Candesartan cilexetil Candesartan cilexetil, Candesartan hexetil, H212/91, TCV-116, Kenzen, Blopress 16 mg Plus, Parapres, Ratacand, Blopress, Amias, Atacand
ATACAND
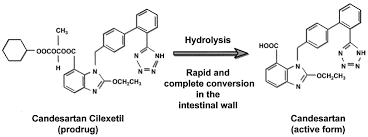
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT1 subtype angiotensin II receptor antagonist. Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1-[p-(o-1H-tetrazol-5ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester). Its empirical formula is C33H34N6O6, and its structural formula is:
 |
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral. ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant. 
Drug Patent Expiration and Exclusivity
| Active Ingredient |
Form |
Dosage |
|
Drug Type |
Application |
Product |
| CANDESARTAN CILEXETIL |
TABLET; ORAL |
4MG |
|
RX |
020838 |
001 |
| CANDESARTAN CILEXETIL |
TABLET; ORAL |
8MG |
|
RX |
020838 |
002 |
| CANDESARTAN CILEXETIL |
TABLET; ORAL |
16MG |
|
RX |
020838 |
003 |
| CANDESARTAN CILEXETIL |
TABLET; ORAL |
32MG |
|
RX |
020838 |
004 |
Patents
There are 6 patent(s) protecting ASTRAZENECA’s ATACAND. The last patent 5534534*PED expires on 2014-01-09.View patent at USPTO
| Patent US |
US |
Expiration |
| 5534534*PED |
|
2014-1-9 |
| 5534534 |
Pharmaceutical compositions for oral use and method of preparing them
A pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) having antagonistic action to angiotensin II ##STR1## (wherein the ring W is an optionally substituted N-containing heterocyclic residue; R.sup.3 is a group capable of forming an anion or a group convertible thereinto; X is a direct bond or a spacer having an atomic length of two or less between the phenylene group and the phenyl group; and n is an integer of 1 or 2) and an oily substance having a lower melting point, and a method for preparing a pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) and an oily substance having a lower melting point, which comprises admixing the compound of the formula (I) with an oily substance having a lower melting point and then subjecting the mixture to molding.
|
2013-7-9(expired) |
| 5196444*PED |
|
2012-12-4(expired) |
| 5196444 |
1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-c arboxylate and compositions and methods of pharmaceutical use thereof
1-(Cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-car boxylate or a pharmaceutically acceptable salt thereof has potent angiotensin II antihypertensive activity, thus being useful as therapeutic agents for treating circulatory system diseases such as hypertensive diseases, heart diseases (e.g. hypercardia, heart failure, cardiac infarction, etc.), strokes, cerebral apoplexy, nephritis, etc.
|
2012-6-4(expired) |
| 7538133*PED |
|
2011-10-18(expired) |
| 5705517*PED |
|
2011-10-18(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ASTRAZENECA.
Approval History
| Date |
Supplement No. |
Action |
Documents |
| 2013-04-26 |
038 |
Labeling Revision |
|
| 2012-04-27 |
035 |
Labeling Revision |
|
| 2012-04-13 |
032 |
Labeling Revision |
|
| 1998-06-04 |
000 |
Approval |
|
| 2011-06-24 |
033 |
Labeling Revision |
|
| 2009-10-22 |
031 |
Patient Population Altered |
|
| 2006-08-17 |
026 |
Labeling Revision |
|
| 2005-05-18 |
022 |
New or Modified Indication |
|
| 2005-02-22 |
024 |
New or Modified Indication |
|
| 2004-12-16 |
023 |
Labeling Revision |
|
| 2000-06-14 |
008 |
Labeling Revision |
|
| 2002-09-13 |
015 |
Comparative Efficacy Claim |
|
| 2003-01-22 |
017 |
Labeling Revision |
|
| 2003-04-23 |
019 |
Labeling Revision |
|
| 2013-02-21 |
037 |
Manufacturing Change or Addition |
|
| 1999-08-11 |
005 |
Package Change |
|
| 2000-12-27 |
009 |
Manufacturing Change or Addition |
|
| 2001-05-24 |
012 |
Manufacturing Change or Addition |
|
| 2001-11-28 |
016 |
Labeling Revision |
|
| 1999-07-28 |
004 |
Control Supplement |
|
| 2001-04-02 |
011 |
Manufacturing Change or Addition |
|
| 2001-10-04 |
014 |
Control Supplement |
|
| 1998-11-16 |
002 |
Manufacturing Change or Addition |
|
| 1999-12-08 |
006 |
Package Change |
|
| 2001-06-07 |
010 |
Manufacturing Change or Addition |
|
| 2001-03-29 |
013 |
Package Change |
|
| 1998-12-07 |
001 |
Manufacturing Change or Addition |
 Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Synthesis
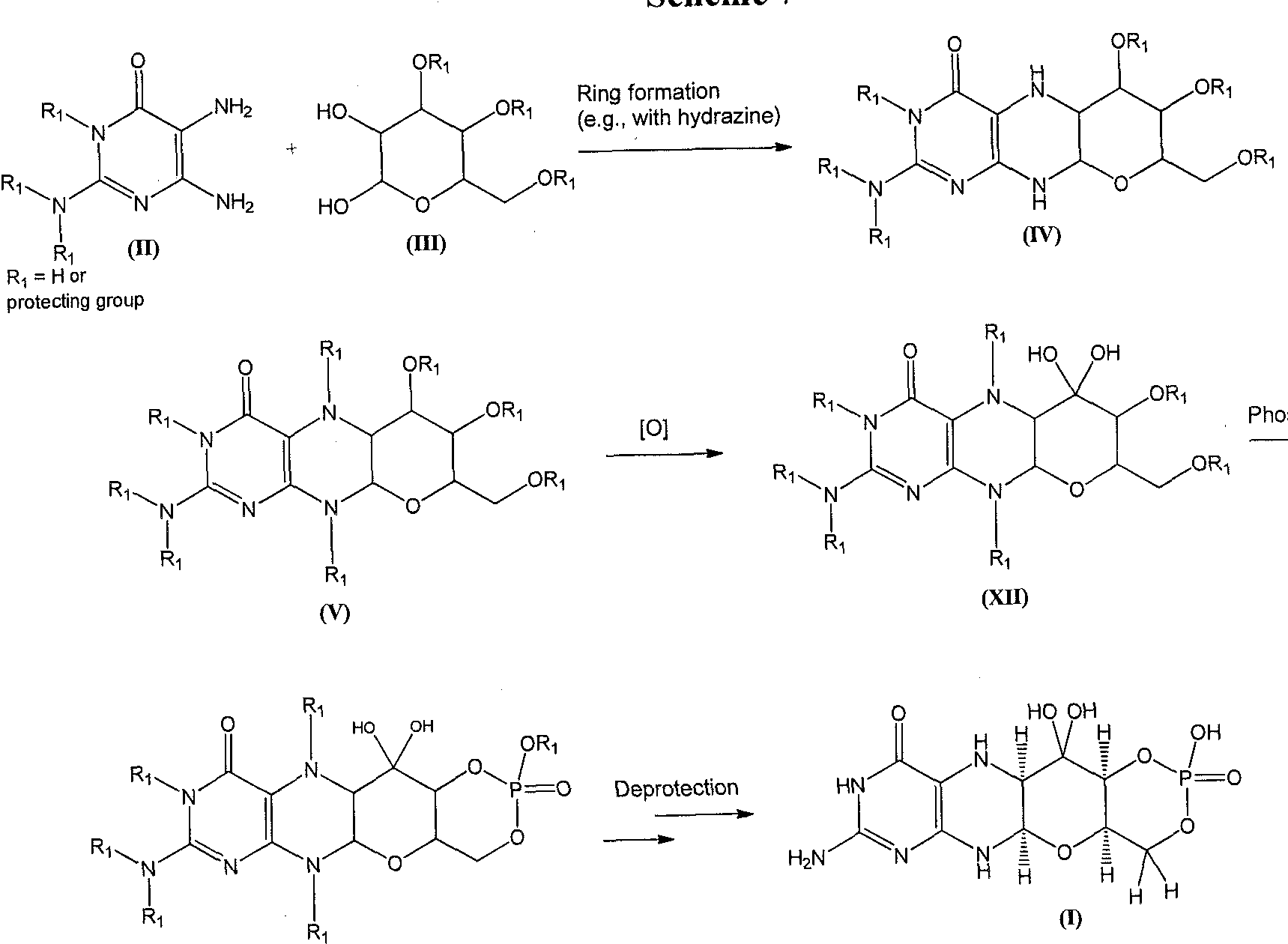
Candesartan is synthesised as follows:  kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1

The method has technical problems as follows: a) the starting material is obtained by a minor reaction, b) its yield is relatively low and its industrial applicability is poor (due to N2 gas formation) because the Curtius rearrangement reaction is involved, and c) materials industrially hard to handle such as SOCI2 or NaH are used. In addition, methods for preparing an intermediate of candesartan cilexetil are disclosed in Organic Process Research & Development 11:490-493(2007), as represented by the following Reaction Scheme 2: Reaction Scheme 2
3:1 s
However, the preparation process has also shortcomings of a) undesired byproducts formed by nitrogenation at ortho- or para-position, b) safety problems from strong acids (sulfuric acid and nitric acid) used twice when introducing and rearranging nitrogen groups, and c) utilization of high-flammable Raney Ni.
cut paste
Novel and Practical Synthesis of Candesartan Cilexetil
Yongjun Mao, Ruisheng Xiong, Zheng Liu, Haihong Li, Jingkang Shen, and Jingshan Shen* *Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Rd., Zhangjiang Hi-Tech Park, Shanghai, 201203, China
Abstract
A novel and convergent synthetic route of candesartan cilexetil (API of Atacand), an effective angiotensin II receptor blocker, is described. Cleavage of the N-Boc and N-trityl protective group are implemented simultaneously and formation of the benzimidazole ring is conducted at the last step of this route, which gives candesartan cilexetil in 55% yield over six steps with 99.1% purity (HPLC).  Full Text HTMLPDF (567KB)PDF with Links (932KB)
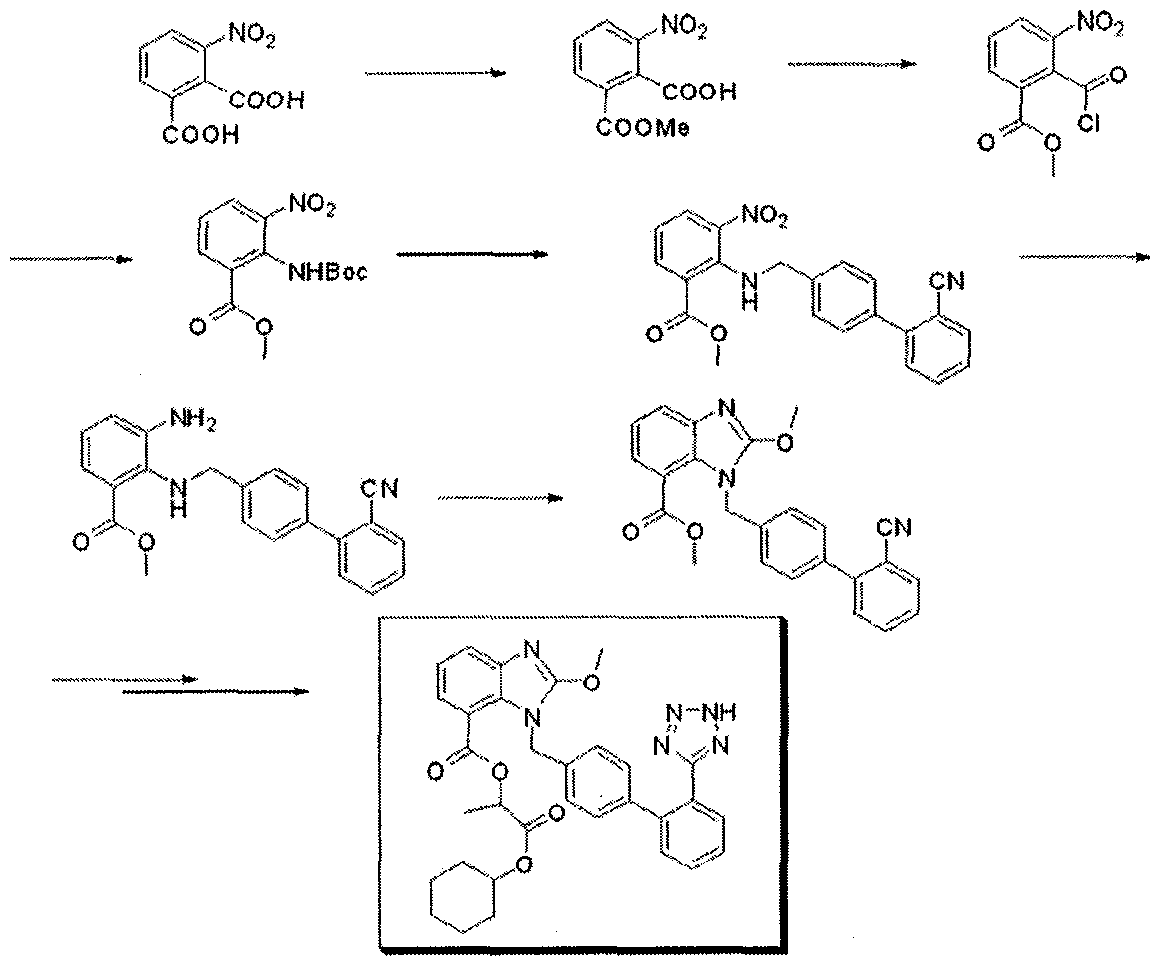
Full Text HTMLPDF (567KB)PDF with Links (932KB)  This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
|
Benzimidazole derivs., their production and use |
|
Naka, T.; Nishikawa, K.; Kato, T. (Takeda Chemical Industries, Ltd.) |
|
EP 0459136; EP 0720982; JP 1992364171; JP 1996099960; US 5196444; US 5328919; US 5401764; US 5703110; US 5705517; US 5962491; US 6004989 |
more info Candesartan cilexetil of Formula I, disclosed in U.S. Patent No. 5,196,444 as crystalline form, i.e., Form-I (C-type crystals), is chemically described as 1- cyclohexyloxycarbonyloxyethyl 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate.

H Formula I It is useful in the treatment of cardiovascular complaints such as hypertension and heart failure. Candesartan cilexetil is poorly soluble in water, which is attributed to its hydrophobic nature. Solubility plays an important role in achieving the desired concentration of a drug in systemic circulation for accomplishing the pharmacological response. Various techniques are known in literature to increase the solubility of poorly-soluble drugs, including decreasing the particle size, complexation, changing the surface characteristics of the particles, and incorporation of drug particles into colloidal systems like nanoparticles and liposomes. Among these, the most commonly used technique to increase the solubility is particle size reduction. Sometimes the rate of dissolution of a poorly-soluble drug is the rate limiting factor in its rate of absorption by the body. These drugs may be more readily bioavailable if administered in a finely divided state. Particle size reduction increases the surface area causing an increase in the dissolution rate of the compound, and hence, its bioavailability. There are certain techniques reported in literature to reduce the particle size of such poorly-soluble drugs. PCT Publication No. WO 2006/122254 discloses stable candesartan cilexetil of fine particle size, wherein the stable micronized candesartan cilexetil is prepared by slurrying a sample of candesartan cilexetil of fine particle size in a suitable solvent for a suitable amount of time. In this application, candesartan cilexetil of fine particle size is obtained directly from the synthesis of candesartan cilexetil or by comminuting candesartan cilexetil using milling. PCT Publication No. WO 2005/123720 describes fine particles of candesartan cilexetil having improved pharmacokinetic profile and a process for their production, wherein fine particle size is obtained by a) dissolving candesartan cilexetil in an organic solvent; b) cooling the solution obtained in step a) under stirring to crystallize candesartan cilexetil from the solution; and c) isolating candesartan cilexetil having a particle size of with d90 not more than about 25 μ. U.S. Patent Application No. 2006/0165806 describes compositions comprising a candesartan, such as candesartan cilexitil. The candesartan particles of the composition have an effective average particle size of less than about 2000 nm. U.S. Patent Application No. 2008/0038359 describes a nanoparticle pharmaceutical formulation comprising a poorly soluble drug substance having an average particle size of less than about 1000 nm, a solid or semisolid dispersion vehicle, and optionally a non-surface modifying excipient. U.S. Patent No. 7,828,996 discloses the methods for forming nanoparticles of a material of narrow polydispersity with ultrasonic waves using a partially submersed sonicator that does not touch any part of the apparatus and the point of addition of organic solvent is in the wave funnel produced by sonication and within the selected distance from the wave-source depending on the desired particle size. U.S. Patent No. 7,780,989 discloses the preparation of a dispersion of nanocrystalline particles in an aqueous medium using ultrasound. U.S. Patent No. 5,314,506 describes a crystallization process in which a jet of a solution containing a substance is impinged with a second jet containing an anti-solvent for the substance. The rapid mixing produced by the impinging jets results in a reduction of the crystals so formed compared to conventional slow crystallization processes. The smallest crystals disclosed are about 3 μ and the majority are in the range of from about 3 μ to about 20 μ. PCT Publication No. WO 00/44468 describes a modification to the apparatus described in U.S. Patent No. 5,314,506, wherein ultrasound energy is applied at the point of impingement of the two jets to further enhance localized mixing and is stated to give direct formation of small crystals with a diameter of less than 1 μ. Generally, the crystalline particles described have an average size of 0.5 μ. Conventional particle size reduction methods such as high energy milling may result in loss of yield, noise and dusting, as well as unwanted exposure to highly potent pharmaceutical compounds. Also, in the case of crystalline compounds, stress generated on crystal surfaces during milling can adversely affect labile compounds. Therefore, there is a need for a process for particle size reduction of candesartan cilexetil, which is industrially advantageous, easy to handle and is cost effective.
-
Candesartan is a potent, selective AT1 subtype angiotensin II receptor antagonist and used for treatment of hypertension. Due to poor absorption of Candesartan in body, the prodrug candesartan cilexetilwas developed. The candesartan cilexetil is rapidly and completely hydrolyzed to candesartan in gastrointestinal tract.
-
[0004]
U.S. Pat. No. 5,196,444 discloses Candesartan cilexetil and a process for its preparation by the reaction of 2-ethoxy-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid with trityl chloride in presence of triethyl amine in methylene chloride and purification by column chromatography gives 2-ethoxy-1-[[2′-(N-triphenylmethyltetrazol-5-yl)-biphenyl-4-yl]methyl] benzimidazole -7-carboxylic acid, which upon condensation with cyclohexyl 1-iodoethyl carbonate in presence of potassium carbonate in DMF followed by purification with column chromatography gives a colorless powder which is recrystallized in ethanol yields ‘C’ type crystals of Candesartancilexitil.
-
[0005]
U.S. Pat. Application No. 2005/131027 discloses a process for preparation of candesartan cilexetil by reaction of trityl candesartanwith cilexetil halide and at least one base in a low boiling solvent in presence of phase transfer catalyst to give Trityl candesartan cilexetil, which upon deprotection with at least one organic acid in at least one organic solvent.
U.S. Pat. Application 2005/131027 further discloses the deprotection of Trityl candesartan cilexetil in methanol without an acid.
-
[0006]
-
[0007]
Chem.Pharm.Bull. 47(2), 182-186 (1999) discloses two novel crystalline forms of Candesartan cilexetil, form-I and form-II.
-
[0008]
PCT publication WO 04/085426 discloses Candesartan cilexetil 1,4-Dioxane solvate and two more crystalline forms, designated as form-III and form-IV. The disclosed process for preparation of form-III involves crystallization of Candesartan cilexetil in toluene and for form-IV involves crystallization in a mixture of methyl tert-butyl ether and methanol.
-
[0009]
PCT publication WO 2005/077941 discloses several crystalline forms, solvates of Candesartan cilexetil along with a process for preparation of form-I (type-C).
-
[0010]
The prior art disclosed methods for preparation of Candesartan cilexetilinvolves purification of Trityl candesartan and Candesartan cilexetil by column chromatography or involves the use of strong acids like IN HCl or the use of organic acids or without an acid in methanol for detrytilation of Trityl candesartan cilexetil.
-
[0011]
There is a requirement of a process for preparation of Candesartancilexetil which yields a pure Candesartan cilexetil without involving the purification by column chromatography and the usage of strong acids for deprotection.
Candesartan cilexetil of formula (I) shown beiow is chemicaily described as (+/-)-1- [[(cyclohexyloxy)carbonyl]oxy]ethyl-2-ethoxy-1 -[[2′-(1 H-tetrazol-5-yl)-1 , 1 ‘-biphenyl- 4-yl]methyl]-1 H-benzimidazoie-7-carboxylate. An alternative designation is (+-)-1- hydroxyethyf 2~Ethoxy-1 -{p-(o-1 H-tetrazo!-5-yIphenyi)benzyJ)-7-benzϊmidazoie~ carboxyiic acid cyclohexyl carbonate (ester), with candesartan being the underlying carboxylic acid, i.e. 2-Ethoxy-1 -(p-(o-1 H-tetrazol-5-ylphenyl)benzy!)-7-benz- imidazolecarboxylic acid.

Because of its ability to inhibit the angiotensin-converting enzyme it is widely used for the treatment of hypertension and related diseases and conditions. As an angiotensin Ii receptor antagonist, candesartan ciiexetil avoids the side-effects of calcium antagonists, and shows high stability and obvious curative effects. Currently candesartan ciiexetil is soid as racemic mixture, it is produced according to published patents, e.g. EP 0 720 982 B1 and EP 0 459 136. in Chem. Pharm. Bull. 47(2), 182-186 (1999) two crystalline forms (Form I and II), together with an amorphous form, are disclosed and characterized by their DSC thermograms, X-ray diffraction patterns and IR spectra. US 5,196,444 disclosed the C-type crystal (Form I) of candesartan cilexetif, and processes for producing it under acidic conditions. WO 04/085426 discloses the dioxane solvate of candesartan ciiexetil, together with two additional crystalline forms. WO 2005/077941 discloses hydrates and solvates of candesartan ciiexetil, together with processes for their preparation. WO 2006/048237 also describes the preparation of new polymorphic forms ofcandesartan ciiexetil, together with processes for their preparation, including the preparation of amorphous candesartan ciiexetil by precipitating it with a liquid cyclic hydrocarbon from a solution of candesartan ciiexetil in a chlorinated solvent. in WO 2005/123721 processes for the preparation of amorphous candesartanciiexetil are provided, comprised of spray-drying and precipitation. HPLC CUT PASTE , READER TO PICK ONLY REQUIRED INFO Candesartan cilexetil (60 g) is dissolved in isopropanol (900 m!_) at 60-65 0C. Solution is hot filtered into reactor and quickly cooled to 35 0C. At this temperature nucleation is provoked with 300 mg of candesartan cilexetil form I and stirring is enforced. Suspension is cooled to 3O0C in 1 hour and rigorous stirring is continued at this temperature for additional 5 hours. Then stirring power is reduced and the suspension is cooled to 2O0C in 8 hours. The product is filtered, washed with isopropanoi and dried for 2 hours at 38°C. Yield: 48.7 g of candesartan cilexetil form I. Area % HPLC: candesartan cilexetil: 99.73%, alky ester of candesartan cilexetil 0.08%, candesartan cilexetii pyran below 0.05%, tritylcandesartan cϋexetil 0.09% Average particle size: 19 /vm, no agglomerates present (see Figure 2) B) Detection of impurities in candesartan cilexetil Example 6 Detection of candesartan cϊlexetil pyran in candesartan cilexetii by HPLC HPLC (external standard method) was performed using the following specifications : Column: Zorbax Eclipse XDB-C18, 50 mm x 4.6 mm i.d.τ 1.8 μm particles Eluent A: 0.01 M NaH2PO4, pH 2.5 Eluent B: acetonitriie Gradient of Eluent:

Flow rate: about 1.2 ml/min Diluent: acetonitriie : water = 70 : 30 (V/V). Detection: UV, wavelength 225 nm injection volume: 5 μl Column temperature : 500C Autosampler temperature: 7°C Example 7 Detection of cilexetil pyran in 1 -chloroethyl cyclohexylcarbonate by GC GC/FID (area percent method) was performed using the following specifications: Column: capillary (fused-silica) AT-WAX or adequate Length: 30 m ID: 0.32 mm Film thickness: 0.25 μm Carrier gas: helium Carrier gas flow rate: 2.0 ml/mi n Split ratio: 10 : 1 Air flow rate: 400 ml/min Hydrogen flow rate: 40 ml/min Make up gas flow ISb rate: 25 ml/min Column temperature 100°C (0 min) → 10°C/min → 2000C (10 min or prolonged if necessary) Injector temperature: 21 O0C Detector temperature: 250OC Injection volume : 1 μl Diluent: Acetonithle: chromatography grade. Chromatographic system suitability Signal/noise of 1 -chloroethyl cyclohexyl carbonate: not less than 10
………………
Seki M * Mitsubishi Tanabe Pharma Corporation, Osaka, Japan An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012; 44: 3231-3237 
Significance
Candesartan cilexetil (Atacand®) is an angiotensin II receptor antagonist that is prescribed for the treatment of hypertension. It is a prodrug that is hydrolyzed to candesartan in the gut. The synthesis depicted, features an efficient protocol for ruthenium-catalyzed C–H arylation of the tetrazole A. Comment A significant challenge in this small-scale synthesis was the final removal of the benzyl protecting group from the tetrazole unit using transfer hydrogenation. Best results were obtained using a ‘thickshell’ Pd/C catalyst from Evonik

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....







































 is the structure on right
is the structure on right