
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
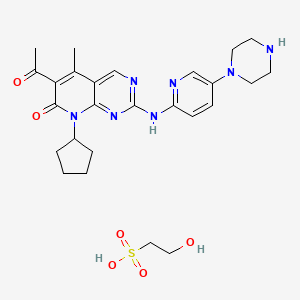
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
recent studies have identified a number of selective CDK4 inhibitors that, as discussed above, may prove useful in treating cancer—either as anti-cancer agents or as chemoprotective agents—and in treating cardiovascular disorders, such as restenosis and atherosclerosis, diseases caused by infectious agents, and autoimmune disorders, including rheumatoid arthritis. For a disclosure of these selective CDK4 inhibitors, see commonly assigned International Patent Application PCT/IB03/00059, filed Jan. 10, 2003 (the ‘059 application), which is herein incorporated by reference in its entirety for all purposes.
The ‘059 application discloses a particularly potent and selective CDK4 inhibitor, 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one:

In standard enzyme assays the compound of Formula 1 exhibits IC50 concentrations for CDK4 and CDK2 inhibition (at 25° C.) of 0.011 μM and >5 μM, respectively. For a discussion of standard CDK4 and CDK2 assays for IC50 determinations, see D. W. Fry et al., J. Biol. Chem. (2001) 16617-16623.
Though the compound of Formula 1 is a potent and selective CDK4 inhibitor, its use in pharmaceutical products presents challenges. For example, the free base has poor water solubility (9 μg/mL) and exhibits low bioavailability in animal studies. A di-HCl salt of the compound of Formula 1 appears to exhibit adequate water solubility. However, moisture uptake studies reveal that, even at low relative humidity (10% RH), the di-HCl salt absorbs water in an amount greater than about 2% of its mass, making it unsuitable for use in a solid drug product. A mono-HCl salt of the compound of Formula 1 is marginally hygroscopic, absorbing more than 2% of its mass at a relative humidity above 80%. However, the process for preparing the mono-HCl salt yields partially crystalline drug substance, indicating potential problems with process scale-up. Other salt forms of the compound of Formula 1 are thus needed.
Pfizer’s breast cancer drug Palbociclib (PD-0332991), a first in the class oral inhibitor of cyclin-dependent kinases (CDK) 4 and 6, is widely seen by investors as Pfizer’s most valuable compound in late-stage development. The FDA awarded Palbociclib “breakthrough therapy designation” in April 2013 based on the preliminary phase 2 data showing palbociclib, combined with Novartis’ drug,Femara (Letrozole), stopped breast tumors progression for more than two years as compared with 7.5 months with letrozole alone. The phase 3 trial started in February 2013 and estimated final completion date is March 2016. Leerink Swann analyst Seamus Fernandez forecasts palbociclib could become a $5 billion drug, with potential for $3 billion in first-line metastatic breast cancer alone.
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases.
6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (also referred to as “Compound 1”),

as well as its intermediates. Compound 1 is described in U.S. Pat. No. 6,936,612, the disclosure of which is hereby incorporated in its entirety. This compound is a protein kinase inhibitor and represents a synthetic, small molecule inhibitor capable of modulating cell cycle control.
A method of preparing Compound 1 is disclosed as Example 36 of U.S. patent application Ser. No. 6,936,612. Methods of preparing the isethionate salt forms of Compound 1 are disclosed in Examples 1-13 of WO 2005/005426. These methods are for synthesis of small quantities of the salt forms of Compound 1 and are not designed for commercial scale-up. Therefore, a preparation of the salt forms for CDK inhibitor 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride which is cost-efficient, scaleable and productive is highly desirable.


USAN (zz-153)
PALBOCICLIB ISETHIONATE
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
MOLECULAR FORMULA C24H29N7O2 . C2H6O4S
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
- PD 0332991-0054
- PF-00080665-73
- UNII-W1NYL2IRDR

SYNTHESIS

:WO2008032157
……………………………….
http://www.google.com/patents/US7781583



COMPARATIVE EXAMPLE 1A Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
COMPARATIVE EXAMPLE 1B Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1A), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
COMPARATIVE EXAMPLE 1C Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in 2005-0059670A1) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 2A Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg (6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6 L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-70° C. and held for 30 hours after which some solids precipitated. Water was added and the reaction cooled to 25° C. over 2 hrs. The resulting slurry was filtered, washed and dried at 45° C. to give 1.2 kg (79% crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (2)
60.0 g of 20% Pd(OH)2/C, 1213.1 g (3.9 moles) of intermediate 2a, and isopropanol were charged and stirred in a Parr reactor, then purged under gas, followed by removal of the catalyst under pressure. The filtrates were concentrated in vacuo at ˜20° C. leaving 917 g of dry brown powder (crude yield ˜84%).
EXAMPLE 3 Preparation of 2-Chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one

EXAMPLE 3A Preparation of 5-bromo-2-chloro-4-cyclopentyl-aminopyrimidine
To 1 g (0.004 mol) of 5-bromo-2,4-dichloropyrimidine in ethanol was added 1.5 kg (0.018 mol) cyclopentylamine under nitrogen. The mixture was stirred at 25° C. for 2 hrs. Water was added to precipitate the product, and the solid was recrystallized using hexane 4:1 to give a white crystalline product (3A).
EXAMPLE 3 Preparation of 2-Chloro-8-Cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one
41.5 g (0.15 mol) of 5-bromo-2-chloro-4-cyclopentylaminopyrimidine 3a and 32.3 g (0.375 mol) of crotonic acid were mixed in 100 L of THF and 105 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred, evacuated and refilled with nitrogen three times, after which 860 mg (0.0022 mol) palladium dichloride dibenzonitrile complex and 685 mg (0.0022 mol) tri-ortho-tolylphosphine were added and the resulting slurry degassed an additional three times. The mixture was then heated and stirred at 70° C. for 16 hrs, after which 35 ml acetic anhydride was added and the mixture stirred for an additional 1.5 hrs. The mixture was cooled and diluted with 100 ml MTBE and then extracted with 1NHCl, then aqueous sodium bicarbonate and brine. The organic phase was dried over magnesium sulfate, filtered, concentrated in vacuo, and recrystallized from IPA to yield 31.2 g (68%) of crude product (3).
EXAMPLE 4 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 4A Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidine-7-one
10 g (0.04 mol) of intermediate 3 and 13 g (0.16 mol) of sodium acetate were mixed with 50 ml of glacial acetic acid and 12 g (0.08 mol) bromine under nitrogen. The solution was heated to 50° C. and stirred for 35 hrs, then cooled to room temperature. Sodium bisulfite solids were added until the bromine color disappeared, then quenched, filtered and washed to provide a solid which was subsequently dissolved in 500 ml hot IPA, filtered hot, and cooled. The resulting crystals were further filtered, and dried in vacuo at 65° C. to yield 8 g (61%) of crude product (4A).
EXAMPLE 4 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
3.78 g (2.10 equiv; 13.6 mmoles) of intermediate 1, 25 ml toluene and lithium bis(trimethylsilyl)amide in 1 M THF (13.6 mmoles; 13.6 mL; 12.1 g) were mixed for 10 min under nitrogen to form a dark solution. In a separate beaker the intermediate 4a (1.00 equiv, 6.47 mmoles; 2.50 g) was slurried in toluene then added to the mixture containing 1 and stirred for 30 min, after which the combined mixture was quenched with 25 ml 1 M sodium bicarbonate and then filtered. Alternatively, the combined mixture can be quenched with ammonium chloride. The filter cake was washed with toluene, then acetone, then water and dried at 60° C. to give 3.5 g (92%) of a grey-yellow solid 4.
EXAMPLE 5 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cycloentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester

768 g (1.3 mol) of intermediate 4, was mixed with 395 g (3.9 mol) of butyl vinyl ether, 4.7 L of n-butanol, and 275 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred and placed under ca. 50 tore vacuum and then refilled with nitrogen; this was repeated 2 more times. To this degassed solution was added 22 g (0.03 mol) Bis-(diphenylphosphinoferrocene)palladium dichloride dichloromethane complex and the resulting slurry was degassed an additional three times as described above. The mixture was then heated and stirred at 95° C. for 20 hrs. The resulting thin red slurry was diluted with 4 L branched octane’s and cooled to about 5° C. after which 1 L saturated aq. potassium carbonate was added and the mixture was filtered and rinsed with 500 ml branched octanes. After drying for 16 hrs at 45° C., 664 g (83%) of gray-solid product (5) was obtained. In addition, column chromatography can be used to further purify the crude product.
EXAMPLE 6 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one

11.6 g (1.00 eq, 19.2 mmol) of intermediate 5, water (10.1 equiv; 193 mmoles; 3.48 mL; 3.48 g) and methanol (3.62 moles; 146 mL; 116 g) were combined and heated to 55-60° C. Isethionic acid was added slowly until a clear solution was obtained; 3.3 g isethionic acid solution was necessary to reach this end point. The resulting clear orange solution was filtered through paper and rinsed through with 20 ml methanol, after which the filtrate was reheated to 55-60° C. and the remaining isethionic acid was added (a total of 9.93 g was added). The reaction mixture precipitated and thickened for 6 hours, after which it was cooled and held at 30-35° C. while triethylamine (2.92 g; 28.8 mmoles) was added slowly as a 10% solution in methanol over 12 hrs. About halfway through the addition of triethylamine, desired polymorphic seeds were added to help formation of the desired polymorph. The resulting slurry was cooled and held at 5° C. for 15 minutes and the crystals were filtered and washed with methanol. The solid product was dried in vacuo at 55° C. to obtain 11 g of yellow crystals of the title compound.

……………………………………………………………………
http://www.google.com/patents/US7345171
EXAMPLES
The following examples are intended to be illustrative and non-limiting, and represent specific embodiments of the present invention.
Example 1 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
Example 2 Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2.3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
Example 3 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in Example 2) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
Example 4 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
To a slurry of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (7.0 g, 15.64 mmol, prepared as in Example 3 following contact with NaOH) dispersed in 250 mL of water was added drop-wise 30 mL of a 0.52 M solution of isethionic acid in MeOH (15.64 mmol) to a pH of 5.2. The solution was filtered through a glass filter (fine) and the clear solution was freeze-dried to give 9.4 g of the amorphous salt. The amorphous salt (3.16 g) was mixed with 25 mL of MeOH and after almost complete dissolution a new precipitate formed. Another 25 mL of MeOH was added and the mixture was stirred at 46° C. to 49° C. for four hours. The mixture was slowly cooled to 32° C. and put in a cold room (+4° C.) overnight. A sample was taken for PXRD, which indicated formation of Form B. The mixture was filtered and the precipitate was dried overnight at 50° C. in a vacuum oven. This furnished 2.92 g of the mono-isethionate salt of the compound of Formula 1 in 92% yield. HPLC-99.25%, PXRD-Form B, CHNS, H-NMR were consistent with the structure.
Example 5 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
MeOH (100 mL) was placed in a 250 mL flask equipped with a mechanical stirrer, thermocouple/controller, condenser, and heating mantle and preheated to 35° C. An amorphous isethionate salt (2 g, prepared as in Example 4) was slowly added in three even portions with a 25 min to 30 min interval between the additions. The reaction mixture was stirred overnight at 35° C. and subsequently cooled. A sample was filtered and examined by PXRD. It was pure Form B. The whole reaction mixture was then used as Form B seeds in a larger scale experiment.
Example 6 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
MeOH (50 mL) was placed in a 250 mL flask equipped with a magnetic stirrer, condenser, thermocouple/controller, and heating mantle, and preheated to 40° C. An amorphous isethionate salt (1 g, prepared as in Example 4) was slowly added in three even portions with 30 min interval between the portions and then stirred overnight at 40° C. The reaction was monitored by in-situ Raman spectroscopy. The sample was taken, filtered and analyzed by PXRD. It was pure Form B by PXRD and Raman spectroscopy. The mixture was cooled to 25° C. at a rate of 3° C./h, cooled to −10° C., filtered, and vacuum dried to furnish 0.85 g of the Form B crystalline product.
Example 7 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
The free base (Formula 1, 0.895 mg, 2 mmol) was mixed with 10 mL of MeOH and seeded with 33 mg of a mono-isethionate salt of the compound of Formula 1 (Form B). Then 5.6 mL of a 0.375 M solution of isethionic acid in MeOH (2.1 mmol) was added in 10 even portions over 75 min time period. The mixture was stirred for an additional hour and a sample was taken for PXRD analysis. It confirmed formation of crystalline Form B. The mixture was stirred at RT overnight and another PXRD was taken. There was no change in the crystal form. The mixture was cooled in a refrigerator at −8° C. overnight, filtered, and dried at 50° C. in a vacuum oven to give 1.053 g (91.8% of theory) of the above-named compound (Form B). HPLC—99.8%, CHNS, H-NMR, IR are consistent with the structure, PXRD-Form B.
Example 8 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form A)
An amorphous isethionate salt (47 mg, prepared as in Example 4) was mixed with 4 mL of EtOH in a 15 mL flask equipped with a magnetic stirrer, thermocouple and condenser. The mixture was heated to reflux, which resulted in the formation of a nearly clear solution. After refluxing for 10-15 min, the mixture became cloudy. It was slowly cooled to 50° C. and was seeded at 69° C. with Form A. The mixture was held at 50° C. for 5 h and was allowed to cool to RT overnight. The mixture was subsequently cooled to 1° C. with an ice bath, held for 1.5 h, filtered, washed with 0.5 mL of cold EtOH, air-dried, and then dried in a vacuum oven at 70° C. overnight to furnished 38.2 mg of a fine crystalline material. The crystalline material was found to be mono-isethionate salt Form A by PXRD. H-NMR was consistent for the mono-isethionate salt and indicated the presence of residual EtOH ca. 5.9 mol % or 0.6 wt %.
Example 9 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form D)
An amorphous isethionate salt (9.0 g, prepared as in Example 4) was mixed with 300 mL of MeOH, stirred and heated to 63.8° C. (at reflux). To the slightly cloudy mixture was added two 50-mL portions of MeOH. The hot mixture was filtered into a 2-L flask equipped with a mechanical stirrer. The mixture was briefly heated to reflux and then cooled to 60° C. IPA (100 mL) was added to the mixture. The mixture was again heated to 60° C. and an additional 110 mL of IPA was added. A precipitate started to form at 59.7° C. The mixture was reheated to 67.5° C., cooled to 50° C., and held overnight. A sample was taken the next morning for PXRD analysis. The mixture was cooled to 25° C. at a rate of 3° C./h and another PXRD sample was taken when the mixture reached 28° C. The mixture was allowed to cool to RT overnight. A precipitate was collected and dried in a vacuum oven at 65° C. and 30 Torr. The procedure produced 7.45 g (82.8% yield) of the crystalline compound (Form D by PXRD analysis). Previously analyzed samples were also Form D. HPLC showed 98.82% purity and CHNS microanalysis was within +/−0.4%. A slurry of isethionate salt Form A, B, and D in MeOH yielded substantially pure Form B in less than three days.
Example 10 Preparation of isethionic acid (2-hydroxy-ethanesulfonic acid)
A 5-L, four-necked, round-bottomed flask, equipped with mechanical stirrer, thermocouple, gas sparger, and an atmosphere vent through a water trap was charged with 748 g (5.05 mol) of sodium isethionate (ALDRICH), and 4 L of IPA. The slurry was stirred at RT. An ice bath was used to keep the internal temperature below 50° C. as 925 g (25.4 mol) of hydrogen chloride gas (ALDRICH) was sparged into the system at a rate such that it dissolved as fast as it was added (as noted by lack of bubbling through the water trap). Sufficient HCl gas was added until the system was saturated (as noted by the start of bubbling through the water trap). During the addition of HCl, the temperature rose to 45° C. The slurry was cooled to RT and filtered over a coarse-fritted filter. The cake was washed with 100 mL of IPA and the cloudy filtrate was filtered through a 10-20μ filter. The resulting clear, colorless filtrate was concentrated under reduced pressure on a rotary evaporator, while keeping the bath temperature below 50° C. The resulting 1.07 kg of clear, light yellow oil was diluted with 50 mL of tap water and 400 mL of toluene and concentrated under reduced pressure on a rotary evaporator for three days, while keeping the bath temperature below 50° C. The resulting 800 g of clear, light yellow oil was diluted with 500 mL of toluene and 250 mL of IPA and concentrated under reduced pressure on a rotary evaporator for 11 days, keeping the bath temperature below 50° C. The resulting 713 g of clear, light yellow oil was titrated at 81 wt % (580 g, 91.1% yield) containing 7.9 wt % water and 7.5 wt % IPA.
Example 11 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A 5-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (300 g, 0.51 mol, prepared as in Example 2), butyl vinyl ether (154 g, 1.54 mol, ALDRICH), n-butanol (1.5 L, ALDRICH), and diisopropyl ethylamine (107 mL, 0.62 mol, ALDRICH). The slurry was placed under approximately 50 Torr vacuum and then refilled with nitrogen 3 times. To this was added 8.3 g (0.01 mol) bis-(diphenylphosphinoferrocene) palladium dichloride dichloromethane (JOHNSON MATTHEY, Lot 077598001) and the resulting slurry was purged an additional three times as described above. The mixture was then heated to 95° C. and stirred for 20 h. The resulting thin red slurry was diluted with 2 L of heptane and cooled to approximately 5° C. At this temperature, 400 mL saturated aqueous potassium carbonate was added and the mixture was filtered and rinsed with 250 mL of heptane. After drying in an oven for 16 h at 45° C., 231.7 g (75% yield) of the title compound was obtained as a yellow solid.
Example 12 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-Pyrido[2,3-d]pyrimidin-7-one (Form B)
A 22-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (725 g, 1.20 mol, prepared as in Example 11) and MeOH (14 L). The slurry was stirred at RT as it was charged with a solution of isethionic acid (530 g, 4.20 mol, prepared as in Example 10), MeOH (1.5 L), and water (70 mL, 3.89 mol). The resulting slurry was heated to 55° C. over 30 minutes and then stirred at 55° C. for 30 minutes. A solution of 175 g (1.73 mol) of Et3N (ALDRICH) in 200 mL of MeOH was charged to the slurry as it was cooled to 30° C. The slurry was held at 30° C. as a solution of 128 g (1.26 mol) of Et3N in 2 L of MeOH was added dropwise over 6 hours. The resulting slurry was sampled to determine crystal form (Form B). The slurry was cooled and held at 5° C. for 15 minutes and was subsequently filtered through a coarse-fritted filter. The resulting filter cake was washed with multiple washes of 200 mL of cold MeOH. The solid product was dried at 55° C. under vacuum to yield 710 g (91% yield) of the title compound as yellow crystals.

1)Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;
2)Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4; Journal of Medicinal Chemistry,2005,48(7),2371-2387;
3)Erdman, David Thomas et al;Preparation of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157
4)Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997
5)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381
6)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.
7)Phase III Study Evaluating Palbociclib (PD-0332991), a Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”;ClinicalTrials.gov number:NCT01864746;currently recruiting participants(as of January 2, 2013)
8)A Randomized, Multicenter, Double-Blind Phase 3 Study Of PD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For Advanced Disease;ClinicalTrials.gov number:NCT01740427;currently recruiting participants(as of January 2, 2013)
9)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy;ClinicalTrials.gov number:NCT01942135;currently recruiting participants(as of January 2, 2013)
| US6936612 | Jan 16, 2003 | Aug 30, 2005 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2005005426A1 | Jun 28, 2004 | Jan 20, 2005 | Vladimir Genukh Beylin | Isethionate salt of a selective cdk4 inhibitor |
| US20030229026 * | Dec 18, 2000 | Dec 11, 2003 | Al-Awar Rima Salim | Agents and methods for the treatment of proliferative diseases |
| US20040006074 * | Dec 2, 2002 | Jan 8, 2004 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| US20040048915 * | Sep 24, 2001 | Mar 11, 2004 | Engler Thomas Albert | Methods and compounds for treating proliferative diseases |
| US20050222163 * | Mar 30, 2005 | Oct 6, 2005 | Pfizer Inc | Combinations of signal transduction inhibitors |
| US20070027147 * | Dec 3, 2004 | Feb 1, 2007 | Takashi Hayama | Biarylurea derivatives |
| WO2008032157A2 * | Aug 27, 2007 | Mar 20, 2008 | David Thomas Erdman | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2010075074A1 | Dec 15, 2009 | Jul 1, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2012098387A1 | Jan 17, 2012 | Jul 26, 2012 | Centro Nacional De Investigaciones Oncológicas (Cnio) | 6, 7-ring-fused triazolo [4, 3 – b] pyridazine derivatives as pim inhibitors |
| US7781583 | Sep 10, 2007 | Aug 24, 2010 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
| US7855211 | Dec 15, 2009 | Dec 21, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| US8247408 * | Oct 9, 2006 | Aug 21, 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755 | Feb 9, 2010 | Sep 25, 2012 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |

old info
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)

| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family


[…] Palbociclib […]
LikeLike
[…] https://newdrugapprovals.org/2014/01/05/palbociclib/ […]
LikeLike