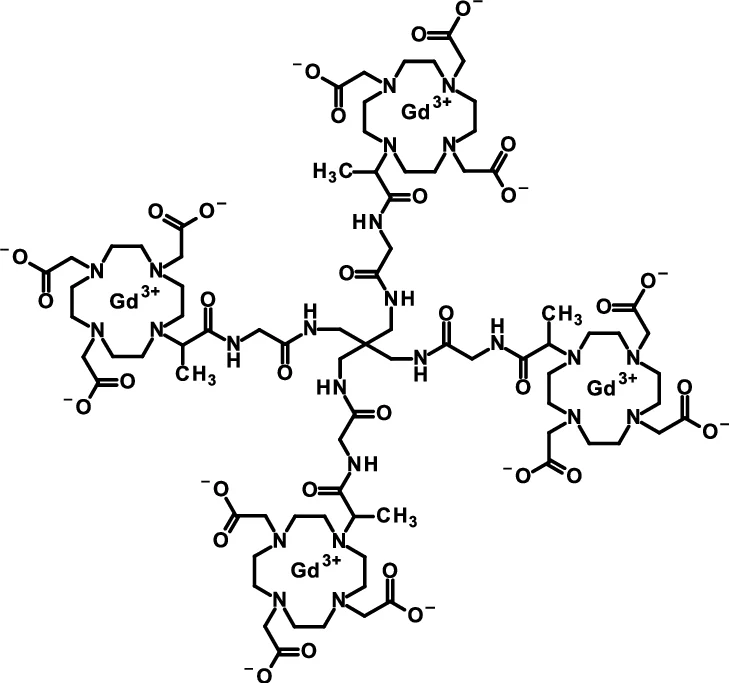
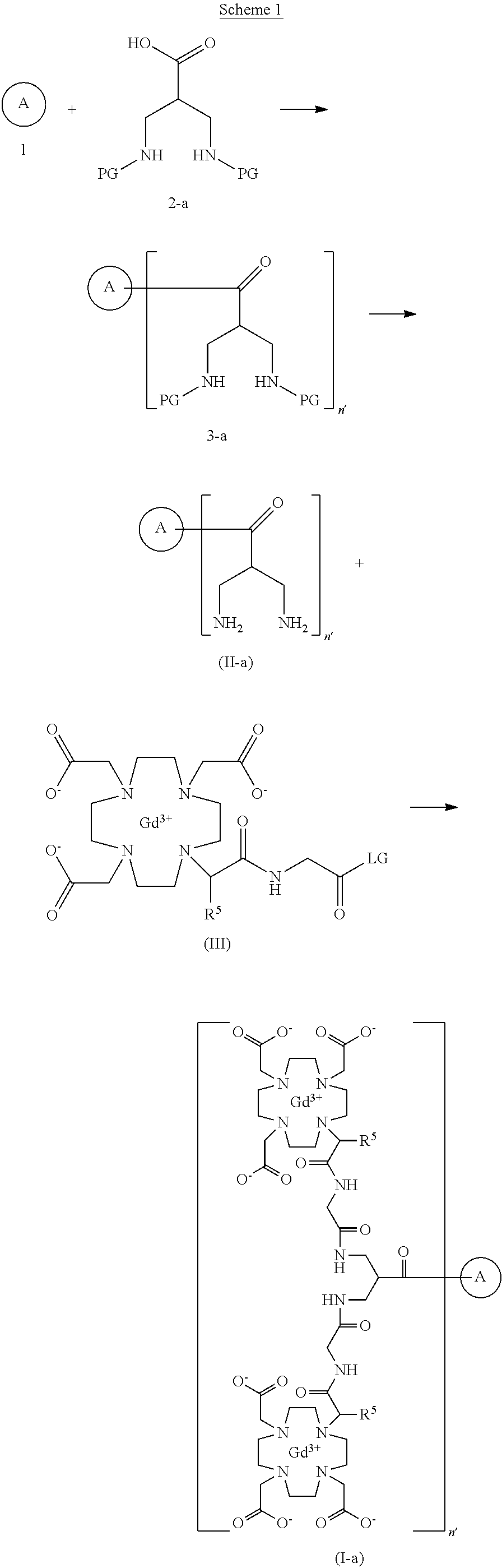
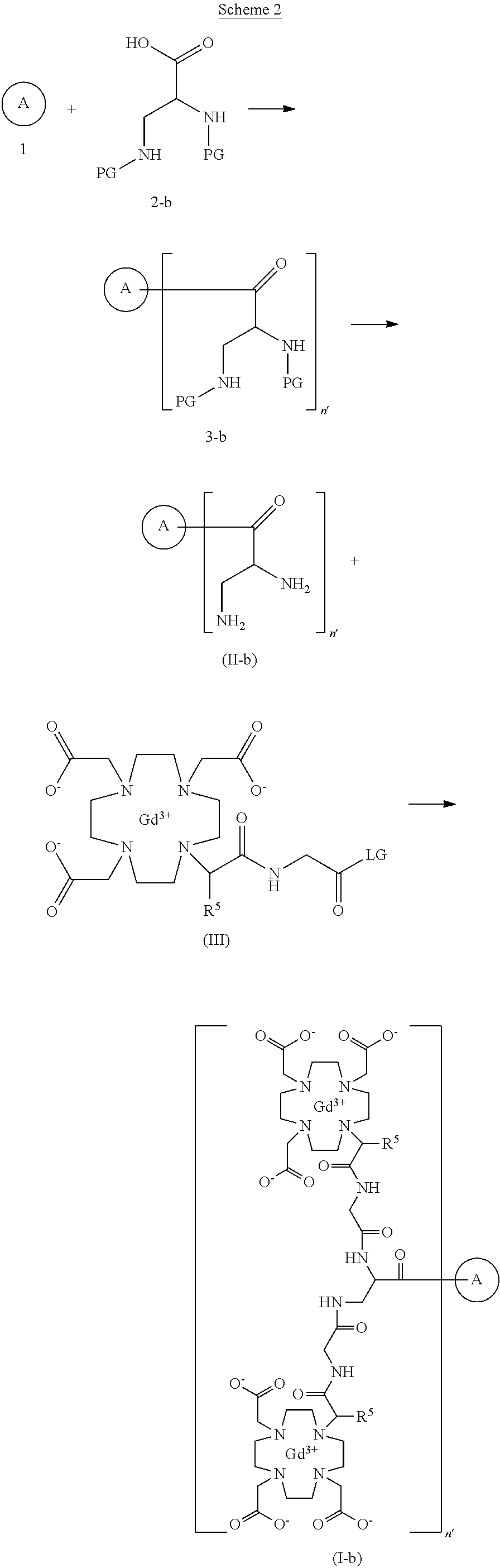
To detect and visualize lesions with abnormal vascularity, in conjunction with MRI
Gadoquatrane (marketed as AMBELVIST®) is a low-dose, macrocyclic gadolinium-based contrast agent (GBCA) developed by Bayer for use in magnetic resonance imaging (MRI). It is designed to enhance the visualization of lesions in the central nervous system (CNS) and other body regions in adult and pediatric patients.
Core Highlights:
Lower Gadolinium Exposure: It requires a dose of 0.04 mmol/kg, which results in 60% less gadolinium exposure compared to standard macrocyclic GBCAs.
Regulatory Approval: The FDA approved it in June 2026 for use in adults and pediatric patients, including term neonates. It was also approved in Japan in March 2026.
Efficacy & Safety: Phase III clinical trials (the QUANTI studies) showed it effectively detects lesions with abnormal vascularity while maintaining an efficacy and safety profile comparable to other standard macrocyclic agents.
Structure: Gadoquatrane features a tetrameric, macrocyclic structure that gives it high relaxivity and stability in the body
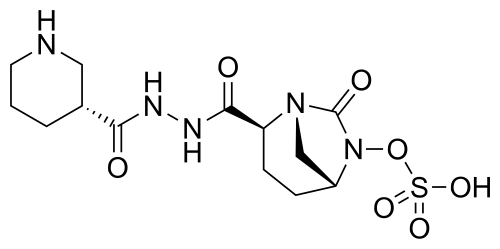
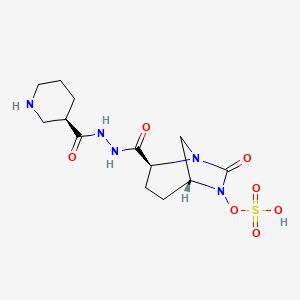
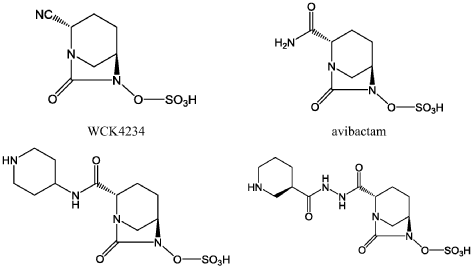
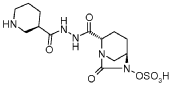
Step-1: Preparation of trans-3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-1 of Example- 1 above, and by using trans-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid (25 gm, 0.084 mol), N,N-dimethyl formamide (625 ml), EDC hydrochloride (24 gm, 0.126 mol), HOBt (16.96 gm, 0.126 mol), (R)-N-tert-butoxycarbonyl-piperidin-3-carboxylic acid hydrazide (21.40 gm , 0.088 mol) to provide the title compound in 17.0 gm quantity, 41% yield as a white solid.
Step-2: Preparation of trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-2 of Example- 1 above, and by using trans-3-[N ‘ -(6-benzyloxy-7-oxo- 1 ,6-diaza-bicyclo [3.2.1 ]octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin-l-carboxylic acid tert-butyl ester (16.5 gm , 0.033 mol), methanol (170 ml) and 10% palladium on carbon (3.5 gm) to provide the title compound in 13.5 gm quantity as a pale pink solid and it was used for the next reaction immediately.
Analysis: MS (ES+) CiglfeNsOe = 411.1 (M+l);
Step-3: Preparation of tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin- 1 -carboxylic acid tert-butyl ester:
By using the procedure described in Step-3 of Example- 1 above, and by using trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-1 -carboxylic acid tert-butyl ester (13.5 gm , 0.033 mol), pyridine (70 ml) and pyridine sulfur trioxide complex (26.11 gm, 0.164 mol), 0.5 N aqueous potassium dihydrogen
phosphate solution (400 ml) and tetrabutylammonium sulphate (9.74 gm, 0.033 mol) to provide the title compound in 25 gm quantity as a yellowish solid, in quantitative yield.
Analysis: MS (ES-) as a salt = 490.0 (M-l) as a free sulfonic acid;
By using the procedure described in Step-4 of Example- 1 above, and by using tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester (24 gm , 0.032 mmol), dichloromethane (60 ml) and trifluoroacetic acid (60 ml) to provide the title compound in 10 gm quantity as a white solid, in 79% yield.
Analysis: MS (ES-)= C13H21N5O7S = 390.2 (M-l) as a free sulfonic acid;
ClassCyclopropanes; General anaesthetics; Phenols; Small molecules
Mechanism of ActionGABA A receptor agonists
RegisteredAnaesthesia; Sedation
10 Apr 2026Sichuan Haisco Pharmaceutical plans a phase III trial for Anesthesia (In Children, In adolescents) (IV) in May 2026 (NCT07510945)
28 Aug 2024No recent reports of development identified for preclinical development in Sedation in USA (IV, Infusion)
01 Aug 2024Zhongda Hospital plans a clinical trial for Sedation (IV) in August 2024 (NCT06538883)
To induce general anesthesia in adults undergoing surgery
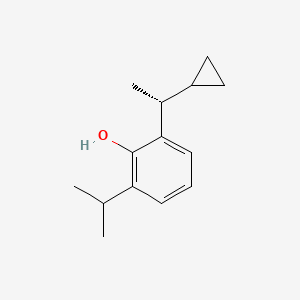
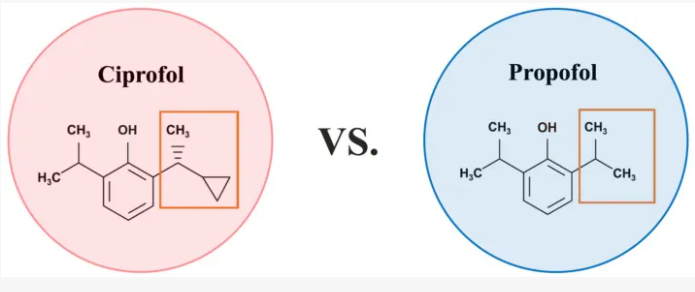
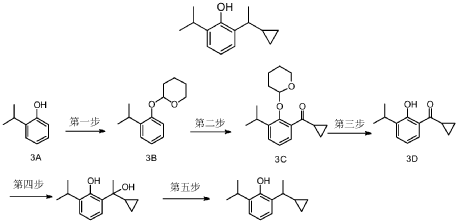
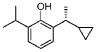
Cipepofol (also known as ciprofol or HSK3486) is a novel, short-acting intravenous anesthetic and sedative. As a structural analog of propofol, it targets \(GABA_{A}\) receptors but is 4 to 6 times more potent. It offers faster recovery, improved cardiovascular stability, and significantly less injection pain than propofol.
Key Clinical Advantages
Superior Efficacy: Requires a lower dose to achieve the same sedative depth as propofol.
Better Safety Profile: Associated with a lower incidence of injection pain, reduced respiratory depression, and better hemodynamic (blood pressure) stability.
Fast Acting: Characterized by rapid onset and quick recovery times, making it ideal for procedures like gastrointestinal endoscopy, bronchoscopy, and general anesthesia induction.
Recent Developments
FDA Approval: Cipepofol (sold under the brand name CYPSEDO) officially received U.S. FDA marketing approval, becoming the first China-originated innovative intravenous anesthetic to enter the global market.
Ongoing Trials: Clinical trials and post-marketing studies are actively evaluating its safety in specific populations, such as elderly patients and children.
Ciprofol is an optically active 2,6-disubstituted alkylphenol with a cyclopropylethyl group incorporated at the second carbon atom. This cyclopropyl group increases the steric effects and introduces stereoselective effects over its anesthetic properties. These properties appear to increase the anesthetic potency of ciprofol, when compared with propofol.[9]
Wang X, Wang X, Liu J, Zuo YX, Zhu QM, Wei XC, et al. (March 2022). “Effects of ciprofol for the induction of general anesthesia in patients scheduled for elective surgery compared to propofol: a phase 3, multicenter, randomized, double-blind, comparative study”. European Review for Medical and Pharmacological Sciences. 26 (5): 1607–1617. PMID35302207.
Zeng Y, Wang DX, Lin ZM, Liu J, Wei XC, Deng J, et al. (February 2022). “Efficacy and safety of HSK3486 for the induction and maintenance of general anesthesia in elective surgical patients: a multicenter, randomized, open-label, propofol-controlled phase 2 clinical trial”. European Review for Medical and Pharmacological Sciences. 26 (4): 1114–1124. PMID35253166.
Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, et al. (May 2017). “Design, Synthesis, and Evaluation of Novel 2,6-Disubstituted Phenol Derivatives as General Anesthetics”. Journal of Medicinal Chemistry. 60 (9): 3606–3617. doi:10.1021/acs.jmedchem.7b00254. PMID28430430.
Qin K, Qin WY, Ming SP, Ma XF, Du XK (July 2022). “Effect of ciprofol on induction and maintenance of general anesthesia in patients undergoing kidney transplantation”. European Review for Medical and Pharmacological Sciences. 26 (14): 5063–5071. PMID35916802.
Liu SB, Yao X, Tao J, Yang JJ, Zhao YY, Liu DW, et al. (March 2023). “Population total and unbound pharmacokinetics and pharmacodynamics of ciprofol and M4 in subjects with various renal functions”. British Journal of Clinical Pharmacology. 89 (3): 1139–1151. doi:10.1111/bcp.15561. PMID36217805. S2CID252818288.
Ding YY, Long YQ, Yang HT, Zhuang K, Ji FH, Peng K (December 2022). “Efficacy and safety of ciprofol for general anaesthesia induction in elderly patients undergoing major noncardiac surgery: A randomised controlled pilot trial”. European Journal of Anaesthesiology. 39 (12): 960–963. doi:10.1097/EJA.0000000000001759. PMID36214498. S2CID252779399.
Bulevirtide-gmod, sold under the brand name Hepcludex, is the first and only FDA-approved medication for treating chronic hepatitis delta virus (HDV) infection in adults. Developed by Gilead Sciences, it received accelerated approval from the U.S. Food and Drug Administration (FDA) on May 22, 2026, filling a critical gap for patients with this severe viral liver disease.
Indication and Clinical Use
Target Patient Profile: Approved for adults with chronic HDV who have compensated cirrhosis or no cirrhosis.
The Clinical Need: HDV only occurs as a co-infection in individuals who already have Hepatitis B (HBV). It is considered the most aggressive form of viral hepatitis, often accelerating liver scarring (fibrosis), liver failure, and liver cancer.
Basis of Approval: The FDA granted accelerated approval based on Phase 3 MYR301 study data, which demonstrated a significant reduction in viral HDV RNA and the normalization of alanine aminotransferase (ALT) liver enzymes.
Mechanism of Action
Bulevirtide-gmod is a first-in-class entry inhibitor. It works by binding to and blocking the sodium taurocholate co-transporting polypeptide (NTCP) receptor on liver cells. Because HDV and HBV rely on this specific receptor to enter hepatocytes, the drug successfully disrupts the viral life cycle and prevents the virus from spreading to healthy liver cells.
Dosage and Administration
Form: Supplied as a lyophilized powder for injection.
Dose: The recommended dose is 8.5 mg once daily.
Administration: Delivered via subcutaneous injection (under the skin).
Safety and Side Effects
Boxed Warning: The drug carries a prominent warning regarding the risk of severe acute exacerbations of hepatitis D and B if treatment is discontinued. Stopping the medication can cause severe, life-threatening viral flares, requiring close medical monitoring for at least 6 months post-treatment.
Common Side Effects: The most frequent adverse reactions of patients) include:
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[8]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[8] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[8]
Bulevirtide was approved for medical use in the European Union in July 2020,[8] and in Canada in August 2025.[5]
Medical uses
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[8][10]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[12][13] that can also dock to NTCP, blocking the virus’s entry mechanism.[14]
Bulevirtide is also effective against hepatitis D because the hepatitis D virus uses the same entry receptor as the hepatitis B virus and is only effective in the presence of a hepatitis B virus infection.[14]
Pre-clinical data in mice suggests that pharmacological inhibition of NTCP-mediated bile salt uptake may also be effective to lower hepatic bile salt accumulation in cholestatic conditions. This reduces hepatocellular damage.[15] An increased ratio of phospholipid to bile salts seen in bile upon NTCP inhibition may further contribute to the protective effect as bile salts are less toxic in presence of phospholipids.[16]
Structural formula
Bulevirtide is a 47-amino acid peptide with the following sequence:[17]
“Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–867. doi:10.1016/j.jhep.2012.12.008. PMID23246506.
Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
Na+ -taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Slijepcevic D, Roscam Abbing RLP, Fuchs CD, Haazen LCM, Beuers U, Trauner M, Oude Elferink RPJ, van de Graaf SFJ. Hepatology. 2018 Sep;68(3):1057-1069. doi: 10.1002/hep.29888
To treat adults with relapsed or refractory mantle cell lymphoma after at least two lines of systemic therapy, including a Bruton’s tyrosine kinase inhibitor
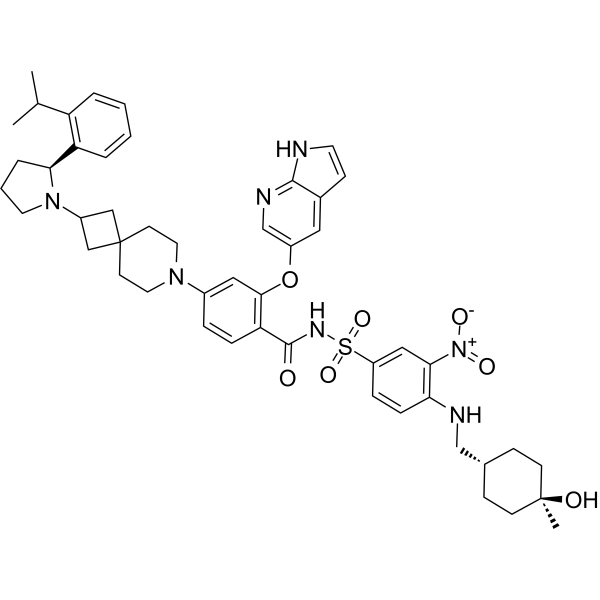
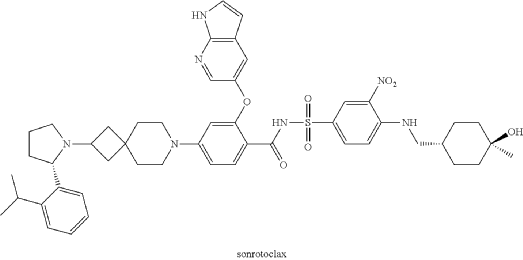
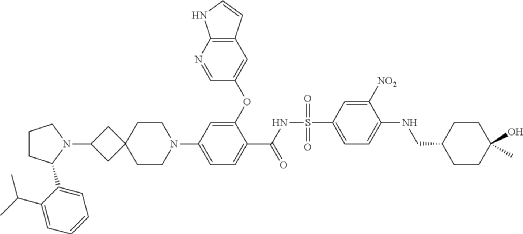
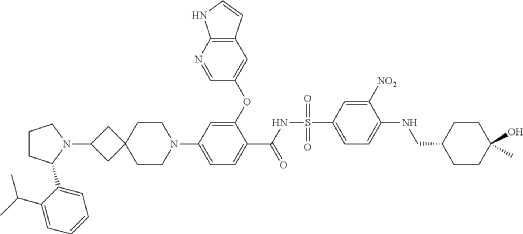
Sonrotoclax is a potent, orally active Bcl2 inhibitor. Sonrotoclax has effective cell killing effect against a variety of lymphoma and leukemia cell lines.
Regulatory Status & Primary Indication
On May 13, 2026, the U.S. Food and Drug Administration (FDA) granted accelerated approval to sonrotoclax for treating adult patients with relapsed or refractory mantle cell lymphoma (MCL). [1]
Eligibility Requirement: Patients must have undergone at least two prior lines of systemic therapy, which must include a Bruton’s tyrosine kinase (BTK) inhibitor.
Clinical Performance: In the supporting Phase 1/2 BGB-11417-201 trial, sonrotoclax demonstrated an overall response rate (ORR) of 52% and a median time to response of 1.9 months
Sonrotoclax is an orally bioavailable inhibitor of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2), with potential pro-apoptotic and antineoplastic activities. Upon oral administration, sonrotoclax specifically binds to and inhibits the activity of the pro-survival protein Bcl-2. This restores apoptotic processes and inhibits cell proliferation in Bcl-2-overexpressing tumor cells. Bcl-2, a protein that belongs to the Bcl-2 family, is overexpressed in various tumor cell types and plays an important role in the negative regulation of apoptosis. Its tumor expression is associated with increased drug resistance and cancer cell survival.
A mixture of (S)-2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(2-(2-(2-isopropylphenyl)pyrrolidin-1-yl)-7-azaspiro[3.5]nonan-7-yl)benzoic acid (44 g, 78 mmol), 4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrobenzenesulfonamide (26.8 g, 78 mmol), TFA (15.7 g, 156 mmol), EDCl (19.4 g, 101 mmol) and DMAP (19 g, 156 mmol) in anhydrous DCM (880 mL) was stirred overnight at room temperature. The reaction was monitored by HPLC. After starting material of (S)-2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(2-(2-(2-isopropylphenyl)pyrrolidin-1-yl)-7-azaspiro[3.5]nonan-7-yl)benzoic acid was consumed completely, the reaction mixture was heated to ˜35° C. and N 1,N 1-dimethylethane-1,2-diamine (17.2 g, 195 mmol) was added in one portion. The reaction was stirred for another 12 hours. The mixture was washed twice with 10 wt % aq. AcOH solution (300 mL×2) and then washed with saturated aq. NaHCO 3 (300 mL×2). The organic layer was collected and concentrated to about 90 mL. 22 g of silica gel was added and stirred for 2 hours. After filtration, 180 mL EA was added into the filtrate at reflux and further stirred for 5 hours. After the mixture was cooled to room temperature, the precipitate was filtered and then the wet cake was washed twice with EA (180 mL). After drying in vacuum at 80-90° C., the desired compound was obtained (48 g, yield: 69.5%). 1H NMR (DMSO-d 6) δ ppm: 11.65 (s, 1H), 11.11 (br, 1H), 8.58-8.39 (m, 2H), 8.00 (d, J=2.8 Hz, 1H), 7.74 (d, J=8.8 Hz, 1H), 7.57-7.37 (m, 4H), 7.30-7.10 (m, 3H), 7.00 (d, J=9.2 Hz, 1H), 6.65 (d, J=1.2 Hz, 1H), 6.35 (s, 1H), 6.17 (s, 1H), 4.24 (s, 1H), 3.39-3.20 (m, 5H), 3.04-2.88 (m, 4H), 2.23 (s, 1H), 1.94-1.47 (m, 11H), 1.44-1.26 (m, 7H), 1.19 (d, J=8.0 Hz, 3H), 1.14 (d, J=8.0 Hz, 3H), 1.10 (s, 4H). MS (ESI, m/e) [M+1] + 889.9.
Example F43: 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-N-((4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrophenyl)sulfonyl)-4-(2-((S)-2-(2-isopropylphenyl)pyrrolidin-1-yl)-7-azaspiro[3.5]nonan-7-yl)benzamide
To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, ESR1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
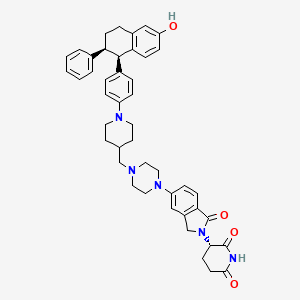
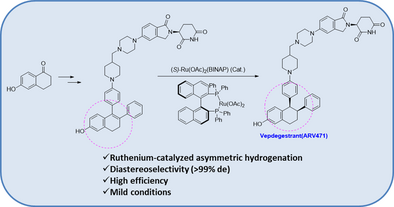
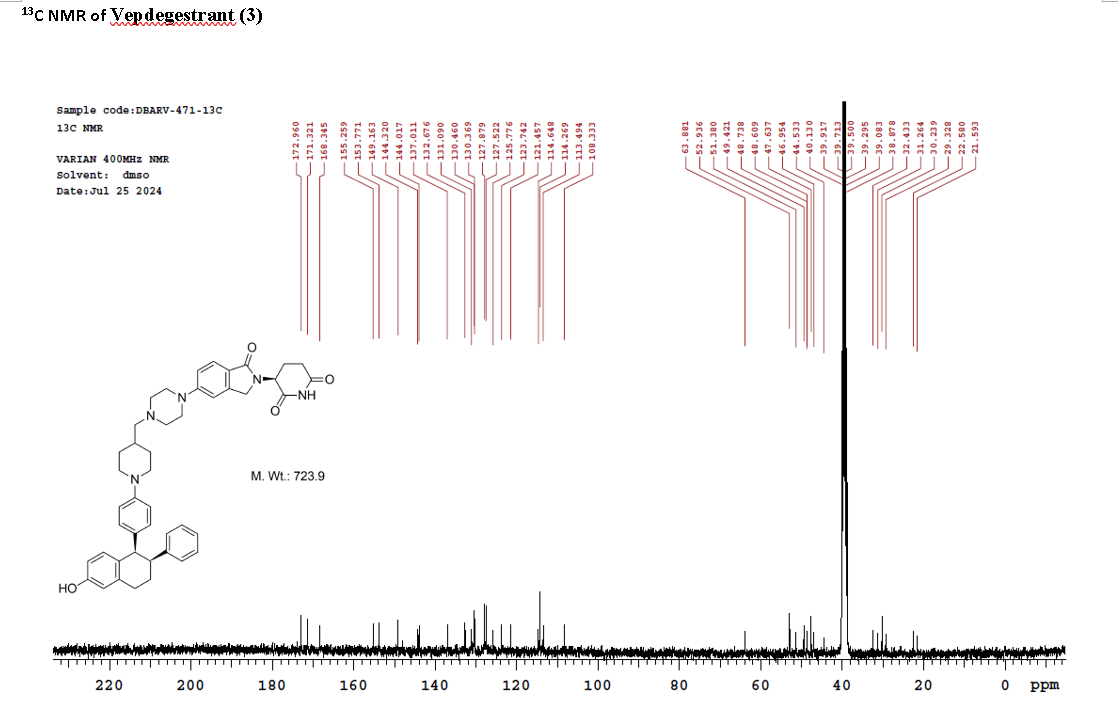
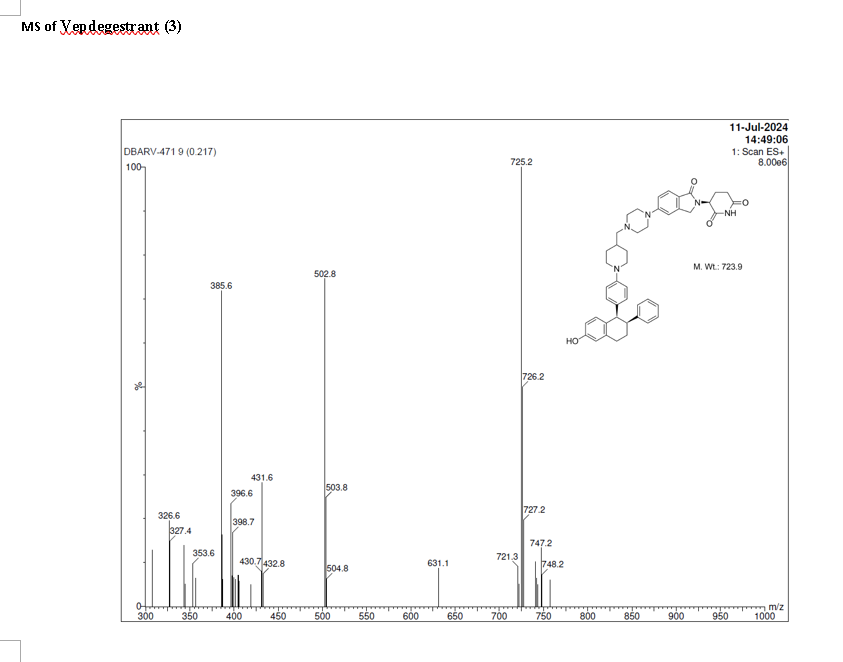
On May 1, 2026, the FDA approved vepdegestrant (Veppanu), a first-in-class oral PROTAC estrogen receptor (ER) degrader developed by Arvinas and Pfizer, for adults with ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer who have progressed on endocrine therapy. It demonstrated significant progression-free survival (PFS) improvements compared to fulvestrant.
Key Details About Vepdegestrant (Veppanu):
Mechanism of Action: As an oral PROTAC (Proteolysis-Targeting Chimera), vepdegestrant targets the estrogen receptor for degradation, designed to be more effective than traditional endocrine therapies, particularly in ESR1-mutated tumors.
Approved Indication: For treating adults with ER+/HER2-, ESR1-mutated advanced/metastatic breast cancer (detected by Guardant360 CDx) after at least one line of endocrine therapy.
Dosage: The recommended dose is 200 mg taken orally once daily with food.
Clinical Efficacy (VERITAC-2): In trials, vepdegestrant showed a significantly longer PFS compared to intramuscular fulvestrant.
Side Effects & Risks: Common side effects include decreased white blood cell counts, increased liver function tests, muscle/bone pain, fatigue, and nausea. Warnings include embryo-fetal toxicity and QTc interval prolongation (heart rhythm issues).
Companion Diagnostic:Guardant360 CDx was approved alongside the drug to identify patients with ESR1 mutations
Vepdegestrant is designed as a PROTAC that recruits the ubiquitin-proteasome system to target the estrogen receptor for degradation.[4] The compound contains both an E3 ubiquitin ligase-binding moiety and an estrogen receptor-binding domain, intended to bring these proteins into proximity to trigger ubiquitination and subsequent proteasomal degradation of the ER protein.[5] In laboratory studies, vepdegestrant demonstrated ER degradation in ER-positive breast cancer cell lines with reported DC50 values of approximately 1-2 nM.[6]
Vepdegestrant is an orally available hetero-bifunctional molecule and selective estrogen receptor (ER) alpha-targeted protein degrader, using the proteolysis targeting chimera (PROTAC) technology, with potential antineoplastic activity. Vepdegestrant is composed of an ER alpha ligand attached to an E3 ligase recognition moiety. Upon oral administration,vepdegestrant targets and binds to the ER ligand binding domain on ER alpha. E3 ligase is recruited to the ER by the E3 ligase recognition moiety and ER alpha is tagged by ubiquitin. This causes ubiquitination and degradation of ER alpha by the proteasome. This decreases ER alpha protein levels, decreases the expression of ER alpha-target genes and halts ER-mediated signaling. This results in an inhibition of proliferation in ER alpha-overexpressing tumor cells. In addition, the degradation of the ER alpha protein releases the ARV-471 and can bind to additional ER alpha target proteins. ER alpha is overexpressed in a variety of cancers and plays a key role in cancer cell proliferation.
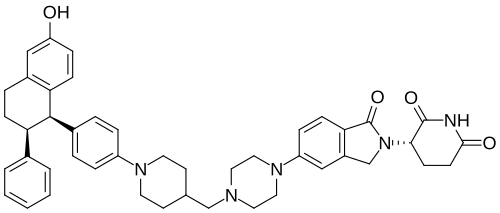
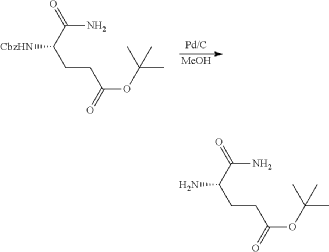
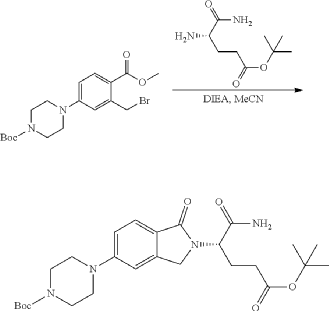
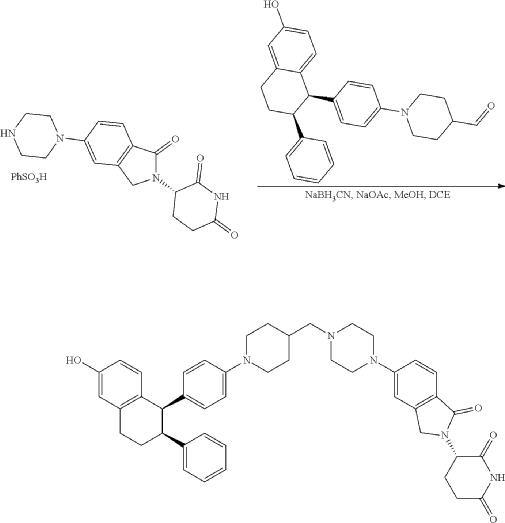
Step 11: Preparation of 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-b))
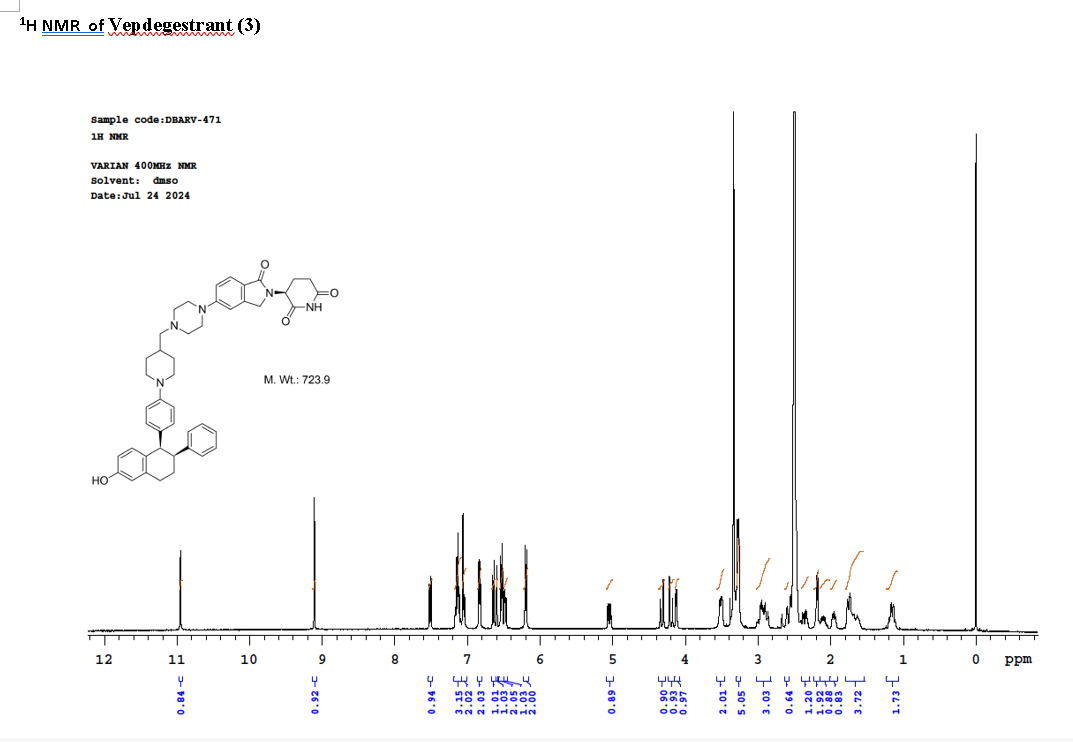
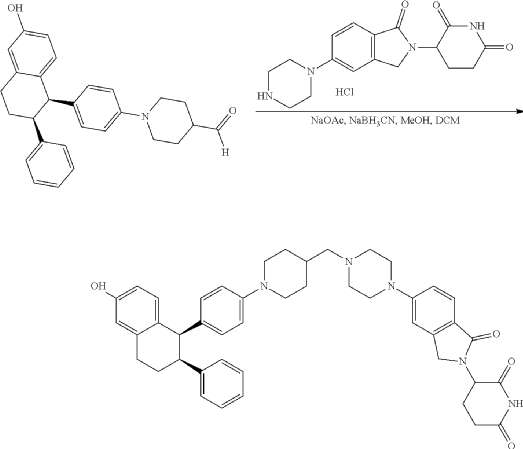
To a solution of 3-(1-oxo-5-piperazin-1-yl-isoindolin-2-yl)piperidine-2,6-dione hydrochloride (319 mg, 0.87 mmol, prepared in Step 17 described for Exemplary Compound 62) in methanol (4 mL) and dichloromethane (4 mL) was added sodium acetate (120 mg, 1.46 mmol, 2 eq). The mixture was stirred at 20° C. for 0.5 h, then to the mixture was added 1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]piperidine-4-carbaldehyde (300 mg, 0.73 mmol, 1 eq) and sodium cyanoborohydride (137 mg, 2.19 mmol, 3 eq). The mixture was stirred at 20° C. for 12 h. LC-MS showed the starting material was consumed completely and one main peak with desired MW was detected. The reaction mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex luna C 18 column, 250×50 mm, 10 um; mobile phase: [water (0.05% HCl)-acetonitrile]; B %: acetonitrile 10%-40% in 30 min). The desired compound 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (288.4 mg, 0.37 mmol, 51% yield) was obtained as a white solid of hydrochloride salt. LC-MS (ESI) m/z: 724.4 [M+1] +; 1H NMR (400 MHz, DMSO-d 6) δ 10.97 (s, 1H), 10.83 (s, 0.9H, HCl), 7.60 (d, J=8.5 Hz, 1H), 7.40 (br s, 2H), 7.22-7.11 (m, 5H), 6.83 (d, J=6.0 Hz, 2H), 6.69-6.63 (m, 2H), 6.58-6.47 (m, 3H), 5.07 (dd, J=5.2, 13.2 Hz, 1H), 4.41-4.30 (m, 2H), 4.28-4.21 (m, 1H), 4.00 (d, J=12.7 Hz, 2H), 3.61 (d, J=11.0 Hz, 2H), 3.54-3.36 (m, 6H), 3.16 (br s, 4H), 3.06-2.84 (m, 3H), 2.76-2.53 (m, 1H), 2.43-2.33 (m, 1H), 2.27 (br s, 1H), 2.16-2.04 (m, 3H), 2.02-1.69 (m, 5H).
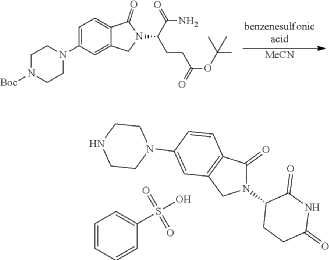
Synthesis of (3S)-3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-c))
To a mixture of (3 S)-3-(1-oxo-5-piperazin-1-yl-isoindolin-2-yl)piperidine-2,6-dione (1.30 g, 3.47 mmol, 1 eq, benzene sulfonate) in dichloromethane (8 mL) and methanol (32 mL) was added sodium acetate (854 mg, 10.41 mmol, 3 eq) in one portion at 20° C. The mixture was stirred at 20° C. for 10 minutes. Then 1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl] piperidine-4-carbaldehyde (1 g, 2.43 mmol, 0.7 eq, prepared as described above in the synthesis of Compound (I-b)) was added. The mixture was stirred at 20° C. for 10 minutes. After that, acetic acid (0.2 mL) and sodium cyanoborohydride (436 mg, 6.94 mmol, 2 eq) was added in one portion. The mixture was stirred at 20° C. for 40 minutes. The mixture was concentrated in vacuum, and 50 mL of tetrahydrofuran and 20 mL of water were added. The mixture was stirred for 20 minutes. Saturated aqueous sodium bicarbonate solution was added to adjust the pH to 8-9. The aqueous phase was extracted with ethyl acetate and tetrahydrofuran (v:v=2:1, 60 mL×3). The combined organic phase was washed with brine (60 mL×1), dried with anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by preparative reverse phase HPLC (column: Phenomenex luna C18 250×50 mm, 10 micron; mobile phase: [water (0.225% formic acid)-acetonitrile]; B %: 20%-50% in 30 min). The product (3S)-3-[5-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl] piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (964 mg, 1.23 mmol, 35% yield, 98% purity, formate) was obtained as a white solid of formic acid salt after lyophilization. Chiral purity was analyzed by chiral SFC (Chiralcel OJ-3 50×4.6 mm, 3 micron; mobile phase: 50% ethanol (0.05% DEA) in CO 2; flow rate: 3 mL/min, wavelength: 220 nm) and observed t p=2.89 min with de over 95%. [α D=−267.5 (c=0.2 in DMF, 25° C.). LC-MS (ESI) m/z: 724.2 [M+1] +. 1H NMR (400 MHz, DMSO-d 6) δ 10.94 (s, 1H), 8.16 (s, 1H, formate), 7.51 (d, J=8.8 Hz, 1H), 7.21-6.98 (m, 5H), 6.83 (d, J=6.4 Hz, 2H), 6.68-6.57 (m, 2H), 6.56-6.44 (m, 3H), 6.20 (d, J=8.8 Hz, 2H), 5.04 (dd, J=5.2, 13.2 Hz, 1H), 4.32 (d, J=16.8 Hz, 1H), 4.19 (d, J=17.2 Hz, 1H), 4.12 (d, J=4.8 Hz, 1H), 3.51 (br d, J=10.0 Hz, 4H), 3.27 (br s, 8H), 3.03-2.82 (m, 3H), 2.63-2.54 (m, 1H), 2.43-2.28 (m, 2H), 2.19 (d, J=6.8 Hz, 2H), 2.15-2.02 (m, 1H), 2.01-1.89 (m, 1H), 1.83-1.51 (m, 4H), 1.28-1.04 (m, 2H).
Iwata, H.; Naito, Y.; Hattori, M.; Yoshimura, A.; Yonemori, K.; Aizawa, M.; et al. (November 2023). “58P Safety and pharmacokinetics (PK) of vepdegestrant in Japanese patients with estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: Results from a Japanese phase I study”. Annals of Oncology. 34: S1488–S1489. doi:10.1016/j.annonc.2023.10.193. S2CID265657144.
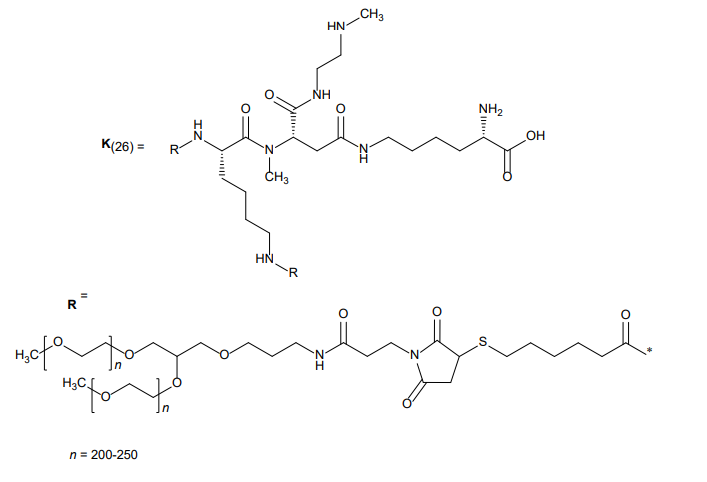
MOLECULAR FORMULA C231H386N64O67S5 + (C2H4O)4n MOLECULAR WEIGHT approx. 45 kDa
The structure of navepegritide (YUVIWEL®) is built using a “prodrug” design. It is not a simple small molecule, but rather a complex conjugate consisting of three distinct components designed to release the active drug slowly over time.
1. The Active Part: C-Type Natriuretic Peptide (CNP)
The core of the molecule is a synthetic 38-amino acid peptide (CNP-38).
Sequence: This peptide mimics the natural human C-type natriuretic peptide, which is essential for bone growth.
Function: Once released, this peptide binds to the natriuretic peptide receptor B (NPR-B) on the surface of chondrocytes (cartilage cells) in the growth plates, stimulating bone formation.
2. The Carrier: Polyethylene Glycol (PEG)
To prevent the body from clearing the small peptide too quickly, it is attached to a large, inert carrier.
Type: It uses a multi-arm, branched 40 kDa Polyethylene Glycol (PEG) molecule.
Purpose: The PEG carrier acts as a shield and a “weight,” making the molecule too large to be filtered out rapidly by the kidneys. This is what allows for once-weekly dosing instead of daily injections.
3. The Linker: TransCon™ Technology
This is the most critical part of the structure. The peptide is attached to the PEG carrier via a cleavable linker.
Mechanism: This linker is designed to break down spontaneously at a predictable rate under physiological conditions (neutral pH and body temperature).
The Result: As the linker slowly breaks, it releases the unmodified, active CNP-38 into the bloodstream. Because the peptide is released in its natural state, it retains its full biological activity.
Summary Table: Structural Components
Component
Description
Role
Peptide
CNP-38 (38 amino acids)
The “payload” that stimulates bone growth.
Linker
pH-sensitive cleavable bond
Controls the slow release of the peptide.
Carrier
40 kDa PEG
Increases the half-life and prevents rapid clearance.
Note: This structure is technically a prodrug because the large PEG-bound version is inactive; only the released CNP-38 peptide performs the therapeutic work.
C-Type natriuretic peptide (CNP), human, (89-126)-fragment (1-38) (CNP-38), conjugated at N6 of Lys26 with four O-methylpoly(ethylene glycol) chains (approx. 10 kDa each) via a cleavable tetra-antennary linker; L-leucyl-L-glutaminyl-L-?-glutamyl-L-histid
Poly(oxy-1,2-ethanediyl), ?-hydro-?-methoxy-, 26,26,26,26-tetraether with L-leucyl-L-glutaminyl-L-?-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-sery
FDA 2026, APPROVALS 2026, 2/27/2026, Yuviwel, Y3BH8M899D, MN-266, TRANSCON CNP, PA (224-233), Influenza, DA-66438, ACP-015, WHO 11981,
To increase linear growth in pediatric patients 2 years and older with achondroplasia with open epiphyses
Navepegritide is a prodrug consisting of a 38-amino acid C-type natriuretic peptide (CNP) moiety conjugated to a multi-arm polyethylene glycol (PEG) carrier via a cleavable linker. This structure allows for the once-weekly dosing approved by the FDA for children with achondroplasia.
Key Details
Purpose: It is designed to increase linear growth by providing continuous exposure to C-type natriuretic peptide (CNP), a protein that helps regulate bone growth.
Mechanism: As a prodrug, it uses Ascendis Pharma’s TransCon technology to release active CNP slowly into the body over a week, maintaining steady levels and avoiding high peaks.
Clinical Benefits: In the pivotal ApproaCH trial, patients treated with navepegritide showed a significant improvement in annualized growth velocity (AGV) compared to those on a placebo. It also showed potential improvements in body proportionality and lower-limb alignment.
Administration: It is administered via a once-weekly subcutaneous injection, offering a less frequent alternative to daily treatments like vosoritide.
Safety: Most common side effects include injection site reactions (redness, itching, or swelling) and a risk of low blood pressure (hypotension).
25 Feb 2026Vanda Pharmaceuticals has patent protection for an improved method of treatment with milsaperidone in USA
25 Feb 2026Vanda Pharmaceuticals has patents pending for an improved method of treatment with milsaperidone in China, Australia, Israel, Mexico and worldwide
56.36 g of boran complex of (3aR, 7R)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1 ,3,2]oxazaborole (1 equivalent) is dissolved under nitrogen in methylenchloride, and the solution is cooled to 0°C. A 1M solution of 1-(4-{3-[4-(6-fluoro-benzo[d]isoxazol-3-yl)-piperidin-1-yl]-propoxy}-3-methoxy-phenyl)-ethanone (iloperidone; 1 equivalent) in methylenchloride is added via a dropping funnel over 90 minutes while the internal temperature is maintained at 0°C ± 2°C. After the addition is complete, the mixture is stirred at 0°C for 20 hours. The reaction mixture is then poured into precooled methanol (0-5°C) during 1 hour. The solution is warmed to room temperature and stirred until the H2 evolution ceases. The solution is concentrated by distillation and the residue dried in vacuum, treated with methanol and stirred for about 1 hour at 50°C and an additional hour at 0CC. The product is isolated by filtration and dried under reduced pressure for 3 hours at 50°C. The title compound is obtained (white crystals).
[α]D20– 19.3° (c=1 in chloroform) Mp: 138.2 – 138.8°C
The boran complex used as starting material can be obtained as follows:
200 ml of a solution of (3aR, 7R)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole (1M in toluene) is stirred at room temperature under nitrogen. 1.2 equivalent borane-dimethylsulfide complex is added with a syringe. The solution is stirred for 2 further hours at room temperature. The borane complex is then crystallised by addition of 4 vol dry hexane and cooling to -12°C for 1.5 hour. The product is isolated by filtration in a sintered glass funnel and dried in vacuum at 40°C. The boran complex is obtained /white crystals).
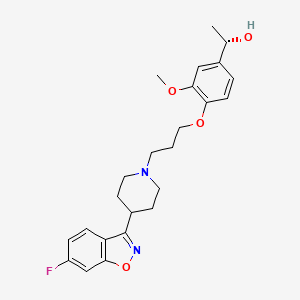
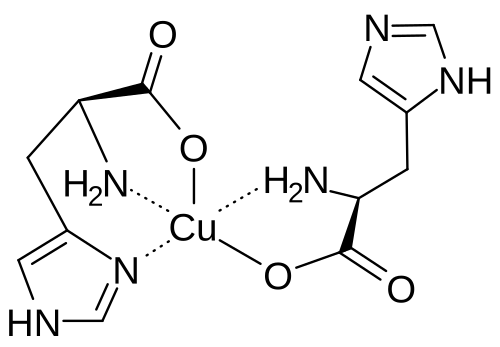
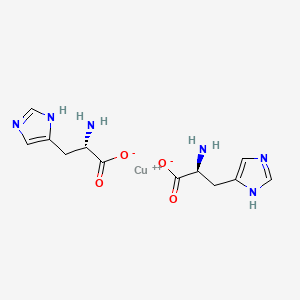
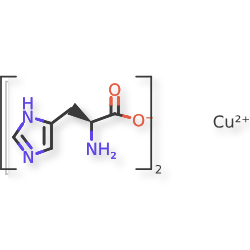
Copper histidinate, sold under the brand name Zycubo, is a medication used for the treatment of Menkes disease.[1] Copper histidinate is a copper replacement therapy given by subcutaneous injection.[1][2]
The most common side effects include infections, respiratory problems, seizures, vomiting, fever, anemia and injection site reactions.[2]
Copper histidinate was approved for medical use in the United States in January 2026.[2]
Menkes disease is a neurodegenerative disorder caused by a genetic defect that impairs a child’s ability to absorb copper.[2] The disease is characterized by seizures, failure to gain weight and grow, developmental delays, and intellectual disability.[2] It leads to abnormalities of the vascular system, bladder, bowel, bones, muscles, and nervous system.[2]
SYN
A275388 — Flores-Pulido AA, Jimenez-Perez VM, Garcia-Chong NR: Sintesis y uso de histidinato de cobre en ninos con enfermedad de Menkes en Mexico. Gac Med Mex. 2019;155(2):191-195. doi: 10.24875/GMM.18004310. [PubMed:31056589]
World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 94”. WHO Drug Information. 39 (3). hdl:10665/383022.
Clinical trial number NCT00811785 for “Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency” at ClinicalTrials.gov

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader









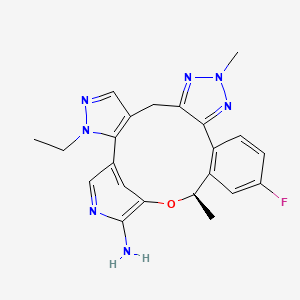
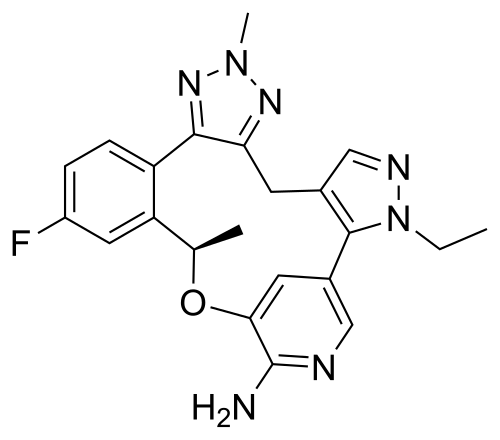
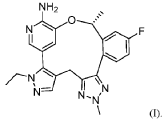
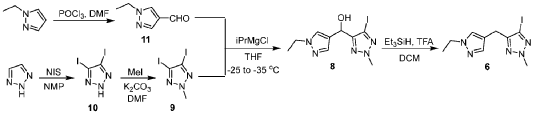










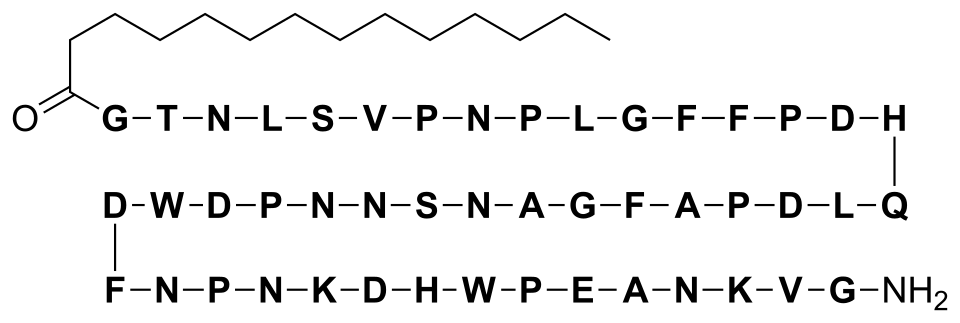

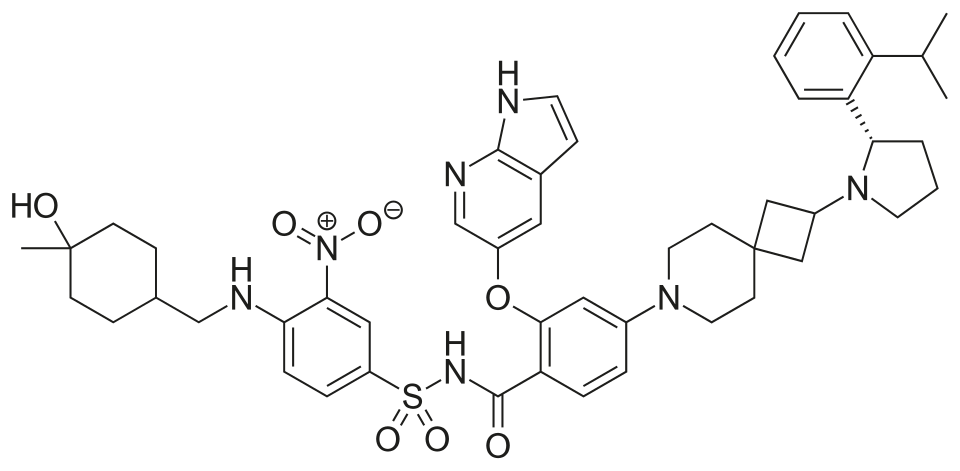
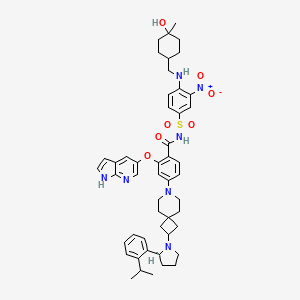


 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}