



Home » Posts tagged 'shionogi'
Tag Archives: shionogi
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ENSITRELVIR
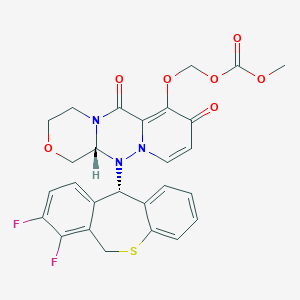
Ensitrelvir
S-217622, S 217622, Xocova, SHIONOGI,
6-[(6-chloro-2-methylindazol-5-yl)amino]-3-[(1-methyl-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-1,3,5-triazine-2,4-dione
CAS 2647530-73-0
| C22H17ClF3N9O2531.9 | |
| Synonyms | BDBM513874bioRxiv20220126.477782, S-217622 |
|---|
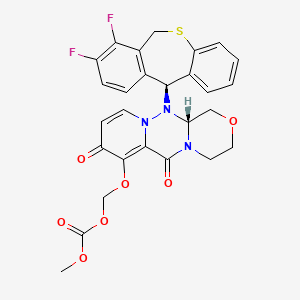
Ensitrelvir fumarate
CAS No. : 2757470-18-9
C22 H17 Cl F3 N9 O2 . C4 H4 O4
1,3,5-Triazine-2,4(1H,3H)-dione, 6-[(6-chloro-2-methyl-2H-indazol-5-yl)imino]dihydro-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-, (6E)-, (2E)-2-butenedioate (1:1)
| Formula: | C26H21ClF3N9O6 |
|---|---|
| M. Wt. : | 647.95 |
FDA 2026, APPROVALS 2026, 5/29/2026, Xocova
To use as post-exposure prophylaxis of coronavirus disease 2019 (COVID-19) following contact with an individual who has COVID-19
A Phase 1 study of S-217622 in healthy adult participants (jRCT2031210202)
Japan Registry of Clinical Trials Web Site 2021, July 16
PMDA APPROVED 2022/11/22, Xocova
Ensitrelvir[1] (code name S-217622, brand name Xocova)[2] is an antiviral drug developed by Shionogi in partnership with Hokkaido University, which acts as an orally active 3C-like protease inhibitor for the treatment of COVID-19 infection.[3][4] It is taken by mouth, and has been successfully tested against the recently emerged Omicron variant.[5]
About S-217622
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID
19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26
Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro
Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo.
Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which
began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl
salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the
conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired
indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The
authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building
block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.;
Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.;
Mukae, H. A phase 2/3 study of S-217622 in participants with SARS
CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024.
(26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura,
T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.;
Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of
ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in
Japanese patients with mild-to-moderate COVID-19 or asymptomatic
SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents
Chemother. 2022, 66, No. 00697.
(27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.;
Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T.
Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants
circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132−
136.
(28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.;
Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.;
et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL
protease inhibitor clinical candidate for treating COVID-19. J. Med.
Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J.
Structural basis for the inhibition of coronaviral main proteases by
ensitrelvir. Structure 2023, 31, 1016.
(30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.;
Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al.
Development of a manufacturing process toward the convergent
synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023,
9, 836−843.
(31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano,
S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In
Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J.,
Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.


Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
View ORCID ProfileYuto Unoh, View ORCID ProfileShota Uehara, View ORCID ProfileKenji Nakahara, View ORCID ProfileHaruaki Nobori, Yukiko Yamatsu, View ORCID ProfileShiho Yamamoto, View ORCID ProfileYuki Maruyama, View ORCID ProfileYoshiyuki Taoda, View ORCID ProfileKoji Kasamatsu, View ORCID ProfileTakahiro Suto, Kensuke Kouki, View ORCID ProfileAtsufumi Nakahashi, Sho Kawashima, View ORCID ProfileTakao Sanaki, Shinsuke Toba, Kentaro Uemura, Tohru Mizutare, View ORCID ProfileShigeru Ando, View ORCID ProfileMichihito Sasaki, View ORCID ProfileYasuko Orba, View ORCID ProfileHirofumi Sawa, View ORCID ProfileAkihiko Sato, View ORCID ProfileTakafumi Sato, View ORCID ProfileTeruhisa Kato, View ORCID ProfileYuki Tachibana
doi: https://doi.org/10.1101/2022.01.26.477782
https://www.biorxiv.org/content/10.1101/2022.01.26.477782v1.full
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry

The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012

Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.

Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.

Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
(6E)-6-[(6-Chloro-2-methyl-2H-indazol-5-yl)imino]-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazinane-2,4-dione (3, S-217622)
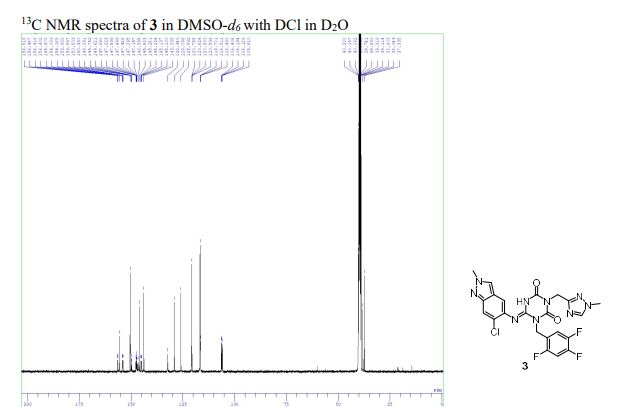
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
- Supporting information[supplements/477782_file02.pdf]
see spectrum at end of page
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ World Health Organization (2021). “International Nonproprietary Names for Pharmaceutical Substances. Proposed INN: List 126” (PDF). WHO Drug Information. 35 (4): 1135.
- ^ Xocova: Powerful New Japanese Pill for Coronavirus Treatment. BioPharma Media, February 2022
- ^ Jump up to:a b Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. (January 2022). “Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19”. bioRxiv. doi:10.1101/2022.01.26.477782. S2CID 246367525.
- ^ “Shionogi presents positive Ph II/III results for COVID-19 antiviral S-217622”. thepharmaletter.com. 31 January 2022.
- ^ Shionogi’s new COVID pill appears to ease omicron symptoms. Nikkei Asia, 21 December 2021
- ^ Japan to consider early approval for Shionogi COVID-19 pill. Japan Times, 8 February 2022
- ^ https://www.reuters.com/business/healthcare-pharmaceuticals/japans-shionogi-seeks-approval-oral-covid-19-drug-2022-02-25/[bare URL]
- ^ “Japan’s Shionogi seeks approval for COVID-19 pill”. Reuters. Reuters. 25 February 2022.
| Clinical data | |
|---|---|
| Other names | S-217622 |
| Identifiers | |
| showIUPAC name | |
| PubChem CID | 162533924 |
| Chemical and physical data | |
| Formula | C22H17ClF3N9O2 |
| Molar mass | 531.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Journal reference:
- Sasaki, M., Tabata, K., Kishimoto, M., et al. (2022). Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19. bioRxiv. doi:10.1101/2022.02.14.480338. https://www.biorxiv.org/content/10.1101/2022.02.14.480338v1.full.
///////////Ensitrelvir, S-217622, S 217622, Xocova, SHIONOGI, CORONA VIRUS, covid 19


JNJ-54861911, Atabecestat , атабецестат , أتابيسيستات ,
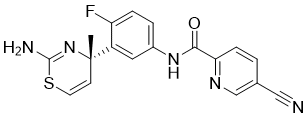
Atabecestat, JNJ-54861911
Cas 1200493-78-2
367.40, C18 H14 F N5 O S
2-Pyridinecarboxamide, N-[3-[(4S)-2-amino-4-methyl-4H-1,3-thiazin-4-yl]-4-fluorophenyl]-5-cyano-
Atabecestat is a beta-secretase inhibitor drug candidate.
(S)-N-(3-(2-amino-4-methyl-4H-1,3-thiazin-4-yl)-4-fluorophenyl)-5-cyanopicolinamide
JNJ-54861911
N-{3-[(4S)-2-Amino-4-methyl-4H-1,3-thiazin-4-yl]-4-fluorophenyl}-5-cyano-2-pyridinecarboxamide
2-Pyridinecarboxamide, N-[3-[(4S)-2-amino-4-methyl-4H-1,3-thiazin-4-yl]-4-fluorophenyl]-5-cyano-
WO 2017111042, 1H-NMR (CDCl3) δ: 1.71 (3H, s), 4.06 (3H, s), 6.29 (2H, d, J = 2.4 Hz), 7.07 (1H, dd, J = 11.3, 8.8 Hz), 7.65 (2H, dd, J = 6.8, 2.8 Hz), 7.86 (1H, ddd, J = 8.8, 4.1, 2.8 Hz), 8.19 (1H, dd, J = 8.1, 2.0 Hz), 8.43 (1H, d, J = 8.1 Hz), 8.89 (1H, d, J = 2.0 Hz), 9.81 (1H, s).
[α]D -11.8±1.0° (DMSO, 23°C, c=0.518)
![]()

Credit: Tien Nguyen/C&EN
Presented by: Yuji Koriyama, associate director at Shionogi & Co.
Target: β-site amyloid presursor protein cleaving enzyme 1 (BACE1), an enzyme whose buildup is implicated in Alzheimer’s disease
Disease: Alzheimer’s disease
Reporter’s notes: Presented by Koriyama, who told the audience he was attending the ACS National Meeting for the first time, JNJ-5486911 joins dozens of clinical candidates from many companies in Phase II and III trials to treat Alzheimer’s disease. Researchers started with a hit that inhibited BACE1 with approximately 2,600 nM affinity and advanced the program until finally reaching a compound with roughly 1 nM affinity. The compound is being jointly developed by Shionogi & Co. and Janssen Pharmaceuticals.
- Originator Shionogi
- Developer Janssen Research & Development
- Class Antidementias; Small molecules
- Mechanism of Action Amyloid precursor protein secretase inhibitors
Highest Development Phases
- Phase II/III Alzheimer’s disease
Most Recent Events
- 16 Jul 2017 Pharmacodynamics data from preclinical trials in Alzheimer’s disease presented at the Alzheimer’s Association International Conference (AAIC-2017)
- 15 Dec 2016 Biomarkers information updated
- 01 Jun 2016 Janssen Research & Development completes a phase I pharmacokinetic interaction trial in Healthy volunteers in Germany (PO) (NCT02611518)

SYNTHESIS


PATENTS
| Applicants: | SHIONOGI & CO., LTD. [JP/JP]; 1-8, Doshomachi 3-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP) (For All Designated States Except US). HORI, Akihiro [JP/JP]; (JP) (For US Only). YONEZAWA, Shuji [JP/JP]; (JP) (For US Only). FUJIKOSHI, Chiaki [JP/JP]; (JP) (For US Only). MATSUMOTO, Sae [JP/JP]; (JP) (For US Only). KOORIYAMA, Yuuji [JP/JP]; (JP) (For US Only). UENO, Tatsuhiko [JP/JP]; (JP) (For US Only). KATO, Terukazu [JP/JP]; (JP) (For US Only) |
| Inventors: | HORI, Akihiro; (JP). YONEZAWA, Shuji; (JP). FUJIKOSHI, Chiaki; (JP). MATSUMOTO, Sae; (JP). KOORIYAMA, Yuuji; (JP). UENO, Tatsuhiko; (JP). KATO, Terukazu; (JP) |
PATENT
PATENT


PATENT
WO 2017111042




Scheme 1-D
[Chem. 27]

Example 1-4
Preparation of Compound 15
[Chem. 31]

Compound 12 (3.0 g, 20.3 mmol) was dissolved in N-methylpyrrolidone (18 mL), and the solution was cooled to 5°C. Thionyl chloride (3.1 g, 26.1 mmol) was added to obtain a solution of Compound 13.
To a suspension of Compound 11 (5.0 g, 16.8 mmol) in ethyl acetate (50 mL) were added sodium bicarbonate (3.5 g, 42.0 mmol) and water (50 mL), and the mixture was stirred for 5 min at 20°C.
The layers were separated, and the organic layer was concentrated to 10 g under reduced pressure. N-Methylpyrrolidone (5 mL) and 35% hydrochloric acid (0.9 g) were added, and the mixture was cooled to 3°C. The solution of Compound 13 and N-methylpyrrolidone (1.5 mL) were added to obtain a solution of Compound 15.
The solution of Compound 15 was added to a mixture of water (15 mL) and ethyl acetate (10 mL). After stirring the mixture for 1 hour, triethylamine (14.8 g, 14.6 mmol), N-methylpyrrolidone (1.5 mL) and water (5 mL) were added and further stirred for 1 hour. Water (45 mL) was added, and the mixture was stirred for 1 hour, filtered and dried to obtain crystals of Compound 15 (Crystalline Form I, 5.71 g, 92.4%).
Compound 15
1H-NMR (CDCl3) δ: 1.71 (3H, s), 4.06 (3H, s), 6.29 (2H, d, J = 2.4 Hz), 7.07 (1H, dd, J = 11.3, 8.8 Hz), 7.65 (2H, dd, J = 6.8, 2.8 Hz), 7.86 (1H, ddd, J = 8.8, 4.1, 2.8 Hz), 8.19 (1H, dd, J = 8.1, 2.0 Hz), 8.43 (1H, d, J = 8.1 Hz), 8.89 (1H, d, J = 2.0 Hz), 9.81 (1H, s).
[α]D -11.8±1.0° (DMSO, 23°C, c=0.518)
Example 1-5
To a suspension of Compound 11 (1831 g, 6.2 mol) in ethyl acetate (18L) were added sodium bicarbonate (1293 g, 15.4 mol) and water (18L), and the mixture was stirred for 5 min at 20°C. The layers were separated, and the organic layer was concentrated to 3.8 kg under reduced pressure to obtain a concentrated solution of Compound 14.
Compound 12 (912 g, 6.2 mol) was dissolved in N-methylpyrrolidone (64L), and the solution was cooled to 4°C. Thionyl chloride (951 g, 8.0 mol) was added, and the mixture was stirred for 30 min. The concentrated solution of Compound 14 was added to obtain a solution of Compound 15.
The solution of Compound 15 and N-methylpyrrolidone (1.6 L) were added to water (18 L), and the mixture was stirred for 40 min at 25°C. 24% sodium hydroxide in water (5 kg), sodium bicarbonate (259 g, 3.1 mmol) and water (2.7 L) were added to the mixture. The mixture was stirred for 1 hour, filtered and dried to obtain crystals (metastable Form II) of Compound 15 (1.93 kg, 85.4%).
Example 1-3
Preparation of Compound 11
[Chem. 30]

A suspension of Compound 9 (20.0 g, 29.0 mmol) in N,N-dimethylacetamide (30 mL) was cooled to 5°C. 1,8-diazabicyclo(5,4,0)-7-undecene (39.7 g, 260.8 mmol) was added, and the mixture was stirred for 22 hours. Water (70 mL) was added to afford a solution of Compound 10.
To a mixture of ethyl acetate (200 mL), water (40 mL) and 62% sulfuric acid (12.7 g) was added the solution of Compound 10, and the mixture was cooled to 10°C. 15% sulfuric acid (3.7 g) was added, and the mixture was warmed to 20°C. The layers were separated, and the organic layer was washed with 5% sodium chloride in water (95 g). The layers were separated, and the organic layer was concentrated in vacuo to 42 mL. Ethyl acetate (20 mL) and 50% potassium carbonate in water (20 g) were added, and the mixture was warmed to 40°C. 4-chlorobenzenethiol (6.29 g, 43.5 mmol) and ethyl acetate (11 mL) were added, and the mixture was stirred for 1 hour. After cooling to 20°C, ethyl acetate (100 mL), water (68 mL) and 15% hydrochloric acid (42.6 g) were added. The layers were separated, and ethyl acetate (149 mL) and 20% potassium carbonate in water (40.5 g) were added to the aqueous layer. The layers were separated, and the organic layer was washed with water (100 mL). The layers were separated, and the organic layer was concentrated to 20 mL. Acetic acid (1.7 g, 29.0 mmol) was added, and the mixture was cooled to 5°C and stirred for 90 min, filtered and dried to afford 7.19 g of crystals of Compound 11 (yield: 83.4%, optical purity of (S)-isomer: 100%).
Compound 11
1H-NMR (DMSO-d6) δ: 6.74 (1H, dd, J=11.86, 8.56 Hz), 6.62 (1H, dd, J=6.97, 2.93 Hz), 6.35-6.40 (2H, m), 6.11 (1H, dd, J=9.60, 4.71 Hz), 1.90 (3H, s), 1.49 (3H, s).
The optical purity was determined as follows.
(Sample Preparation)
25 mg of Compound 11 was weighed and dissolved in a solvent to prepare a 50 mL sample solution.
(Method)
Using liquid chromatography, the peak area was determined by automatic integration method for each of (R)- and (S)-isomers of Compound 11.
(Conditions)
Detector: ultraviolet absorptiometer (wave length: 230 nm)
Column: CHIRALCEL OD-RH, φ4.6×150 mm, 5 μm, (Daicel Corporation)
Column Temp.: constant at around 40°C
Mobile Phase: water/acetonitrile (LC grade)/methanol (LC grade)/triethylamine (1320:340:340:1)
Flow Rate: 1.0 mL/min (retention time of Compound 11: about 8 min for (R)-isomer, about 9 min for (S)-isomer)
Time span of measurement: over 15 min from the sample injection
Injection Volume: 10 μL
Sample Cooler Temp.: constant at around 25°C
Autoinjector Rinse Solution: water/acetonitrile (1:1)

//////////////JNJ-54861911, Atabecestat , атабецестат , أتابيسيستات ,Phase III , Alzheimer’s disease, DEMENTIA, Shionogi, Developer, Janssen Research & Development
C[C@]1(C=CSC(N)=N1)c3cc(NC(=O)c2ccc(C#N)cn2)ccc3F
Baloxavir marboxil, バロキサビルマルボキシル , балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 ,


Baloxavir marboxil
バロキサビルマルボキシル
балоксавир марбоксил [Russian] [INN]
بالوكسافير ماربوكسيل [Arabic] [INN]
玛巴洛沙韦 [Chinese] [INN]
Carbonic acid, [[(12aR)-12-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl]oxy]methyl methyl ester
({(12aR)-12-[(11S)-7,8-Difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-6,8-dioxo-3,4,6,8,12,12a-hexahydro-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl}oxy)methyl methyl carbonate
- (((12aR)-12-((11S)-7,8-Difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-6,8-dioxo-3,4,6,8,12,12ahexahydro-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl carbonate
- Carbonic acid, (((12aR)-12-((11S)-7,8-difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl ester
Antiviral
In Japan the product is indicated for treatment influenza types A and B in adults and children
RG-6152
- Originator Shionogi
- Developer Roche; Shionogi
- Class Antivirals; Dibenzothiepins; Esters; Pyridines; Small molecules; Triazines
- Mechanism of Action Endonuclease inhibitors
Highest Development Phases
- Marketed Influenza A virus infections; Influenza B virus infections
- Phase III Influenza virus infections
- Preclinical Influenza A virus H5N1 subtype
|
Xofluza (TN)
Antiviral
|
|
| Formula |
C27H23F2N3O7S
|
|---|---|
| Cas |
1985606-14-1
|
| Mol weight |
571.5492
|
| 2018/2/23 | PMDA | JAPAN | APPROVED | Baloxavir marboxil | Xofluza | Shionogi |

| バロキサビル マルボキシル Baloxavir Marboxil  C27H23F2N3O7S : 571.55 [1985606-14-1] |
![]()

https://chem.nlm.nih.gov/chemidplus/sid/1985606141
Baloxavir marboxil (trade name Xofluza, compound code S-033188/S-033447) is a medication being developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was in late-stage trials in Japan and the United States as of early 2018, with collaboration from Roche AG.[1].
It was approved for sale in Japan on February 23, 2018.[2]
Baloxavir marboxil is a medication developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was approved for use in Japan in February 2018 and is in late phase trials in the United States as of early 2018. Roche, which makes Tamiflu, has acquired the license to sell Xofluza internationally, but it may not be until 2019 that it could be available in the United States [7]. Interestingly, a study has determined that administering Baloxavir marboxil with neuraminidase inhibitors leads to a synergistic effect in influenza treatment

It is an influenza therapeutic agent (cap-dependent endonuclease inhibitor), characterized by only taking one dose. Unlike neuraminidase inhibitors such as oseltamivir (Tamiflu) and zanamivir (Relenza) that inhibit the action of neuraminidase, which liberates viruses from the infected cells surface, baloxavir marboxil may prevent replication by inhibiting the cap-dependent endonuclease activity of the viral polymerase.[3]
In October 2015, the Japanese Ministry of Health, Labour and Welfare granted Sakigake status to Shionogi’s baloxavir marboxil for A type or B -type influenza virus infection . In October 2015, the drug was designated for Priority Review by the Ministry of Health, Labour and Welfare, presumably for the treatment of A type or B -type influenza virus infection .
This drug is a CAP endonuclease inhibitor [1]. The influenza endonuclease is an essential subdomain of the viral RNA polymerase enzyme. CAP endonuclease processes host pre-mRNAs to serve as primers for viral mRNA and therefore has been a common target for studies of anti-influenza drugs.
Viral gene transcription is primed by short-capped oligonucleotides that are cleaved from host cell pre mRNA by endonuclease activity. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription.The N-terminal domain of PA subunit (PAN) has been confirmed to accommodate the endonuclease activity residues, which is highly preserved among subtypes of influenza A virus and is able to fold functionally [4]. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription. The endonuclease domain binds the N-terminal half of PA (PAN) and contains a two-metal (Mn2+) active site that selectively cleaves the pre-mRNA substrate at the 3′ end of a guanine [3].
The administration of a CAP endonuclease inhibitor, such as Baloxavir marboxil, prevents the above process from occurring, exhibiting its action at the beginning of the pathway before CAP endonuclease may exert its action
It achieves this by inhibiting the process known as cap snatching[4], which is a mechanism exploited by viruses to hijack the host mRNA transcription system to allow synthesis of viral RNAs.
Shionogi, in collaboration with licensee Roche (worldwide except Japan and Taiwan), have developed and launched baloxavir marboxil
In March 2018, Shionogi launched baloxavir marboxil for the treatment of influenza types A and B in Japan . In September 2017, Shionogi was planning to file an NDA in the US; in February 2018, the submission remained in preparation
By September 2016, baloxavir marboxil had been awarded Qualified Infectious Disease Product (QIDP) designation in the US
In March 2017, a multicenter, randomized, double-blind, parallel-group, phase III study (NCT02954354; 1601T0831; CAPSTONE-1) was initiated in the US, Canada and Japan to compare a single dose of baloxavir marboxil versus placebo or oseltamivir bid for 5 days in influenza patients aged from 12 to 64 years of age (n = 1494). The primary endpoint was the time to alleviation of symptoms (TTAS).
PATENTS
JP 5971830
Kawai, Makoto; Tomita, Kenji; Akiyama, Toshiyuki; Okano, Azusa; Miyagawa, Masayoshi
PATENTS
WO 2017104691
Shishido, Takao; Noshi, Takeshi; Yamamoto, Atsuko; Kitano, Mitsutaka
In Japanese Patent Application No. 2015-090909 (Patent No. 5971830, issued on Aug. 17, 2016, Registered Publication), a compound having a CEN inhibitory action and represented by the formula:
[Chemical Formula 2]
is described. Anti-influenza agents of six mechanisms are enumerated as drugs that can be used together with the above compounds. However, no specific combinations are described, nor is it disclosed nor suggested about the combined effect.
Synthesis Example 2
[formula 39]
Compound III-1 (1.00g, 2.07mmol) to a suspension of DMA (5 ml) of chloromethyl methyl carbonate (0.483 g, 3.10 mmol) and potassium carbonate (0 .572 g, 4.14 mmol) and potassium iodide (0.343 g, 2.07 mmol) were added, the temperature was raised to 50 ° C. and the mixture was stirred for 6 hours. Further, DMA (1 ml) was added to the reaction solution, and the mixture was stirred for 6 hours. The reaction solution was cooled to room temperature, DMA (6 ml) was added, and the mixture was stirred at 50 ° C. for 5 minutes and then filtered. 1 mol / L hydrochloric acid water (10 ml) and water (4 ml) were added dropwise to the obtained filtrate under ice cooling, and the mixture was stirred for 1 hour. The precipitated solid was collected by filtration and dried under reduced pressure at 60 ° C. for 3 hours to obtain compound II-4 (1.10 g, 1.93 mmol, yield 93%).
1 H-NMR (DMSO-D 6) δ: 2.91-2.98 (1 H, m), 3.24-3.31 (1 H, m), 3.44 (1 H, t, J = 10.4 Hz) J = 10.8, 2.9 Hz), 4.06 (1 H, d, J = 14.3 Hz), 4.40 (1 H, dd, J = 11.5, 2.8 Hz), 3.73 (3 H, s), 4.00 , 5.67 (1 H, d, J = 6.5 Hz), 5.72 (1 H, d, J = 11.8 Hz), 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 J = 8.0, 1.1 Hz), 7.14 – 7.18 (1 H, m ), 7.23 (1 H, d, J = 7.8 Hz), 7.37 – 7.44 (2 H, m)
PATENTS
JP 6212678
PATENTS
JP 6249434
JP 5971830
SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF FINAL PRODUCT

Japan’s New Drug: One Pill May Stop The Flu in Just One Day
Isao Teshirogi, president and chief executive officer of Shionogi & Co., speaks during an interview in Tokyo, Japan. Photographer: Kiyoshi Ota/Bloomberg
One day, you may be able to stop flu viruses in your body in just one day with just one pill. Based on an announcement yesterday, that day may be someday very soon in May in Japan.
On Friday, Japanese pharmaceutical company Shionogi announced that the flu medication that they have developed, Xofluza, otherwise known as baloxavir marboxil (which sounds a bit like a Klingon General), has been approved to be manufactured and sold in Japan. Beginning in October 2015, the medication underwent priority review by Japan’s Ministry of Health, Labor, and Welfare. Shionogi filed for approval in the autumn of 2017. Compared to Tamiflu, which requires two doses each day for five days, apparently only a single dose of Xofluza will be needed to treat the flu. Even though Xofluza has received approval, people will have to wait until the Japanese national insurance sets a price for the medication, which according to Preetika Rana writing for the Wall Street Journal, may not occur until May.
Xofluza works via a different mechanism from neuroaminidase inhibitors like Tamiflu (oseltamivir) and Relenza (zanamivir). Flu viruses are like squatters in your home that then use the furniture and equipment in your home to reproduce. Yes, I know, that makes for a lovely picture. A flu infection begins when flu viruses reach your lungs. Each flu virus will enter a cell in your lungs and then use your cell’s genetic material and protein production machinery to make many, many copies of itself. In order to do this, the flu virus uses “cap-snatching”, which has nothing to do with bottle caps or Snapchat. The virus employs an endonuclease enzyme to clip off and steal the caps or ends of your messenger RNA and then re-purposes these caps to reproduce its own genetic material. After the virus has made multiple copies of itself, the resulting viruses implement another enzyme called a neuroaminidase to separate themselves from parts of the host cell and subsequently spread throughout the rest of your body to cause havoc. While Tamiflu, Relenza, and other neuroaminidase inhibitors try to prevent the neuroaminidase enzyme from working, Xofluza acts at an earlier step, stopping the “cap-snatching” by blocking the endonuclease enzyme.

In a clinical trial, Xofluza stopped an infected person from shedding flu virus sooner than Tamiflu. (Photo Illustration by Ute Grabowsky/Photothek via Getty Images)
By acting at an earlier step before the virus has managed to replicate, Xofluza could stop a flu virus infection sooner than neuroaminidase inhibitors. The results from Shionogi’s Phase III CAPSTONE-1 clinical trial compared Xofluza (then called Cap-dependent Endonuclease Inhibitor S-033188, which doesn’t quite roll off the tongue) with oseltamivir and placebo, with results being published in Open Forum Infectious Diseases. The study found that baloxavir marboxil (or Xofluza) stopped an infected person from shedding flu virus earlier (median 24 hours) than oseltamivir (median 72 hours). Those taking baloxavir marboxil also had lower measured amounts of viruses than those taking oseltamivir throughout the first 3 days of the infection. Baloxavir marboxil also seemed to shorten the duration of flu symptoms (median 53.7 hours compared to a median of 80.2 hours for those taking placebo). Since symptoms are largely your body’s reaction to the flu virus, you can begin shedding virus before you develop symptoms, and symptoms can persist even when you are no longer shedding the virus.
The key with any of these flu medications is early treatment, especially within the first 24 to 48 hours of infection, which may be before you notice any symptoms. Once the virus has replicated and is all over your body, your options are limited. The vaccine still remains the best way to prevent an infection.
In the words of Alphaville, this new drug could be big in Japan. While Xofluza won’t be available in time to help with the current flu season, this year’s particularly harsh flu season has highlighted the need for better ways to treat the flu. But will the United States see Xofluza anytime soon? Similar to Pokemon, Xofluza may need a year or two to reach the U.S. market. But one day, one pill and one day may be a reality in the U.S.
http://www.shionogi.co.jp/en/company/news/2018/pmrltj0000003nx1-att/e180223.pdf
XOFLUZA TM (Baloxavir Marboxil) Tablets 10mg/20mg Approved for the Treatment of Influenza Types A and B in Japan Osaka, Japan, February 23, 2018 – Shionogi & Co., Ltd. (Head Office: Osaka; President & CEO: Isao Teshirogi, Ph.D.; hereafter “Shionogi”) announced that XOFLUZATM (generic name: baloxavir marboxil) tablets 10mg/20mg was approved today by the Ministry of Health, Labour and Welfare for the treatment of Influenza Types A and B. As the cap-dependent endonuclease inhibitor XOFLUZATM suppresses the replication of influenza viruses by a mechanism different from existing anti-flu drugs, XOFLUZATM was designated for Sakigake procedure with priority review by the Ministry of Health, Labour, and Welfare of Japan in October 2015. Shionogi filed for approval to manufacture and sell XOFLUZATM in October 25, 2017. As the treatment with XOFLUZATM requires only a single oral dose regardless of age, it is very convenient, and is expected to improve adherence. XOFLUZATM is expected to be a new treatment option that can improve the quality of life in influenza patients. Shionogi will launch the product immediately after the National Health Insurance (NHI) price listing. Shionogi’s research and development targets infectious disease as one of its priority areas, and Shionogi have positioned “protecting people from the threat of infectious diseases” as one of its social mission targets. Shionogi strives constantly to bring forth innovative drugs for the treatment of infectious diseases, to protect the health of patients we serve.
References
- Jump up^ Rana, Preetika (10 February 2018). “Experimental Drug Promises to Kill the Flu Virus in a Day”. Wall Street Journal.
- Jump up^ “XOFLUZA (Baloxavir Marboxil) Tablets 10mg/20mg Approved For The Treatment Of Influenza Types A And B In Japan”. 23 February 2018 – via http://www.publicnow.com.
- Jump up^ Dias, Alexandre; Bouvier, Denis; Crépin, Thibaut; McCarthy, Andrew A.; Hart, Darren J.; Baudin, Florence; Cusack, Stephen; Ruigrok, Rob W. H. (2009). “The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit”. Nature. 458(7240): 914–918. doi:10.1038/nature07745. ISSN 0028-0836.
- Jump up^ “Cap snatching”.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H23F2N3O7S |
| Molar mass | 571.55 g·mol−1 |
| 3D model (JSmol) | |

Shionogi & Company, Limited(塩野義製薬株式会社 Shionogi Seiyaku Kabushiki Kaisha) is a Japanesepharmaceutical company best known for developing Crestor. Medical supply and brand name also uses Shionogi (“シオノギ”).
Shionogi has business roots that date back to 1878, and was incorporated in 1919. Among the medicines produced are for hyperlipidaemia, antibiotics, and cancer medicines.
In Japan it is particularly known as a producer of antimicrobial and antibiotics. Because of antibiotic resistance and slow growth of the antibiotic market, it has teamed up with US based Schering-Plough to become a sole marketing agent for its products in Japan.
Shionogi had supported the initial formation of Ranbaxy Pharmaceuticals, a generic manufacturer based in India. In 2012 the company became a partial owner of ViiV Healthcare, a pharmaceutical company specialising in the development of therapies for HIV.[3]
The company is listed on the Tokyo Stock Exchange and Osaka Securities Exchange and is constituent of the Nikkei 225 stock index.[4]
Medicines
- Claritin, An anti-histamine marketed in alliance with Schering-Plough.
- Crestor, cholesterol drug
- Nitrazepam, a short-term treatment for insomnia.
- Differin, a topical retinoid for acne.
- Moxifloxacin, antibacterial antiseptic that treats a number of infections
- Cymbalta, an SNRI class anti-depressant, marketed in alliance with Eli Lilly
- Osphena, an estrogen receptor agonist
Media
- Shionogi has a close relationship with Fuji Television Network, Inc., because Shionogi is the sponsor of “Music Fair” (as of 2018, aired on 17 TV stations including TV Oita System Co.) started in 1964.
- Shionogi was a main sponsor of Team Lotus during the age 1991/1994.[5]
References
- “Shionogi Company Profile”. Retrieved March 18, 2014.
- “Shionogi Annual Report 2013” (PDF). Retrieved March 18, 2014.
- “Shionogi and ViiV Healthcare announce new agreement to commercialise and develop integrase inhibitor portfolio”. viivhealthcare.com. Retrieved 18 March 2014.
- “Components:Nikkei Stock Average”. Nikkei Inc. Retrieved March 11,2014.
- Perry, Alan. “Sponsor Company Profiles”. Retrieved 25 April 2012.
External links
- Official Website (in English)
/////////Baloxavir marboxil, バロキサビルマルボキシル, JAPAN 2018, Xofluza, S-033188, S-033447, RG-6152, Qualified Infectious Disease Product, Priority Review, SAKIGAKE, балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 , Shionogi, roche
COC(=O)OCOC1=C2C(=O)N3CCOCC3N(N2C=CC1=O)C4C5=C(CSC6=CC=CC=C46)C(=C(C=C5)F)F
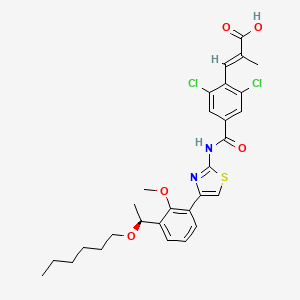
Lusutrombopag….Oral thrombopoietin (TPO) mimetic
Lusutrombopag
Update…..FDA approved july2018
(E)-3-[2,6-dichloro-4-[[4-[3-[(1S)-1-hexoxyethyl]-2-methoxyphenyl]-1,3-thiazol-2-yl]carbamoyl]phenyl]-2-methylprop-2-enoic acid
(S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid
(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylacrylic acid
UNII 6LL5JFU42F, CAS 1110766-97-6,
D10476, MW591.546 , [US2010267783], MF C29H32Cl2N2O5S, S-888711
Shionogi & Co., Ltd., 塩野義製薬株式会社 INNOVATOR
Optically active compound (C-3B) Melting point: 142-145°C…………….EP2184279B1
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)………….EP2184279B1
Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C), NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs)………EP2184279B1
Thrombopoietin receptor agonist, Oral thrombopoietin (TPO) mimetic
- 24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
- 31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
- 08 Nov 2013 Phase II development is ongoing in the US and the Europe
Process for preparing intermediates of an optically active 1,3-thiazole containing thrombopoietin receptor agonist Also claims crystalline forms of lusutrombopag intermediates and a process for preparing lusutrombopag. Shionogi is developing lusutrombopag, a small-molecule thrombopoietin mimetic, as an oral tablet formulation for treating thrombocytopenia.
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.

The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, “(E) -3- [2,6-Dichloro-4- [4- [3 – [(S) -1-hexyloxyethyl] – 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid “. lusutrombopag is represented by the following chemical structural formula.

Eltrombopag is represented by the following chemical structural formula.

Avatrombopag is represented by the following chemical structural formula.

Totrombopag choline is represented by the following chemical structural formula.


C 3B IS THE COMPD OF ROT (-) AND S, E FORM

Example 2
Synthesis of (R)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3A) (not included in the present invention) and (S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (C-3B)
According to the same method as in Example 1, an optically active compound (C-3A) and an opticallly active compound (C-3B) were synthesized from (RS)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid (B-3) obtained in Reference Example 3.Optically active compound (C-3A)Melting point: 139-141°C UNDESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82 – 0.87 (m, 3H) Optical rotaion +4.5 degrees (DMSO, c = 1.001, 25°C)
Optically active compound (C-3B)Melting point: 142-145°C DESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)

First step: Synthesis of (S)-1-(3-bromo-2-methyloxyphenyl)ethane-1-ol (17)
Using the same method as that of the first step of Example 3, the compound (17) was obtained from the compound (16) at a yield 77%.
-
-
Optical rotation: -23.5 ± 0.6 degrees (CHCl3, c = 1.050, 21°C)
NMR (CDCl3) θ ppm: 1.49 (3H, d, J = 6.6 Hz), 2.33 (1H, brs), 3.88 (3H, s), 5.19 (1H, q, J = 6.4 Hz), 7.01 (1H, t, J = 7.9 Hz), 7.40 (1H, dd, J = 7.7 Hz, J = 1.1 Hz), 7.46 (1H, dd, J = 8.0 Hz, J = 1.4 Hz)
-
Second step: Synthesis of (S)-1-bromo-3-(1-hexyloxyethyl)-2-methyloxybenzene (18)
-
-
Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
-
Third step and fourth step: Synthesis of (S)-4-(3-(1-hexyloxyethyl)-2-methyloxyphenyl)thiazole-2-amine (20)
-
-
Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
-
Compound (19)
-
-
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2-1.4 (6H, m), 1.45 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 3.29 (2H, m), 3.78 (3H, s), 4.73 (2H, m), 4.80 (1H, q, J = 6.4 Hz), 7.24 (1H, t, J = 7.8Hz), 7.52 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.65 (1H, dd, J = 7.7 Hz, J = 1.8 Hz)
-
Compound (20)
-
Optical rotation: -4.2 ± 0.4 degrees (DMSO, c = 1.025, 21°C)
NMR (CDCl3) δ ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, m), 1.35 (3H, d, J = 6.5 Hz), 1.48 (2H, m), 3.25 (2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) -
Fifth step: Synthesis of ethyl (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylate (21)
-
-
Using the same method as that of the fifth step of Example 3, the compound (21) was obtained from the compound (20) at a yield of 94%.
Optical rotation: +4.7 ± 0.4 degrees (CHCl3, c = 1.07, 21°C)
NMR (CDCl3 ) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2 – 1.35 (6H, m), 1.38 (3H, t, J = 7.1
Hz), 1.44 (3H, d, J = 6.4 Hz), 1.57 (2H, m), 1.77 (3H, d, J = 1.4 Hz), 3.30 (2H, m), 3.59 (3H, s), 4.31 (2H, q, J = 7.1 Hz), 4.83 (1H, q, J = 6.4 Hz), 7.17 (1H, t, J = 7.7 Hz), 7.42 (1H, d, J = 1.7 Hz), 7.42 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.51 (1H, s), 7.67 (1H, dd, J = 7.6 Hz, J = 1.7 Hz), 7.89 (2H, s), 10.30 (1H, brs)
-
Sixth step: Synthesis of (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylic acid (C-3B)
-
Using the same method as that of the sixth step of Example 3, the compound (C-3B) was obtained from the compound (21) at a yield of 80%.Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) -
Results of powder X-ray deffraction are shown in Fig. 5.
-
Diffraction angle of main peak: 2θ = 17.8, 21.1, 22.5, 23.3, 24.1, and 24.4 degrees
WO2005014561/EP1655291A1
https://www.google.co.in/patents/EP1655291A1?cl=en
WO2014003155, claiming a composition comprising lusutrombopag, useful for treating thrombocytopenia.
https://www.google.co.in/patents/US20150148385?cl=en
.
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
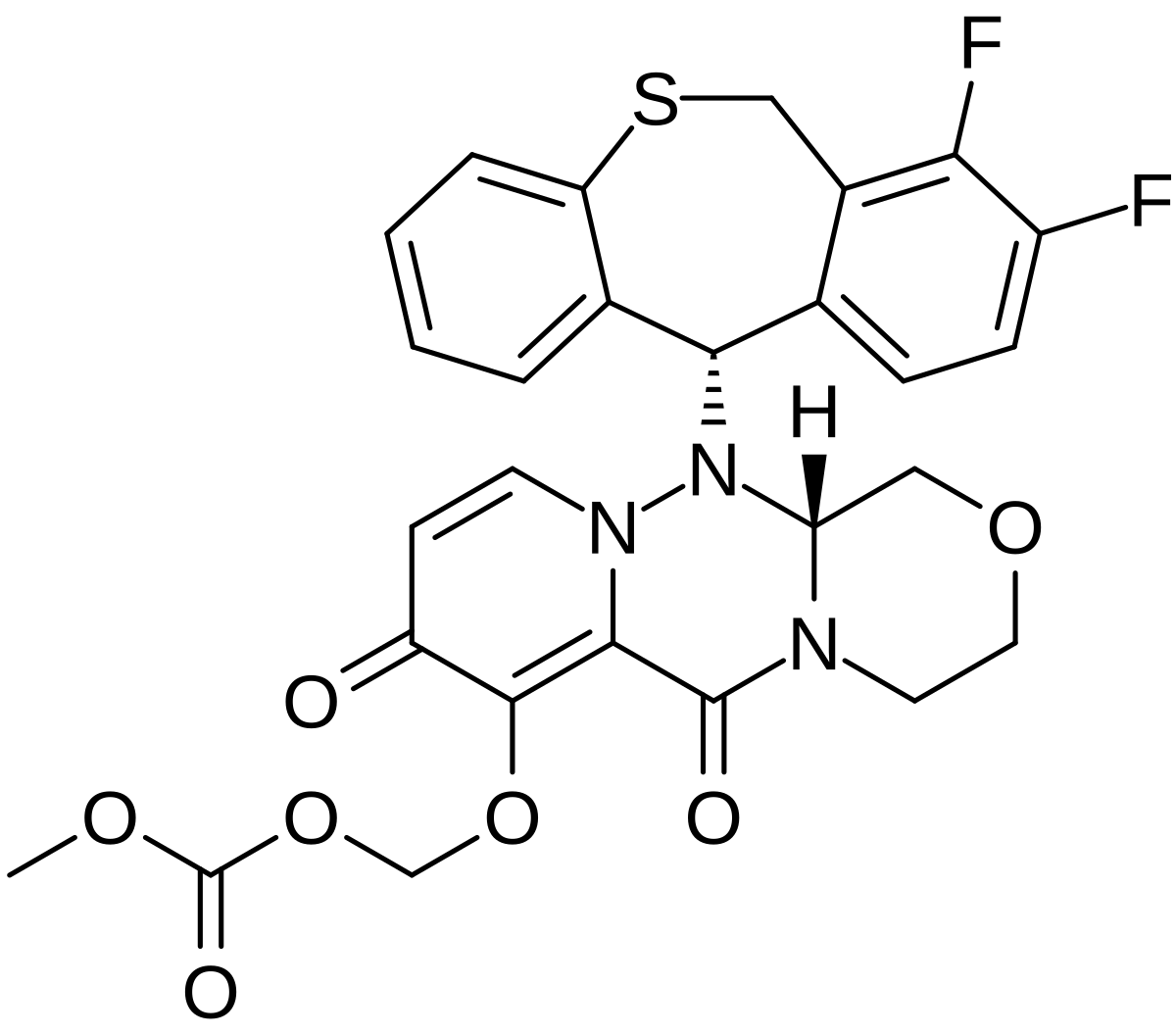
(Step 1) Synthesis of compound (VII ‘) under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran – heptane – ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ‘) (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI ‘))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ‘) under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ‘)). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ‘)). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ‘) (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ‘)).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II ‘). Next, the above-mentioned compound (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound ‘Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ‘) (1.50kg) and compound (VII’) (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ‘) (2.98kg, 95.2% yield).
1H-NMR (CDCl 3 ) delta: 0.87 (t, J = 6.8 Hz, 3H), 1.20-1.34 (M, 6H), 1.37 (t, J = 7.1 Hz, 3H), 1.44 (D, J = 6.5 Hz , 3H), 1.52-1.59 (m, 2H), 1.77 (d, J = 1.3Hz, 3H), 2.62 (s, 6H), 3.28-3.34 (m, 2H), 3.59 (s, 3H), 4.31 ( q, J = 7.1Hz, 2H), 4.83 (q, J = 6.5Hz, 1H), 7.16 (t, J = 7.7Hz, 1H), 7.40-7.43 (m, 2H), 7.51 (s, 1H), 7.68 (dd, J = 7.7, 1.8Hz, 1H), 7.92 (d, J = 1.3Hz, 2H), 10.58 (s, 1H). The results of the powder X-ray diffraction and I are shown in Figure 9 and Table 7. [Table 7]
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ‘) The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ‘) and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ‘) (1.99kg, 93.9% yield).
NMR (CDCl 3 ) delta ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, M), 1.48 (3H, D, J = 6.4 Hz), 1.52 – 1.64 (2H, M), 1.86 (3H, d, J = 1.4Hz), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) I is shown in Figure 15 the results of powder X-ray diffraction.
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (…. Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
| 101 | Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama, Hajime Yamada, Hiroshi Takemoto, Takeshi Shiota, Yoshikazu Tanaka, Noriko Yamane, Kouji Takahashi, Naoki Oyabu, Kenji Kuwabara, Itsuki Oshima, Kenzo Koizumi, Hiroshi Yoshida, Ayumu Nogami, Tomomi Yamada, Yutaka Yoshida, Takami Murashi, Shinichiro Hara. |
| 101 – Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia
Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). |
update………..
FDA approves lusutrombopag for thrombocytopenia in adults with chronic liver disease
https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm615348.htm
synthesis………..https://newdrugapprovals.org/2015/08/20/lusutrombopag-oral-thrombopoietin-tpo-mimetic/
On July 31, 2018, the Food and Drug Administration approved lusutrombopag (Mulpleta, Shionogi Inc.) for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure.
Approval was based on two randomized, double-blind, placebo-controlled trials (L-PLUS 1 and L-PLUS 2, NCT02389621) involving 312 patients with chronic liver disease and severe thrombocytopenia who were undergoing an invasive procedure and had a platelet count less than 50 x 109/L. Patients were randomized 1:1 to receive 3 mg of lusutrombopag or placebo once daily for up to 7 days.
In L-PLUS 1, 78% of patients (38/49) receiving lusutrombopag required no platelet transfusion prior to the primary invasive procedure, compared with 13% (6/48) who received placebo (95% CI for treatment difference: 49%, 79%; p<0.0001). In L-PLUS 2, 65% (70/108) of patients who received lusutrombopag required no platelet transfusion prior to the primary invasive procedure or rescue therapy for bleeding from randomization through 7 days after the procedure, compared with 29% (31/107) receiving placebo (95% CI for treatment difference: 25%, 49%; p<0.0001).
The most common adverse reaction in ≥ 3% of patients was headache.
The recommended lusutrombopag dosage is 3 mg orally once daily with or without food for 7 days.
View full prescribing information for Mulpleta.
FDA granted this application priority review and fast track designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
Follow the Oncology Center of Excellence on Twitter @FDAOncology.
Check out recent approvals at the OCE’s podcast, Drug Information Soundcast in Clinical Oncology.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
//////
phase 3, shionogi, japan, lusutrombopag, S 888711
CCCCCCOC(C)C1=CC=CC(=C1OC)C2=CSC(=N2)NC(=O)C3=CC(=C(C(=C3)Cl)C=C(C)C(=O)O)Cl
Ospemifene ….EMA accepts MAA submission of Shionogi’s ospemifene for the treatment of VVA

Ospemifene
CAS Number: 128607-22-7
OSPHENA is indicated for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause
Also known as:
- CCRIS 9205
- Deamino-hydroxytoremifene
- Fc-1271
- FC-1271a
- Ospemifene
- Osphena
- UNII-B0P231ILBK
Molecular Formula: C24H23ClO2
Molecular Weight: 378.89 g.mol-1
Ospemifene, FC-1271a
2-[4-[4-Chloro-1,2-diphenyl-1(Z)-butenyl]phenoxy]ethanol
2-(P-((Z)-4-Chloro-1,2-diphenyl-1-butenyl)phenoxy)ethanol
2-(4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy)ethanol
Orion Corp. (Originator), Hormos (Codevelopment)
Marja Sodervall, Maire Eloranta, Arja Kalapudas, Brian Kearton, Michael McKenzie, “METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE.” U.S. Patent US20080214860, issued September 04, 2008.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 6245819 | 2013-02-26 | 2020-07-21 |
| United States | 8236861 | 2013-02-26 | 2026-08-11 |
Data
| Patent No
US |
PatentExpireyDate | patent use code |
|---|---|---|
| 6245819 | Jul 21, 2020 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1370 |
| Exclusivity Code | ExclusivityDate |
|---|---|
| NCE | Feb 26, 2018 |
UPDATE……….ON OCT 2015
| Date of issue ofmarketing authorisation valid throughout the European Union | 15/01/2015 |
|---|
NMR……http://file.selleckchem.com/downloads/nmr/S428501-Ospemifene-HNMR-Selleck.pdf
HPLC….http://file.selleckchem.com/downloads/hplc/S428501-Ospemifene-HPLC-Selleck.pdf
Ospemifene appears as a white to almost white, non-hygroscopic crystalline powder. It is insoluble in water, soluble in ethanol and propanol, very slightly soluble in isopropanol. The partition coefficient was found 4.43 and the pKa was calculated 14.26. The molecule has two geometrical isomeric forms. The active substance ospemifene is the Z-isomer. Polymorphism was not observed.
The chemical name of the active substance ospemifene is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl) phenoxy]ethanol, corresponding to the molecular formula C24H23O2Cl and has a relative molecular mass of 378.9.
Ospemifene is a new selective non-hormonal estrogen receptor modulator (SERM) that is used for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause. FDA approved on February 26, 2013.
Bone Diseases, Treatment of, ENDOCRINE DRUGS, Gynecological Disorders, Treatment of , Hormone Replacement Therapy, METABOLIC DRUGS, Treatment of Osteoporosis, Treatment of Postmenopausal Syndrome , Selective Estrogen Receptor Modulators (SERM)
Article
 OSPEMIFINE
OSPEMIFINEArticle 27 March 2013
Shionogi Limited, the London-based European subsidiary of Shionogi & Co., Ltd announced today that on 26th March 2013 the European Medicines Agency (EMA) accepted its Marketing Authorisation Application (MAA) submission for ospemifene for the treatment of vulvar and vaginal atrophy (VVA) in post-menopausal women.
this is already approved by FDA
“We are pleased to announce the MAA submission for ospemifene to the EMA following the US Food and Drug Administration (FDA) approval last month. The acceptance of the MAA submission for ospemifene not only represents an important step forward in expanding the treatment options for women living in Europe with this condition, but it is also an important milestone for Shionogi as it continues to build its business in Europe” said Takashi Takenoshita, CEO of Shionogi Limited.
Osphena (ospemifene) to treat women experiencing moderate to severe dyspareunia (pain during sexual intercourse), a symptom of vulvar and vaginal atrophy due to menopause.
Dyspareunia is a condition associated with declining levels of estrogen hormones during menopause. Less estrogen can make vaginal tissues thinner, drier and more fragile, resulting in pain during sexual intercourse.
Osphena, a pill taken with food once daily, acts like estrogen on vaginal tissues to make them thicker and less fragile, resulting in a reduction in the amount of pain women experience with sexual intercourse.
“Dyspareunia is among the problems most frequently reported by postmenopausal women,” said Victoria Kusiak, M.D., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Osphena provides an additional treatment option for women seeking relief.”
Osphena’s safety and effectiveness were established in three clinical studies of 1,889 postmenopausal women with symptoms of vulvar and vaginal atrophy. Women were randomly assigned to receive Osphena or a placebo. After 12 weeks of treatment, results from the first two trials showed a statistically significant improvement of dyspareunia in Osphena-treated women compared with women receiving placebo. Results from the third study support Osphena’s long-term safety in treating dyspareunia.
Common side effects reported during clinical trials included hot flush/flashes, vaginal discharge, muscle spasms, genital discharge and excessive sweating.
Osphena is marketed by Florham Park, N.J.-based Shionogi, Inc.
- Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy – May 9, 2012
- END OF ARTICLE
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
INTRODUCTION
Ospemifene (commercial name Osphena produced by Shionogi) is an oral medication indicated for the treatment of dyspareunia – pain during sexual intercourse – encountered by some women, more often in those who are post-menopausal. Ospemifene is aselective estrogen receptor modulator (SERM)[1] acting similarly to an estrogen on thevaginalepithelium, building vaginal wall thickness which in turn reduces the pain associated with dyspareunia. Dyspareunia is most commonly caused by “vulval and vaginal atrophy.”[2]
The medication was approved by the FDA in February 2013.[3]
Ospemifene is used to treat dyspareunia. It is available as a 60 mg tablet that is taken by mouth once a day. The fact that ospemifene can be taken orally is an advantage over other products that are used to treat dyspareunia, because these are generally in a topical dosage form and have to be applied locally.[2] The oral dosage form is much easier and more convenient for patients to administer.
It is “an agonist/antagonist that makes vaginal tissue thicker and less fragile resulting in a reduction in the amount of pain women experience with sexual intercourse.”[2] This drug should be used for the shortest amount of time possible due to associated adverse effects.[2]
Approval process
Hormos Medical Ltd., which is a part of QuatRx Pharmaceuticals, filed a patent on January 19, 2005 for a solid dosage form of ospemifene.[5] In March of 2010, QuatRX Pharmaceuticals licensed ospemifene to Shionogi & Co., Ltd. for them to develop it into a product and put it on the market.[6] A New Drug Application (NDA) was submitted to the FDA on April 26, 2012.[7] Amendments to the NDA were submitted in June, July, August, October, and November 2012, and January and February of 2013.[7] It was ultimately approved by the FDA on February 26, 2013.[6]
Preclinial and clinical trials
Preclinical trials were performed in ovariectomized rats to model menopause.[8] Oral ospemifene was compared with raloxifene (another SERM), its metabolites 4-hydroxy ospemifene and 4′-hydroxy ospemifene, estradiol, and ospemifene administered as an intravaginal suppository.[8] Estradiol was used as a positive control and raloxifene was used because it is in the same drug class as ospemifene.[8]Multiple doses of oral ospemifene were tested.[8] 10 mg/kg/day of Ospemifene was found to cause a greater increase in vaginal weight and vaginal epithelial height than 10 mg/kg/day of raloxifene.[8] Vaginal weight had a 1.46x increase after a two week treatment of 10mg/kg/day of ospemifene.[8] The number of progesterone receptors was increased in the vaginal stroma and epithelium, which indicates that ospemifene has “estrogenic activity.”[8]
A binding assay was also performed to measure the affinity of ospemifene for the estrogen receptor (ERα and ERβ).[8] The study showed that ospemifene bound ERα and ERβ with similar affinity.[8] Ospemifene bound the estrogen receptors with a lower affinity than estradiol.[8] Ospemifene was shown to be an antagonist of “ERE-mediated transactivation on MCF-7 cells,” which the authors concluded indicates “anti-estrogenic activity in breast cancer cells.”[8]
Two 12 week phase 3 clinical trials were performed for ospemifene.[9] To evaluate the efficacy of the drug, 4 signs and symptoms of dyspareunia were measured. These included the “change in percent parabasal cells,” “change in percent superficial cells,” “change in vaginal pH,” and “change in most bothersome symptom (vaginal dryness and vaginal pain associated with sexual activity.”[9]Ospemifene was more effective than placebo in all four of these categories.[9] A dose-response was also seen in the trial; ospemifene 60 mg had greater efficacy than ospemifene 30 mg.[9] Safety was also evaluated in these phase 3 trials. There was a 5.2% increase in the incidence of hot flushes, 1.6% increase in urinary tract infections, and 0.5% increase in the incidence of headache with ospemifene over placebo.[9] One of the phase 3 trials was double-blinded and randomized and involved 826 women who were post-menopausal.[10]The women in the study were required to have one or more vulvovaginal atrophy (VVA) symptom that was moderate or severe in nature, no more than 5% of cells that were superficial when given a vaginal smear, and have a vaginal pH of at least 5.0.[10] Another phase 3 trial involved 605 women who were between the ages of 40 and 80, were diagnosed with VVA, and whose worst symptom was dyspareunia.[11]
 OSPEMIFENE
OSPEMIFENE
In the first half of the 2013 fiscal year, Osphena® generated 0.1 B yen in revenue, which is probably roughly equivalent to $974, 944 U.S. dollars.[12] When Osphena® was put onto the market, it was predicted to earn $495 million in 2017.[13]
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
“SERM”s (selective estrogen receptor modulators) have both estrogen-like and antiestrogenic properties (Kauffman & Bryant, 1995). The effects may be tissue-specific as in the case of tamoxifen and toremifene which have estrogen-like effects in the bone, partial estrogen-like effect in the uterus and liver, and pure antiestrogenic effect in breast cancer.
Raloxifene and droloxifen are similar to tamoxifen and toremifene, except that their antiestrogenic properties dominate. Based on the published information, many SERMs are more likely to cause menopausal symptoms than to prevent them. They have, however, other important benefits in elderly women: they decrease total and LDL cholesterol, thus deminishing the risk of cardiovascular diseases, and they may prevent osteoporosis and inhibit breast cancer growth in postmenopausal women.
Ospemifene is the Z-isomer of the compound of formula (I)

and it is one of the main metabolites of toremifene, is known to be an estrogen agonist and antagonist (Kangas, 1990; International patent publications WO 96/07402 and WO 97/32574 ). The compound is also called (deaminohydroxy)toremifene and it is also known under the code FC-1271a. Ospemifene has relatively weak estrogenic and antiestrogenic effects in the classical hormonal tests (Kangas, 1990). It has anti-osteoporosis actions and it decreases total and LDL cholesterol levels in both experimental models and in human volunteers (International patent publications WO 96/07402 and WO 97/32574 ). It also has antitumor activity in an early stage of breast cancer development in an animal breast cancer model.
Ospemifene is also the first SERM which has been shown to have beneficial effects in climacteric syndromes in healthy women. The use of ospemifene for the treatment of certain climacteric disorders in postmenopausal women, namely vaginal dryness and sexual dysfunction, is disclosed in WO 02/07718 . The published patent application WO 03/103649 describes the use of ospemifene for inhibition of atrophy and for the treatment or prevention of atrophyrelated diseases or disorders in women, especially in women during or after the menopause.
SYNTHESIS

credit chemdrug
The condensation of desoxybenzoin (I) with 2-(benzyloxy)ethyl bromide (II) by means of aqueous 48% NaOH containing triethylbenzylammonium chloride (TEBAC) gives 4-(benzyloxy)-1,2-diphenyl-1-butanone (III), which by reaction with the Grignard reagent (IV) – prepared from 4-(tetrahydropyranyloxy)phenyl bromide (V) and Mg in THF – yields the triphenylbutanol derivative (VI). Elimination of the THP-protecting group of compound (VI) by means of H2SO4 in ethanol/water at room temperature affords the triphenylbutanol derivative (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol to provide the butane-1,4-diol derivative (VIII). Cyclization of the butane-1,4-diol (VIII) by means of H2SO4 in hot ethanol/water gives 2-(4-hydroxyphenyl)-2,3-diphenyltetrahydrofuran (IX), which is treated with 48% HBr in refluxing AcOH to yield a mixture of (E)- and (Z)-4-(4-hydroxyphenyl)-3,4-diphenyl-3-buten-1-ol (X), which is separated by chemical work up. The phenolic OH group of the desired (Z)-isomer (X) is condensed with 2-(benzyloxy)ethyl bromide (II) by means of NaOH and tetrabutylammonium bromide in refluxing toluene/ water to afford the benzyloxyethyl ether (XII). Reaction of the aliphatic OH group of ether (XII) with PPh3 and CCl4 in acetonitrile provides the corresponding chloro derivative (XIII), which is finally debenzylated with H2 over Pd/C in ethyl acetate/ethanol.
Sorbera, L.A.; Castar, J.; Bay
Ospemifene. Drugs Fut 2004, 29, 1, 38
………………………………………………..
SYNTHESIS
US 4996225; US 5491173,
WO 9732574, WO 9607402,

The condensation of desoxybenzoin (I) with tetrahydropyranyl ether (II) in aq. 48% NaOH containing TEBAC gives 1,2-diphenyl-4-(tetrahydropyranyloxy)-1-butanone (III), which by a Grignard condensation with 4-methoxyphenylmagnesium bromide (IV) in THF yields the monoprotected triphenylbutanediol (V). The deprotection of (V) with H2SO4 in ethanol/water at room temperature affords the triphenylbutane-1,4-diol (VI), which is cyclized with H2SO4 in hot ethanol/water to provide 2-(4-methoxyphenyl)-2,3-diphenyltetrahydrofuran (VII). The reaction of (VII) with 48% HBr in refluxing acetic acid gives a mixture of (E)- and (Z)-4-(4-hydroxyphenyl)-3,4-diphenyl-1-butanol that is separated by chemical working up to obtain the desired (Z)-isomer (VIII). The condensation of the phenolic OH of (VIII) with benzyl protected 2-bromoethanol (IX) by means of NaOH and tetrabutylammonium bromide in refluxing toluene/water gives the benzyloxyethyl ether (X). The reaction of the aliphatic OH group of (X) with PPh3 and CCl4 in acetonitrile yields the corresponding chloro derivative (XI), which is finally debenzylated by hydrogenation with H2 over Pd/C in ethyl acetate/ethanol.
……………………………………………………………
SYNTHESIS
Ospemifene simple structure, its point is to control the synthesis of the product cis-trans isomerization of the double bond. Chloride 1 and benzene ( 2 ) occurs pay – acylation reaction 3 . Ester4 aluminum trichloride under the action of Fries rearrangement of 5 , 5 on the propylene oxide under alkaline conditions to obtain 6 , 6 and 3 McMurry coupling occurs directly generated Ospemifene.
……………………………………………
SYNTHESIS
https://www.google.com/patents/EP2121553B1
-
Ospemifene is the Z-isomer of the compound of formula (Ib)

-
The common starting material in the syntheses of (Ib), namely compound (II), is previously known (Toivola, 1990; EP 0095875 ). According to a method disclosed in EP 095875 , this compound was prepared by dealkylation of a corresponding ether to give (II). The method may be used to produce a mixture of isomers of compounds (Ib), but most preferably is used to prepare the pure E- and Z-isomers of this compound.
-
Particularly in case the Z-isomer of the compound (Ib) is desired, a preferable method for the synthesis of compound (II) is a McMurry reaction of commercially available starting materials, 4-hydroxybenzophenone with 3-chloropropiophenone. The McMurry reaction is a well-known reductive coupling of ketones involving two steps: (1) a single electron transfer to the carbonyl groups from an alkali metal, followed by (2) deoxygenation of the 1,2-diol with low-valent titanium to yield the alkene. This reaction produces mainly the Z-isomer of compound (II)

-
The alkylation in step a) is carried out in an organic solvent, preferably carried out in tetrahydrofuran. It is also preferable to add a base to the solvent, most preferably sodium hydride
-
Zinc (15.0 g, 0.23 mol) and tetrahydrofuran (THF) (180 ml) was added to the reaction vessel and cooled to -10 °C. Titan tetrachloride was added dropwise to the mixture (21.6 g, 0.114 mol) at about -10 °C. After the addition was completed the mixture was refluxed for two hours. Then the mixture was cooled to 40 °C and 4-hydroxybenzophenone (7.68 g, 0.039 mol) and 3-chloropropiophenone (6.48 g, 0.039 mol) dissolved in THF (75 ml) were added to the mixture. Refluxing was continued for additional 3.5 hours. The cooled reaction mixture was poured in aqueous potassium carbonate solution (21 g K2CO3 + 210 ml water) and allowed to stand overnight at the ambient temperature. The mixture was filtered and the precipitate was washed with THF. The filtrate was evaporated to dryness. The residue was dissolved in ethyl acetate and washed with water. Ethyl acetate phase was evaporated to dryness and the residue was crystallized first from methanol-water (8:2) and then from methanol-water (9:1). Yield 5.4 g.
-
Z-isomer:1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 6.48 (d, 2H, aromatic proton ortho to hydroxy), 6.75 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.4 (m, 10H, aromatic protons)
- EXAMPLE 14-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (Compound II)
EXAMPLE 2
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (0.23 g, 0.689 mmol) was dissolved in tetrahydrofuran (3 ml) under nitrogen atmosphere. Sodium hydride (0.025 g, 1.03 mmol) was added to the solution and the mixture was stirred at room temperature for an hour. 2-(2-iodo-ethoxy)-tetrahydropyran (0.3 g, 1.17 mmol) was added and the mixture was refluxed for 2 hours. Additional portions of 2-(2-iodo-ethoxy)-tetrahydro-pyran (0.5 g, 2 mmol) were added to the mixture during seven hours. After cooling and adding water, THF was evaporated and the mixture was extracted three times with ethyl acetate. The organic phase was washed with 2 N aqueous sodium hydroxide and water, dried with sodium sulphate and evaporated to dryness. The residue (which is Compound (IV) where Pr is tetrahydropyranyl) was dissolved in ethanol and acidified with 2 N aqueous hydrogen chloride solution. The mixture was stirred at room temperature over night, evaporated and extracted with dichloromethane. After washing with water the organic phase was dried (Na2SO4) and evaporated. The residue was purified by flash chromatography with dichloromethane/methanol 9.5/0.5 as eluent. Yield 0.17 g, 59 %.
-
Z-isomer, 1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
EXAMPLE 3
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
The compound was prepared by the same method as described in Example 2 using 2-(2-iodo-ethoxymethyl)-benzene as a reagent and removing the benzylic protecting group using the method described in Example (e) ofUS Patent No. 6,891,070 B2 . Briefly, the removal is carried out under a nitrogen atmosphere, in the presence of Zn powder and acetyl chloride.
- EXAMPLE 5
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-acetic acid ethyl ester (Example 4) was dissolved in tetrahydrofuran at room temperature under nitrogen atmosphere. Lithium aluminium hydride was added to the solution in small portions until the reaction was complete. The reaction was quenched by adding saturated ammonium chloride solution to the mixture. The product was extracted into toluene, which was dried and evaporated in vacuo. The yield 100 mg, 43 %.
-
1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
PATENT
https://www.google.com/patents/US6891070
e) 2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol:
Z-1-{4-[2-(2-Benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene (3.8 g, 7.4 mmol) is dissolved in ethyl acetate under nitrogen atmosphere, Zn powder (0.12 g, 1.85 mmol) and acetyl chloride (1.27 g, 16.3 mmol) are added and the mixture is stirred at 50° C. for 3 h (Bhar, 1995). The reaction mixture is cooled to room temperature, water (10 ml) is added and stirring is continued for additional 10 min. The aqueous layer is separated and the organic phase is washed with 1 M aqueous hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in methanol (16 ml) and water (4 ml). The acetate ester of the product is hydrolysed by making the mixture alkaline with sodium hydroxide (1 g) and stirring the mixture at room temperature for 1 h. Methanol is evaporated, water is added and the residue is extracted in ethyl acetate and washed with 1 M hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in toluene (25 ml), silica gel (0.25 g) is added and mixture is stirred for 15 min. Toluene is filtered and evaporated to dryness.
The residue is crystallised from heptane-ethyl acetate (2:1). The yield is 71%.
Z-isomer: 1H NMR (CDCl3): 2.92 (t, 2H), 3.41 (t, 2H), 3.58-3.63 (m, 2H), 3.69-3.80 (m, 4H), 3.96-4.01 (m, 2H), 6.56 (d, 2H), 6.78 (d, 2H), 7.10-7.40 (m, 10H).
E-2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol is prepared analogously starting from E-1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene. The product is purified by flash chromatography with toluene-methanol (10:0.5) as eluent.
E-isomer: 1H NMR (CDCl3): 2.97 (t, 2H), 3.43 (t, 2H), 3.65-3.79 (m, 4H), 3.85-3.90 (m, 2H), 4.13-4.17 (m, 2H), 6.85-7.25 (m, 2H).
Debenzylation of 1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene is also carried out by hydrogenation with Pd on carbon as a catalyst in ethyl acetate-ethanol solution at room temperature.
PATENT
http://www.google.com/patents/WO2014060639A1?cl=en
EXAMPLE 5. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospemifene) by base hydrolysis of pivaloyl-groiip
; . (Z)-2-(4-(4-Chloro- l ,2-diphenylbut-l-en- l-yl)phenqxy)ethyl pivalate ( 1 g, 2.16 mmol) was dissolved in THF (8 ml) followed by addition of MeOH (1 ml) and water (1 ml). Sodium hydroxide (0.1 g, 2.5 mmol) was added in orie portion and the reaction was stirred at room temperature for 12 h. After completion of the reaction the mixture was partitioned between water (20 ml) and EtOAc (20 ml). Organic phase was washed with water (20 ml) and brine (20 ml); dried (Na2S04), filtered, and concentrated: The residue was crystallized from -PrOH yielding ospernifene (0:29 g, 35 %) as a white solid.
1H-NMR (400 MHz, CDC13) δ (ppm): 7.37 (2H, t, 7=8Hz, ArH), 7.29 (3Η, t, J=7.2Hz, ArH), 7.20 (2Η, t,7=7.6Hz, ArH), 7.16-7.13 (3Η, m, ArH), 6.80 (2Η, d, J=8.8Hz, ArH), 6.57 (2Η, d, 7=8.8Hz, ArH), 3.94 (2Η, t, y=4.4Hz, ArOCH2CH2OH), 3.87 (2H, m, ArOCH2CH OH), 3.42 (2H, t, J=7.2Hz, C1CH2CH2), 2.92 (2H, t, 7=7.2Hz, C1CH2CH2), 1.95 (1Η, t, 7=6.4Hz, OH).
13C- NMR (100 MHz, CDC13) δ (ppm): 157.2, 143.2, 142.1 , 141.3, 2 x 135.7, 132.2, 130.0, 129.8, 128.8, 128.7, 127.4, 127.0, 113.9, 69.3, 61.8, 43.3, 39.0.
EXAMPLE 6. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospernifene) by reductive cleavage of pivaloyl-grou
(Z)-2-(4-(4-Chloro- 1 ,2-diphenylbut- 1 -en- 1 -yl)phenoxy)ethyl pivalate (3.5 g, 7.56 mmol) was dissolved in toluene (35 ml) and stirred under nitrogen for 5 min at room temperature. Lithium aluminium hydride solution (1 M in THE) (7.56 ml, 7.56 n mbi) was added dropwise to the reaction and the mixture was stirred at room temperature for 30 min. After HPLC indicated completion, the reaction was quenched by addition of saturated NH4Cl-sblution (75 ml). Additional amount of toluene (30 ml) was added and the phases were separated. The organic phase was washed with water (50 ml), brine (50 ml), dried (Na2S04), filtered and concentrated in vacuo. The residue was crystallized from 90 % MeOH yielding ospernifene (1 ,75 g, 61 9c) as a white solid.
1H NMR PREDICT

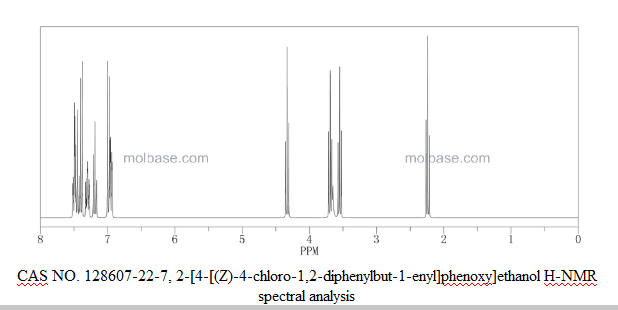
13C NMR PREDICT


References
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O (2003). “Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial”. Menopause10 (5): 433–9.doi:10.1097/01.GME.0000063609.62485.27.PMID14501605.
- Tanzi MG (April 2013). “Ospemifene: New treatment for postmenopausal women.”. Pharmacy Today. American Pharmacists Association.
- “FDA approves Osphena for postmenopausal women experiencing pain during sex”. FDA News Release (U.S. Food and Drug Administration). 2013-02-26.
- “Ospemifene: Indications, Side Effects, Warnings”. Drugs.com.
- EP application 2286806, Lehtola V-M, Halonen K, “Solid formulations of ospemifene”, published 2011-02-23, assigned to Hormos Medical Ltd.
- “Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy”. Drugs.com.
- Kusiak V (2013-02-13). “NDA Approval” (PDF). U.S. Food and Drug Administration.
- Unkila M, Kari S, Yatkin E, Lammintausta R (November 2013). “Vaginal effects of ospemifene in the ovariectomized rat preclinical model of menopause”. J. Steroid Biochem. Mol. Biol.138: 107–15.doi:10.1016/j.jsbmb.2013.04.004. PMID23665515.
- Center for Drug Evaluation and Research (2013-02-26). “Clinical Pharmacology and Biopharmaceutics Review Application Number 203505Orig1s000” (PDF). Office of Clinical Pharmacology Review. U.S. Food and Drug Administration.
- Bachmann GA, Komi JO (2010). “Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study”. Menopause17 (3): 480–6.doi:10.1097/gme.0b013e3181c1ac01. PMID20032798.
- Portman DJ, Bachmann GA, Simon JA (June 2013). “Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy”. Menopause20 (6): 623–30.doi:10.1097/gme.0b013e318279ba64. PMID23361170.
- http://www.shionogi.co.jp/en/ir/pdf/e_p131101.pdf. First Half of Fiscal 2013 Financial Results. Nov. 1, 2013.
- http://www.thepharmaletter.com/article/fda-approves-shionogi-s-osphena-for-postmenopausal-women-experiencing-pain-during-sex. ThePharmaLetter
PATENTS
|
8-8-2012
|
Method for enhancing the bioavailablity of ospemifene
|
|
|
1-21-2011
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
11-5-2010
|
METHODS FOR THE INHIBITION OF ATROPHY OR FOR TREATMENT OR PREVENTION OF ATROPHY-RELATED SYMPTOMS IN WOMEN
|
|
|
10-13-2010
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
3-18-2009
|
METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE
|
|
|
5-11-2007
|
Novel oral formulations of ospemifene
|
|
|
5-11-2007
|
Formulations of fispemifene
|
|
|
1-11-2006
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
12-9-2005
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
8-26-2005
|
Solid formulations of ospemifene
|
|
8-26-2005
|
Method for treatment or prevention of osteoporosis in individuals with high bone turnover
|
|
|
4-6-2005
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-11-2003
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-13-2001
|
Method for the treatment of vaginal dryness and sexual dysfunction in women during or after the menopause
|
Japan, Shionogi Receives Marketing and Manufacturing Approval of a Drug for Lipodystrophy,METRELEPTIN for Subcutaneous Injection

Metreleptin, an analog of the human hormone leptin, is a unique potential therapy for certain metabolic disorders in patients with rare forms of inherited or acquired lipodystrophy. Lipodystrophy is a very rare condition characterized by loss of subcutaneous fat.
Shionogi & Co., Ltd. today announced that it received marketing and manufacturing approval of recombinant human leptin, “METRELEPTIN for subcutaneous injection 11.25 mg ‘SHIONOGI'”
(generic name: metreleptin) for lipodystrophy on March 25,2013 in Japan.
Metreleptin was in-licensed by Shionogi from US-based AmylinPharmaceuticals, LLC., a subsidiary of Bristol-Myers Squibb Company currently.
Metreleptin is being studied as a potential therapy for certain metabolic disorders in patients with inherited or acquired lipodystrophy. Metreleptin is believed to work by reducing fat accumulation in organs, caused by the disease, thereby improving insulin sensitivity. Clinical studies have been conducted by investigators at the National Institutes of Health (NIH) and other academic institutions in the US, Europe, and Japan to determine whether metreleptin can improve glycemic control and hypertriglyceridemia in patients with lipodystrophy.
In April 2012, Amylin completed its Biologics License Application (BLA) for metreleptin to treat diabetes and/or hypertriglyceridemia (high levels of triglycerides in the bloodstream) in patients with rare forms of lipodystrophy and requested Priority Review by the FDA.
If approved, metreleptin would be the first therapy indicated specifically for the treatment of diabetes and/or hypertriglyceridemia in patients with inherited or acquired lipodystrophy, and the first approved therapeutic use of a leptin analog.
About Lipodystrophy
Lipodystrophy is a life-threatening, “ultra orphan” rare disease that is estimated to impact a few thousand people worldwide, often with an early age of onset, for which there is a significant unmet medical need. There are currently no approved drugs that treat the underlying cause of the disease.
Fat tissue is a major endocrine organ producing important metabolic hormones such as leptin. People with lipodystrophy lack the required fat tissue for normal metabolic function. This can be partial, affecting select areas of the body, or generalized, affecting nearly the entire body. A lack of fat tissue can lead to relative deficiency of leptin.
Without adequate leptin function, the metabolic system, which regulates food intake and the storage and break-down of dietary fat and carbohydrates, falls out of balance. As a result, fat accumulates in the blood and organs such as liver and muscle, which can lead to life-threatening complications including insulin-resistant diabetes, hypertriglyceridemia (high levels of triglycerides in the bloodstream), acute pancreatitis, and hepatic steatosis or steatohepatitis, also known as fatty liver disease. There are no approved drugs that address the underlying relative leptin deficiency that is believed to contribute in large part to the metabolic abnormalities that occur in lipodystrophy. Currently available therapies for diabetes and hypertriglyceridemia are often rendered marginally effective due to the severity of the condition.
FDA Approves Osphena,Ospemifene for Postmenopausal Women Experiencing Dyspareunia
Ospemifene
CAS Number: 128607-22-7
Molecular Formula: C24H23ClO2
Molecular Weight: 378.89 g.mol-1
February 26, 2013 — The U.S. Food and Drug Administration today approved Osphena (ospemifene) to treat women experiencing moderate to severe dyspareunia (pain during sexual intercourse), a symptom of vulvar and vaginal atrophy due to menopause.
Dyspareunia is a condition associated with declining levels of estrogen hormones during menopause. Less estrogen can make vaginal tissues thinner, drier and more fragile, resulting in pain during sexual intercourse.
Osphena, a pill taken with food once daily, acts like estrogen on vaginal tissues to make them thicker and less fragile, resulting in a reduction in the amount of pain women experience with sexual intercourse.
“Dyspareunia is among the problems most frequently reported by postmenopausal women,” said Victoria Kusiak, M.D., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Osphena provides an additional treatment option for women seeking relief.”
Osphena’s safety and effectiveness were established in three clinical studies of 1,889 postmenopausal women with symptoms of vulvar and vaginal atrophy. Women were randomly assigned to receive Osphena or a placebo. After 12 weeks of treatment, results from the first two trials showed a statistically significant improvement of dyspareunia in Osphena-treated women compared with women receiving placebo. Results from the third study support Osphena’s long-term safety in treating dyspareunia.
Common side effects reported during clinical trials included hot flush/flashes, vaginal discharge, muscle spasms, genital discharge and excessive sweating.
Osphena is marketed by Florham Park, N.J.-based Shionogi, Inc.
Ospemifene, FC-1271a
2-[4-[4-Chloro-1,2-diphenyl-1(Z)-butenyl]phenoxy]ethanol
Orion Corp. (Originator), Hormos (Codevelopment)
Bone Diseases, Treatment of, ENDOCRINE DRUGS, Gynecological Disorders, Treatment of , Hormone Replacement Therapy, METABOLIC DRUGS, Treatment of Osteoporosis, Treatment of Postmenopausal Syndrome , Selective Estrogen Receptor Modulators (SERM)
- Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy – May 9, 2012
credit chemdrug
The condensation of desoxybenzoin (I) with 2-(benzyloxy)ethyl bromide (II) by means of aqueous 48% NaOH containing triethylbenzylammonium chloride (TEBAC) gives 4-(benzyloxy)-1,2-diphenyl-1-butanone (III), which by reaction with the Grignard reagent (IV) – prepared from 4-(tetrahydropyranyloxy)phenyl bromide (V) and Mg in THF – yields the triphenylbutanol derivative (VI). Elimination of the THP-protecting group of compound (VI) by means of H2SO4 in ethanol/water at room temperature affords the triphenylbutanol derivative (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol to provide the butane-1,4-diol derivative (VIII). Cyclization of the butane-1,4-diol (VIII) by means of H2SO4 in hot ethanol/water gives 2-(4-hydroxyphenyl)-2,3-diphenyltetrahydrofuran (IX), which is treated with 48% HBr in refluxing AcOH to yield a mixture of (E)- and (Z)-4-(4-hydroxyphenyl)-3,4-diphenyl-3-buten-1-ol (X), which is separated by chemical work up. The phenolic OH group of the desired (Z)-isomer (X) is condensed with 2-(benzyloxy)ethyl bromide (II) by means of NaOH and tetrabutylammonium bromide in refluxing toluene/ water to afford the benzyloxyethyl ether (XII). Reaction of the aliphatic OH group of ether (XII) with PPh3 and CCl4 in acetonitrile provides the corresponding chloro derivative (XIII), which is finally debenzylated with H2 over Pd/C in ethyl acetate/ethanol.
Sorbera, L.A.; Castar, J.; Bay
Ospemifene. Drugs Fut 2004, 29, 1, 38

{kind=link}