
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Aztreonam
- Molecular FormulaC13H17N5O8S2
- Average mass435.433 Da
(2S,3S)-3-{[(2Z)-2-(2-Ammonio-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-2-methyl-4-oxo-1-azetidinesulfonate2-[[(Z)-[1-(2-amino-4-thiazolyl)-2-[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]amino]-2-oxoethylidene]amino]oxy]-2-methyl-propanoic acid
278-839-9[EINECS]
5159
78110-38-0[RN]
UA2451400
азтреонам [Russian] [INN]
أزتريونام [Arabic] [INN]
氨曲南 [Chinese] [INN]
AztreonamCAS Registry Number: 78110-38-0
CAS Name: [2S-[2a,3b(Z)]]-2-[[[1-(2-Amino-4-thiazolyl)-2-[(2-methyl-4-oxo-1-sulfo-3-azetidinyl)amino]-2-oxoethylidene]amino]oxy]-2-methylpropanoic acid
Additional Names: azthreonam
Manufacturers’ Codes: SQ-26776
Trademarks: Azactam (BMS); Primbactam (Menarini)
Molecular Formula: C13H17N5O8S2, Molecular Weight: 435.43
Percent Composition: C 35.86%, H 3.94%, N 16.08%, O 29.40%, S 14.73%
Literature References: The first totally synthetic monocyclic b-lactam (monobactam) antibiotic. It has a high degree of resistance to b-lactamases and shows specific activity vs aerobic gram-negative rods.
Prepn: R. B. Sykes et al.,NL8100571 (1981 to Squibb), C.A.96, 181062x (1982).
Fast-atom-bombardment mass spectra: A. I. Cohen et al.,J. Pharm. Sci.71, 1065 (1982). Activity vs gram-negative bacteria: R. B. Sykes et al.,Antimicrob. Agents Chemother.21, 85 (1982). Series of articles on structure-activity, in vitro and in vivo properties, pharmacokinetics: J. Antimicrob. Chemother.8, Suppl. E, 1-148 (1981).
Toxicology: G. R. Keim et al.,ibid. 141. Mechanism of action study: A. D. Russell, J. R. Furr, ibid.9, 329 (1982). Comparative stability to renal dipeptidase: H. Mikami et al.,Antimicrob. Agents Chemother.22, 693 (1982). Human pharmacokinetics: E. A. Swabb et al.,ibid.21, 944 (1982).
Clinical evaluation in urinary tract infection: C. Donadio et al.,Drugs Exp. Clin. Res.13, 167 (1987). Clinical efficacy in neonatal sepsis: S. Sklavunu-Tsurutsoglu et al.,Rev. Infect. Dis.13, Suppl. 7, S591 (1991). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.17, 1-39 (1988).
Properties: White crystalline, odorless powder, dec 227°. Very slightly sol in ethanol, slightly sol in methanol, sol in DMF, DMSO. Practically insol in toluene, chloroform, ethyl acetate.
Derivative Type: Disodium salt
Molecular Formula: C13H15N5Na2O8S2, Molecular Weight: 479.40
Percent Composition: C 32.57%, H 3.15%, N 14.61%, Na 9.59%, O 26.70%, S 13.38%
Properties: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim).
Toxicity data: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Monobactams.
Aztreonam is a beta-lactam antibiotic used to treat select aztreonam sensitive gram negative bacteria.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Aztreonam lysine | XNM7LT65NP | 827611-49-4 | KPPBAEVZLDHCOK-JHBYREIPSA-N |
A monocyclic beta-lactam antibiotic originally isolated from Chromobacterium violaceum. It is resistant to beta-lactamases and is used in gram-negative infections, especially of the meninges, bladder, and kidneys. It may cause a superinfection with gram-positive organisms.
Aztreonam, sold under the brand name Azactam among others, is an antibiotic used primarily to treat infections caused by gram-negative bacteria such as Pseudomonas aeruginosa.[1][2] This may include bone infections, endometritis, intra abdominal infections, pneumonia, urinary tract infections, and sepsis.[1] It is given by intravenous or intramuscular injection or by inhalation.[1]
Common side effects when given by injection include pain at the site of injection, vomiting, and rash.[1] Common side effects when inhaled include wheezing, cough, and vomiting.[1] Serious side effects include Clostridium difficile infection and allergic reactions including anaphylaxis.[1] Those who are allergic to other β-lactam have a low rate of allergy to aztreonam.[1] Use in pregnancy appears to be safe.[1] It is in the monobactam family of medications.[1] Aztreonam inhibits cell wall synthesis by blocking peptidoglycan crosslinking to cause bacterial death.[1]
Aztreonam was approved for medical use in the United States in 1986.[1] It was removed from the World Health Organization’s List of Essential Medicines in 2019.[3][4] It is available as a generic medication.[1] It is a manufactured version of a chemical from the bacterium Chromobacterium violaceum.[5]
Medical uses
Nebulized forms of aztreonam are used to treat infections that are complications of cystic fibrosis and are approved for such use in Europe and the US; they are also used off-label for non-CF bronchiectasis, ventilator-associated pneumonia, chronic obstructive pulmonary disease, mycobacterial disease, and to treat infections in people who have received lung transplants.[6]
Aztreonam has strong activity against susceptible Gram-negative bacteria, including Pseudomonas aeruginosa. It is resistant to some beta-lactamases, but is inactivated by extended-spectrum beta-lactamases.
It has no useful activity against Gram-positive bacteria or anaerobes. It is known to be effective against a wide range of bacteria including Citrobacter, Enterobacter, E. coli, Haemophilus, Klebsiella, Proteus, and Serratia species.[7] The following represents minimum inhibitory concentration (MIC) susceptibility data for a few medically significant microorganisms.
- Staphylococcus aureus 8 – >128 μg/ml
- Staphylococcus epidermidis 8 – 32 μg/ml
- Streptococcus pyogenes 8 – ≥128 μg/ml
Synergism between aztreonam and arbekacin or tobramycin against P. aeruginosa has been suggested.[9]
SYN
ACS Medicinal Chemistry Letters, 11(2), 162-165; 2020
https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00534

Aztreonam, first discovered in 1980, is an FDA approved, intravenous, monocyclic beta-lactam antibiotic. Aztreonam is active against Gram-negative bacteria and is still used today. The oral bioavailability of aztreonam in humans is less than 1%. Herein we describe the design and synthesis of potential oral prodrugs of aztreonam.
A stirring mixture of CES1 (20 mg, 2200 Units) in a 15 mM solution of sodium phosphate monobasic (enzyme grade) in acetonitrile-d3 / D2O (1 mL; ratio of 2.5 : 97.5) was heated at 37 °C for 5 min. 2-(((Z)-(1-(2-aminothiazol-4-yl)-2-(((2S,3S)-1-((2,2- dimethyl-4-(pivaloyloxy)butoxy)sulfonyl)-2-methyl-4-oxoazetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)-2-methylpropanoic acid TFA salt 28c (10 mg, 14 µmol) was added, and the suspension was stirred for 70 min at 37 °C. Over the course of the reaction a fine precipitate formed. The mixture was filtered through a 25 mm, 0.45 µM glass fiber syringe filter (Pall Corporation Acrodisc). The filtrate was analyzed by 1H-NMR spectroscopy to reveal that 3,3-dimethyltetrahydrofuran was released as one of the products. The presence of 3,3-dimethyltetrahydrofuran was confirmed by 1H-NMR analysis of the same sample spiked with 2 µL of authentic materialSYN Faming Zhuanli Shenqing, 106520857,
SYN
Synthesis Reference
Neal G. Anderson, Carl F. Anderson, “Delta form of aztreonam and preparation thereof.” U.S. Patent US4826973, issued January, 1983.
SYN
https://patents.google.com/patent/US7145017B2/enAztreonam is a monobactam antibiotic disclosed in U.S. Pat. No. 4,775,670, which is incorporated by reference herein in its entirety. Aztreonam has the chemical name (Z)-2-[[[(2-amino-4-thiazolyl)[[(2S,-3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]carbamoyl]methylene]amino]oxy]-2-methylpropionic acid. Aztreonam is also known as [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid and (2S, 3S)-3-[[2-[2-amino-4-thiazolyl]-(Z)-2[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidine-1-sulfonic acid.Aztreonam has the structure:

Aztreonam is known to exist in various polymorphic forms including the α, β, δ, and γ forms.U.S. Pat. No. 4,775,670 discloses a process for making Aztreonam, a compound of formula I:

The process includes acylating a compound of formula IV:

The acylation entails reacting a compound of formula IV with a carboxylic acid or the corresponding carboxylic acid halide or carboxylic acid anhydride (R1—OH) in the presence of a carbodiimide such as dicyclohexylcarbodiimide and a substance capable of forming an active ester in situ such as N-hydroxybenzotriazole. U.S. Pat. No. 4,775,670 discloses that when the acyl group (R1) contains reactive functional groups, such as amino or carboxyl groups, it may be necessary to first protect those functional groups, then carry out the acylation reaction, and finally deprotect the resulting product. The deprotection is carried out by reaction of the acylation product with trifluoroacetic acid in the presence of anisole under anhydrous conditions.Similarly, U.S. Pat. No. 4,946,838 discloses a process for making crystalline anhydrous Aztreonam comprising reacting the diphenylmethyl ester of Aztreonam ([3S-[3β(Z),4α]]-3-[[(2-amino-4-thiazolyl)[(1-diphenylmethoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid) with trifluoroacetic acid in the presence of anisole under anhydrous conditions to produce the α-form of Aztreonam. The α-form is recrystallized from an anhydrous organic solvent to produce the β-form of Aztreonam. The β-form is anhydrous, substantially non-hygroscopic and more stable than the α-form.U.S. Pat. No. 5,254,681 discloses a process for preparing monobactams of formula (I):

wherein R is acyl. The process comprises acylating azetidin with 2-(2-amino-4-thiazolyl)-2-(Z)-(alkoxyimino) acetic acid in the presence of 1-hydroxy-benzotriazole and dicyclohexylcarbodiimide.U.S. Pat. No. 5,194,604 discloses a process and intermediates for making beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechains of formula (I), such as Aztreonam. The process comprises acylating a compound of formula III:

with a compound of formula (II):

in which R7 is

wherein

is a 4, 5, 6 or 7 membered heterocyclic ring having at least one nitrogen atom in the ring or such a group fused to a phenyl or substituted phenyl ring, to form a compound of formula (I):

wherein R1–R6 are as defined in U.S. Pat. No. 5,194,604.U.S. Pat. No. 4,652,651, which is incorporated by reference herein in its entirety, discloses a process for making 1-sulpho-2-oxoazetidine derivatives of the formula (I):

in which Het is an optionally amino-substituted, 5- or 6-membered, aromatic heterocycle containing 1 or 2 nitrogen atoms and optionally also an oxygen or sulphur atom, R1 may be lower alkoxycarbonyl-lower alkyl and R2 may be lower alkyl. The process entails acylating a compound of formula (II):

in which R20 equals R2 and R3 is hydrogen or sulpho, with a thioester of the formula (III):

in which Het is as above and R10 has any of the values of R1. U.S. Pat. No. 4,652,651 discloses that where R10 is a lower alkoxycarbonyl-lower alkyl group, for example the t-butoxycarbonylmethyl group, this can be converted, if desired, into the corresponding carboxylower alkyl group by treatment with a strong acid such as trifluoroacetic acid (optionally in the presence of anisole), hydrochloric acid or p-toluenesulphonic acid at a low temperature such as −10° C. to room temperature.There remains a need in the art for a process of making Aztreonam which does not require anhydrous reaction conditions and which also enables high yield and high purity. The present invention answers this need.
SUMMARY OF THE INVENTIONThe invention is based on the discovery that Aztreonam can be produced by reacting [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid with aqueous acid. The process of the invention, enables yields of between 70–75% and purities above 98%, preferably above 99%. The inventive aqueous process is advantageous over the prior art anhydrous processes in that the reaction conditions are more mild, there is no need to clean the final product and there is no need to keep the system dry. Thus, the aqueous process is less expensive than the anhydrous processes.The present invention is directed to a process for preparing [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid by hydrolyzing the ester group of [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid. The hydrolysis may be effected by reacting the ester with aqueous acid, at elevated temperatures.One reaction scheme for carrying out the process is shown below:

EXAMPLE 15.4 g Azetidin is dissolved in 20 ml acetonitrile (or dimethyl formamide) with the assistance of 5 ml of triethylamine at room temperature. The solution is cooled to 0° C. A solution of 4 g TAEM in 25 ml THF is added with magnetic stirring. If the color disappears, 8 g TAEM in 50 ml THF is added. After 10 minutes, another 4.1 g TAEM in 25 ml THF is added. The solution is stirred at 0° C. for an additional hour. The pH is adjusted to about 4–5 with a freshly prepared TFA solution (TFA-THF 1:4, V/V). Being careful not to evaporate the acetonitrile, the THF is evaporated (weight loss is about 90 g) at 30° C. under vacuum. The remaining residue is diluted with 200 ml ethylacetate and then extracted with 100 ml and then 50 ml of distilled water. The aqueous extracts are combined and washed twice with 50 ml ethylacetate after readjustment of the pH to about 4–5. The dissolved ethylacetate is removed from the aqueous phase by vacuum at 30° C. 10–15 g KCl (or NaCl) is dissolved. The solution is acidified with HCl solution (cc. HCl-distilled water 1:4, V/V) with stirring (approx. 10 ml). The solution is cooled to 0° C. with slow stirring and crystallization occurs. The resulting suspension is refrigerated overnight (at about 5° C.). The suspension is filtered on a glass filter, and the crystals are washed with chilled water. The washed crystals are dried at room temperature. The product, Aztreonam t-butyl ester, is about 12.5–13 g white solid, which is sufficiently pure for the next step.
EXAMPLE 265 g Azetidine is dissolved in a mixture of 240 ml acetonitrile and 60 ml triethylamine. When dissolution is complete, TAEM is added in four portions. The suspension is stirred for 20–30 min, then diluted with 500 ml EtOAc and 500 ml water and stirred for 5–10 min. The pH of the emulsion is set to 5 with 2.4 M HCl solution. After the phases separate, the pH of the aqueous phase is checked. If the pH is between 4.20 and 5.30, the two phases are filtered and separated, otherwise more HCl is added. The upper phase is diluted with 900 ml ethylacetate and extracted with 2×500 ml water (faster phase separation). The combined aqueous phase is diluted with 500 ml water and washed with 2×500 ml ethylacetate. The dissolved ethylacetate is removed from the aqueous phase by vacuum. The aqueous phase is acidified further to pH 2 with 2.4 M HCl solution. The solution is stirred and cooled. Crystallization starts soon. The suspension is stirred and cooled to 0° C., stirring at this temperature overnight. The suspension is filtered, washed with chilled water, dried at 38° C. in air-circulated oven for 3 h. The yield is approx. 116–120 g of Aztreonam t-butyl ester.
EXAMPLE 3Aztreonam t-butyl ester (113.6 g, 0.231 mol) is suspended in 975 ml water at 60° C. with stirring and 325 ml trifluoroacetic acid is added. The solution is stirred for 60 min., then it is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 240 ml chilled water and filtered again. The filtrate is re-suspended in 360 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 61.6 g Aztreonam (water content: 15–16%).
EXAMPLE 4Aztreonam t-butyl ester (18.0 g, 0.0366 mol) is suspended in 144 ml water at 60° C. with stirring and 40 ml aqueous hydrochloric acid (1:1, V/V) is added. The solution is stirred for 60 min, then 37 ml 5.4 M NaOH solution is added. The solution is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 50 ml chilled water and filtered again. The filtrate is re-suspended in 70 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 8.3 g Aztreonam (water content: 15–16%). The crude Aztreonam is crystallized.
EXAMPLE 5Aztreonam t-butyl ester (100.00 g, Assay as is: 97.2%, 0.19796 mol)) is suspended in a mixture of 450 ml water and 5 ml trifluoroacetic acid. The suspension, which slowly becomes clear, is heated to 58° C. with stirring and 100 ml trifluoroacetic acid is added. The solution is stirred for 105 min at 60–63° C. The solution is added to chilled water (450 ml) with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 18 hours. The product is filtered on a glass-filter and washed with 300 ml chilled water. The product is suspended in 650 ml chilled water, then filtered and washed with 300 ml cold acetone. The product is suspended in 400 ml cold acetone and filtered and dried in an air-ventilation oven at 30° C. for 30 min. Yield: 66.6 g (63%, according to assays) Aztreonam (Assay: 100.5%, water content: 18.0%).HPLC Impurity Profile:
- Aztreonam: 99.22%
- Aztreonam t-butyl ester: 0.44%
HPLC Impurity Profile of Sample from Reaction Mixture: - Aztreonam: 82.20%
- Aztreonam t-butyl ester: 0.43%
- Aztreonam, open-chained: 7.22%
- Other main degradation product (RRT=0.56): 5.24%
EXAMPLE 6Aztreonam t-butyl ester (27.11 g, Assay as is: 96.5%, 0.05328 mol) is suspended in a mixture of 122 ml water and 1.35 ml cc. HCl. The suspension is heated to 62° C. with stirring and 30 ml cc. HCl is added. The suspension, which becomes clear after approx. 15 min, (then the product starts to precipitate), is stirred for 30 min at 63–65° C. Chilled water (162 ml) is added with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 2 hours. The product is filtered on a glass-filter, washed twice with 120 ml chilled water, twice with 125 ml cold acetone and filtered. The product is dried at room temperature overnight. Yield: 19.44 g (72%, according to assays) Aztreonam (Assay: 100.1%, water content: 14.4%).HPLC Impurity Profile:
- Aztreonam: 99.65%
- Aztreonam t-butyl ester: 0.21%
HPLC Impurity Profile of Sample from Reaction Mixture: - Aztreonam: 89.43%
- Aztreonam t-butyl ester: 0.26%
- Aztreonam, open-chained: 4.70%
- Other main degradation product (RRT=0.56): 1.47%
SYN
Manufacturing Process
This mixture was sterilized for 15 minutes at 121°C at 15 lbs/inch2 steam pressure prior to use. The fermentation flasks were incubated at 25°C for 40 to 45 hours on a of rotary shaker. A 250 liter batch of Agrobacterium radiobacter A.T.C.C. No. 31700 is fermented in a 100 gallon steel vessel with a media and operating conditions described below. Culture of Agrobacterium radiobacter grown out on agar slants, pH 7.3 consisted of yeast extract (1 g), beef extract (1 g), NZ amine A (2 g), glucose (10 g), agar (15 g) in 1000 ml distilled water. Loopful of surface growth from agar slant was used as the source of incolumn. Medium of oatmeal (20 g), tomato paste (20 g) tapped water to 1000 ml, pH 7, was sterilized for 15 min at 121°C at 15 lbs/inch2 steam pressure prior to use. 100 ml of the medium, containing incolumn is incubated at 25°C for about 24 hours on a rotary shaker. It was added to a mixture of yeast extract (5 g), glucose (10 g) in 1 L distilled water and incubated for about 42 hours at 25°C in 100 gallon stainless steel fermentation vessel.
During incubation, the broth is agitated at 155 r.p.m. and aerated at rate of 10.0 cubic feet per minute. An antifoam agent (Ucon LB625, Union Carbide) was added as needed. The fermentation beer was adjusted to pH 4 with aqueous HCl and calls separated by centrifugation. The supernatante (200 L) was extracted with 40 L of 0.05 m cetyldimethylbenzyl ammonium chloride in dichloromethane and extract concentrated in vacuo to 5.5 L. The concentrate was then extracted with solution of 177 g of sodium thiocyanate in 2 L of water, adjusting the mixture of pH 4.35 with phosphoric acid. The aqueous extract was concentrated in vacuo to 465 ml and added to 1840 ml of methanol. Solids are filtrated yielded 194 g of crude solid product. It was dissolved and chromatographed on a 5×106.5 cm column of Sephadex G-10 three times and after concentrating in vacuo gave 3.5 g of crude antibiotic M53 (azetreonam) which was chromatographed at first on QAE Sephadex A- 25 (liner gradient, prepared from 2.5 L of water and 2.5 L of 0.25 M sodium nitrate). Then the residue (fractions 26-75) gave M53 (natrium salt) after evaporation. It was triturated with methanol and the souble fraction, 0.40 g was chromatographed on a 2.5×20 cm column of Diaion HP20AG, eluting at 2 ml per minute with water and collecting 20 ml fractions. Fractions 26-75 gave 51.9 mg of antibiotic M53 (sodium salt).
Chemical Synthesis
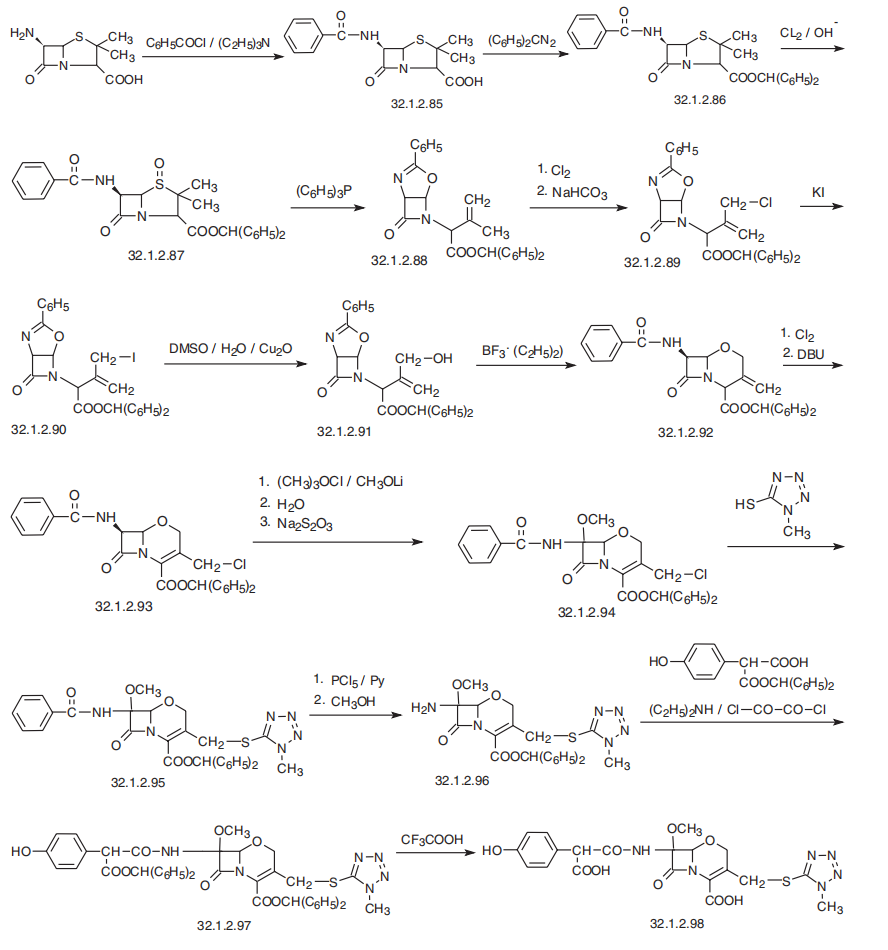
Aztreonam, (Z)-2[[[(2-amino-4-thiazolyl)[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]cabamoyl]methylen]amino]oxy]-2-methylpropionoic acid (32.1.4.9), is synthesized from tert-butyloxycarbonylthreonine, which is reacted with O-benzylhydroxylamine in the presence of dicyclohexylcarbodimide and 1-hydroxybenzotriazole, to form the benzyl hydroxamide derivative (32.1.4.1). This product undergoes a reaction with triphenylphosphine and ethyl azodicarboxylate, which results in the cyclodehydration of the product to (3S-trans)-N-benzyloxy-3-tert-butyloxycarbonylamino-4-methyl-azetidinone (32.1.4.2). Debenzylating this by hydrogen reduction using a palladium on carbon catalyst forms (3S-trans)-N-hydroxy-3-tertbutyloxycarbonyl-amino-4-methyl-azetidinone (32.1.4.3). The hydroxyl group in this compound is removed by reducing it with titanium trichloride, which forms azetidinone (32.1.4.4). Removing the tert-butyloxycarbonyl protection using trifluoroacetic acid and subsequent acylation of the resulting product with the benzyl chloroformate gives (3S-trans)-benzyloxycarbonylamino-4-methylazetidinone (32.1.4.5). Sulfonating this product with a mixture of sulfur trioxide and dimethylformamide gives the corresponding N-sulfonic acid. Turning the resulting Nsulfonic acid into a potassium salt by reacting it with potassium hydrophosphate, followed by replacing the potassium cation with a tetrabutylammonium cation by reacting it with tetrabutylammonium sulfate gives the product (32.1.4.6). Reducing this with hydrogen using a palladium on carbon catalyst gives 3-amino-4-methyl-monobactamic acid (32.1.4.7). Acylating this with (Z) 2-amino-α-[[2-(diphenylmethoxy)-1,1-dimethyl-2-oxoethoxy]imino] 4-thiazoleacetic acid in the presence of dicyclohexylcarbodiimide and 1-hydroxy-benzotriazole gives the diphenylmethyl ester of the desired aztreonam (32.1.4.8), which is hydrolyzed to aztreonam (32.1.4.9) using trifluoroacetic acid.
It is believed that the methyl group at position 4 increases the stability of the beta-lactam ring with respect to most beta-lactamases, and at the same time it does not induce formation of beta-lactamase as cephalosporins and imipenems do.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Spectrum of activity
Acinetobacter anitratus, Escherichia coli, Pseudomonas aeruginosa, and Proteus mirabilis are generally susceptible to aztreonam, while some staphylococci, Staphylococcus aureus, Staphylococcus haemolyticus and Xanthomonas maltophilia are resistant to it. Furthermore, Aeromonas hydrophila, Citrobacter koseri (Citrobacter diversus), Pantoea agglomerans (Enterobacter agglomerans), Haemophilus spp. and Streptococcus pyogenes have developed resistance to aztreonam to varying degrees.[10]
Aztreonam is often used in people who are penicillin allergic or who cannot tolerate aminoglycosides.[medical citation needed]
Administration[edit]
Aztreonam is poorly absorbed when given orally, so it must be administered as an intravenous or intramuscular injection (trade name Azactam ), or inhaled (trade name Cayston) using an ultrasonic nebulizer. In the United States, the Food and Drug Administration (FDA) approved the inhalation form on 22 February 2010, for the suppression of P. aeruginosa infections in patients with cystic fibrosis.[11] It received conditional approval for administration in Canada and the European Union in September 2009,[11] and has been fully approved in Australia.[12]
Side effects
Reported side effects include injection site reactions, rash, and rarely toxic epidermal necrolysis. Gastrointestinal side effects generally include diarrhea and nausea and vomiting. There may be drug-induced eosinophilia. Because of the unfused beta-lactam ring there is somewhat lower cross-reactivity between aztreonam and many other beta-lactam antibiotics, and it may be safe to administer aztreonam to many patients with hypersensitivity (allergies) to penicillins and nearly all cephalosporins.[13] There is a much lower risk of cross-sensitivity between aztreonam and other beta-lactam antibiotics than within other beta-lactam antibiotics. However, there is a higher chance of cross-sensitivity if a person is specifically allergic to ceftazidime, a cephalosporin. Aztreonam exhibits cross-sensitivity with ceftazidime due to a similar side chain.[14]
Mechanism of action
Aztreonam is similar in action to penicillin. It inhibits synthesis of the bacterial cell wall, by blocking peptidoglycan crosslinking. It has a very high affinity for penicillin-binding protein-3 and mild affinity for penicillin-binding protein-1a. Aztreonam binds the penicillin-binding proteins of Gram-positive and anaerobic bacteria very poorly and is largely ineffective against them.[13] Aztreonam is bactericidal, but less so than some of the cephalosporins.[medical citation needed]
References
- ^ Jump up to:a b c d e f g h i j k l “Aztreonam”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 381. ISBN 9780857111562.
- ^ World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). The selection and use of essential medicines: report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children). Geneva: World Health Organization. hdl:10665/330668. ISBN 9789241210300. ISSN 0512-3054. WHO technical report series;1021.
- ^ Yaffe SJ, Aranda JV (2010). Neonatal and Pediatric Pharmacology: Therapeutic Principles in Practice. Lippincott Williams & Wilkins. p. 438. ISBN 9780781795388.
- ^ Quon BS, Goss CH, Ramsey BW (March 2014). “Inhaled antibiotics for lower airway infections”. Annals of the American Thoracic Society. 11 (3): 425–34. doi:10.1513/annalsats.201311-395fr. PMC 4028738. PMID 24673698.
- ^ Mosby’s Drug Consult 2006 (16th ed.). Mosby, Inc. 2006.
- ^ “Aztreonam Susceptibility and Minimum Inhibitory Concentration (MIC) Data” (PDF). toku-e.com. 3 February 2020.
- ^ Kobayashi Y, Uchida H, Kawakami Y (December 1992). “Synergy with aztreonam and arbekacin or tobramycin against Pseudomonas aeruginosa isolated from blood”. The Journal of Antimicrobial Chemotherapy. 30 (6): 871–2. doi:10.1093/jac/30.6.871. PMID 1289363.
- ^ “Aztreonam spectrum of bacterial susceptibility and Resistance” (PDF). Retrieved 15 May 2012.
- ^ Jump up to:a b Larkin C (22 February 2010). “Gilead’s Inhaled Antibiotic for Lungs Wins Approval”. BusinessWeek. Archived from the original on 2 March 2010. Retrieved 5 March 2010.
- ^ “FDA approves Gilead cystic fibrosis drug Cayston”. BusinessWeek. 23 February 2010. Retrieved 5 March 2010.
- ^ Jump up to:a b AHFS Drug Information 2006 (2006 ed.). American Society of Health-System Pharmacists. 2006.
- ^ Terico, AT; Gallagher, JC (December 2014). “Beta-lactam hypersensitivity and cross-reactivity”. Journal of Pharmacy Practice. 27 (6): 530–44. doi:10.1177/0897190014546109. PMID 25124380. S2CID 19275020.
External links
- “Aztreonam”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Azactam, Cayston, others |
| AHFS/Drugs.com | Monograph |
| License data | EU EMA: by INN |
| Pregnancy category | AU: B1 |
| Routes of administration | Intravenous, intramuscular, inhalation |
| ATC code | J01DF01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-onlyEU: Rx-only |
| Pharmacokinetic data | |
| Bioavailability | 100% (IM) 0.1% (by mouth in rats) Unknown (by mouth in humans) |
| Protein binding | 56% |
| Metabolism | Liver (minor %) |
| Elimination half-life | 1.7 hours |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78110-38-0 |
| PubChem CID | 5742832 |
| DrugBank | DB00355 |
| ChemSpider | 4674940 |
| UNII | G2B4VE5GH8 |
| KEGG | D00240 |
| ChEBI | CHEBI:161680 |
| ChEMBL | ChEMBL158 |
| CompTox Dashboard (EPA) | DTXSID0022640 |
| ECHA InfoCard | 100.071.652 |
| Chemical and physical data | |
| Formula | C13H17N5O8S2 |
| Molar mass | 435.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 227 °C (441 °F) (dec.) |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0070024A11981-07-131983-01-19E.R. Squibb & Sons, Inc.The crystalline anhydrous form of (3S-(3 alpha(z),4 beta))-3-(((2-amino-4-thiazolyl)(1-carboxy-1-methylethoxy)-imino)-acetyl)-amino)-4-methyl-2-oxo-1-azetidinesulfonic acid, method for its preparation, mixture and pharmaceutical composition containing itUS4529698A1981-01-191985-07-16E. R. Squibb & Sons, Inc.Process for preparing a 2-oxo-1-azetidinesulfonic acid saltUS4652651A1983-05-311987-03-24Hoffmann-La Roche Inc.Process for the manufacture of 1-sulpho-2-oxoazetidine carboxylic acid intermediates via catalytic ester cleavageUS4775670A1980-09-291988-10-04E. R. Squibb & Sons, Inc.2-oxo-1-azetidinesulfonic acid saltsEP0297580A11987-07-011989-01-04E.R. Squibb & Sons, Inc.Amorphous form of aztreonamUS4826973A1984-07-201989-05-02E. R. Squibb & Sons, Inc.Delta form of aztreonam and preparation thereofUS4923998A1977-03-141990-05-08Fujisawa Pharmaceutical Company, Ltd.Cephem and cepham compounds and processes for preparation thereofUS4946838A1981-07-131990-08-07E. R. Squibb & Sons, Inc.Crystalline anhydrous aztreonamUS5194604A1990-06-291993-03-16E. R. Squibb & Sons, Inc.Process and intermediates for beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechainsUS5254681A1989-08-021993-10-19Consiglio Nazionale Delle RicercheProcess for preparing monobactames and their intermediate productPL165700B11991-10-151995-01-31PanMethod of obtaining z2/2-aminothiazolyl-4/-2/-t-butoxycarbonyl-1-methylethoxyimine/ acetic acidWO2002051356A22000-12-272002-07-04Salus Pharma, Inc.Inhalable aztreonam for treatment and prevention of pulmonary bacterial infectionsWO2003018578A12001-08-272003-03-06Aurobindo Pharma Ltd.Method for producing beta form of crystalline anhydrous aztreonamUS20040062721A12000-12-272004-04-01Montgomery Alan BruceInhalable aztreonam lysinate formulation for treatment and prevention of pulmonary bacterial infectionsWO2004052333A12002-12-112004-06-24Pari GmbhPharmaceutical compositions for the pulmonary delivery of aztreonamUS20050014739A12003-05-152005-01-20Viktor GyollaiAztreonam beta polymorph with very low residual solvent contentUS20050032775A12003-07-022005-02-10Viktor GyollaiAztreonam L-lysine and methods for the preparation thereof
Publication numberPriority datePublication dateAssigneeTitle
Family To Family CitationsCN1802371A2003-05-152006-07-12特瓦药厂有限公司Aztreonam beta-polymorph with very low residual solvent contentAU2004256124B2 *2003-07-022011-04-28Corus Pharma, Inc.Aztreonam L-lysine and methods for the preparation thereofWO2006122253A1 *2005-05-092006-11-16Sicor, Inc.Process for making aztreonamWO2007083187A2 *2006-01-162007-07-26Orchid Chemicals & Pharmaceuticals LimitedAn improved process for the preparation of monobactam antibioticCN101412715B *2008-12-162010-04-14海南百那医药发展有限公司Aztreonam compound and preparation thereofCN102127068B *2010-12-312012-08-29山西普德药业股份有限公司Method for synthesizing aztreonam compoundCN102311431B *2011-08-302014-12-10海南海药股份有限公司Method for preparing anhydrous beta-aztreonamCN105017241B *2015-06-242018-03-06山东罗欣药业集团股份有限公司A kind of aztreonam compound and its preparation
////////////////Aztreonam, SQ 26776, antibacterial, lactam, monobactam, UA2451400, азтреонам , أزتريونام , 氨曲南 ,
C[C@H]1[C@H](NC(=O)C(=N/OC(C)(C)C(=O)O)\C2=CSC([NH3+])=N2)C(=O)N1S([O-])(=O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}