
Home » Posts tagged 'manufacturing'
Tag Archives: manufacturing
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Welcome Scientific update to Pune, India 2-3 and 4-5 Dec 2014 for celebrating Process chemistry
WEBSITE http://www.scientificupdate.co.uk/
SCIENTIFIC UPDATE HAS A REPUTATION FOR ITS HIGH QUALITY EVENTS, BOTH FOR THE SCIENTIFIC CONTENT AND ALSO FOR THE EFFICIENCY OF ITS ORGANISATION. KEEP YOUR SKILLS UP TO DATE AND INVEST IN YOUR CONTINUING PERSONAL PROFESSIONAL DEVELOPMENT.



TRAINING COURSE 2-3 DEC 2014
Process Development for Low Cost Manufacturing
When:02.12.2014 – 03.12.2014
Tutors:
Where: National Chemical Laboratory – Pune, India
Brochure:View Brochure
Register http://scientificupdate.co.uk/training/scheduled-training-courses.html
DESCRIPTION
Chemical process research and development is recognised as a key function during the commercialisation of a new product particularly in the generic and contract manufacturing arms of the chemical, agrochemical and pharmaceutical industries.
The synthesis and individual processes must be economic, safe and must generate product that meets the necessary quality requirements.
This 2-day course presented by highly experienced process chemists will concentrate on the development and optimisation of efficient processes to target molecules with an emphasis on raw material cost, solvent choice, yield improvement, process efficiency and work up, and waste minimisation.
Process robustness testing and reaction optimisation via stastical methods will also be covered.
A discussion of patent issues and areas where engineering and technology can help reduce operating costs.
The use of engineering and technology solutions to reduce costs will be discussed and throughout the course the emphasis will be on minimising costs and maximising returns.


Conference 4-5 DEC 2014
TITLE . Organic Process Research & Development – India
Subtitle:The 32nd International Conference and Exhibition
When:04.12.2014 – 05.12.2014
Where:National Chemical Laboratory – Pune, India
Brochure:View Brochure
Register..http://scientificupdate.co.uk/conferences/conferences-and-workshops.html

for
- Process Research & Development Chemists
- Chemical Engineers in Industry
- Heads of Departments & Team Leaders
Benefits
- Invest in yourself: keeping up to date on current developments and future trends could mean greater job security.
- Learn from a wide range of industrial case studies given by hand-picked industrial speakers.
- Take home relevant ideas and information that are directly applicable to your own work with the full proceedings and a CD of the talks.
- Save time. Our intensive, commercial-free programme means less time away from work.
- Meet and network with the key people in the industry in a relaxed and informal atmosphere.

Do you want to improve efficiency and innovation in your synthetic route design, development and optimisation?
The efficient conversion of a chemical process into a process for manufacture on tonnage scale has always been of importance in the chemical and pharmaceutical industries. However, in the current economic and regulatory climate, it has become increasingly vital and challenging to do so efficiently. Indeed, it has never been so important to keep up to date with the latest developments in this dynamic field.
At this Organic Process Research & Development Conference, you will hear detailed presentations and case studies from top international chemists. The hand-picked programme of speakers has been put together specifically for an industrial audience. They will discuss the latest issues relating to synthetic route design, development and optimisation in the pharmaceutical, fine chemical and allied fields. Unlike other conferences, practically all our speakers are experts from industry, which means the ideas and information you take home will be directly applicable to your own work.
The smaller numbers at our conferences create a more intimate atmosphere. You will enjoy plenty of opportunities to meet and network with speakers and fellow attendees during the reception, sit-down lunches and extended coffee breaks in a relaxed and informal environment. Together, you can explore the different strategies and tactics evolving to meet today’s challenges.
This is held in Pune, close proximity to Mumbai city, very convenient to stay and travel to either in Pune or Mumbai. I feel this should be an opportunity to be grabbed before the conference is full and having no room
Hurry up rush


References
1 https://newdrugapprovals.org/scientificupdate-uk-on-a-roll/
2 http://scientificupdate.co.uk/conferences/conferences-and-workshops.html
3 http://en.wikipedia.org/wiki/Pune
PROFILES
Will Watson

Dr Will Watson gained his PhD in Organic Chemistry from the University of Leeds in 1980. He joined the BP Research Centre at Sunbury-on-Thames and spent five and a half years working as a research chemist on a variety of topics including catalytic dewaxing, residue upgrading, synthesis of novel oxygenates for use as gasoline supplements, surfactants for use as gasoline detergent additives and non-linear optical compounds.
In 1986 he joined Lancaster Synthesis and during the next 7 years he was responsible for laboratory scale production and process research and development to support Lancaster’s catalogue, semi-bulk and custom synthesis businesses.
In 1993 he was appointed to the position of Technical Director, responsible for all Production (Laboratory and Pilot Plant scale), Process Research and Development, Engineering and Quality Control. He helped set up and run the Lancaster Laboratories near Chennai, India and had technical responsibility for the former PCR laboratories at Gainesville, Florida.
He joined Scientific Update as Technical Director in May 2000. He has revised and rewritten the ‘Chemical Development and Scale Up in the Fine Chemical & Pharmaceutical Industries’ course and gives this course regularly around the world. He has been instrumental in setting up and developing new courses such as ‘Interfacing Chemistry with Patents’ and ‘Making and Using Fluoroorganic Molecules’.
He is also involved in an advisory capacity in setting up conferences and in the running of the events. He is active in the consultancy side of the business and sits on the Scientific Advisory Boards of various companies.
………………………………………………………………………………………………….
John Knight

Dr John Knight gained a first class honours degree in chemistry at the University of Southampton, UK. John remained at Southampton to study for his PhD in synthetic methodology utilizing radical cyclisation and dipolar cyloaddition chemistry.
After gaining his PhD, John moved to Columbia University, New York, USA where he worked as a NATO Postdoctoral Fellow with Professor Gilbert Stork. John returned to the UK in 1987 joining Glaxo Group Research (now GSK) as a medicinal chemist, where he remained for 4 years before moving to the process research and development department at Glaxo, where he remained for a further 3½ years.
During his time at Glaxo, John worked on a number of projects and gained considerable plant experience (pilot and manufacturing). In 1994 John moved to Oxford Asymmetry (later changing its name to Evotec and most recently to Aptuit) when it had just 25 staff. John’s major role when first at Oxford Asymmetry was to work with a consultant project manager to design, build and commission a small pilot plant, whilst in parallel developing the chemistry PRD effort at Oxford Asymmetry.
The plant was fully operational within 18 months, operating to a 24h/7d shift pattern. John continued to run the pilot plant for a further 3 years, during which time he had considerable input into the design of a second plant, which was completed and commissioned in 2000. After an 18-month period at a small pharmaceutical company, John returned to Oxford in 2000 (by now called Evotec) to head the PRD department. John remained in this position for 6.5 years, during which time he assisted in its expansion, established a team to perform polymorph and salt screening studies and established and maintained high standards of development expertise across the department.
John has managed the chemical development and transfer of numerous NCE’s into the plant for clients and been involved in process validations. He joined Scientific Update in January 2008 as Scientific Director.
Pune images

From top:1 Fergusson College, 2 Mahatma Gandhi Road, Shaniwarwada 3 the HSBC Global Technology India Headquarters, and the 4National War Memorial Southern Command



NCL PUNE
 The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.
The National Chemical Laboratory is located in the state of Maharashtra in India. Maharashtra state is the largest contributor to India’s GDP. The National Chemical Laboratory is located in Pune city, and is the cultural capital of Maharashtra. Pune city is second only to Mumbai (the business capital of India) in size and industrial strength. Pune points of interest include: The tourist places in Pune include: Lal Deval Synagogue, Bund Garden, Osho Ashram, Shindyanchi Chhatri and Pataleshwar Cave Temple.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 


KEYWORDS
JOHN KNIGHT, WILL WATSON, SCIENTIFIC UPDATE, PROCESS, COURSE, CONFERENCE, INDIA, PUNE, PROCESS DEVELOPMENT, LOW COST, MANUFACTURING, SCALEUP
AVOSENTAN

AVOSENTAN
N-[6-Methoxy-5-(2-methoxyphenoxy)-2-(4-pyridyl)pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]-amide,
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide,
Endothelin ETA Receptor Antagonists
M.Wt: 479.51
Formula: C23H21N5O5S
Roche (Originator)
CAS No.: 290815-26-8
- RO 67-0565
- SPP 301
- UNII-L94KSX715K
PHASE 3
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=spp301+OR+Avosentan
SPP-301 is an oral, once-daily, second-generation endothelin ETA receptor antagonist which had been in phase III clinical development at Speedel for the treatment of diabetic nephropathy. In December 2006, the company reported that the phase III trial had been stopped based on the recommendation from the trial’s Data Safety Monitoring Board (DSMB) to stop the trial following incidence of a significant imbalance in fluid retention in patients in the study arms. Speedel reported that the compound will be evaluated for potential new clinical development for the treatment of diabetic kidney disease and other indications.
Originally developed by Roche and specifically optimized for improved liver safety, SPP-301 was licensed to Speedel in October 2000. In 2003, Speedel exercised its option to license from Roche all rights to SPP-301, including exclusive worldwide rights for the full development and commercialization of the ETA antagonist. SPP-301 has fast track designation and has undergone a special protocol assessment (SPA) by the FDA. Speedel had been studying the drug for the treatment of hypertension.
AVOSENTAN
290815-26-8 CAS
PATENTS
2. WO 2004078104
3. WO 2005113543
4. WO 2007031501
5. WO 2008077916
Dutzler R, Ernstb B, Hediger MA, Keppler D, Mohr P, Neidhart W, Märki HP.Chimia (Aarau). 2010;64(9):662-6.
………………………
INTRODUCTION
-
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide corresponding to the formula

is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2.
-
Own investigations have shown that there exist two distinct crystalline forms, hereinafter referred to as form A and form B, as well as a number of further solvates, in particular the methanol, ethanol, isopropanol, dichloromethane, acetone, methyl ethyl ketone and tetrahydrofuran solvates.
-
It was further surprisingly found that the thermodynamically stable crystalline form – form B – can be prepared under controlled conditions and that said form B can be prepared with a reliable method in an industrial scale, which is easy to handle and to process in the manufacture and preparation of formulations.

………………..

4,6-Dichloro-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine (described in EP 0 799 209) can be transformed to the intermediate of formula (III)—according to scheme 1—on reaction with an appropriate sulfonamide of formula (II), wherein R1 is as defined in claim 1, in a suited solvent such as DMSO or DMF at room temperature or at elevated temperature and in the presence of a suited base such as potassium carbonate.


EXAMPLE 1
[0064] a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
[0065] C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
[0066] Preparation of the starting material:
[0067] b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide ( CHLORO STARTING MATERIAL) as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
……………………………….
http://www.google.com/patents/US6417360
EXAMPLE 1
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
Preparation of the starting material:
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
…………………….
http://www.google.com/patents/EP0799209B1
SYNTHESIS OF
4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine
A BASIC STARTING MATERIAL FOR AVOSENTAN
- Preparation of the starting material
-
- b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hours 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- c) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
- d) A suspension of 154.6 g of 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCl3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2Cl2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2Cl2. The combined CH2Cl2 extracts are dried with MgSO4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2Cl2remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine, melting point 178-180°C.
…………………………
http://www.google.com/patents/WO2000052007A1
Preparation of the starting material:
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide IE THE 6 CHLORO COMPD
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 1 .66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40°C until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60 °C for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as yellow crystals. Melting point 177-179 °C. ISN mass spectrum, m/e 482.2 (M-l calculated for C22Hi8ClN5O5Sι: 482).

………………………………………………………………………………………….
NEXT

Example 1AVOSENTAN
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl- pyrimidin-4-yl] -amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg SO , concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120 °C to give the desired 5-methyl-pyridine-2-sulfonic acid [6- methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as white crystals. Melting point 225-226 °C. ISN mass spectrum, m/e 478.2 (M-l calculated for
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
…………………………………………….

IS DESCRIBED IN
http://www.google.com/patents/EP2331513A1?cl=en
ALSO

-
Diabetic nephropathy is the principle cause of end stage renal disease in the western world. It is a major cause of morbidity and mortality in Type-I Diabetes, but is an increasing problem in Type-II Diabetes and because the incidence of this is five times that of Type-I Diabetes, it contributes at least 50% of diabetics with end stage renal disease.
-
The initial stage of subtle morphologic changes in the renal glomeruli is followed by microalbuminuria. This is associated with a modestly rising blood pressure and an increased incidence of cardiovascular disease. There follows a continued increase in urinary protein excretion and declining glomerular filtration rate. Diabetic nephropathy has many possible underlying pathophysiological causes including metabolic, glycosylation of proteins, haemodynamics, altered flow/pressure in glomeruli, the development of hypertension and cytokine production; all of these are associated with the development of extracellular matrix and increased vascular permeability leading to glomerular damage and proteinuria.
| WO2005113543A1 * | May 12, 2005 | Dec 1, 2005 | Alexander Bilz | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO2007031501A2 * | Sep 11, 2006 | Mar 22, 2007 | Speedel Pharma Ag | Pyridylsulfonamidyl-pyrimidines for the prevention of blood vessel graft failure |
| WO2008077916A1 * | Dec 21, 2007 | Jul 3, 2008 | Ovidiu Baltatu | Pharmaceutical composition using aliskiren and avosentan |
| EP1454625A1 * | Mar 6, 2003 | Sep 8, 2004 | Speedel Development AG | Pyridylsulfonamidyl-pyrimidines for the treatment of diabetic nephropathies |
| EP1595880A1 * | May 13, 2004 | Nov 16, 2005 | Speedel Pharma AG | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| EP1938812A1 * | Dec 22, 2006 | Jul 2, 2008 | Speedel Pharma AG | Pharmaceutical composition using aliskiren and avosentan |
| US6951856 | Jul 10, 2001 | Oct 4, 2005 | Actelion Pharmaceuticals Ltd. | Arylethene-sulfonamides |
| US7402587 | May 12, 2005 | Jul 22, 2008 | Speedel Pharma Ag | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
| EP0713875A1 * | Nov 13, 1995 | May 29, 1996 | F. Hoffmann-La Roche AG | Sulfonamides |
| EP0897914A1 * | Aug 10, 1998 | Feb 24, 1999 | F. Hoffmann-La Roche Ag | Process for the preparation of 2,5-disubstitued pyridines |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html

Palbociclib
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
recent studies have identified a number of selective CDK4 inhibitors that, as discussed above, may prove useful in treating cancer—either as anti-cancer agents or as chemoprotective agents—and in treating cardiovascular disorders, such as restenosis and atherosclerosis, diseases caused by infectious agents, and autoimmune disorders, including rheumatoid arthritis. For a disclosure of these selective CDK4 inhibitors, see commonly assigned International Patent Application PCT/IB03/00059, filed Jan. 10, 2003 (the ‘059 application), which is herein incorporated by reference in its entirety for all purposes.
The ‘059 application discloses a particularly potent and selective CDK4 inhibitor, 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one:

In standard enzyme assays the compound of Formula 1 exhibits IC50 concentrations for CDK4 and CDK2 inhibition (at 25° C.) of 0.011 μM and >5 μM, respectively. For a discussion of standard CDK4 and CDK2 assays for IC50 determinations, see D. W. Fry et al., J. Biol. Chem. (2001) 16617-16623.
Though the compound of Formula 1 is a potent and selective CDK4 inhibitor, its use in pharmaceutical products presents challenges. For example, the free base has poor water solubility (9 μg/mL) and exhibits low bioavailability in animal studies. A di-HCl salt of the compound of Formula 1 appears to exhibit adequate water solubility. However, moisture uptake studies reveal that, even at low relative humidity (10% RH), the di-HCl salt absorbs water in an amount greater than about 2% of its mass, making it unsuitable for use in a solid drug product. A mono-HCl salt of the compound of Formula 1 is marginally hygroscopic, absorbing more than 2% of its mass at a relative humidity above 80%. However, the process for preparing the mono-HCl salt yields partially crystalline drug substance, indicating potential problems with process scale-up. Other salt forms of the compound of Formula 1 are thus needed.
Pfizer’s breast cancer drug Palbociclib (PD-0332991), a first in the class oral inhibitor of cyclin-dependent kinases (CDK) 4 and 6, is widely seen by investors as Pfizer’s most valuable compound in late-stage development. The FDA awarded Palbociclib “breakthrough therapy designation” in April 2013 based on the preliminary phase 2 data showing palbociclib, combined with Novartis’ drug,Femara (Letrozole), stopped breast tumors progression for more than two years as compared with 7.5 months with letrozole alone. The phase 3 trial started in February 2013 and estimated final completion date is March 2016. Leerink Swann analyst Seamus Fernandez forecasts palbociclib could become a $5 billion drug, with potential for $3 billion in first-line metastatic breast cancer alone.
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases.
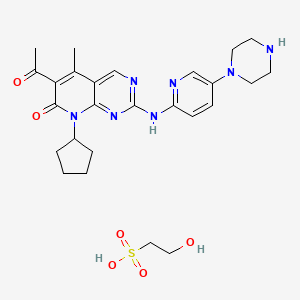
6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (also referred to as “Compound 1”),

as well as its intermediates. Compound 1 is described in U.S. Pat. No. 6,936,612, the disclosure of which is hereby incorporated in its entirety. This compound is a protein kinase inhibitor and represents a synthetic, small molecule inhibitor capable of modulating cell cycle control.
A method of preparing Compound 1 is disclosed as Example 36 of U.S. patent application Ser. No. 6,936,612. Methods of preparing the isethionate salt forms of Compound 1 are disclosed in Examples 1-13 of WO 2005/005426. These methods are for synthesis of small quantities of the salt forms of Compound 1 and are not designed for commercial scale-up. Therefore, a preparation of the salt forms for CDK inhibitor 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride which is cost-efficient, scaleable and productive is highly desirable.


USAN (zz-153)
PALBOCICLIB ISETHIONATE
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
MOLECULAR FORMULA C24H29N7O2 . C2H6O4S
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
- PD 0332991-0054
- PF-00080665-73
- UNII-W1NYL2IRDR

SYNTHESIS

:WO2008032157
……………………………….
http://www.google.com/patents/US7781583



COMPARATIVE EXAMPLE 1A Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
COMPARATIVE EXAMPLE 1B Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1A), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
COMPARATIVE EXAMPLE 1C Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in 2005-0059670A1) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 2A Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg (6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6 L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-70° C. and held for 30 hours after which some solids precipitated. Water was added and the reaction cooled to 25° C. over 2 hrs. The resulting slurry was filtered, washed and dried at 45° C. to give 1.2 kg (79% crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (2)
60.0 g of 20% Pd(OH)2/C, 1213.1 g (3.9 moles) of intermediate 2a, and isopropanol were charged and stirred in a Parr reactor, then purged under gas, followed by removal of the catalyst under pressure. The filtrates were concentrated in vacuo at ˜20° C. leaving 917 g of dry brown powder (crude yield ˜84%).
EXAMPLE 3 Preparation of 2-Chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one

EXAMPLE 3A Preparation of 5-bromo-2-chloro-4-cyclopentyl-aminopyrimidine
To 1 g (0.004 mol) of 5-bromo-2,4-dichloropyrimidine in ethanol was added 1.5 kg (0.018 mol) cyclopentylamine under nitrogen. The mixture was stirred at 25° C. for 2 hrs. Water was added to precipitate the product, and the solid was recrystallized using hexane 4:1 to give a white crystalline product (3A).
EXAMPLE 3 Preparation of 2-Chloro-8-Cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one
41.5 g (0.15 mol) of 5-bromo-2-chloro-4-cyclopentylaminopyrimidine 3a and 32.3 g (0.375 mol) of crotonic acid were mixed in 100 L of THF and 105 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred, evacuated and refilled with nitrogen three times, after which 860 mg (0.0022 mol) palladium dichloride dibenzonitrile complex and 685 mg (0.0022 mol) tri-ortho-tolylphosphine were added and the resulting slurry degassed an additional three times. The mixture was then heated and stirred at 70° C. for 16 hrs, after which 35 ml acetic anhydride was added and the mixture stirred for an additional 1.5 hrs. The mixture was cooled and diluted with 100 ml MTBE and then extracted with 1NHCl, then aqueous sodium bicarbonate and brine. The organic phase was dried over magnesium sulfate, filtered, concentrated in vacuo, and recrystallized from IPA to yield 31.2 g (68%) of crude product (3).
EXAMPLE 4 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 4A Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidine-7-one
10 g (0.04 mol) of intermediate 3 and 13 g (0.16 mol) of sodium acetate were mixed with 50 ml of glacial acetic acid and 12 g (0.08 mol) bromine under nitrogen. The solution was heated to 50° C. and stirred for 35 hrs, then cooled to room temperature. Sodium bisulfite solids were added until the bromine color disappeared, then quenched, filtered and washed to provide a solid which was subsequently dissolved in 500 ml hot IPA, filtered hot, and cooled. The resulting crystals were further filtered, and dried in vacuo at 65° C. to yield 8 g (61%) of crude product (4A).
EXAMPLE 4 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
3.78 g (2.10 equiv; 13.6 mmoles) of intermediate 1, 25 ml toluene and lithium bis(trimethylsilyl)amide in 1 M THF (13.6 mmoles; 13.6 mL; 12.1 g) were mixed for 10 min under nitrogen to form a dark solution. In a separate beaker the intermediate 4a (1.00 equiv, 6.47 mmoles; 2.50 g) was slurried in toluene then added to the mixture containing 1 and stirred for 30 min, after which the combined mixture was quenched with 25 ml 1 M sodium bicarbonate and then filtered. Alternatively, the combined mixture can be quenched with ammonium chloride. The filter cake was washed with toluene, then acetone, then water and dried at 60° C. to give 3.5 g (92%) of a grey-yellow solid 4.
EXAMPLE 5 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cycloentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester

768 g (1.3 mol) of intermediate 4, was mixed with 395 g (3.9 mol) of butyl vinyl ether, 4.7 L of n-butanol, and 275 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred and placed under ca. 50 tore vacuum and then refilled with nitrogen; this was repeated 2 more times. To this degassed solution was added 22 g (0.03 mol) Bis-(diphenylphosphinoferrocene)palladium dichloride dichloromethane complex and the resulting slurry was degassed an additional three times as described above. The mixture was then heated and stirred at 95° C. for 20 hrs. The resulting thin red slurry was diluted with 4 L branched octane’s and cooled to about 5° C. after which 1 L saturated aq. potassium carbonate was added and the mixture was filtered and rinsed with 500 ml branched octanes. After drying for 16 hrs at 45° C., 664 g (83%) of gray-solid product (5) was obtained. In addition, column chromatography can be used to further purify the crude product.
EXAMPLE 6 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one

11.6 g (1.00 eq, 19.2 mmol) of intermediate 5, water (10.1 equiv; 193 mmoles; 3.48 mL; 3.48 g) and methanol (3.62 moles; 146 mL; 116 g) were combined and heated to 55-60° C. Isethionic acid was added slowly until a clear solution was obtained; 3.3 g isethionic acid solution was necessary to reach this end point. The resulting clear orange solution was filtered through paper and rinsed through with 20 ml methanol, after which the filtrate was reheated to 55-60° C. and the remaining isethionic acid was added (a total of 9.93 g was added). The reaction mixture precipitated and thickened for 6 hours, after which it was cooled and held at 30-35° C. while triethylamine (2.92 g; 28.8 mmoles) was added slowly as a 10% solution in methanol over 12 hrs. About halfway through the addition of triethylamine, desired polymorphic seeds were added to help formation of the desired polymorph. The resulting slurry was cooled and held at 5° C. for 15 minutes and the crystals were filtered and washed with methanol. The solid product was dried in vacuo at 55° C. to obtain 11 g of yellow crystals of the title compound.

……………………………………………………………………
http://www.google.com/patents/US7345171
EXAMPLES
The following examples are intended to be illustrative and non-limiting, and represent specific embodiments of the present invention.
Example 1 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
Example 2 Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2.3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
Example 3 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in Example 2) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
Example 4 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
To a slurry of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (7.0 g, 15.64 mmol, prepared as in Example 3 following contact with NaOH) dispersed in 250 mL of water was added drop-wise 30 mL of a 0.52 M solution of isethionic acid in MeOH (15.64 mmol) to a pH of 5.2. The solution was filtered through a glass filter (fine) and the clear solution was freeze-dried to give 9.4 g of the amorphous salt. The amorphous salt (3.16 g) was mixed with 25 mL of MeOH and after almost complete dissolution a new precipitate formed. Another 25 mL of MeOH was added and the mixture was stirred at 46° C. to 49° C. for four hours. The mixture was slowly cooled to 32° C. and put in a cold room (+4° C.) overnight. A sample was taken for PXRD, which indicated formation of Form B. The mixture was filtered and the precipitate was dried overnight at 50° C. in a vacuum oven. This furnished 2.92 g of the mono-isethionate salt of the compound of Formula 1 in 92% yield. HPLC-99.25%, PXRD-Form B, CHNS, H-NMR were consistent with the structure.
Example 5 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
MeOH (100 mL) was placed in a 250 mL flask equipped with a mechanical stirrer, thermocouple/controller, condenser, and heating mantle and preheated to 35° C. An amorphous isethionate salt (2 g, prepared as in Example 4) was slowly added in three even portions with a 25 min to 30 min interval between the additions. The reaction mixture was stirred overnight at 35° C. and subsequently cooled. A sample was filtered and examined by PXRD. It was pure Form B. The whole reaction mixture was then used as Form B seeds in a larger scale experiment.
Example 6 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
MeOH (50 mL) was placed in a 250 mL flask equipped with a magnetic stirrer, condenser, thermocouple/controller, and heating mantle, and preheated to 40° C. An amorphous isethionate salt (1 g, prepared as in Example 4) was slowly added in three even portions with 30 min interval between the portions and then stirred overnight at 40° C. The reaction was monitored by in-situ Raman spectroscopy. The sample was taken, filtered and analyzed by PXRD. It was pure Form B by PXRD and Raman spectroscopy. The mixture was cooled to 25° C. at a rate of 3° C./h, cooled to −10° C., filtered, and vacuum dried to furnish 0.85 g of the Form B crystalline product.
Example 7 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
The free base (Formula 1, 0.895 mg, 2 mmol) was mixed with 10 mL of MeOH and seeded with 33 mg of a mono-isethionate salt of the compound of Formula 1 (Form B). Then 5.6 mL of a 0.375 M solution of isethionic acid in MeOH (2.1 mmol) was added in 10 even portions over 75 min time period. The mixture was stirred for an additional hour and a sample was taken for PXRD analysis. It confirmed formation of crystalline Form B. The mixture was stirred at RT overnight and another PXRD was taken. There was no change in the crystal form. The mixture was cooled in a refrigerator at −8° C. overnight, filtered, and dried at 50° C. in a vacuum oven to give 1.053 g (91.8% of theory) of the above-named compound (Form B). HPLC—99.8%, CHNS, H-NMR, IR are consistent with the structure, PXRD-Form B.
Example 8 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form A)
An amorphous isethionate salt (47 mg, prepared as in Example 4) was mixed with 4 mL of EtOH in a 15 mL flask equipped with a magnetic stirrer, thermocouple and condenser. The mixture was heated to reflux, which resulted in the formation of a nearly clear solution. After refluxing for 10-15 min, the mixture became cloudy. It was slowly cooled to 50° C. and was seeded at 69° C. with Form A. The mixture was held at 50° C. for 5 h and was allowed to cool to RT overnight. The mixture was subsequently cooled to 1° C. with an ice bath, held for 1.5 h, filtered, washed with 0.5 mL of cold EtOH, air-dried, and then dried in a vacuum oven at 70° C. overnight to furnished 38.2 mg of a fine crystalline material. The crystalline material was found to be mono-isethionate salt Form A by PXRD. H-NMR was consistent for the mono-isethionate salt and indicated the presence of residual EtOH ca. 5.9 mol % or 0.6 wt %.
Example 9 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form D)
An amorphous isethionate salt (9.0 g, prepared as in Example 4) was mixed with 300 mL of MeOH, stirred and heated to 63.8° C. (at reflux). To the slightly cloudy mixture was added two 50-mL portions of MeOH. The hot mixture was filtered into a 2-L flask equipped with a mechanical stirrer. The mixture was briefly heated to reflux and then cooled to 60° C. IPA (100 mL) was added to the mixture. The mixture was again heated to 60° C. and an additional 110 mL of IPA was added. A precipitate started to form at 59.7° C. The mixture was reheated to 67.5° C., cooled to 50° C., and held overnight. A sample was taken the next morning for PXRD analysis. The mixture was cooled to 25° C. at a rate of 3° C./h and another PXRD sample was taken when the mixture reached 28° C. The mixture was allowed to cool to RT overnight. A precipitate was collected and dried in a vacuum oven at 65° C. and 30 Torr. The procedure produced 7.45 g (82.8% yield) of the crystalline compound (Form D by PXRD analysis). Previously analyzed samples were also Form D. HPLC showed 98.82% purity and CHNS microanalysis was within +/−0.4%. A slurry of isethionate salt Form A, B, and D in MeOH yielded substantially pure Form B in less than three days.
Example 10 Preparation of isethionic acid (2-hydroxy-ethanesulfonic acid)
A 5-L, four-necked, round-bottomed flask, equipped with mechanical stirrer, thermocouple, gas sparger, and an atmosphere vent through a water trap was charged with 748 g (5.05 mol) of sodium isethionate (ALDRICH), and 4 L of IPA. The slurry was stirred at RT. An ice bath was used to keep the internal temperature below 50° C. as 925 g (25.4 mol) of hydrogen chloride gas (ALDRICH) was sparged into the system at a rate such that it dissolved as fast as it was added (as noted by lack of bubbling through the water trap). Sufficient HCl gas was added until the system was saturated (as noted by the start of bubbling through the water trap). During the addition of HCl, the temperature rose to 45° C. The slurry was cooled to RT and filtered over a coarse-fritted filter. The cake was washed with 100 mL of IPA and the cloudy filtrate was filtered through a 10-20μ filter. The resulting clear, colorless filtrate was concentrated under reduced pressure on a rotary evaporator, while keeping the bath temperature below 50° C. The resulting 1.07 kg of clear, light yellow oil was diluted with 50 mL of tap water and 400 mL of toluene and concentrated under reduced pressure on a rotary evaporator for three days, while keeping the bath temperature below 50° C. The resulting 800 g of clear, light yellow oil was diluted with 500 mL of toluene and 250 mL of IPA and concentrated under reduced pressure on a rotary evaporator for 11 days, keeping the bath temperature below 50° C. The resulting 713 g of clear, light yellow oil was titrated at 81 wt % (580 g, 91.1% yield) containing 7.9 wt % water and 7.5 wt % IPA.
Example 11 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A 5-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (300 g, 0.51 mol, prepared as in Example 2), butyl vinyl ether (154 g, 1.54 mol, ALDRICH), n-butanol (1.5 L, ALDRICH), and diisopropyl ethylamine (107 mL, 0.62 mol, ALDRICH). The slurry was placed under approximately 50 Torr vacuum and then refilled with nitrogen 3 times. To this was added 8.3 g (0.01 mol) bis-(diphenylphosphinoferrocene) palladium dichloride dichloromethane (JOHNSON MATTHEY, Lot 077598001) and the resulting slurry was purged an additional three times as described above. The mixture was then heated to 95° C. and stirred for 20 h. The resulting thin red slurry was diluted with 2 L of heptane and cooled to approximately 5° C. At this temperature, 400 mL saturated aqueous potassium carbonate was added and the mixture was filtered and rinsed with 250 mL of heptane. After drying in an oven for 16 h at 45° C., 231.7 g (75% yield) of the title compound was obtained as a yellow solid.
Example 12 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-Pyrido[2,3-d]pyrimidin-7-one (Form B)
A 22-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (725 g, 1.20 mol, prepared as in Example 11) and MeOH (14 L). The slurry was stirred at RT as it was charged with a solution of isethionic acid (530 g, 4.20 mol, prepared as in Example 10), MeOH (1.5 L), and water (70 mL, 3.89 mol). The resulting slurry was heated to 55° C. over 30 minutes and then stirred at 55° C. for 30 minutes. A solution of 175 g (1.73 mol) of Et3N (ALDRICH) in 200 mL of MeOH was charged to the slurry as it was cooled to 30° C. The slurry was held at 30° C. as a solution of 128 g (1.26 mol) of Et3N in 2 L of MeOH was added dropwise over 6 hours. The resulting slurry was sampled to determine crystal form (Form B). The slurry was cooled and held at 5° C. for 15 minutes and was subsequently filtered through a coarse-fritted filter. The resulting filter cake was washed with multiple washes of 200 mL of cold MeOH. The solid product was dried at 55° C. under vacuum to yield 710 g (91% yield) of the title compound as yellow crystals.

1)Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;
2)Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4; Journal of Medicinal Chemistry,2005,48(7),2371-2387;
3)Erdman, David Thomas et al;Preparation of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157
4)Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997
5)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381
6)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.
7)Phase III Study Evaluating Palbociclib (PD-0332991), a Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”;ClinicalTrials.gov number:NCT01864746;currently recruiting participants(as of January 2, 2013)
8)A Randomized, Multicenter, Double-Blind Phase 3 Study Of PD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For Advanced Disease;ClinicalTrials.gov number:NCT01740427;currently recruiting participants(as of January 2, 2013)
9)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy;ClinicalTrials.gov number:NCT01942135;currently recruiting participants(as of January 2, 2013)
| US6936612 | Jan 16, 2003 | Aug 30, 2005 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2005005426A1 | Jun 28, 2004 | Jan 20, 2005 | Vladimir Genukh Beylin | Isethionate salt of a selective cdk4 inhibitor |
| US20030229026 * | Dec 18, 2000 | Dec 11, 2003 | Al-Awar Rima Salim | Agents and methods for the treatment of proliferative diseases |
| US20040006074 * | Dec 2, 2002 | Jan 8, 2004 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| US20040048915 * | Sep 24, 2001 | Mar 11, 2004 | Engler Thomas Albert | Methods and compounds for treating proliferative diseases |
| US20050222163 * | Mar 30, 2005 | Oct 6, 2005 | Pfizer Inc | Combinations of signal transduction inhibitors |
| US20070027147 * | Dec 3, 2004 | Feb 1, 2007 | Takashi Hayama | Biarylurea derivatives |
| WO2008032157A2 * | Aug 27, 2007 | Mar 20, 2008 | David Thomas Erdman | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2010075074A1 | Dec 15, 2009 | Jul 1, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2012098387A1 | Jan 17, 2012 | Jul 26, 2012 | Centro Nacional De Investigaciones Oncológicas (Cnio) | 6, 7-ring-fused triazolo [4, 3 – b] pyridazine derivatives as pim inhibitors |
| US7781583 | Sep 10, 2007 | Aug 24, 2010 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
| US7855211 | Dec 15, 2009 | Dec 21, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| US8247408 * | Oct 9, 2006 | Aug 21, 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755 | Feb 9, 2010 | Sep 25, 2012 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |

old info
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)

| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Zosuquidar

LY335979, RS-33295-198 (Zosuquidar)
Roche Palo Alto (Originator)
LY335979 (Zosuquidar) is a selective Pgp (P-glycoprotein) inhibitor with a Ki of 59 nM. LY335979 significantly enhanced the survival of mice implanted with Pgp-expressing murine leukemia (P388/ADR) when administered in combination with either daunorubicin, doxorubicin or etoposide.
LY335979 (Zosuquidar)
M.Wt: 636.99
Formula: C32H31F2N3O2.3HCl
Name: Zosuquidar trihydrochloride
Elemental Analysis: C, 60.34; H, 5.38; Cl, 16.70; F, 5.97; N, 6.60; O, 5.02
CAS : 167465-36-3
167354-41-8 (free base)
Roche Bioscience (Originator), Eli Lilly and Company (Licensee).
US5654304, WO1994024107A1, WO2000075121, US6570016
Drug Des Discov 1992, 9(1): 69, Bioorg Med Chem Lett 1995, 5(21): 2473, Drugs Fut 2003, 28(2): 125
Zosuquidar is currently under development. It is now in “Phase 3” of clinical tests in the United States. Its action mechanism consists of the inhibition of P-glycoproteins; other drugs with this mechanism include tariquidar and laniquidar. P-glycoproteins are proteins which convert the energy derived from the hydrolysis of ATP to structural changes in protein molecules, in order to perform coupling, thus discharging medicine from cells. If P-glycoprotein coded with the MDR1 gene manifests itself in cancer cells, it discharges much of the antineoplastic drugs from the cells, making cancer cells medicine tolerant, and rendering antineoplastic drugs ineffective. This protein also manifests itself in normal organs not affected by the cancer (such as the liver, small intestine, and skin cells in blood vessels of the brain), and participates in the transportation of medicine. The compound Zosuquidar inhibits this P-glycoprotein, causing the cancer cells to lose their medicine tolerance, and making antineoplastic drugs effective
Clinicial trials: Clinical report published in 2010 showed that zosuquidar did not improve outcome in older acute myeloid leukemia, in part, because of the presence P-gp independent mechanisms of resistance. (Blood. 2010 Nov 18;116(20):4077-85.)
Zosuquidar is a potent P-glycoprotein inhibitor, which binds with high affinity to P-glycoprotein and inhibits P-glycoprotein-mediated multidrug resistance (MDR). P-glycoprotein, encoded by the MDR-1 gene, is a member of the ATP-binding cassette superfamily of transmembrane transporters and prevents the intracellular accumulation of many natural product-derived cytotoxic agents
Zosuquidar
U.S. Patent No. 5,112,817 to Fukazawa et al. discloses certain quinoline derivatives useful as anticancer drug potentiators for the treatment of multidrug resistance. One of the initially promising active agents there-disclosed is MS-073, which has the following structure:

MS-073
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11- methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. One such derivative having good activity, oral bioavailability, and stability, is zosuquidar, a compound of formula (2R)-anti-5-
3 – [4-( 10, 11 -difluoromethanodibenzosuber-5-yl)piperazin- 1 -yl]-2-hydroxypropoxy) quinoline.

Zosuquidar
Given the limitations of previous generations of MDR modulators, three preclinical critical success factors were identified and met for zosuquidar: 1) it is a potent inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (Kj = 59 nM) and is among the most active modulators of P-gp-associated resistance described to date. Zosuquidar has also demonstrated good in vivo activity in preclinical animal studies. In addition, the compound does not appear to be a substrate for P-gp efflux, resulting in a relatively long duration of reversal activity in resistant cells even after the modulator has been withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal pharmacokinetic (PK) interactions with several oncolytics tested in preclinical models. Such minimal PK interaction permits normal doses of oncolytics to be administered and also a more straightforward interpretation of the clinical results.
Zosuquidar is generally administered in the form of the trihydrochloride salt. Conventional zosuquidar trihydrochloride formulations include those containing zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg) dissolved in enough water for injection, to yield a free base concentration of 5 mg/mL. The formulation is filled into vials and lyophilized to give a vial containing 50 mg of free base. For such formulations, a 30 mL vial size is necessary to contain 50 mg of thezosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50 mg vials are needed to contain the formulation, greatly increasing manufacturing costs and reducing convenience for the end user {e.g., a pharmacist). Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion complexes with any drug whose molecule can fit into the lipophile-seeking cavities of the cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of various cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include α-, β-, and χ-cyclodextrins. The α-cyclodextrins include six glucopyranose units, the β- cyclodextrins include seven glucopyranose units, and the χ-cyclodextrins include eight glucopyranose units. The β -cyclodextrins are generally preferred as having a suitable cavity size for zosuquidar. Cyclodextrin can be in any suitable form, including amorphous and crystalline forms, with the amorphous form generally preferred. Cyclodextrins suitable for use in the formulations of preferred embodiments include the hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives of β- cyclodextrin, carboxyamidomethyl-β-cyclodextrin, carboxymethyl-β-cyclodextrin, and diethylamino-β-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin derivatives are disclosed in the following United States patents: U.S. Pat. No. 4,024,223; U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat. No. 4,351,846; U.S. Pat. No. 4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No. 4,424,209; U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881; U.S. Pat. No. 4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No. 4,497,803; U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504; U.S. Pat. No. 4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No. 4,603,123; U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641; U.S. Pat No. 4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No. 4,728,510; and U.S. Pat. No. 4,751,095.
Chemically modified and substituted α-, β-, and χ-cyclodextrins are generally preferred over unmodified α-, β-, and χ-cyclodextrins due to improved toxicity and solubility properties. The degree of substitution of the hydroxy 1 groups of the glucopyranose units of the cyclodextrin ring can affect solubility. In general, a higher average degree of substitution of substituent groups in the cyclodextrin molecule yields a cyclodextrin of higher solubility.
Examples for Pgp inhibitors are cyclosporine A, valpodar, elacridar, tariquidar, zosuquidar, laniquidar, biricodar, S-9788, MS-209, BIBW-22 (BIBW-22-BS) , toremifene, verapamil, dexverapamil , quinine, quinidine, trans- flupentixol, chinchonine and others (J. Roberts, C. Jarry (2003) : J. Med. Chem. 46, 4805 – 4817) . The list of inhibitors of P-glycoprotein is increasing (e.g. Wang et al . (2002) : Bioorg. Med. Chem. Lett. 12, 571 – 574) .
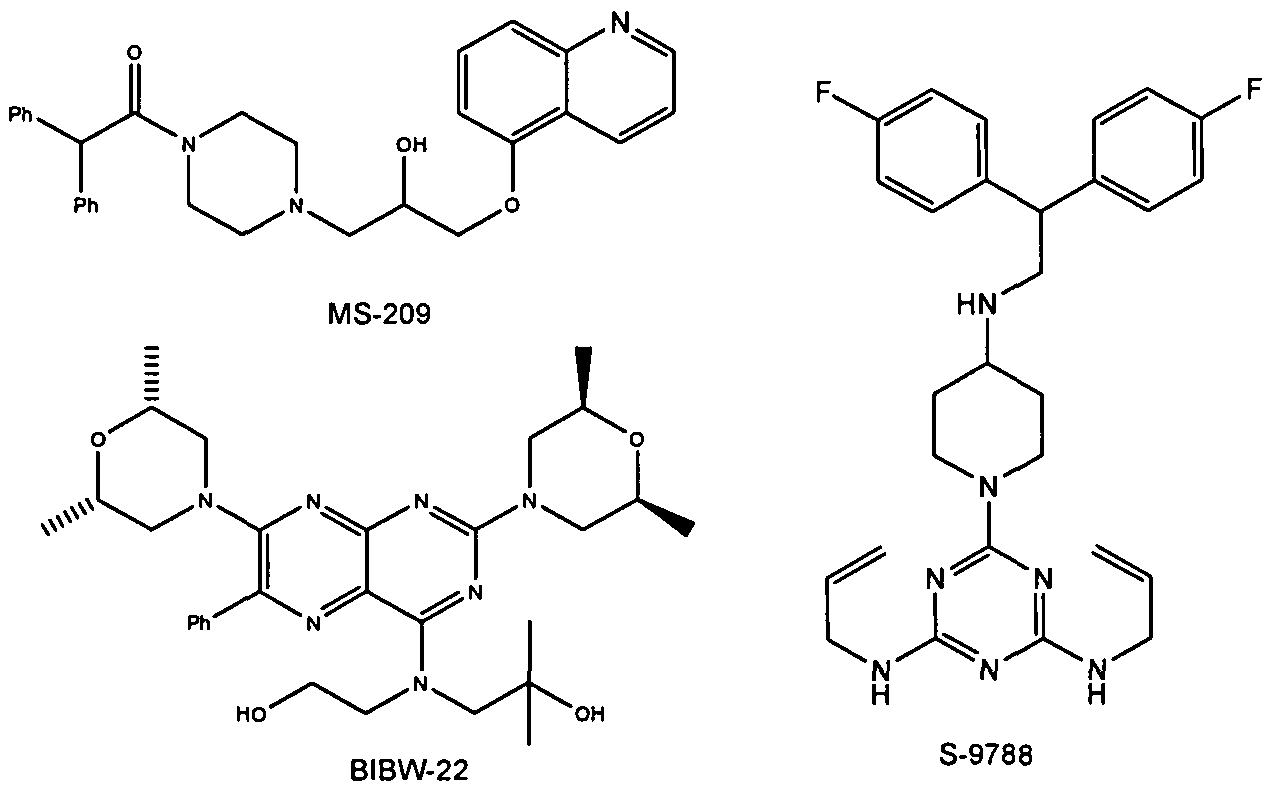
Figure 2: Structures of BIBW-22, MS-209 and S-9788
|
7-12-2000
|
10,11-methanodibenzosuberane derivatives
|
|
|
10-17-2007
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline
|
|
|
9-2-2009
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-YL)piperazin-1-YL]-2-hydroxypropoxy}quinoline
|
……………………

U.S. Pat. Nos. 5,643,909 and 5,654,304, incorporated herein by reference, disclose a series of 10,11-methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride disclosed therein, is currently under development as a pharmaceutical agent.
U.S. pat. No. 5,654,304 (‘304), incorporated by reference herein, discloses a series of 10,11-(optionally substituted)methanodibenzosuberane derivatives useful in enhancing, the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-Difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride is disclosed in ‘304 and is currently under development as a pharmaceutical agent. WO00/75121 discloses Form I, a crystalline form of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride.
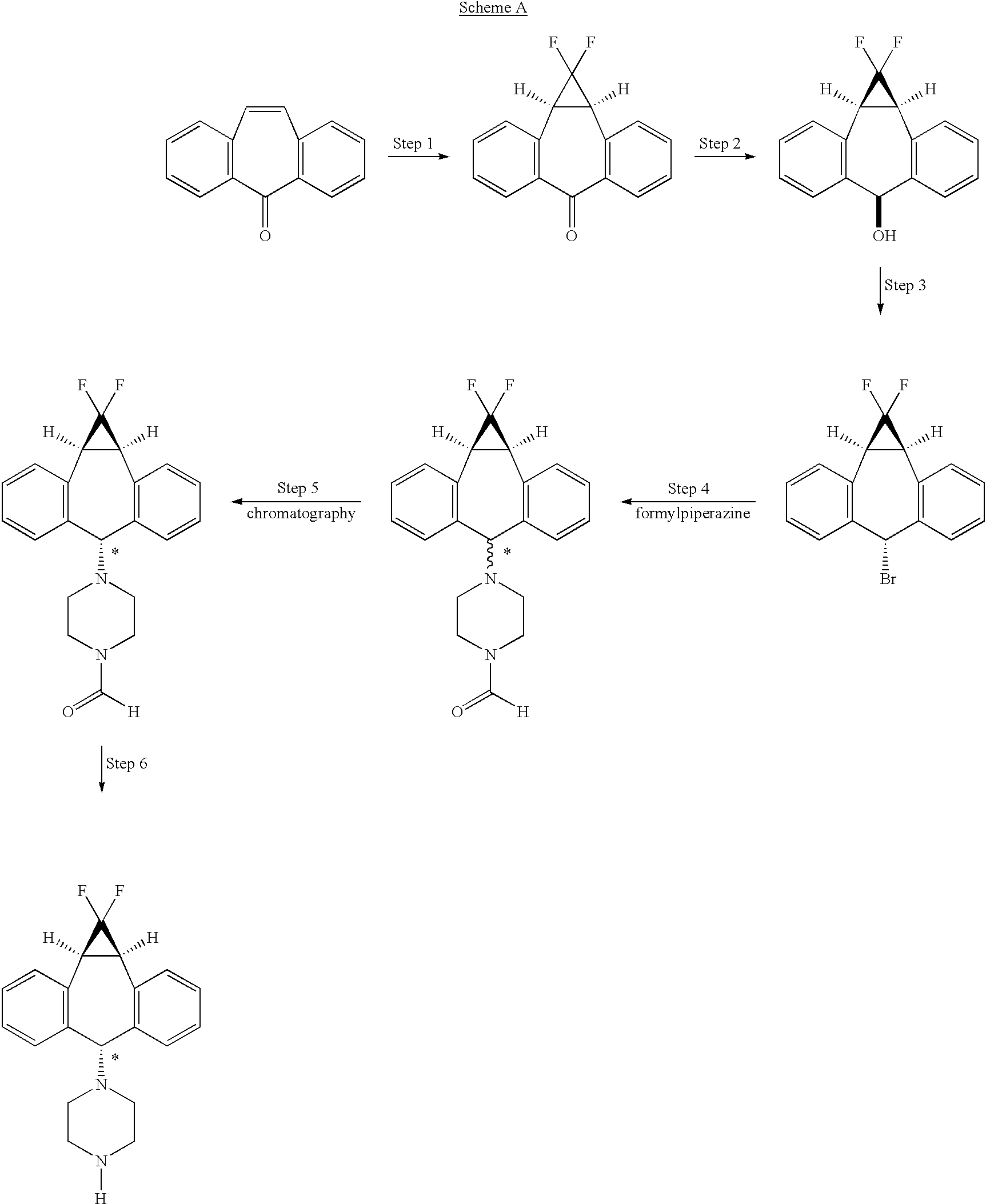
The art disclosed in U.S. Pat. No. 5,776,939, and U.S. Pat. No. 5,643,909 both incorporated herein by reference, and PCT Patent Applications (Publication numbers WO 94/24107 and 98/22112) teach the use of 1-formylpiperazine to introduce the piperazine group of the compound of formula II

Compound II is a mixture of syn isomer (III)

and anti isomer (IV)

The process as disclosed in U.S. Pat. Nos. 5,643,909 and 5,654,304 (represented by scheme A, below) involves (a) chromatographic separation(s) of the formyl piperazine compound; and (b) deformylation of the formyl piperazine compound to provide compound IV.https://www.google.co.in/patents/US6570016?cl=en
The process of the present invention uses piperazine to react with the (1aα,6α,10bα)-6-halo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cycloheptene compound or derivative, instead of formylpiperazine.
The process of the present invention is advantageous because piperazine is readily available in commercial quantities whereas 1-formylpiperazine, which was utilized in the process disclosed in U.S. Pat. No. 5,643,909 is often not readily available in commercial quantities. Additionally piperazine enjoys a significant cost advantage over 1-formylpiperazine.
The use of piperazine instead of 1-formylpiperazine is a significant advancement over the prior art because it obviates the need to deformylate or hydrolyze off the formyl group (step 6, scheme A), thereby providing fewer operational steps. U.S. Pat. No. 5,643,909 teaches the separation of the 1-formylpiperazine compounds by chromatography or repeated crystallization. The present invention obviates the need for chromatographic separations of the formylpiperazine diastereomeric addition compounds (see step 4, scheme A)
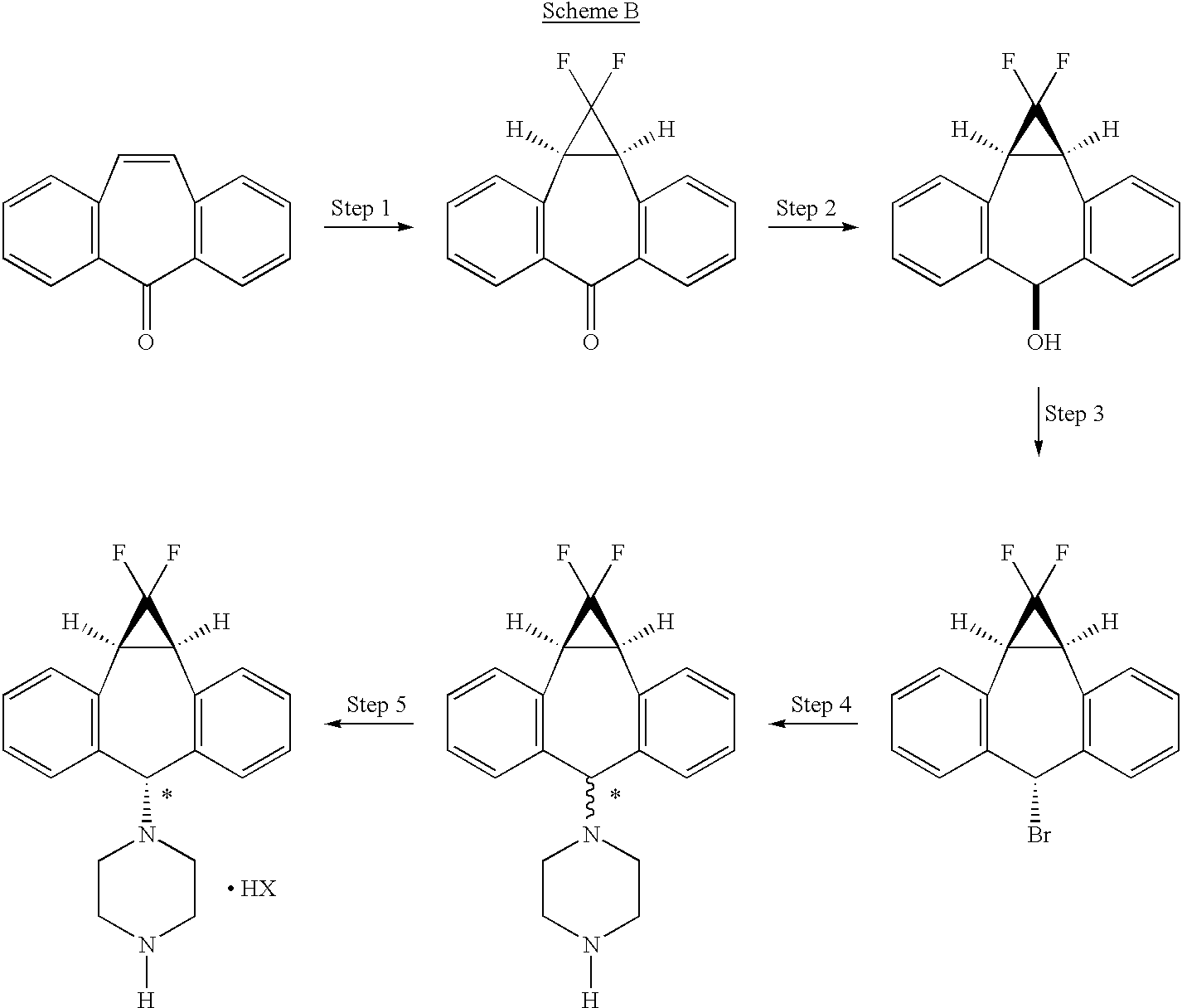
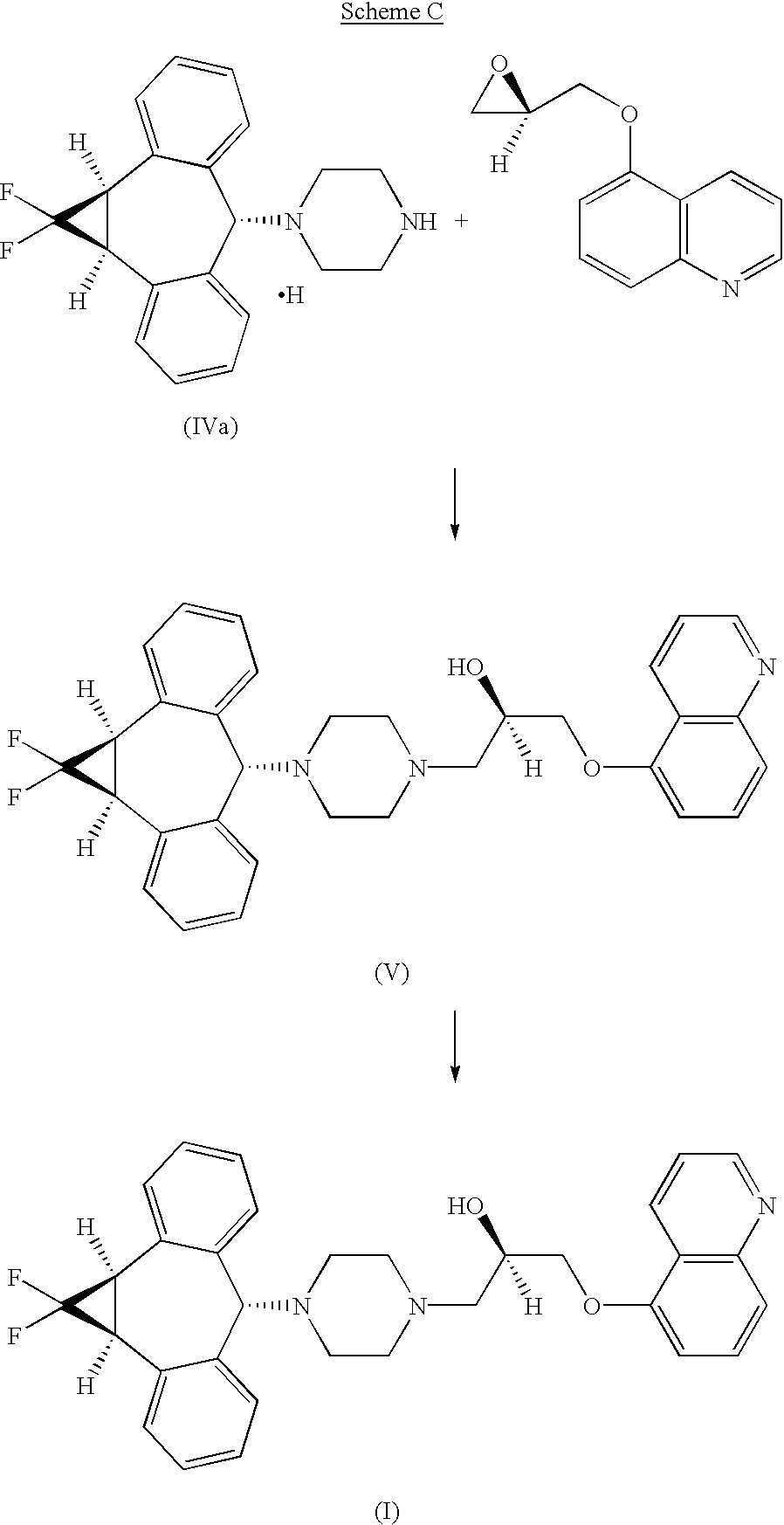
EXAMPLES
The following examples and preparations are illustrative only and are not intended to limit the scope of the invention in any way.
Preparation 1 R-1-(5-Quinolinyloxy)-2,3-epoxypropane
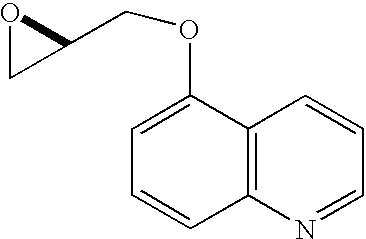
A mixture of 5-hydroxyquinoline (5.60 g, 38.6 mmol), R-glycidyl nosylate (10.0 g, 38.6 mmol), powdered potassium carbonate (11.7 g, 84.9 mmol), and N,N-dimethylformamide (100 mL) was stirred at ambient temperature until HPLC analysis (40% acetonitrile/60% of a 0.5% aqueous ammonium acetate solution, 1 mL/min, wavelength=230 nm, Zorbax RX-C8 25 cm×4.6 mm column) indicated complete disappearance of glycidyl nosylate (approximately 6 hours). The reaction mixture was filtered through paper and the filter cake was washed with 200 mL of a 3:1 mixture of MTBE and methylene chloride. The filtrate was washed with 200 mL of water and the aqueous layer was extracted four times with 100 mL of 3:1 MTBE/methylene chloride. The combined organic layers were dried over 30 grams of magnesium sulfate and the dried solution was then stirred with 50 grams of basic alumina for 30 minutes. The alumina was removed by filtration and the filter cake was washed with 200 mL of 3:1 MTBE/methylene chloride. The filtrate was concentrated to a volume of 100 mL, 300 mL of MTBE were added, and the solution was again concentrated to 80 mL. After heating to 50° C., the solution was treated with 160 mL of heptane dropwise over 15 minutes, allowed to cool to 40° C., and seeded, causing the formation of a crystalline precipitate. The mixture was stirred for two hours at ambient temperature and then at 0-5° C. for an additional 2 hours. The crystals were filtered, washed with cold heptane, and dried to provide 5.68 g (73.2%) of (2R)-1-(5-quinolinyloxy)-2,3-epoxypropane as white needles.
mp 79-81° C.;
[α]25 D−36.4° (c 2.1, EtOH);
1H NMR (500 MHz, CDCl3)δ 2.83 (dd, J=4.8, 2.7 Hz, 1H), 2.97 (m, 1H), 3.48 (m, 1H), 4.10 (dd, J=11.0, 6.0 Hz, 1H), 4.43 (dd, J=11.0, 2.7 Hz, 1H), 6.85 (d, J=7.8 Hz, 1H), 7.38 (dd, J=8.5 Hz, 4.1 Hz, 1H), 7.59 (m, 1H), 7.71 (d, J=8.5 Hz, 1H), 8.61 (m, 1H), 8.90 (m, 1H).
Example 1 (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride

Preparation of the above compound is exemplified in the following preparative steps.
Step 1 1,1-Difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6 (1H)-one

A solution of sodium chlorodifluoroacetate (350 g) in diglyme (1400 mL) was added dropwise over 4 to 8 hours, preferably over 6 hours, to a solution of 5H-dibenzo[a,d]cyclo-hepten-5-one (25 g) in diglyme (500 mL), with stirring, and under nitrogen, maintaining the reaction temperature at 160°-165° C. The cooled reaction mixture was poured into water (1.8 L) and extracted with ether (1.8 L). The organic phase was washed with water, dried over sodium sulfate (Na2SO4), and evaporated. The residue was recrystallized from ethanol, then from acetone/hexane to give 14 g of 1,1-difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6(1H)-one.
mp 149.6° C.
Flash chromatography of the combined mother liquors on silica gel, eluting with 20% acetone/hexane, gave an additional 6.5 g of the target compound.
Step 2 (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

A solution of 1,1-difluoro-1a,10b-dihydro-dibenzo[a,e]cyclopropa[c]cyclohepten-6(1H)-one (20.4 g) in tetrahydrofuran/methanol (1:2, 900 mL) was cooled in an ice bath. Sodium borohydride (12 g) was added in portions. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 2 hours, then poured into water. The product was filtered off, washed with water, and dried to give 20 g of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol (ii).
mp 230.1°-230.6° C.
Step 2A Combined Steps 1 and 2 Procedure (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

To a solution of 103.1 g (0.500 mol) of 5H-dibenzo[a,d]cyclohepten-5-one (2) in 515 mL of triethylene glycol dimethyl ether heated to between 180° C. and 210° C. was added over 7 hours, 293.3 g (2.15 mol) of chlorodifluoroacetic acid lithium salt (as a 53% by weight solution in ethylene glycol dimethyl ether). The ethylene glycol dimethyl ether was allowed to distill from the reaction as the salt addition proceeded. The GC analysis of an aliquot indicated that all of the 5H-dibenzo[a,d]cyclohepten-5-one had been consumed. The reaction was cooled to ambient temperature and then combined with 400 mL of ethyl acetate and 75 g of diatomaceous earth. The solids were removed by filtration and washed with 300 mL of ethyl acetate. The washes and filtrate were combined and the ethyl acetate was removed by concentration under vacuum leaving 635 g of dark liquid. The dark liquid was cooled to 18° C. and to this was added, over 15 minutes, 6.62 g (0.175 mol) of sodium borohydride (as a 12% by wt solution in 14 M NaOH). After stirring for 2 h the reaction was quenched by careful addition of 900 mL of a 1:3.5:4.5 solution of conc. HCl-methanol-water. The suspension was stirred for 30 min and the crude product was collected by filtration, washed with 600 mL of 1:1 methanol-water and dried to 126.4 g of dark brown solid. The crude product was slurried in 600 mL of methylene chloride, filtered, washed twice with 150 mL portions of methylene chloride, and dried to 91.6 g (71%) of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol. Gas Chromatography (GC) Conditions; Column: JW Scientific DB-1, Initial Temperature 150° C. for 5 min, 10° C./min ramp, Final temp 250° C. for 5 min. tR: intermediate, 11.5 min; reaction product (alcohol), 11.9 min; starting material, 12.3 minutes.
Step 3 Preparation of (1aα,6α,10b)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cycloheptene

A slurry of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol (3.0 g, 11.6 mmol, 1.0 equiv) in heptane (24 mL) was treated with 48% HBr (1.58 mL, 14.0 mmol, 1.2 equiv) and the reaction was heated at reflux with vigorous stirring for 2.5 hr. Solvent was then removed by atmospheric distillation (bp 95-98° C.) until approximately 9 mL of distillate was collected. The reaction was cooled and treated with EtOAc (15 mL), Na2SO4 and activated charcoal. The mixture was stirred at RT for 15 min and filtered through hyflo. The filter cake was washed with 50:50 EtOAc:heptane and the filtrate was concentrated in vacuo to provide the title product as a crystalline solid.
mp 119° C. (3.46 g corr., 93%);
1H NMR (500 MHz CDCl3) δ 7.20-7.41 (8H, m), 5.81 (1H, s), 3.41 (2H, d, J 12.5 Hz);
13CNMR (126 MHz CDCl3) δ 141.3, 141.2, 133.5, 130.1, 129.8, 128.3, 128.2, 112.9, 110.6, 110.5, 108.3, 53.6, 30.2, 30.1, 30.0.
Anal. Calcd. For C16H11BrF2: C, 59.84; H, 3.45. Found: C, 60.13; H, 3.50.
Step 3A Preparation of (1aα,6α,10bα)-6-Bromo-1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene

To a stirred suspension of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol, (18.4 g, 71.2 mmol) in 151 mL of methylene chloride which had been cooled to 10-17° C. was added phosphorous tribromide (9.6 g, 35.6 mmol) dropwise over 15 minutes. The cooling bath was removed and the reaction was stirred for 2 hours at ambient temperature. Analysis by gas chromatography indicated complete consumption of starting material. Cold water (92 mL) and activated carbon (1.84 g) were added and the resulting mixture was stirred for 30 minutes. The activated carbon was removed by filtration through Hyflo brand filter aid and the two phases were separated. The organic phase was washed with water (184 mL×2), brine (184 ml), dried over magnesium sulfate and concentrated to dryness under vacuum, affording 21.7 g (94.8%) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene.
1H NMR (CDCl3, 300 MHz) δ 3.36 (s, 1H), 3.40 (s, 1H), 5.77 (s, 1H), 7.16-7.38 (m, 8H).
Steps 4 and 5 (1aα,6α,10bα)-1-(1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl)-piperazine, Hydrobromide Salt

To a solution of 237.5 g (0.739 mol) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]-cyclopropa[c]cycloheptene in 3.56 L of acetonitrile was added 207.7 g (2.41 mol) of piperazine and the mixture was heated to reflux for 2 hours, at which time analysis by gas chromatography showed complete consumption of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene (iii) and formation of a mixture of syn and anti piperazine compounds (III and IV) in an anti-syn ratio of 55:45. The reaction was cooled to about 7° C. and stirred for 30 minutes at that temperature. The reaction mixture was filtered to remove the precipitated syn-isomer (III) and the filter cake was washed with 250 mL of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 262.4 grams of a foam which was dissolved in 450 mL of acetonitrile with heating. The solution was cooled to about 12° C. in an ice bath and stirred for 1 hour at that temperature. The precipitated syn-piperazine compound of formula (III) was filtered and washed with 125 ml of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 194.1 g and dissolved in 1.19 L of ethyl acetate. The organic solution was washed sequentially with 500 mL portions of 1N sodium hydroxide, water, and saturated sodium chloride. The ethyl acetate solution was dried over sodium sulfate and concentrated to give 137.0 grams of residue which was dissolved in 1.37 L of methylene chloride and seeded with (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrobromide salt, followed by the addition of 70.8 grams of 48% aqueous hydrobromic acid. The mixture was stirred for about 45 minutes, causing the anti-isomer to crystallize as its hydrobromide salt. The crystals were filtered, washed with methylene chloride, and dried to provide purified hydrobromide salt of compound (IVa), shown by HPLC to have an anti-syn ratio of 99.3:0.7. Treatment of the isolated hydrobromide salt of compound (IVa) with aqueous sodium hydroxide, extraction into methylene chloride, separation of the aqueous layer and concentration to dryness gave 80.1 grams (33.2% yield based on starting material) of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine as the free base. Acidification of a solution of the free base in 800 mL of methylene chloride by addition of 41.2 g of 48% hydrobromic acid as described above afforded 96.4 g of pure hydrobromide salt (title compound) with an anti-syn ratio of 99.8:0.2 (HPLC), mp 282-284° C. 1H NMR (DMSO-d6) δ 2.41 (m, 4H), 3.11 (m, 4H), 3.48 (d, J=12.4 Hz, 2H), 4.13 (s, 1H), 7.2 (m, 8H), 8.65 (bs, 2H). 13C NMR (DMSO-d6) δ 28.0, 42.9, 48.0, 75.1, 108.5, 112.9, 117.3, 127.5, 128.0, 128.6, 129.6, 132.4, 141.3. IR: (KBr) 3019, 2481, 1587, 1497, 1298 cm−1. Anal. Calcd for C20H21BrF2N2: C, 58.98; H, 5.20; N, 6.88. Found: C, 58.75; H, 5.29; N, 7.05.
Step 6 Preparation of (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)propan-2-ol Trihydrochloride
A suspension of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrochloride compound of formula IVa (5.41 g, 14.9 mmol) and powdered sodium carbonate (3.16 g, 29.8 mmol) in 54 mL of 3A ethanol was stirred at ambient temperature for 1 hour. R-1-(5-quinolinyloxy)-2,3-epoxypropane (3.00 g, 14.9 mmol) was added in one portion and the reaction mixture was heated to 65° C. for 19 hours. HPLC analysis (Gradient system with solvent A (acetonitrile) and solvent B (0.02M sodium monophosphate buffer containing 0.1% triethylamine adjusted to pH 3.5 with phosphoric acid) as follows: 0-12 min, 30% solvent A/70% solvent B; 12-30 min, linear gradient from 30% to 55% solvent A/70% to 45% solvent B; 30-35 min, 55% solvent A/45% solvent B, 1 mL/min, 1=240 nm, Synchropak SCD-100 25 cm×4.6 mm column) indicated the total consumption of the piperazinyl compound of formula (IV). The mixture was allowed to cool to room temperature, filtered through a plug of silica gel, and eluted with an additional 90 mL of ethanol. The eluent was concentrated to a volume of approximately 60 mL and heated to 65° C. with stirring. A solution of HCl in ethanol (16.1 g at 0.135 g/g of solution, 59.6 mmol) was added dropwise over 10 minutes and the resultant product solution was seeded, causing the trihydrochloride salt to precipitate. The mixture was allowed to cool to ambient temperature and stirred slowly (less than 100 RPM) for 2 hours. The precipitate was filtered, washed with ethanol, and dried in vacuo at 50° C. to give the crude trihydrochloride salt which was further purified by recrystallization from methanol/ethyl acetate to provide 7.45 g (78.4%) of (2R)-anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)-propan-2-ol trihydrochloride.
Step 6a
The syn isomer compound of formula (III) isolated as described supra (combined steps 4 and 5), can be utilized to produce the corresponding syn-5-{3-[4-(10,11-difluoromethano-dibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (XII) essentially as shown below for the free base of the anti isomer (IVa)in step 6.
https://www.google.co.in/patents/US6570016?cl=en
………………………………………
http://www.google.it/patents/WO1994024107A1?cl=en
REACTION SCHEME 1

FormuIa 1
Formula 1

Formula 2 Formula 2

Formula 3
Formula 3

Formula 4

Formula I
……………………………………….
http://www.google.com/patents/WO2000075121A3

1HNMR (500 MHz DMSO-d6) δ9.41 (2H, br. s), 7.17-7.31 (8H, m), 4.17 (1H, s), 3.52 (2H, d, J=12.4 Hz), 3.11 (4H, br. s), 2.48-2.51 (4H, m)
13CNMR (126 MHz DMSO-d6) δ142.3, 133.4, 130.5, 129.6, 129.0, 128.4, 115.9, 113.6, 111.3, 76.2, 49.0, 43.6, 29.2, 29.1, 29.0; FD MS: m/e 326 (M+).
Anal. Calcd. For C20H21ClF2N2: C, 66.20; H, 5.83; N, 7.72.
Found: C, 66.08; H, 5.90; N, 7.72.
…………………………………………..
http://www.google.com/patents/US6570016?cl=fr


(2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
……………….
Chemical Shift Data and Peak Assignments for the Crystal Forms.

Form II has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 29.9, 50.1, 55.3, 62.0, 66.5, 72.0, 75.8, 104.8, 107.5, 108.2, 109.1, 110.2, 112.0, 118.4, 119.5, 120.1, 123.1, 128.7, 131.1, 133.0, 134.8, 136.4, 136.9, 139.9, 140.0, 142.3, 144.5, 146.6, 149.0, 144.2, 153.0 and 153.6 ppm.
Form III has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 30.3, 50.4, 59.1, 63.2, 72.8, 77.2, 109.1, 110.2, 112.2, 112.8, 118.7, 119.5, 119.9, 121.0, 122.2, 123.0, 128.9, 130.6, 132.7, 134.0, 136.4, 140.0, 141.0, 141.8, 142.5, 143.3, 146.1, 153.1, 153.8 and 154.7 ppm.
Daiichi Sankyo anticoagulant edoxaban succeeds in Phase III

Edoxaban, DU-176b
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
NOV20, 2013
Daiichi Sankyo will file edoxaban on both sides of the Atlantic shortly after the bloodthinner proved as effective and safer than warfarin in a Phase III trial of patients with atrial fibrillation.
The company has presented data on edoxaban, a once-daily oral factor Xa inhibitor, at the American Heart Association meeting in Dallas, from a study involving 21,105 patients across 46 countries. The drug, evaluated in 60mg and 30mg doses, met its primary endpoint of non-inferiority compared to warfarin for the prevention of stroke or systemic embolic events in patients with non-valvular AF.http://www.pharmatimes.com/Article/13-11-20/Daiichi_Sankyo_anticoagulant_edoxaban_succeeds_in_Phase_III.aspx
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]
- “First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost. 6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID 18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis 104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID 20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost. 104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID 23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
-
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
-


-
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
-
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
-

-
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
-
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
SIMILAR

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:

formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
FDA approves second brain imaging drug Vizamyl (flutemetamol F 18 injection)to help evaluate patients for Alzheimer’s disease, dementia
 is the structure on right
is the structure on right
Vizamyl (flutemetamol F 18 injection)
http://jnm.snmjournals.org/content/50/8/1251/F1.expansion.html get structure

| Chemical name: | 2-{3-[18F]fluoro-4-(methylamino)phenyl}-1,3-benzothiazol-6-ol diagnostic aid |
| Cas Number | 765922-62-1 |
| INN name | flutemetamol |
| Molecular Formula: |  |
GE Healthcare
FDA PRESS RELEASE
For Immediate Release: Oct. 25, 2013
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm
The U.S. Food and Drug Administration today approved Vizamyl (flutemetamol F 18 injection), a radioactive diagnostic drug for use with positron emission tomography (PET) imaging of the brain in adults being evaluated for Alzheimer’s disease (AD) and dementia.
Dementia is associated with diminishing brain functions such as memory, judgment, language and complex motor skills. The dementia caused by AD is associated with the accumulation in the brain of an abnormal protein called beta amyloid and damage or death of brain cells. However, beta amyloid can also be found in the brain of patients with other dementias and in elderly people without neurologic disease.
Vizamyl works by attaching to beta amyloid and producing a PET image of the brain that is used to evaluate the presence of beta amyloid. A negative Vizamyl scan means that there is little or no beta amyloid accumulation in the brain and the cause of the dementia is probably not due to AD. A positive scan means that there is probably a moderate or greater amount of amyloid in the brain, but it does not establish a diagnosis of AD or other dementia. Vizamyl does not replace other diagnostic tests used in the evaluation of AD and dementia.
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm
About GE Healthcare
GE Healthcare provides transformational medical technologies and services to meet the demand for increased access, enhanced quality and more affordable healthcare around the world. GE GE -0.23% works on things that matter – great people and technologies taking on tough challenges. From medical imaging, software & IT, patient monitoring and diagnostics to drug discovery, biopharmaceutical manufacturing technologies and performance improvement solutions, GE Healthcare helps medical professionals deliver great healthcare to their patients.
For our latest news, please visit http://newsroom.gehealthcare.com
Flutemetamol [ 18 F] Injection is a diagnostic positron emission tomography (PET) agent for the imaging of β-amyloid plaques in the brain. The synthesis of the agent can be performed using automated synthesis platforms with or without using specially-tailored cassettes. For example, the synthesis can be performed using either the TRACERlab FX F-N platform or the FASTlab™ platform, commercially available from GE Healthcare a division of General Electric Company in conjunction with auxiliary preparative high pressure liquid
chromatography equipment. After synthesis, the bulk agent is transferred to high pressure liquid chromatography (HPLC) equipment to separate the physico-chemically similar compounds [ 18 F] flutemetamol from its deprotected precursor, AHl 11832 (6-hydroxy-2-(4′-(N- methyl)amino-3′-nitro)phenylbenzothiazole) and hence obtain purified [ 18 F] flutemetamol.
However there still exists a need in the art for alternative purification methods for the preparation of [ 18 F] flutemetamol. The invention as described below answers such a need. Specifically, Applicants have now found a process that eliminates the use of preparative HPLC equipment. SUMMARY OF THE INVENTION
As [ 18 F]flutemetamol and its deprotected precursor, AH111832 (6-hydroxy-2-(4′-(N- methyl)amino-3′-nitro)phenylbenzothiazole) are physico-chemically very similar, preparative HPLC is required to separate them. However, Applicants have now found that it is possible to replace the preparative HPLC equipment in previous purification processes with low cost, single-use solid phase extraction (SPE) cartridges for purification of [ 18 F]flutemetamol.
Accordingly, the present invention provides a purification process comprising the following steps:
(a) passing a diluted crude product reaction mixture comprising flutemetamol through a first reverse phase SPE cartridge;
(b) washing said first reverse phase SPE cartridge with a water/acetonitrile,
tetrahydrofuran(THF)/water, methanol(MeOH)/water or isopropanol/water mixture; preferably, a water/acetonitrile mixture;
(c) rinsing said first reverse phase SPE cartridge with water once step (b) is completed; (d) eluting said first reverse phase SPE cartridge with acetonitrile or tetrahydrofuran; preferably, acetonitrile;
(e) directly passing the mixture from said eluting step (d) through a normal phase SPE cartridge to give an acetonitrile or tetrahydrofuran solution; preferably, an acetonitrile solution, comprising purified flutemetamol; (f) diluting said acetonitrile or tetrahydrofuran solution; preferably, an acetonitrile solution, comprising purified flutemetamol, with water to form a diluted water/acetonitrile or a diluted water/tetrahydrofuran solution; preferably, a diluted water/acetonitrile solution, comprising purified flutemetamol, wherein said water/acetonitrile solution contains about 40- 70% (v/v) water; preferably at least about 40% (v/v) water; more preferably at least about 50% (v/v) water;
(g) passing the diluted water/acetonitrile or diluted water/tetrahydrofuran solution; preferably, diluted water/acetonitrile solution, comprising purified flutemetamol of step (f) through a second reverse phase SPE cartridge and trapping the flutemetamol on said cartridge second reverse phase SPE cartridge;
(h) rinsing said second reverse phase SPE cartridge with water; and
(i) eluting the trapped purified flutemetamol from second reverse phase SPE cartridge with an injectable organic solvent; preferably, ethanol or DMSO; preferably with ethanol.
According to the invention, the purified flutemetamol can be collected after step (i).
The present invention also provides a purification process of the present invention, wherein the process is automated.
yr 2011 clip
Alzheimer’s disease (AD) is defined histologically by the presence of extracellular β-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles in the cerebral cortex. The diagnosis of dementia, along with the prediction of who will develop dementia, has been assisted by magnetic resonance imaging and positron emission tomography (PET) by using [18F]fluorodeoxyglucose (FDG). These techniques, however, are not specific for AD. Based on the chemistry of histologic staining dyes, several Aβ-specific positron-emitting radiotracers have been developed to image neuropathology of AD. Among these, [11C]PiB is the most studied Aβ-binding PET radiopharmaceutical in the world. The histologic and biochemical specificity of PiB binding across different regions of the AD brain was demonstrated by showing a direct correlation between Aβ-containing amyloid plaques and in vivo [11C]PiB retention measured by PET imaging. Because 11C is not ideal for commercialization, several 18F-labeled tracers have been developed. At this time, [18F]3′-F-PiB (Flutemetamol), 18F-AV-45 (Florbetapir), and 18F-AV-1 (Florbetaben) are undergoing extensive phase II and III clinical trials. This article provides a brief review of the amyloid biology and chemistry of Aβ-specific 11C and 18F-PET radiopharmaceuticals. Clinical trials have clearly documented that PET radiopharmaceuticals capable of assessing Aβ content in vivo in the brains of AD subjects and subjects with mild cognitive impairment will be important as diagnostic agents to detect in vivo amyloid brain pathology. In addition, PET amyloid imaging will also help test the amyloid cascade hypothesis of AD and as an aid to assess the efficacy of antiamyloid therapeutics currently under development in clinical trials.
LASTACAFT, ALCAFTADINE.. Drug Patent Expiration, 21st Nov 2013

ALCAFTADINE
Alcaftadine is used to prevent eye irritation brought on by allergic conjunctivitis. It is a H1histamine receptor antagonist.
It was approved by the U.S. Food and Drug Administration in 2010 under the trade name Lastacaft.
LASTACAFT, ALLERGAN
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| ALCAFTADINE | SOLUTION/DROPS; OPHTHALMIC | 0.25% | RX | 022134 | 001 |
Patents
There are 1 patent(s) protecting ALLERGAN’s LASTACAFT.
The last patent expires on 2013-11-21.
| Patent | Expiration | |
|---|---|---|
| US5468743 | Imidazo[2,1-b]benzazepine derivatives, compositions and method of use
The present invention is concerned with novel imidazo[2, 1-b][3]benzazepines of formula ##STR1## the pharmaceutically acceptable addition salts and stereochemically isomeric forms thereof, wherein each of the dotted lines independently represents an optional bond; R.sup.1 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.2 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.3 represents hydrogen, C.sub.1-4 alkyl, ethenyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, hydroxyC.sub.1-4 alkyl, formyl or hydroxycarbonyl; R.sup.4 represents hydrogen, C.sub.1-4 alkyl, hydroxyC.sub.1-4 alkyl, phenyl or halo; R.sup.5 represents hydrogen, C.sub.1-4 alkyl or halo; L represents hydrogen; C.sub.1-6 alkyl; C.sub.1-6 alkyl substituted with one substituent selected from the group consisting of hydroxy, halo, C.sub.1-4 alkyloxy, hydroxycarbonyl, C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyloxycarbonyl-C.sub.1-4 alkyloxy, hydroxycarbonylC.sub.1-4 alkyloxy, C.sub.1-4 alkyloxycarbonylamino, C.sub.1-4 alkylaminocarbonyl, C.sub.1-4 alkylaminocarbonylamino, C.sub.1-4 alkylaminothiocarbonylamino, aryl, aryloxy and arylcarbonyl; C.sub.1-6 alkyl substituted with both hydroxy and aryloxy; C.sub.3-6 alkenyl; C.sub.3-6 alkenyl substituted with aryl; or, L represents a radical of formula –Alk–Y–Het.sup.1 (a-1),–Alk–NH–CO–Het.sup.2 (a-2)or –Alk–Het.sup.3 (a-3); provided that 6,11-dihydro-11-(4-piperidinylidene)-5H-imidazo[2,1-b][3]benzazepine is ecxluded, which are useful antiallergic compounds.Compositions comprising said compounds, methods of using and processes for preparing the same.
|
2013-11-21 |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ALLERGAN.
Exclusivity ends on 2015-07-28.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2010-07-28 | 000 | Approval |

NovoEight (turoctocog alfa) Receives Approval from the FDA

Bagsværd, Denmark, 16 October 2013 – Today, Novo Nordisk announced that the U.S. Food and Drug Administration (FDA) has approved its Biologics License Application (BLA) for recombinant coagulation factor VIII, Novoeight.
The FDA approved Novoeight for use in adults and children with hemophilia A for:
- Control and prevention of bleeding
- Perioperative management
- Routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
http://www.drugs.com/newdrugs/novoeight-turoctocog-alfa-receives-approval-fda-3931.html

turoctocog alfa (NN7008)
old clip
Novo Nordisk recently announced the company has submitted the regulatory application to the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for its turoctocog alfa (NN7008) for prevention and treatment of bleeding in people with hemophilia A.
The decision for submission was based on results from the guardian trials consisting of over 200 people with hemophilia A, making guardian the largest pre-registration clinical trial program for hemophilia A. The trials contained previously treated adults and children with severe hemophilia A.
Turoctocog alfa is a third-generation recombinant coagulation factor VIII drug, designed to increase reliability, safety and portability for patients with hemophilia A.
“We are very excited about having reached this goal. Turoctocog alfa represents a new treatment alternative for people with hemophilia A and is one of the first important outcomes of the hemophilia research strategy we embarked upon in 2006,” Mads Krogsgaard Thomsen, executive vice president and chief science officer of Novo Nordisk said in a news release.
The company said that in the next few months, it plans to submit applications for regulatory approval in other countries as well.
Hemophilia A is estimated to affect 500,000 people worldwide, and is extremely under-diagnosed in developing countries.
ema clip
On 19 September 2013, the Committee for Medicinal Products for Human Use(CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product NovoEight 250, 500, 100, 1500, 2000 or 3000 IU, powder and solvent for solution for injection, intended for the treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor-VIII deficiency).The applicant for this medicinal product is Novonordisk. It may request are-examination of the CHMP opinion, provided it notifies the European Medicines Agency in writing of its intention within 15 days of receipt of the opinion.
The active substance of NovoEight is turoctocog alfa, human recombinant factor VIII that enables the temporary substitution of the endogenous coagulation factor VIII in haemophilia A patients. The benefits with NovoEight are its ability to prevent and treat the bleeds in previously treated patients with severe haemophilia A. The most common side effects are increase in hepatic enzymes and injection-site reaction.
A pharmacovigilance plan for NovoEight will be implemented as part of themarketing authorisation.
The approved indication is:
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor-VIII deficiency).
It is proposed that NovoEight be prescribed by physicians experienced in the treatment of haemophilia A. It is proposed that treatment should be initiated under the supervision of a doctor experienced in the treatment of haemophilia.
Detailed recommendations for the use of this product will be described in thesummary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
The CHMP, on the basis of quality, safety and efficacy data submitted, considers there to be a favourable benefit-to-risk balance for NovoEight and therefore recommends the granting of the marketing authorisation.
Monoclonal antibody (mAbs) 2013

2013——-29 monoclonal antibody (mAbs) drugs are in Phase III clinical development.
While around 350 therapeutic mAbs are currently in clinical development globally, only 28 had entered active Phase 2/3 or Phase 3 studies as of January 2013, Additionally one mAb mixture was under evaluation in Phase III.
Historically, mAbs that target antigens relevant to cancer have comprised approximately 50% of the mAb clinical pipeline,
but in 2013 the picture has changed: 66% or 19 of the antibodies to watch in 2013 are for non-cancer indications.

The non-cancer mAbs include alirocumab (Regeneron; Sanofi, hypercholesterinemia);
AMG 145 (Amgen, hypercholesterinemia),
epratuzumab (UCB, SLE),
gantenerumab (Roche; Alzheimer’s disease),
gevokizumab (Xoma/Servier, Non-infectious uveitis),
itolizumab (Biocon, Plaque psoriasis), ixekizumab (Eli Lilly and Co., psoriasis),
lebrikizumab (Roche/Genentech, rheumatoid arthritis),
mepolizumab (GSK, Asthma, COPD etc.),
ocrelizumab (Roche/Genentech, multiple sclerosis),
reslizumab (Teva, Eosinophilic asthma), romosozumab (Amgen, Postmenopausal osteoporosis),
sarilumab (Regeneron; Sanofi, rheumatoid arthritis),
secukinumab (Novartis, rheuma, psoriasis),
sirukumab (Janssen R&D LLC, rheumatoid arthritis),
solanezumab (Eli Lilly and Co., Alzheimer’s disease),
tabalumab (Eli Lilly and Co., rheuma, SLE)
and
vedolizumab (Millenium, Ulcerative colitis; Crohn disease).
The mixture of actoxumab and bezlotoxumab (MK-3415A, Merck & Co.) is being evaluated in two Phase 3 studies as a treatment for Clostridium difficile infection.
The ten cancer mAbs are:
elotuzumab (Bristol-Myers Squibb, Abbott, multiple myeloma),
farletuzumab (Morphotek, ovarian cancer),
inotuzumab ozogamicin (Pfizer; UCB, ALL, NHL),
naptumomab estafenatox (Active Biotech, renal cell carcinoma),
necitumumab (ImClone LLC, NSCL),
nivolumab (Bristol-Myers Squibb, NSCL, renal cell carcinoma),
obinutuzumab (Roche/Genetech, Diffuse large B cell lymphoma, CLL, NHL),
onartuzumab (Roche/Genetech, NSCL cancer; gastric cancer),
racotumomab (CIMAB; Laboratorio Elea S.A.C.I.F. y A, NSCL),
and ramucirumab (ImClone LLC, Gastric; liver, breast, colorectal, NSCL cancers).
