Home » Posts tagged 'world drug tracker'
Tag Archives: world drug tracker
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Myself, NIPER-G and NDTL Collaborate to Synthesize ‘Methandienone LTM’ for Global Anti-Doping Efforts
A proud moment for me [ ANTHONY MELVIN CRASTO ] as Scientific Advisor at Niper-G Dept Pharma Ministry of Chemicals and Fertilizers Govt of India 
Congrats team Niper-G and team National Dope Testing Lab Govt of India Prof. (Dr.) P. L. Sahu and Myself being a part of it as Scientific Advisor Niper-G for few years in Medicinal chem dept. Launched by Union minister Mansukh Mandaviya on 4th sept 2025 in Delhi
will be distributed across the world via World Anti-Doping agency WADA
Methandienone LTM is high purity rare reference material
Hope my interactions and guidance has given fruitful results. Thanks to Dr Murty for appointing me as advisor. Methandienone LTM will make India proud across the World 
A great achievement for India as nation and advanced capability demonstration
https://www.pib.gov.in/PressReleasePage.aspx?PRID=2163812
India Develops Rare Reference Material for Enhanced Anti-Doping Testing in Sports
NIPER Guwahati and NDTL Collaborate to Synthesize ‘Methandienone Long-Term Metabolite’ for Global Anti-Doping Efforts

//////////NIPER-G, NDTL, Methandienone LTM, Global Anti-Doping, INDIA
Myself Anthony Crasto up for grabs as Advisor API & INT, Chem.
Admin note for myself . I am up for Grabs
I myself Dr Anthony Melvin Crasto Looking for a post retirement assignment as Advisor API & INT, Chem.
With 36 yrs rich experience, about dozen patents, 10000plus steps covered, 200 API targets, 30 plus products commercialization in plant in full career. Hands on knowledge of Synthesis, Process, scaleup, cost reduction, DOE , softwares etc
Kindly contact me
Dr Anthony Melvin Crasto
+919321316780
amcrasto@gmail.com
About myself
Dr Anthony Crasto
click on my website to know about me
Read http://amcrasto.weebly.com/
Also http://amcrasto.weebly.com/awards.html
Also
http://amcrasto.weebly.com/felicitations.html
1000 lakh google hits, 100lakh blog views, 10 lakh viewers in USA alone, all in 7 continents, 226 countries, 30 Indian and International awards, helping millions across the world

‘Top 10 Prominent & Great Personalities of the year 2022’ campaign by Fame Finders. ANTHONY CRASTO


‘Top 10 Prominent & Great Personalities of the year 2022’ campaign by Fame Finders.
Feeling great to be selected in the ‘Top 10 Prominent & Great Personalities of the year 2022’ campaign by Fame Finders. It is an online campaign to honour inspiring personalities and feature them in the upcoming edition of the top news sites, including – Deccan Chronicle/Asian Age/Deccan Herald, ANI, Zee5, Latestly, Lokmat Times, DailyHunt, Google News, JioNews, MSN and 70+ sites.Jan 16 2023

[youtube https://www.youtube.com/watch?v=x98E01m9anw&w=560&h=315]

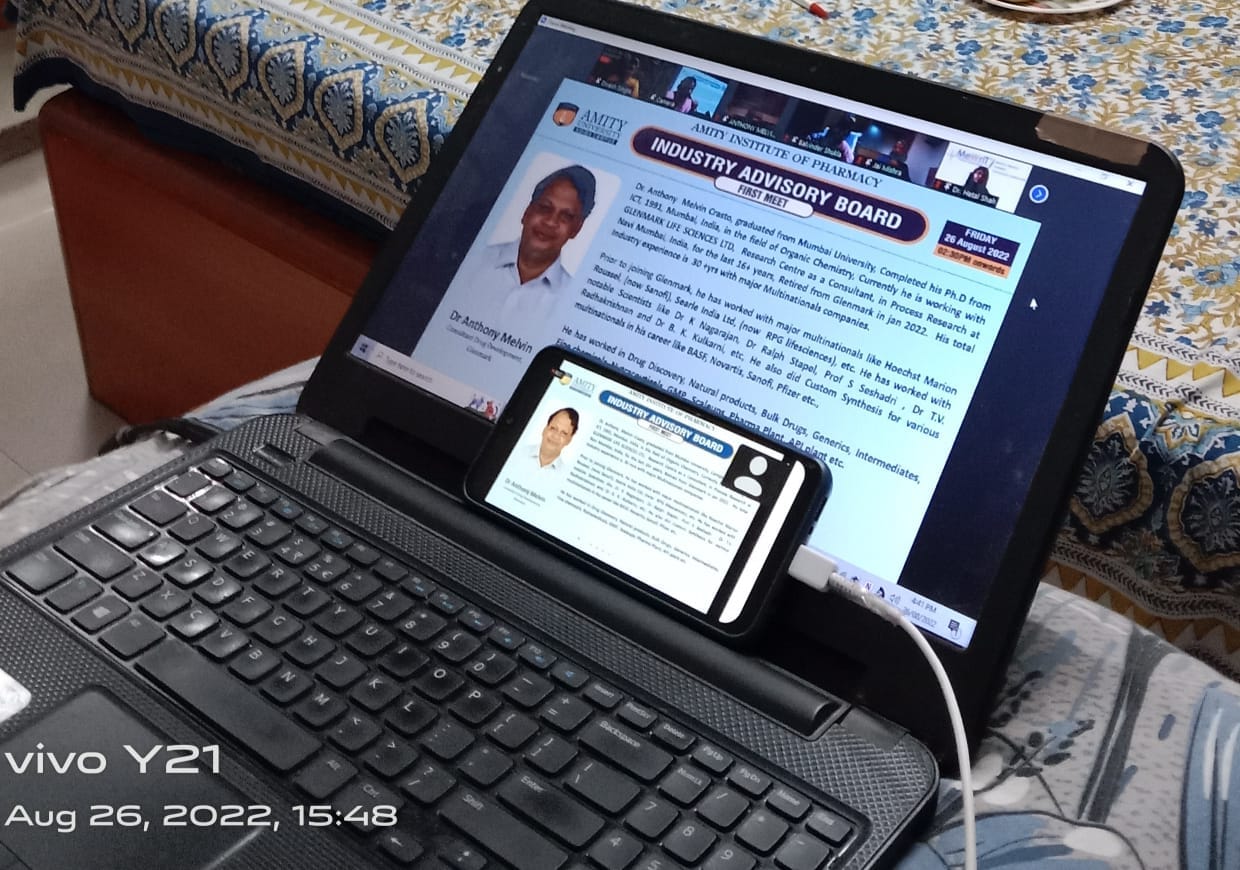
Chief Advisor Industry Advisory Board Amity University Noida, India. 26th Aug 2022

DR ANTHONY MELVIN CRASTO is Chief Advisor Industry Advisory Board Amity Univ. Noida, INDIA
Journey begins with online meet. 26th aug 2022
Service to education is service to humanity
Brochure for First Industry Advisory Board meeting at Amity Institute of Pharmacy is attached. Date 26th Aug 2022
it as hybrid-Online/offline.


Thnx
Dr Sandeep Arora
#amity
@amity #education #pharmacy
Amity University
Amity University, Greater Noida Campus


DIFLUPREDNATE

(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluor-11-hydroxy-3,20-dioxopregna-1,4-dien-17-ylbutyrat[German][ACD/IUPAC Name]
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate[ACD/IUPAC Name]
23674-86-4[RN]
245-815-4[EINECS]
2652
6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
DIFLUPREDNATE
CAS# 23674-86-4
- Molecular FormulaC27H34F2O7
- Average mass508.552 Da
- W 6309
- W-6309
- DFBA
- Difluoroprednisolone butyrate acetate
S8A06QG2QE
TU3831500
дифлупреднат[Russian][INN]
ديفلوبريدنات[Arabic][INN]
二氟泼尼酯[Chinese][INN]
(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate
23674-86-4[RN], 245-815-4[EINECS], 2652, 6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
Depositor-Supplied Patent Identifiers
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020325543-A1 | Diagnostic method | 2017-11-20 | |
| WO-2012088044-A2 | Compositions and methods for improving ocular surface health, corneal clarity, optical function and maintaining visual acuity | 2010-12-20 | |
| US-7790905-B2 | Pharmaceutical propylene glycol solvate compositions | 2002-02-15 | 2010-09-07 |
| US-7927613-B2 | Pharmaceutical co-crystal compositions | 2002-02-15 | 2011-04-19 |
PATENT
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A

SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A

SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).

SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).

PATENT
https://patents.google.com/patent/CN103509075A/en

Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
Embodiment 2:4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound)
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
Embodiment 3:3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters (formula V)
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
Embodiment 4:4, fluoro-17 α of 9 (11)-diene-6-, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters
10g3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters are dissolved in 60mL acetonitrile, and under nitrogen protection ,-4 ℃ are stirred half an hour.Slowly drip the acetonitrile suspension 40mL of Selecfluor in reaction flask, under nitrogen protection, react 2 hours, TLC (sherwood oil: ethyl acetate=3: 1) monitoring reaction, raw material reaction is complete.Stopped reaction, adds water in reaction flask, ethyl acetate extraction three times, saturated common salt water washing twice, anhydrous sodium sulfate drying.Vacuum concentration is removed organic solvent, obtain faint yellow solid 4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI), productive rate 85%. 1H-NMR(500MHz,CDC1 3):δ(ppm)5.90(1H,d,J=4.5Hz,4-H),5.59(1H,s,11-H),5.07(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.46(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),0.73(3H,s,19-CH 3).
Embodiment 5:4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII)
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
Embodiment 6:6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula VIII)
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
Embodiment 7:6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula IX)
14g 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in 500mL eggplant type bottle, adds 200mL acetone, stirs raw material is fully dissolved, and adds afterwards 3g Potassium ethanoate, is warming up to 60 ℃ of return stirring 13h.TLC (sherwood oil: ethyl acetate=2: 1) monitoring finds that new product occurs.Stop heating, in reaction solution, add water, ethyl acetate extraction, anhydrous sodium sulfate drying organic phase, after standing 30min, steams except organic solvent, obtains yellow oil, productive rate 96%.Column chromatography is purified, and obtains white solid powder, and nuclear-magnetism confirmation structure is 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester. 1H-NMR(300MHz,CDC1 3):δ(ppm)6.11(1H,d,J=4.5Hz,4-H),5.31(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),0.94(3H,s,18-CH 3),0.97(3H,t,J=7.5Hz),1.55(3H,s,19-CH 3),3.52(1H,s,11-H);ESI-MS?m/z:491.2[M+H +],513.2[M+Na +].
Embodiment 8:6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula X)
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
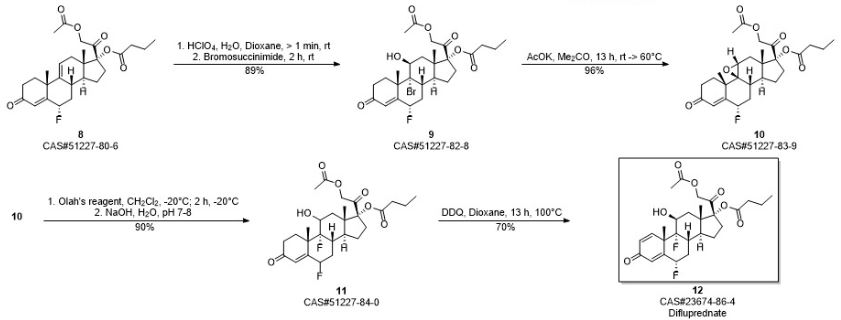
Embodiment 9:6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (difluprednate) (formula I)
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:

Patent
Publication numberPriority datePublication dateAssigneeTitle
US3780177A *1967-06-161973-12-18Warner Lambert Co17-butyrate,21-ester derivatives of 6alpha,9alpha-difluoroprednisolone,compositions and use
US4525303A *1982-06-211985-06-25Dainippon Ink And Chemicals Inc.Process for preparation of steroids
CN101397321A *2007-09-292009-04-01天津药业研究院有限公司Preparation of hydrocortisone and derivatives thereof
CN102076344A *2008-05-282011-05-25瓦利杜斯生物医药有限公司Non-hormonal steroid modulators of nf-kb for treatment of disease
CN102134266A *2010-12-302011-07-27北京市科益丰生物技术发展有限公司Preparation method of melengestrol acetate
Publication numberPriority datePublication dateAssigneeTitle
CN102964412A *2012-11-272013-03-13山东省医药工业研究所Novel crystal form and preparation method of difluprednate
CN103965277A *2014-05-192014-08-06中国科学院上海有机化学研究所Method for synthesizing difluprednate from sterol fermentation product
CN106632561A *2016-12-162017-05-10广州仁恒医药科技股份有限公司Method for preparing difluprednate
CN106749464A *2016-12-292017-05-31奥锐特药业有限公司Steroidal epoxide carries out open loop, the method for fluorination reaction and its device
CN107915766A *2016-10-112018-04-17江苏福锌雨医药科技有限公司A kind of preparation method of fludrocortison acetate
CN108503679A *2018-04-032018-09-07广州仁恒医药科技股份有限公司A kind of purification process of Difluprednate intermediate
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]

NEW DRUG APPROVALS
TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
References
- ^ “Sirion Therapeutics Announces FDA Approval of Durezol for Treatment of Postoperative Ocular Inflammation and Pain” (Press release). Sirion Therapeutics, Inc. 2008-06-24. Retrieved 2008-06-30.
- ^ Clinical trial number NCT00501579 for “Study of Difluprednate in the Treatment of Uveitis” at ClinicalTrials.gov
- ^ Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS (May 2014). “Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study”. Investigative Ophthalmology & Visual Science. 55 (5): 2993–3002. doi:10.1167/iovs.13-12660. PMC 4581692. PMID 24677110.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609025 |
| License data | US FDA: Difluprednate |
| Routes of administration | eye drops |
| ATC code | D07AC19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 23674-86-4 |
| PubChem CID | 32037 |
| DrugBank | DB06781 |
| ChemSpider | 391990 |
| UNII | S8A06QG2QE |
| KEGG | D01266 |
| ChEBI | CHEBI:31485 |
| ChEMBL | ChEMBL1201749 |
| CompTox Dashboard (EPA) | DTXSID0046773 |
| ECHA InfoCard | 100.041.636 |
| Chemical and physical data | |
| Formula | C27H34F2O7 |
| Molar mass | 508.559 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////DIFLUPREDNATE, W 6309, W-6309, DFBA, Difluoroprednisolone butyrate acetate, S8A06QG2QE, TU3831500, дифлупреднат , ديفلوبريدنات , 二氟泼尼酯 , OCCULAR, PAIN
CCCC(=O)OC1(CCC2C1(CC(C3(C2CC(C4=CC(=O)C=CC43C)F)F)O)C)C(=O)COC(=O)C
Anthony crasto is now Consultant Glenmark Lifesciences at Glenmark Life Sciences!

I’m happy to share that I’m starting a new position as Consultant Glenmark Lifesciences at Glenmark Life Sciences!
17th Jan 2022, A new innings
I retired 16th Jan 2022 at 58 yrs from Glenmark . completed 16 yrs 2 months
30 plus years in the field of Process research

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////////////
MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES

MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES
Service to education is service to humanity, Taking free education to the world, Every difficulty an opportunity,In the dark but yet in light,proud of my disabilty, Taking industry experience to students, See the ability not the disabilty, The only disability in life is a bad attitude !!!, Dreamt a billion when hit by million, Mentor to the world
![]() All my awards, https://lnkd.in/fGxx9VF
All my awards, https://lnkd.in/fGxx9VF
Click on above links
Sept 2021, This blog New Drug Approvals approaching 34 lakh views
NEW DRUG APPROVALS FAST APPROACING 34 LAKH VIEWS in 225 countries 7 continents. 5 lakh viewers in USA alone
#worlddrugtracker#worldpeaceambassador#india#helpingmillions#amcrasto#medchem#blog#education#service#free#divyang#everydifficultyanopportunity#opensuperstar#pharmadon#qbdking#ict#ictian#pharma


NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Anthony crasto’s blog New drug approvals touches 3 lakh views…….Helping millions

link is https://newdrugapprovals.org/
All about Drugs, live, by DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Helping millions, 7 million hits on google, pushing boundaries, one lakh plus connections worldwide, 3 lakh plus VIEWS on this blog in 193 countries

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Matthew A. J. Duncton† *
Renovis, Inc. (a wholly-owned subsidiary of Evotec AG), Two Corporate Drive, South San Francisco, CA 94080, United States. E-mail: mattduncton@yahoo.com; Tel: +1 917-345-3183
First published on the web 22nd August 2011
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
The addition of a radical to a heteroaromatic base is commonly referred to as a Minsici reaction. Such reactions constitute a broad-set of selective CH-functionalization processes. This review describes some of the major applications of Minisci reactions and related processes to medicinal or biological chemistry, and highlights some potential developments within this area.
Introduction
The aim of this review is to summarize the use of Minisci reactions within medicinal chemistry, and to highlight some future opportunities to continue progression of this chemistry. As such, it is not an aim that detailed mechanistic information, or a comprehensive list of examples be described. For this, the reader is directed to excellent articles from Minisci, Harrowven and Bowman.1–3 Rather, the review is written to show that Minisci reactions are extremely valuable CH-functionalization processes within medicinal chemistry. However, their use has been somewhat under-utilized when compared with other well-known selective transformations (e.g. palladium-catalysed cross-couplings). Therefore, it is hoped that in the future, Minisci chemistry will continue to develop, such that the reactions become a staple-set of methods for medicinal and biological chemists alike.
To aid discussion, the review is divided in to several sections. First, some historical perspective is given. This is followed by a discussion of scope and limitations. The main-body of the review describes some specific examples of Minisci reactions and related processes, with a focus on their use within medicinal, or biological chemistry. Finally, brief mention is given to potential future applications, some of which may be beneficial in providing ‘high-content’ diverse libraries for screening.
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
…………………….
WIKI
The Minisci reaction is a named reaction in organic chemistry. It is a radical substitution to an aromatic compound, in particular to a heteroaromatic base, that introduces an alkyl group. The reaction was published about in 1971 by F. Minisci.[1] The aromatic compound is generally electron-deficient and with N-aromatic compounds the nitrogen atom is protonated.[2] A typical reaction is that between pyridine and pivalic acid to 2-tert-butylpyridine with silver nitrate, sulfuric acid and ammonium persulfate. The reaction resembles Friedel-Crafts alkylation but with opposite reactivity and selectivity.[3]
The Minisci reaction proceeds regioselectively and enables the introduction of a wide range of alkyl groups.[4] A side-reaction is acylation.[5] The ratio between alkylation and acylation depends on the substrate and the reaction conditions. Due to the simple raw materials and the simple reaction conditions the reaction has many applications in heterocyclic chemistry.[6][7]

Mechanism
A free radical is formed from the carboxylic acid in an oxidative decarboxylation with silver salts and an oxidizing agent. The oxidizing agent reoxidizes the silver salt. The radical then reacts with the aromatic compound. The ultimate product is formed by rearomatisation. The acylated product is formed from the acyl radical.[4][5]

References
- F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo: Nucleophilic character of alkyl radicals—VI : A new convenient selective alkylation of heteroaromatic bases, in: Tetrahedron 1971, 27, 3575–3579.
- Minisci reaction Jie Jack Li in Name Reactions 2009, 361-362, doi:10.1007/978-3-642-01053-8_163
- Strategic applications of named reactions in organic synthesis: background and detailed mechanisms László Kürti, Barbara Czakó 2005
- F. Fontana, F. Minisci, M. C. N. Barbosa, E. Vismara: Homolytic acylation of protonated pyridines and pyrazines with α-keto acids: the problem of monoacylation, in: J. Org. Chem. 1991, 56, 2866–2869; doi:10.1021/jo00008a050.
- M.-L. Bennasar, T. Roca, R. Griera, J. Bosch: Generation and Intermolecular Reactions of 2-Indolylacyl Radicals, in: Org. Lett. 2001, 3, 1697–1700; doi:10.1021/ol0100576.
- P. B. Palde, B. R. McNaughton, N. T. Ross, P. C. Gareiss, C. R. Mace, R. C. Spitale, B. L. Miller: Single-Step Synthesis of Functional Organic Receptors via a Tridirectional Minisci Reaction, in: Synthesis 2007, 15, 2287–2290; doi:10.1055/s-2007-983792.
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.

{kind=link}
{kind=link}