







Home » Posts tagged 'drugs'
Tag Archives: drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Myself Anthony Crasto up for grabs as Advisor API & INT, Chem.
Admin note for myself . I am up for Grabs
I myself Dr Anthony Melvin Crasto Looking for a post retirement assignment as Advisor API & INT, Chem.
With 36 yrs rich experience, about dozen patents, 10000plus steps covered, 200 API targets, 30 plus products commercialization in plant in full career. Hands on knowledge of Synthesis, Process, scaleup, cost reduction, DOE , softwares etc
Kindly contact me
Dr Anthony Melvin Crasto
+919321316780
amcrasto@gmail.com
About myself
Dr Anthony Crasto
click on my website to know about me
Read http://amcrasto.weebly.com/
Also http://amcrasto.weebly.com/awards.html
Also
http://amcrasto.weebly.com/felicitations.html
1000 lakh google hits, 100lakh blog views, 10 lakh viewers in USA alone, all in 7 continents, 226 countries, 30 Indian and International awards, helping millions across the world

DIFLUPREDNATE

(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluor-11-hydroxy-3,20-dioxopregna-1,4-dien-17-ylbutyrat[German][ACD/IUPAC Name]
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate[ACD/IUPAC Name]
23674-86-4[RN]
245-815-4[EINECS]
2652
6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
DIFLUPREDNATE
CAS# 23674-86-4
- Molecular FormulaC27H34F2O7
- Average mass508.552 Da
- W 6309
- W-6309
- DFBA
- Difluoroprednisolone butyrate acetate
S8A06QG2QE
TU3831500
дифлупреднат[Russian][INN]
ديفلوبريدنات[Arabic][INN]
二氟泼尼酯[Chinese][INN]
(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate
23674-86-4[RN], 245-815-4[EINECS], 2652, 6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
Depositor-Supplied Patent Identifiers
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020325543-A1 | Diagnostic method | 2017-11-20 | |
| WO-2012088044-A2 | Compositions and methods for improving ocular surface health, corneal clarity, optical function and maintaining visual acuity | 2010-12-20 | |
| US-7790905-B2 | Pharmaceutical propylene glycol solvate compositions | 2002-02-15 | 2010-09-07 |
| US-7927613-B2 | Pharmaceutical co-crystal compositions | 2002-02-15 | 2011-04-19 |
PATENT
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A

SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A

SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).

SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).

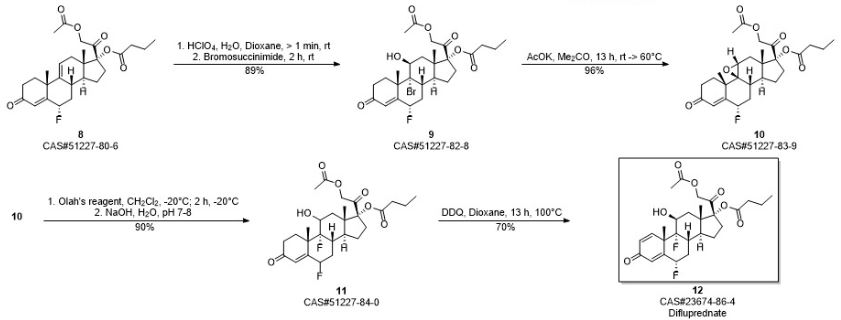
PATENT
https://patents.google.com/patent/CN103509075A/en

Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
Embodiment 2:4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound)
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
Embodiment 3:3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters (formula V)
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
Embodiment 4:4, fluoro-17 α of 9 (11)-diene-6-, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters
10g3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters are dissolved in 60mL acetonitrile, and under nitrogen protection ,-4 ℃ are stirred half an hour.Slowly drip the acetonitrile suspension 40mL of Selecfluor in reaction flask, under nitrogen protection, react 2 hours, TLC (sherwood oil: ethyl acetate=3: 1) monitoring reaction, raw material reaction is complete.Stopped reaction, adds water in reaction flask, ethyl acetate extraction three times, saturated common salt water washing twice, anhydrous sodium sulfate drying.Vacuum concentration is removed organic solvent, obtain faint yellow solid 4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI), productive rate 85%. 1H-NMR(500MHz,CDC1 3):δ(ppm)5.90(1H,d,J=4.5Hz,4-H),5.59(1H,s,11-H),5.07(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.46(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),0.73(3H,s,19-CH 3).
Embodiment 5:4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII)
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
Embodiment 6:6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula VIII)
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
Embodiment 7:6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula IX)
14g 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in 500mL eggplant type bottle, adds 200mL acetone, stirs raw material is fully dissolved, and adds afterwards 3g Potassium ethanoate, is warming up to 60 ℃ of return stirring 13h.TLC (sherwood oil: ethyl acetate=2: 1) monitoring finds that new product occurs.Stop heating, in reaction solution, add water, ethyl acetate extraction, anhydrous sodium sulfate drying organic phase, after standing 30min, steams except organic solvent, obtains yellow oil, productive rate 96%.Column chromatography is purified, and obtains white solid powder, and nuclear-magnetism confirmation structure is 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester. 1H-NMR(300MHz,CDC1 3):δ(ppm)6.11(1H,d,J=4.5Hz,4-H),5.31(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),0.94(3H,s,18-CH 3),0.97(3H,t,J=7.5Hz),1.55(3H,s,19-CH 3),3.52(1H,s,11-H);ESI-MS?m/z:491.2[M+H +],513.2[M+Na +].
Embodiment 8:6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula X)
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
Embodiment 9:6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (difluprednate) (formula I)
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:

Patent
Publication numberPriority datePublication dateAssigneeTitle
US3780177A *1967-06-161973-12-18Warner Lambert Co17-butyrate,21-ester derivatives of 6alpha,9alpha-difluoroprednisolone,compositions and use
US4525303A *1982-06-211985-06-25Dainippon Ink And Chemicals Inc.Process for preparation of steroids
CN101397321A *2007-09-292009-04-01天津药业研究院有限公司Preparation of hydrocortisone and derivatives thereof
CN102076344A *2008-05-282011-05-25瓦利杜斯生物医药有限公司Non-hormonal steroid modulators of nf-kb for treatment of disease
CN102134266A *2010-12-302011-07-27北京市科益丰生物技术发展有限公司Preparation method of melengestrol acetate
Publication numberPriority datePublication dateAssigneeTitle
CN102964412A *2012-11-272013-03-13山东省医药工业研究所Novel crystal form and preparation method of difluprednate
CN103965277A *2014-05-192014-08-06中国科学院上海有机化学研究所Method for synthesizing difluprednate from sterol fermentation product
CN106632561A *2016-12-162017-05-10广州仁恒医药科技股份有限公司Method for preparing difluprednate
CN106749464A *2016-12-292017-05-31奥锐特药业有限公司Steroidal epoxide carries out open loop, the method for fluorination reaction and its device
CN107915766A *2016-10-112018-04-17江苏福锌雨医药科技有限公司A kind of preparation method of fludrocortison acetate
CN108503679A *2018-04-032018-09-07广州仁恒医药科技股份有限公司A kind of purification process of Difluprednate intermediate
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]

NEW DRUG APPROVALS
TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
References
- ^ “Sirion Therapeutics Announces FDA Approval of Durezol for Treatment of Postoperative Ocular Inflammation and Pain” (Press release). Sirion Therapeutics, Inc. 2008-06-24. Retrieved 2008-06-30.
- ^ Clinical trial number NCT00501579 for “Study of Difluprednate in the Treatment of Uveitis” at ClinicalTrials.gov
- ^ Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS (May 2014). “Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study”. Investigative Ophthalmology & Visual Science. 55 (5): 2993–3002. doi:10.1167/iovs.13-12660. PMC 4581692. PMID 24677110.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609025 |
| License data | US FDA: Difluprednate |
| Routes of administration | eye drops |
| ATC code | D07AC19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 23674-86-4 |
| PubChem CID | 32037 |
| DrugBank | DB06781 |
| ChemSpider | 391990 |
| UNII | S8A06QG2QE |
| KEGG | D01266 |
| ChEBI | CHEBI:31485 |
| ChEMBL | ChEMBL1201749 |
| CompTox Dashboard (EPA) | DTXSID0046773 |
| ECHA InfoCard | 100.041.636 |
| Chemical and physical data | |
| Formula | C27H34F2O7 |
| Molar mass | 508.559 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////DIFLUPREDNATE, W 6309, W-6309, DFBA, Difluoroprednisolone butyrate acetate, S8A06QG2QE, TU3831500, дифлупреднат , ديفلوبريدنات , 二氟泼尼酯 , OCCULAR, PAIN
CCCC(=O)OC1(CCC2C1(CC(C3(C2CC(C4=CC(=O)C=CC43C)F)F)O)C)C(=O)COC(=O)C
Anthony crasto is now Consultant Glenmark Lifesciences at Glenmark Life Sciences!

I’m happy to share that I’m starting a new position as Consultant Glenmark Lifesciences at Glenmark Life Sciences!
17th Jan 2022, A new innings
I retired 16th Jan 2022 at 58 yrs from Glenmark . completed 16 yrs 2 months
30 plus years in the field of Process research

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////////////
Sreeni Labs Private Limited, Hyderabad, India ready to deliver New, Economical, Scalable Routes to your advanced intermediates & API’s in early Clinical Drug Development Stages

Sreeni Labs Private Limited, Hyderabad, India is ready to take up challenging synthesis projects from your preclinical and clinical development and supply from few grams to multi-kilo quantities. Sreeni Labs has proven route scouting ability to design and develop innovative, cost effective, scalable routes by using readily available and inexpensive starting materials. The selected route will be further developed into a robust process and demonstrate on kilo gram scale and produce 100’s of kilos of in a relatively short time.
Accelerate your early development at competitive price by taking your route selection, process development and material supply challenges (gram scale to kilogram scale) to Sreeni Labs…………
INTRODUCTION
Sreeni Labs based in Hyderabad, India is working with various global customers and solving variety of challenging synthesis problems. Their customer base ranges from USA, Canada, India and Europe. Sreeni labs Managing Director, Dr. Sreenivasa Reddy Mundla has worked at Procter & Gamble Pharmaceuticals and Eli Lilly based in USA.
The main strength of Sreeni Labs is in the design, development of innovative and highly economical synthetic routes and development of a selected route into a robust process followed by production of quality product from 100 grams to 100s of kg scale. Sreeni Labs main motto is adding value in everything they do.
They have helped number of customers from virtual biotech, big pharma, specialty chemicals, catalog companies, and academic researchers and drug developers, solar energy researchers at universities and institutions by successfully developing highly economical and simple chemistry routes to number of products that were made either by very lengthy synthetic routes or by using highly dangerous reagents and Suzuki coupling steps. They are able to supply materials from gram scale to multi kilo scale in a relatively short time by developing very short and efficient synthetic routes to a number of advanced intermediates, specialty chemicals, APIs and reference compounds. They also helped customers by drastically reducing number of steps, telescoping few steps into a single pot. For some projects, Sreeni Labs was able to develop simple chemistry and avoided use of palladium & expensive ligands. They always begin the project with end in the mind and design simple chemistry and also use readily available or easy to prepare starting materials in their design of synthetic routes
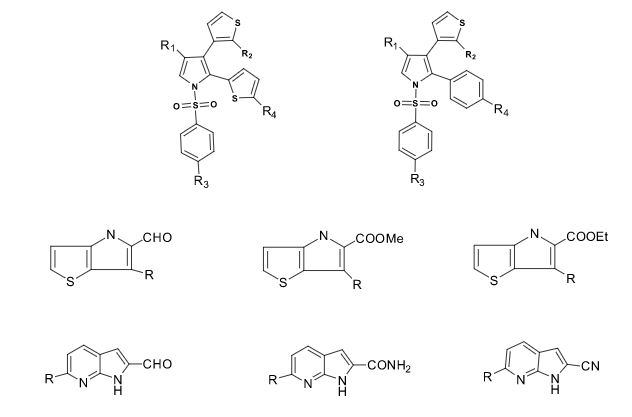
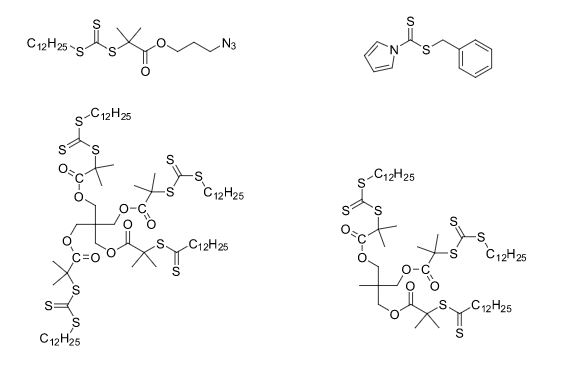
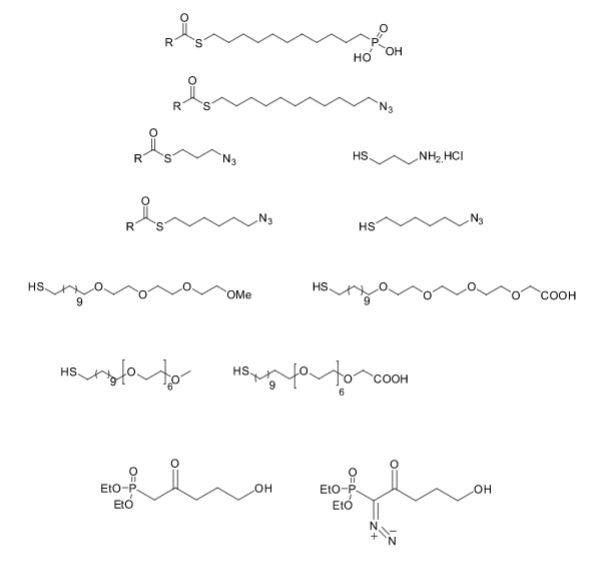
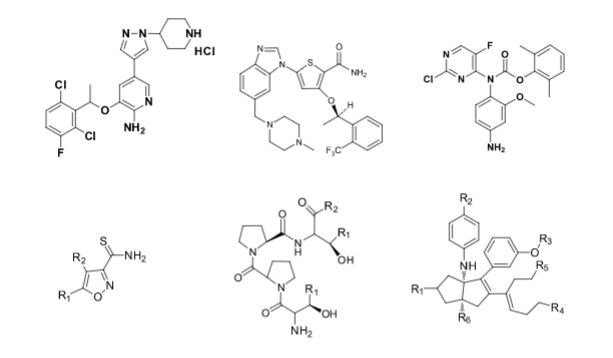
Over the years, Sreeni labs has successfully made a variety of products ranging from few mg to several kilogram scale. Sreeni labs has plenty of experience in making small select libraries of compounds, carbocyclic compounds like complex terpenoids, retinal derivatives, alkaloids, and heterocyclic compounds like multi substituted beta carbolines, pyridines, quinolines, quinolones, imidazoles, aminoimidazoles, quinoxalines, indoles, benzimidazoles, thiazoles, oxazoles, isoxazoles, carbazoles, benzothiazoles, azapines, benzazpines, natural and unnatural aminoacids, tetrapeptides, substituted oligomers of thiophenes and fused thiophenes, RAFT reagents, isocyanates, variety of ligands, heteroaryl, biaryl, triaryl compounds, process impurities and metabolites.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They can also take up custom synthesis and scale up of medchem analogues and building blocks. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving couple of PO based (fee for service) projects.
Some of the compounds prepared by Sreeni labs;



See presentation below
LINK ON SLIDESHARE
Sreeni Labs Profile from Sreenivasa Reddy
Managing Director at Sreeni Labs Private Limited\
Few Case Studies : Source SEEENI LABS
QUOTE………….
One virtual biotech company customer from USA, through a common friend approached Sreeni Labs and told that they are buying a tetrapeptide from Bachem on mg scale at a very high price and requested us to see if we can make 5g. We accepted the challenge and developed solution phase chemistry and delivered 6g and also the process procedures in 10 weeks time. The customer told that they are using same procedures with very minor modifications and produced the tetrapeptide ip to 100kg scale as the molecule is in Phase III.
One East coast customer in our first meeting told that they are working with 4 CROs of which two are in India and two are in China and politely asked why they should work with Sreeni Labs. We told that give us a project where your CROs failed to deliver and we will give a quote and work on it. You pay us only if we deliver and you satisfy with the data. They immediately gave us a project to make 1.5g and we delivered 2g product in 9 weeks. After receiving product and the data, the customer was extremely happy as their previous CRO couldn’t deliver even a milligram in four months with 3 FTEs.
One Midwest biotech company was struggling to remove palladium from final API as they were doing a Suzuki coupling with a very expensive aryl pinacol borane and bromo pyridine derivative with an expensive ligand and relatively large amount of palldium acetate. The cost of final step catalyst, ligand and the palladium scavenging resin were making the project not viable even though the product is generating excellent data in the clinic. At this point we signed an FTE agreement with them and in four months time, we were able to design and develop a non suzuki route based on acid base chemistry and made 15g of API and compared the analytical data and purity with the Suzuki route API. This solved all three problems and the customer was very pleased with the outcome.
One big pharma customer from east coast, wrote a structure of chemical intermediate on a paper napkin in our first meeting and asked us to see if we can make it. We told that we can make it and in less than 3 weeks time we made a gram sample and shared the analytical data. The customer was very pleased and asked us to make 500g. We delivered in 4 weeks and in the next three months we supplied 25kg of the same product.
Through a common friend reference, a European customer from a an academic institute, sent us an email requesting us to quote for 20mg of a compound with compound number mentioned in J. med. chem. paper. It is a polycyclic compound with four contiguous stereogenic centers. We gave a quote and delivered 35 mg of product with full analytical data which was more pure than the published in literature. Later on we made 8g and 6g of the same product.
One West coast customer approached us through a common friend’s reference and told that they need to improve the chemistry of an advanced intermediate for their next campaign. At that time they are planning to make 15kg of that intermediate and purchased 50kg of starting raw material for $250,000. They also put five FTEs at a CRO for 5 months to optimize the remaining 5 steps wherein they are using LAH, Sodium azide, palladium catalyst and a column chromatography. We requested the customer not to purchase the 50kg raw material, and offered that we will make the 15kg for the price of raw material through a new route in less than three months time. You pay us only after we deliver 15 kg material. The customer didn’t want to take a chance with their timeline as they didn’t work with us before but requested us to develop the chemistry. In 7 weeks time, we developed a very simple four step route for their advanced intermediate and made 50g. We used very inexpensive and readily available starting material. Our route gave three solid intermediates and completely eliminated chromatographic purifications.
One of my former colleague introduced an academic group in midwest and brought us a medchem project requiring synthesis of 65 challenging polyene compounds on 100mg scale. We designed synthetic routes and successfully prepared 60 compounds in a 15 month time.
UNQUOTE…………
The man behind Seeni labs is Dr. Sreenivasa Reddy Mundla

Dr. Sreenivasa Reddy Mundla.
Managing Director at Sreeni Labs Private Limited
Sreeni Labs Private Limited
Road No:12, Plot No:24,25,26
- IDA, Nacharam
Hyderabad, 500076
Telangana State, India
Links
LINKEDIN https://in.linkedin.com/in/sreenivasa-reddy-10b5876
FACEBOOK https://www.facebook.com/sreenivasa.mundla
RESEARCHGATE https://www.researchgate.net/profile/Sreenivasa_Mundla/info
EMAIL mundlasr@hotmail.com, Info@sreenilabs.com, Sreeni@sreenilabs.com
Dr. Sreenivasa Reddy Mundla

Dr. M. Sreenivasa Reddy obtained Ph.D from University of Hyderabad under the direction Prof Professor Goverdhan Mehta in 1992. From 1992-1994, he was a post doctoral fellow at University of Wisconsin in Professor Jame Cook’s lab. From 1994 to 2000, worked at Chemical process R&D at Procter & Gamble Pharmaceuticals (P&G). From 2001 to 2007 worked at Global Chemical Process R&D at Eli Lilly and Company in Indianapolis.
In 2007 resigned to his job and founded Sreeni Labs based in Hyderabad, Telangana, India and started working with various global customers and solving various challenging synthesis problems.
The main strength of Sreeni Labs is in the design, development of a novel chemical route and its development into a robust process followed by production of quality product from 100 grams to 100’s of kg scale.
They have helped number of customers by successfully developing highly economical simple chemistry routes to number of products that were made by Suzuki coupling. they are able to shorten the route by drastically reducing number of steps, avoiding use of palladium & expensive ligands. they always use readily available or easy to prepare starting materials in their design of synthetic routes.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving PO based projects
Experience
Founder & Managing Director
Sreeni Labs Private Limited
August 2007 – Present (8 years 11 months)
Sreeni Labs Profile
Principal Research Scientist
Eli Lilly and Company
March 2001 – August 2007 (6 years 6 months)
Senior Research Scientist
Procter & Gamble
July 1994 – February 2001 (6 years 8 months)
Education
University of Hyderabad
Doctor of Philosophy (Ph.D.),
1986 – 1992

With Sreenivasa Mundla, Narahara sastry, Ram Kishan Rao, Jagadeesh Bharatam, Jagadish Gunjur and Jagadish Bharatham.
PUBLICATIONS
Jianye Zhang · Zhiqian Dong · Sreenivasa Reddy Mundla · X Eric Hu · William Seibel ·Ruben Papoian · Krzysztof Palczewski · Marcin Golczak
Article: ChemInform Abstract: Regioselective Synthesis of 4Halo ortho-Dinitrobenzene Derivative
Aug 2010 · ChemInform
Hong-yu Li · William T. McMillen · Charles R. Heap · Denis J. McCann · Lei Yan · Robert M. Campbell · Sreenivasa R. Mundla · Chi-Hsin R. King · Elizabeth A. Dierks · Bryan D. Anderson · Karen S. Britt · Karen L. Huss
Apr 2008 · Journal of Medicinal Chemistry
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Feb 2008 · ChemInform
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Nov 2007 · Tetrahedron
Hong-yu Li · Yan Wang · Charles R Heap · Chi-Hsin R King · Sreenivasa R Mundla · Matthew Voss · David K Clawson · Lei Yan · Robert M Campbell · Bryan D Anderson · Jill R Wagner ·Karen Britt · Ku X Lu · William T McMillen · Jonathan M Yingling
Apr 2006 · Journal of Medicinal Chemistry

Hui Cao · Sreenivasa R. Mundla · James M. Cook
Aug 2003 · Tetrahedron Letters
Article: ChemInform Abstract: A New Method for the Synthesis of 2,6-Dinitro and 2Halo6-nitrostyrenes
Nov 2000 · ChemInform
Article: ChemInform Abstract: A Novel Method for the Efficient Synthesis of 2-Arylamino-2-imidazolines
-
Nov 2000 · ChemInformAug 2000 · Tetrahedron LettersAug 2000 · Tetrahedron Letters


TGF-β inhibitors
The present invention provides 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl) -5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole monohydrate, i.e., Formula I.

EXAMPLE 1 Preparation of 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl-5,6-dihydro-4H -pyrrolo[1,2-b]pyrazole monohydrate

Galunisertib
1H NMR (CDCl3): δ=9.0 ppm (d, 4.4 Hz, 1H); 8.23-8.19 ppm (m, 2H); 8.315 ppm (dd, 1.9 Hz, 8.9 Hz, 1H); 7.455 ppm (d, 4.4 Hz, 1H); 7.364 ppm (t, 7.7 Hz, 1H); 7.086 ppm (d, 8.0 Hz, 1H); 6.969 ppm (d, 7.7 Hz, 1H); 6.022 ppm (m, 1H); 5.497 ppm (m, 1H); 4.419 ppm (t, 7.3 Hz, 2H); 2.999 ppm (m, 2H); 2.770 ppm (p, 7.2 Hz, 7.4 Hz, 2H); 2.306 ppm (s, 3H); 1.817 ppm (m, 2H). MS ES+: 370.2; Exact: 369.16

ABOVE MOLECULE IS
https://newdrugapprovals.org/2016/05/04/galunisertib/
Galunisertib
Phase III
A TGF-beta receptor type-1 inhibitor potentially for the treatment of myelodysplastic syndrome (MDS) and solid tumours.
LY-2157299
CAS No.700874-72-2
READ MY PRESENTATION ON
KEYWORDS Sreenivasa Mundla Reddy, Managing Director, Sreeni Labs Private Limited, Hyderabad, Telangana, India, new, economical, scalable routes, early clinical drug development stages, Custom synthesis, custom manufacturing, drug discovery, PHASE 1, PHASE 2, PHASE 3, API, drugs, medicines
WCK 2349 in phase II trials by Wockhardt
 . CH3SO3H
. CH3SO3HWCK 2349
Cas 948895-94-1 methane sulfonate
Base..706809-20-3
527.563., C22 H26 F N3 O5 . C H4 O3 S
8-[4-(L-Alanyloxy)piperidin-1-yl]-9-fluoro-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic acid methanesulfonate
S-(-)-9-fluoro-6,7-dihydro-8-(4-L-alaninyloxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonate
(2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt
Wockhardt Research Centre INNOVATOR

Oral broad-spectrum antibiotic
WO 2000068229, WO 2002009758, WO 2007102061, WO 2008053295, Indian (2015), IN 267210 , IN 2008MU00864,
Shetty, N.M.; Nandanwar, M.B.; Kamalavenkatesh, P.; et al.
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5 (S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic acid of structural Formula I can be used to treat bacterial Gram-positive, Gram-negative and anaerobic infections; especially infections caused by resistant Gram-positive organism and Gram-negative organism, mycobacterial infections and emerging nosocomial pathogen infections.
Formula I
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
PATENT
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en

Scheme 1
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC
PATENT
WO 2000068229
A S-(-)-optically pure benzoquinolizine carboxylic acid, its derivatives, its pharmaceutically acceptable salts, derivatives, pseudopolymorphs, polymorphs or hydrates thereof of formula I,

Formula I
Patent
WO 2011101710
PATENT
The tablets may optionally be coated with film forming agents and/or pharmaceutically acceptable excipients. Particularly suitable for use are commercially available coating compositions comprising film-forming polymers marketed under various trade names, such as Opadry® and Eudragit®. The coating layers over the tablet may be applied as solution/dispersion of coating ingredients using conventional techniques known in the art.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1

Procedure: The compound of Formula I or pharmaceutically acceptable salts, esters or products thereof, lactose and croscannellose sodium were sifted and dry mixed in a rapid mixer granulator. The above mass was granulated by spraying aqueous solution of povidone. The granules were dried in a fluidized bed drier, sifted and oversize granules were milled in a Quadra mill. The resultant granules were mixed with talc, croscarmellose sodium, microcrystalline cellulose and sodium stearyl fumarate in a double cone blender. The lubricated granules were compressed into tablets using suitable tooling. Tablets were coated with aqueous dispersion of opadry.
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data

//////////////////////////////

aChemical name: S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate. bChemical name: S-(–)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3, 3-dimethylpiperidin-1-yl)-1,4 dihydro-4-oxo-quinoline-3-carboxylic acid hydrochloride monohydrate. cChemical name: R-(+)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3,3-dimethylpiperidin-1-yl)-1,4 dihydro-4-oxo-quinoline-3-carboxylic acid hydrochloride monohydrate.

31 Aug, 2014,
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
Read more at:
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
Levonadifloxacin arginine salt, WCK 771
RN: 306748-89-0
RN: 306748-89-0
-
C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
-
L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)

WCK 771………..S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate
(-)-9-Fluoro-8-(4-hydroxypiperidin-1-yl)-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic acid L-arginine salt hydrate
L-arginine salt of (S)-nadifloxacin
A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus
J Med Chem 2005, 48(16): 5232
J Med Chem 2005, 48(16): 5232
CN 102532131, WO 2005023805, WO 2002009758, WO 2001085095, WO 2000068229
| WO1991012815A1 * | Feb 25, 1991 | Sep 5, 1991 | Squibb Bristol Myers Co | COMPOSITIONS AND METHODS FOR TREATING INFECTIONS CAUSED BY ORGANISMS SENSITIVE TO β-LACTAM ANTIBIOTICS |
| WO2000068229A2 * | May 8, 2000 | Nov 16, 2000 | S K Agarwal | (s)-benzoquinolizine carboxylic acids and their use as antibacterial agents |
| WO2001085095A2 * | May 3, 2001 | Nov 15, 2001 | Shiv Kumar Agarwal | Chiral fluoroquinolizinone arginine salt forms |
| WO2002009758A2 * | Jul 31, 2001 | Feb 7, 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| EP2062582A1 * | Aug 14, 2007 | May 27, 2009 | Tianjin Hemey Bio-Tech Co., Ltd. | The antibiotics composition comprising beta-lactam antibiotics and buffers |
| US4524073 * | Jul 20, 1983 | Jun 18, 1985 | Beecham Group P.1.C. | β-Lactam compounds |
| US6465428 * | Aug 25, 2000 | Oct 15, 2002 | Aventis Pharma S.A. | Pharmaceutical combinations based on dalfopristine and quinupristine, and on cefepime |
| US20040254381 * | Aug 15, 2003 | Dec 16, 2004 | Day Richard A. | Antibiotic compositions and methods of using the same |
| US20050148571 * | Nov 29, 2002 | Jul 7, 2005 | Nancy Niconovich | Method of treating bacterial infections using gemifloxacin or a salt thereof and a betha-Lactam antibiotic |
| US20090148512 * | Apr 17, 2008 | Jun 11, 2009 | Lannett Co Inc | Novel uses of chloramphenicol and analogous thereof |
| US20090232744 * | Feb 26, 2009 | Sep 17, 2009 | Pari Pharma Gmbh | Macrolide compositions having improved taste and stability |
| WO2002009758A2 * | 31 Jul 2001 | 7 Feb 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| US6750224 | 17 Aug 2000 | 15 Jun 2004 | Wockhardt Limited | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

///////////keywords USFDA, Qualified Infectious Disease Product status, Wockhardt, drugs, WCK 2349, QIDP
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
Etelcalcetide, AMG 416, KAI-4169, velcalcetide
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2

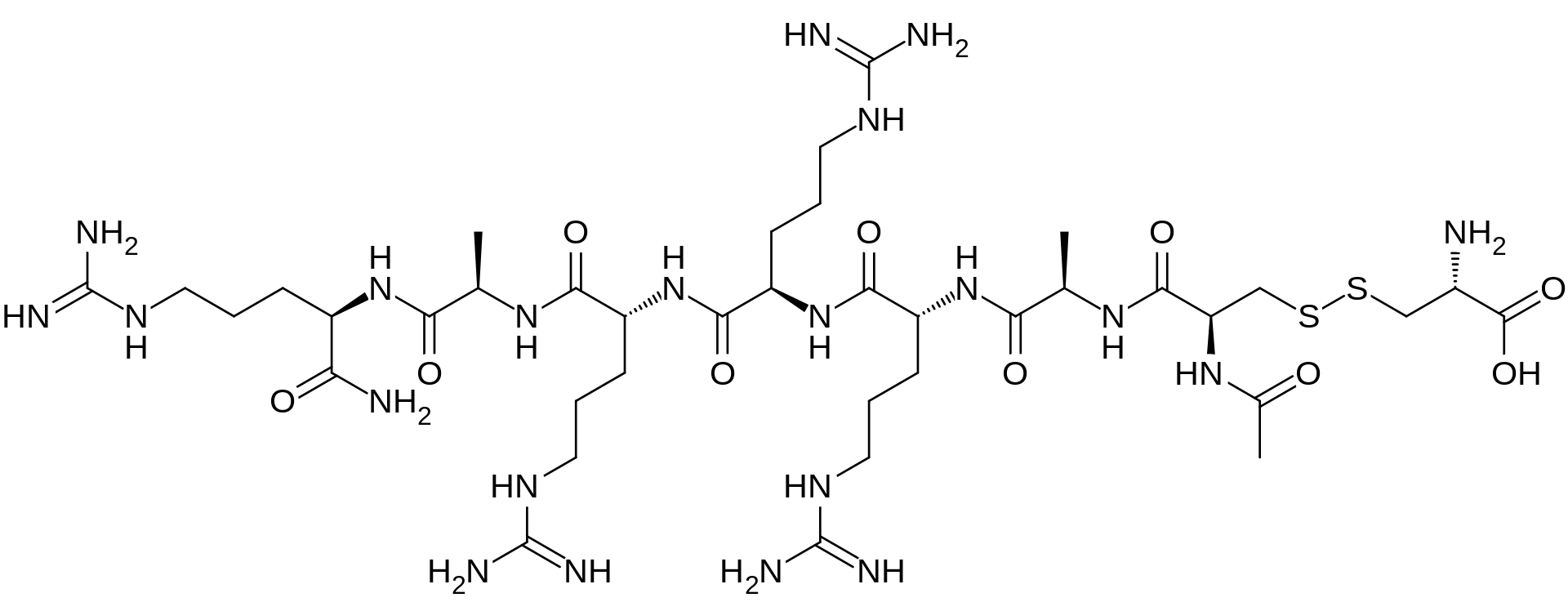
AMG 416 IS (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2)
Etelcalcetide (AMG 416, KAI-4169, velcalcetide)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
http://www.amgenpipeline.com/pipeline/
WO 2011/014707. , the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH.
Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution.
A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Generic Name:Etelcalcetide
Synonym:KAI-4169
CAS Number:1262780-97-1
N-acetyl-D-cysteinyl-S-(L-cysteine disulfide)-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
HYDROCHLORIDE
Generic Name:Etelcalcetide Hydrochloride
AMG 416, KAI-4169, previously also known as velcalcetide hydrochloride
CAS :1334237-71-6
Chemical Name:N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
Method for preparing etelcalcetide and its salts, particularly hydrochloride. See WO2014210489, for a prior filing claiming stable liquid formulation of etelcalcetide. Amgen, following its acquisition of KAI Pharmaceuticals, and Japanese licensee Ono Pharmaceuticals are developing etelcalcetide, a long-acting iv isozyme-selective peptide-based protein kinase C epsilon inhibitor and agonist of the calcium-sensing receptor, for treating secondary hyperparathyroidism (SHPT) in patients with end-stage renal disease receiving dialysis.
In August 2015, an NDA was submitted seeking approval of the drug for SHPT in patients with chronic kidney disease (CKD) on hemodialysis (HD) in the US.
In September 2015, Amgen filed an MAA under the centralized procedure in the EU for the approval of etelcalcetide for treating SHPT in patients with CKD on HD therapy.
KAI is also investigating a transdermal patch formulation of the drug for treating primary HPT.
Secondary hyperparathyroidism in patients with chronic kidney disease receiving dialysis
AMG 416 is a peptide agonist of the human cell surface calcium-sensing receptor (CaSR). It is being investigated as a treatment for secondary hyperparathyroidism in patients with chronic kidney disease receiving dialysis.
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary HyperparathyroidismSHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
Milestones
- 25 Aug 2015 Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015 Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015 Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
PATENT
WO2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
PATENT
WO 2015154031
The hydrochloride salt of AMG 416 has the chemical structure:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
(SEQ ID NO:l)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
AMG 416 and a method for its preparation are described in International Pat. Publication No. WO 2011/014707, which is incorporated herein by reference for any purpose. As described in International Pat. Publication No. WO 2011/014707, AMG 416 may be assembled by solid-phase synthesis from the corresponding Fmoc-protected D-amino acids. After cleavage from the resin, the material may be treated with Boc-L-Cys(NPyS)-OH to form the disulfide bond. The Boc group may then be removed with trifluoroacetate (TFA) and the resulting product purified by reverse-phase high pressure liquid chromatography (HPLC) and isolated as the TFA salt form by lyophilization. The TFA salt can be converted to a pharmaceutically acceptable salt by carrying out a subsequent salt exchange procedure. Such procedures are well known in the art and include, e.g., an ion exchange technique, optionally followed by purification of the resultant product (for example by reverse phase liquid chromatography or reverse osmosis).
There is a need for an efficient method of producing AMG 416, or a pharmaceutically acceptable salt thereof (e.g., AMG 416 HC1), and particularly one appropriate for commercial scale manufacturing.
In a first aspect, provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support; and activating the side chain of the D-Cys residue of the cleaved peptide.
In a second aspect, provided is a method for preparing AMG 416, the method comprising: providing a peptide having a structure of Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 (SEQ ID NO:4); and contacting the peptide with L-Cys to produce a conjugated product.
In yet a third aspect provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support, i.e., to provide an unsupported peptide, and activating the side chain of the D-Cys residue of the unsupported peptide to generate an AMG 416 SPy intermediate (where SPy is 2-pyridinesulfenyl or S-Pyr), dissolving the AMG 416 SPy intermediate in an aqueous 0.1% TFA (trifluoroacetic acid solution), and purifying the AMG 416 SPy derivative by HPLC.
The term “AMG 416”, also known as etelcalcetide, formerly known as velcalcetide or KAI-4169, refers to a compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine, which has the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Reference to AMG 416, or to any compound or AMG 416 fragment, intermediate, or precursor as described herein, is intended to encompass neutral, uncharged forms thereof, as well as pharmaceutically acceptable salts, hydrates and solvates thereof.
The terms “AMG 416 hydrochloride” and “AMG 416 HC1” are interchangeable and refer to a hydrochloride salt form of AMG 416 having the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 shows the chemical structure of AMG 416 (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO: l).
FIG. 2 shows the chemical structure of Rink Amide AM resin and Ac-D-Cys(Trt)- D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-Resin (SEQ ID NO:3).
FIG. 3 shows a reaction scheme in which the SPy intermediate product (Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO:4) is formed from the peptidyl-resin (Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-NH-Resin) (SEQ ID NO:3).
FIG. 4 shows a reaction scheme in which a TFA salt of AMG 416 is formed from the SPy intermediate (AA1_7(SPy)).
FIG. 5 shows a reaction scheme in which the HC1 salt of AMG 416 is formed from the TFA salt of AMG 416.
FIG. 6 shows a reaction scheme in which Boc-D-Arg(Pbf)-OH is formed from Boc-D-Arg-OH.
FIG. 7 shows a reaction scheme in which D-Arg(Pbf)-OH is formed from Boc-D-Arg(Pbf)-OH.
EXAMPLE 5
Purification of the SPy Intermediate and Production of AMG 416 HC1
An alternative method for preparation of AMG 416 HC1 salt is described here. As described in Example 2 above, the SPy intermediate product was dried at 20°C under full vacuum after cleavage from the resin, precipitation and filtration. The precipitate was then dissolved in a 0.1% TFA aqueous solution and loaded onto a C-18 column for HPLC purification. The column was run at <60 bar and the solution temperature was 15-25 °C throughout. The eluents were 0.1% TFA in acetonitrile and 0.1% TFA in water. The fractions were stored at 5°C, they were sampled and then fractions were pooled. The combined pools from two runs were diluted and a concentration/purification run was performed using the same HPLC column to decrease the total volume and remove additional impurities. The fractions were stored at 5°C.
The fractions containing the AMG 416 SPy intermediate were subjected to azeotropic distillation to change the solvent from the 0.1% TFA to a 15% water in IPA solution, charging with IPA as needed. To the resultant AMG 416 SPy intermediate in IPA solution was then added L-Cysteine 1.15 eq and the reaction was allowed to proceed at room temperature for conjugation to occur and to form the AMG 416 TFA salt as described above in Example 4. The AMG 416 TFA solution was added to a solution of 12M aqueous HC1, 0.27 L/kg and IPA 49.4 L/kg over 3 hours via subsurface addition, resulting in direct precipitation of the AMG 416 4.5 HC1 salt. The batch was aged for 3 hours and sampled for analysis.
The material was filtered and slurry washed with 96 wt% IPA, 10 L/kg. The cake was then re-slurried for 4 hours in 10 L/kg of 96% wt% IPA. The material was filtered and further slurry washed with 96% IPA, 10 L/kg and then IPA 10 L/kg. The material was dried under full vacuum at 25°C. The dry cake was dissolved in water 8 L/kg and the batch was concentrated via distillation to remove residual IPA and achieve the desired concentration. The solution temperature was kept below 25 °C throughout the distillation.
PATENT
WO2014210489
SEE
EXAMPLE 1
Solubility of AMG 416 in Succinate Buffered Saline
In this study, the solubility of AMG 416 in succinate buffered-saline was investigated. AMG 416 HC1 (103 mg powder, 80 mg peptide) was dissolved in 200 iL of sodium succinate buffered saline (25 mM succinate, 0.9% saline, pH 4.5). After briefly vortexing, a clear solution was obtained with a nominal concentration of 400 mg/mL. Because expansion of the solution volume was not determined, the solubility of AMG 416 can be conservatively stated as at least 200 mg/mL. Although the maximal solubility was not determined in this experiment, AMG 416 is soluble in pH 4.5 succinate buffered saline to concentrations of at least 200 mg/mL.
REFERENCES
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
//////////////Etelcalcetide, AMG 416, KAI-4169, velcalcetide, peptide drugs
Indian pharma’s struggle to tighten standards paves way for M&A deals

People walk past a chemist shop at a market in Mumbai. Photo: Reuters
MUMBAI – India’s smaller generic drugmakers, struggling to cope with a bruised reputation and tougher regulation in the United States, are under pressure to consider branching out to new, less-profitable markets or sell out to larger rivals.
Two years after its most high-profile regulatory setback to date in the United States – Ranbaxy’s $500 million U.S. fine for drug safety violations – India’s $15 billion a year generic drug industry is still rebuilding its image in its biggest market.
Many of its top firms are facing sanctions at some of their factories, as the U.S. Food and Drug Administration (FDA) tightens checks and its approvals process.
Combined with government-mandated price controls on drugs at home, that is piling pressure on smaller players.
“If they want to have a presence globally, they have to make investments. If they can’t, then they’ll have to focus on other markets or scale back their ambition outside of India, and that’s probably what will happen,” said Subhanu Saxena, CEO of Cipla , India’s fourth-largest drugmaker by revenue.
Ashok Anand, president of Hikal Ltd , a Mumbai-based drugmaker with a market value of $167 million, said some peers were putting themselves on the block.
“If they cannot deal with the stricter regulations, they might just prefer to sell out,” he said.
Pressure on U.S. sales has been felt across the Indian industry, with all drugmakers hit by delays in FDA approvals as the U.S. safety body overhauls its review process. Growth in U.S. revenue for drugmakers slowed to 14 percent in the year to March 2015, less than half what it was in the year to March 2012, according to brokerage Edelweiss.

But for larger players who want to plug gaps or, for the likes of Glenmark and Aurobindo who aim to grow in the United States, this pressure has lowered prices and could pave the way for attractive deals, bankers said.
“Now that some of the smaller companies are reeling under intensive regulatory scrutiny and want to cash out on their investments, valuations would be much more realistic,” said the head of India M&A at a large European bank in Mumbai.
SPENDING SPREE
Indian manufacturers say they have spent millions in high-end testing equipment, improved training and have hired larger teams in quality control since Ranbaxy was fined for manipulating clinical data.
Some consultants estimate spending on compliance has more than doubled to reach about 6 to 7 percent of sales for the larger companies.
But while the number of U.S. export bans issued to Indian companies fell to eight in 2014 from 21 in 2013, according to FDA data, the agency continues to find manufacturing violations at the plants of some of the biggest drugmakers in the country, an indication of the pervasiveness of the problem.
Sun Pharmaceutical Industries , Wockhardt , Dr Reddy’s Laboratories and Cadila Healthcarehave all faced FDA rebukes over the past year.
Smaller firms Ipca and Aarti Drugs faced FDA bans on their plants this year.
These failures – which executives blame on India’s “quick fix” culture and consultants blame on a failure to prioritize compliance – have clouded short-term growth prospects and added to pressure on smaller players, pushing some to look elsewhere.
“They can choose to be in lesser-regulated markets, such as Latin America, where there is a lot of demand. But they will have to live with much thinner margins,” said the finance director of a small Indian drugmaker, who did not want to be named. “It’s survival of the fittest.” REUTERS
http://m.todayonline.com/business/indian-pharmas-struggle-tighten-standards-paves-way-ma-deals
///////
USFDA grants Qualified Infectious Disease Product status to two Wockhardt drugs WCK 771, WCK 2349.
31 Aug, 2014,
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
Read more at:
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
Levonadifloxacin arginine salt, WCK 771
RN: 306748-89-0
RN: 306748-89-0
-
C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
-
L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)
WCK 771………..S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate
(-)-9-Fluoro-8-(4-hydroxypiperidin-1-yl)-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic acid L-arginine salt hydrate
L-arginine salt of (S)-nadifloxacin
A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus
J Med Chem 2005, 48(16): 5232
J Med Chem 2005, 48(16): 5232
CN 102532131, WO 2005023805, WO 2002009758, WO 2001085095, WO 2000068229
WCK 2349

cas 948895-94-1 methane sulfonate
base..706809-20-3
8-[4-(L-Alanyloxy)piperidin-1-yl]-9-fluoro-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic acid methanesulfonate
WO 2000068229, WO 2002009758, WO 2007102061, WO 2008053295
Shetty, N.M.; Nandanwar, M.B.; Kamalavenkatesh, P.; et al.
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
keywords USFDA, Qualified Infectious Disease Product status, Wockhardt, drugs, WCK 771, WCK 2349, QIDP
aChemical name: S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate. bChemical name: S-(–)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3, 3-dimethylpiperidin-1-yl)-1,4 dihydro-4-oxo-quinoline-3-carboxylic acid hydrochloride monohydrate. cChemical name: R-(+)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3,3-dimethylpiperidin-1-yl)-1,4 dihydro-4-oxo-quinoline-3-carboxylic acid hydrochloride monohydrate.
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
DOI: 10.1039/C1MD00134E (Review Article) Med. Chem. Commun.,2011, 2, 1135-1161
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Matthew A. J. Duncton† *
Renovis, Inc. (a wholly-owned subsidiary of Evotec AG), Two Corporate Drive, South San Francisco, CA 94080, United States. E-mail: mattduncton@yahoo.com; Tel: +1 917-345-3183
Received 24th May 2011 , Accepted 3rd July 2011
First published on the web 22nd August 2011
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
The addition of a radical to a heteroaromatic base is commonly referred to as a Minsici reaction. Such reactions constitute a broad-set of selective CH-functionalization processes. This review describes some of the major applications of Minisci reactions and related processes to medicinal or biological chemistry, and highlights some potential developments within this area.
Introduction
The aim of this review is to summarize the use of Minisci reactions within medicinal chemistry, and to highlight some future opportunities to continue progression of this chemistry. As such, it is not an aim that detailed mechanistic information, or a comprehensive list of examples be described. For this, the reader is directed to excellent articles from Minisci, Harrowven and Bowman.1–3 Rather, the review is written to show that Minisci reactions are extremely valuable CH-functionalization processes within medicinal chemistry. However, their use has been somewhat under-utilized when compared with other well-known selective transformations (e.g. palladium-catalysed cross-couplings). Therefore, it is hoped that in the future, Minisci chemistry will continue to develop, such that the reactions become a staple-set of methods for medicinal and biological chemists alike.
To aid discussion, the review is divided in to several sections. First, some historical perspective is given. This is followed by a discussion of scope and limitations. The main-body of the review describes some specific examples of Minisci reactions and related processes, with a focus on their use within medicinal, or biological chemistry. Finally, brief mention is given to potential future applications, some of which may be beneficial in providing ‘high-content’ diverse libraries for screening.
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
…………………….
WIKI
The Minisci reaction is a named reaction in organic chemistry. It is a radical substitution to an aromatic compound, in particular to a heteroaromatic base, that introduces an alkyl group. The reaction was published about in 1971 by F. Minisci.[1] The aromatic compound is generally electron-deficient and with N-aromatic compounds the nitrogen atom is protonated.[2] A typical reaction is that between pyridine and pivalic acid to 2-tert-butylpyridine with silver nitrate, sulfuric acid and ammonium persulfate. The reaction resembles Friedel-Crafts alkylation but with opposite reactivity and selectivity.[3]
The Minisci reaction proceeds regioselectively and enables the introduction of a wide range of alkyl groups.[4] A side-reaction is acylation.[5] The ratio between alkylation and acylation depends on the substrate and the reaction conditions. Due to the simple raw materials and the simple reaction conditions the reaction has many applications in heterocyclic chemistry.[6][7]

Mechanism
A free radical is formed from the carboxylic acid in an oxidative decarboxylation with silver salts and an oxidizing agent. The oxidizing agent reoxidizes the silver salt. The radical then reacts with the aromatic compound. The ultimate product is formed by rearomatisation. The acylated product is formed from the acyl radical.[4][5]

References
- F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo: Nucleophilic character of alkyl radicals—VI : A new convenient selective alkylation of heteroaromatic bases, in: Tetrahedron 1971, 27, 3575–3579.
- Minisci reaction Jie Jack Li in Name Reactions 2009, 361-362, doi:10.1007/978-3-642-01053-8_163
- Strategic applications of named reactions in organic synthesis: background and detailed mechanisms László Kürti, Barbara Czakó 2005
- F. Fontana, F. Minisci, M. C. N. Barbosa, E. Vismara: Homolytic acylation of protonated pyridines and pyrazines with α-keto acids: the problem of monoacylation, in: J. Org. Chem. 1991, 56, 2866–2869; doi:10.1021/jo00008a050.
- M.-L. Bennasar, T. Roca, R. Griera, J. Bosch: Generation and Intermolecular Reactions of 2-Indolylacyl Radicals, in: Org. Lett. 2001, 3, 1697–1700; doi:10.1021/ol0100576.
- P. B. Palde, B. R. McNaughton, N. T. Ross, P. C. Gareiss, C. R. Mace, R. C. Spitale, B. L. Miller: Single-Step Synthesis of Functional Organic Receptors via a Tridirectional Minisci Reaction, in: Synthesis 2007, 15, 2287–2290; doi:10.1055/s-2007-983792.
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.
Design, construction and start-up of a pilot plant for active pharmaceutical ingredients
- Linde-KCA-Dresden built a pilot plant 16 times the size of the laboratory apparatus for Novo Nordisk A/S
- The building layouts show different areas for the pilot plant, the utility systems and the technical rooms with hazardous and non-hazardous proof conditions (top). The engineering documents include


The construction of a pilot plant constitutes an important milestone in the development cycle of a new active pharmaceutical ingredient. Pilot plants are used to gain the technological experience needed for the scale-up process and are required for producing sufficient quantities of the active pharmaceutical ingredient for clinical trials and other types of tests. As a result, even these testing facilities must comply with GMP regulations.
http://www.cpp-net.com/pharma/-/article/5829537/15910929/Challenging+task/

{kind=link}
{kind=link}