Home » Posts tagged 'medicinal chemistry'
Tag Archives: medicinal chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Myself Anthony Crasto up for grabs as Advisor API & INT, Chem.
Admin note for myself . I am up for Grabs
I myself Dr Anthony Melvin Crasto Looking for a post retirement assignment as Advisor API & INT, Chem.
With 36 yrs rich experience, about dozen patents, 10000plus steps covered, 200 API targets, 30 plus products commercialization in plant in full career. Hands on knowledge of Synthesis, Process, scaleup, cost reduction, DOE , softwares etc
Kindly contact me
Dr Anthony Melvin Crasto
+919321316780
amcrasto@gmail.com
About myself
Dr Anthony Crasto
click on my website to know about me
Read http://amcrasto.weebly.com/
Also http://amcrasto.weebly.com/awards.html
Also
http://amcrasto.weebly.com/felicitations.html
1000 lakh google hits, 100lakh blog views, 10 lakh viewers in USA alone, all in 7 continents, 226 countries, 30 Indian and International awards, helping millions across the world

DIFLUPREDNATE

(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluor-11-hydroxy-3,20-dioxopregna-1,4-dien-17-ylbutyrat[German][ACD/IUPAC Name]
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate[ACD/IUPAC Name]
23674-86-4[RN]
245-815-4[EINECS]
2652
6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
DIFLUPREDNATE
CAS# 23674-86-4
- Molecular FormulaC27H34F2O7
- Average mass508.552 Da
- W 6309
- W-6309
- DFBA
- Difluoroprednisolone butyrate acetate
S8A06QG2QE
TU3831500
дифлупреднат[Russian][INN]
ديفلوبريدنات[Arabic][INN]
二氟泼尼酯[Chinese][INN]
(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate
23674-86-4[RN], 245-815-4[EINECS], 2652, 6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
Depositor-Supplied Patent Identifiers
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020325543-A1 | Diagnostic method | 2017-11-20 | |
| WO-2012088044-A2 | Compositions and methods for improving ocular surface health, corneal clarity, optical function and maintaining visual acuity | 2010-12-20 | |
| US-7790905-B2 | Pharmaceutical propylene glycol solvate compositions | 2002-02-15 | 2010-09-07 |
| US-7927613-B2 | Pharmaceutical co-crystal compositions | 2002-02-15 | 2011-04-19 |
PATENT
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A

SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A

SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).

SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).

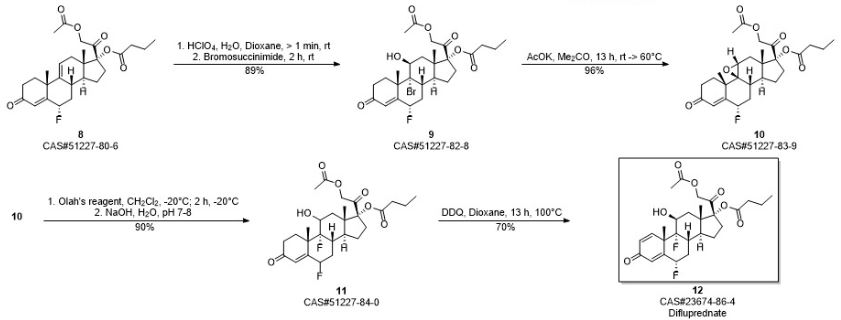
PATENT
https://patents.google.com/patent/CN103509075A/en

Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
Embodiment 2:4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound)
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
Embodiment 3:3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters (formula V)
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
Embodiment 4:4, fluoro-17 α of 9 (11)-diene-6-, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters
10g3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters are dissolved in 60mL acetonitrile, and under nitrogen protection ,-4 ℃ are stirred half an hour.Slowly drip the acetonitrile suspension 40mL of Selecfluor in reaction flask, under nitrogen protection, react 2 hours, TLC (sherwood oil: ethyl acetate=3: 1) monitoring reaction, raw material reaction is complete.Stopped reaction, adds water in reaction flask, ethyl acetate extraction three times, saturated common salt water washing twice, anhydrous sodium sulfate drying.Vacuum concentration is removed organic solvent, obtain faint yellow solid 4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI), productive rate 85%. 1H-NMR(500MHz,CDC1 3):δ(ppm)5.90(1H,d,J=4.5Hz,4-H),5.59(1H,s,11-H),5.07(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.46(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),0.73(3H,s,19-CH 3).
Embodiment 5:4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII)
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
Embodiment 6:6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula VIII)
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
Embodiment 7:6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula IX)
14g 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in 500mL eggplant type bottle, adds 200mL acetone, stirs raw material is fully dissolved, and adds afterwards 3g Potassium ethanoate, is warming up to 60 ℃ of return stirring 13h.TLC (sherwood oil: ethyl acetate=2: 1) monitoring finds that new product occurs.Stop heating, in reaction solution, add water, ethyl acetate extraction, anhydrous sodium sulfate drying organic phase, after standing 30min, steams except organic solvent, obtains yellow oil, productive rate 96%.Column chromatography is purified, and obtains white solid powder, and nuclear-magnetism confirmation structure is 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester. 1H-NMR(300MHz,CDC1 3):δ(ppm)6.11(1H,d,J=4.5Hz,4-H),5.31(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),0.94(3H,s,18-CH 3),0.97(3H,t,J=7.5Hz),1.55(3H,s,19-CH 3),3.52(1H,s,11-H);ESI-MS?m/z:491.2[M+H +],513.2[M+Na +].
Embodiment 8:6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula X)
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
Embodiment 9:6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (difluprednate) (formula I)
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:

Patent
Publication numberPriority datePublication dateAssigneeTitle
US3780177A *1967-06-161973-12-18Warner Lambert Co17-butyrate,21-ester derivatives of 6alpha,9alpha-difluoroprednisolone,compositions and use
US4525303A *1982-06-211985-06-25Dainippon Ink And Chemicals Inc.Process for preparation of steroids
CN101397321A *2007-09-292009-04-01天津药业研究院有限公司Preparation of hydrocortisone and derivatives thereof
CN102076344A *2008-05-282011-05-25瓦利杜斯生物医药有限公司Non-hormonal steroid modulators of nf-kb for treatment of disease
CN102134266A *2010-12-302011-07-27北京市科益丰生物技术发展有限公司Preparation method of melengestrol acetate
Publication numberPriority datePublication dateAssigneeTitle
CN102964412A *2012-11-272013-03-13山东省医药工业研究所Novel crystal form and preparation method of difluprednate
CN103965277A *2014-05-192014-08-06中国科学院上海有机化学研究所Method for synthesizing difluprednate from sterol fermentation product
CN106632561A *2016-12-162017-05-10广州仁恒医药科技股份有限公司Method for preparing difluprednate
CN106749464A *2016-12-292017-05-31奥锐特药业有限公司Steroidal epoxide carries out open loop, the method for fluorination reaction and its device
CN107915766A *2016-10-112018-04-17江苏福锌雨医药科技有限公司A kind of preparation method of fludrocortison acetate
CN108503679A *2018-04-032018-09-07广州仁恒医药科技股份有限公司A kind of purification process of Difluprednate intermediate
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]

NEW DRUG APPROVALS
TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
References
- ^ “Sirion Therapeutics Announces FDA Approval of Durezol for Treatment of Postoperative Ocular Inflammation and Pain” (Press release). Sirion Therapeutics, Inc. 2008-06-24. Retrieved 2008-06-30.
- ^ Clinical trial number NCT00501579 for “Study of Difluprednate in the Treatment of Uveitis” at ClinicalTrials.gov
- ^ Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS (May 2014). “Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study”. Investigative Ophthalmology & Visual Science. 55 (5): 2993–3002. doi:10.1167/iovs.13-12660. PMC 4581692. PMID 24677110.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609025 |
| License data | US FDA: Difluprednate |
| Routes of administration | eye drops |
| ATC code | D07AC19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 23674-86-4 |
| PubChem CID | 32037 |
| DrugBank | DB06781 |
| ChemSpider | 391990 |
| UNII | S8A06QG2QE |
| KEGG | D01266 |
| ChEBI | CHEBI:31485 |
| ChEMBL | ChEMBL1201749 |
| CompTox Dashboard (EPA) | DTXSID0046773 |
| ECHA InfoCard | 100.041.636 |
| Chemical and physical data | |
| Formula | C27H34F2O7 |
| Molar mass | 508.559 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////DIFLUPREDNATE, W 6309, W-6309, DFBA, Difluoroprednisolone butyrate acetate, S8A06QG2QE, TU3831500, дифлупреднат , ديفلوبريدنات , 二氟泼尼酯 , OCCULAR, PAIN
CCCC(=O)OC1(CCC2C1(CC(C3(C2CC(C4=CC(=O)C=CC43C)F)F)O)C)C(=O)COC(=O)C
MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES

MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES
Service to education is service to humanity, Taking free education to the world, Every difficulty an opportunity,In the dark but yet in light,proud of my disabilty, Taking industry experience to students, See the ability not the disabilty, The only disability in life is a bad attitude !!!, Dreamt a billion when hit by million, Mentor to the world
![]() All my awards, https://lnkd.in/fGxx9VF
All my awards, https://lnkd.in/fGxx9VF
Click on above links
Sept 2021, This blog New Drug Approvals approaching 34 lakh views
NEW DRUG APPROVALS FAST APPROACING 34 LAKH VIEWS in 225 countries 7 continents. 5 lakh viewers in USA alone
#worlddrugtracker#worldpeaceambassador#india#helpingmillions#amcrasto#medchem#blog#education#service#free#divyang#everydifficultyanopportunity#opensuperstar#pharmadon#qbdking#ict#ictian#pharma


NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
TENELIGLIPTIN

TENELIGLIPTIN
| Teneligliptin; 760937-92-6; UNII-28ZHI4CF9C; Teneligliptin (INN); 28ZHI4CF9C | |
| MF | C22H30N6OS |
|---|---|
| MW | 426.5782 g/mol |
Teneligliptin (INN; trade name Tenelia) is a pharmaceutical drug for the treatment of type 2 diabetes mellitus. It is approved for use in Japan.[1] It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or “gliptins”.[2] {(2S,4S)-4-[4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)-1-piperazinyl]-2-pyrrolidinyl}(1,3-thiazolidin-3-yl)methanone
Teneligliptin was launched in Japan in 2012 by Mitsubishi Pharma and Daiichi Sankyo for the treatment of type 2 diabetes mellitus. In 2013, the indication was partially changed to include it as a combination therapy with existing oral hypoglycemic agents, such as biganides, alpha-glucosidaseinhibitors, rapid-acting insulin secretagogues, and insulin preparations, as well as sulfonylureas and thiazolidines that had been approved for the combination.
In 2014, the product was registered in KR for the treatment of type 2 diabetes mellitus.
In 2013, Mitsubishi Tanabe Pharma filed for approval in Japan for use of the compound as combination therapy for the treatment of diabetes type 2.
| CAS | 760937-92-6 |
|---|
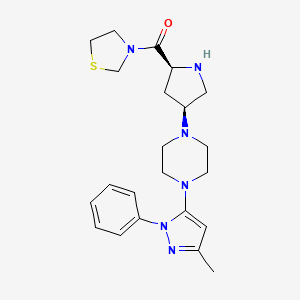
3-{(2S,4S)-4-[4-(3-methyl-l -phenyl- 1 H- pyrazol-5-yl)- l-piperazinyl]-2-pyrrolidinylcarbonyl}-l , 3-thiazolidine is represented structurally by a compound of formula (I):

Teneligliptin (CAS 760937-92-6) is a novel, potent and long-lasting dipeptidyl peptidase-4 inhibitor in treatment of type 2 diabetes. Dipeptidyl-peptidase-4 (DPP- 4) inhibitor has been demonstrated to improve glycemic control, in particular postparandial hyperglycemic control.
Despite of their common mechanism of action, DPP-4 inhibitors show marked structural heterogeneity. DPP-4 inhibitors may be classified into peptidomimetic (i.e. sitagliptin, vildagliptin, saxagliptin, and anagliptin) and non-peptidomimetic (i.e. alogliptin and linagliptin) subtypes.
Teneligliptin, is chemically known as a 3- {((2S,4S)-4-(4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-yl 25 carbonyl}thiazolidine hemipentahydrobromide hydrate and is peptidomimetic with the molecular formula of C22H30N6OS.2½HBr.xH2O and molecular weight of 642.88 g/mol for hemipentahydrobromide. The hydrate can be from mono to dihydrate.
U.S. Patent No. 7,074,794 B2 (the US ‘794) discloses teneligliptin as L-proline derivative and its pharmaceutically acceptable salts which exhibits a Dipeptidyl 5 peptidase IV (DPP-IV) inhibitory activity, which is useful for the treatment or prophylaxis of diabetes, obesity, HIV infection, cancer metastasis, dermopathy, prostatic hyperplasia, periodontitis, autoimmune diseases and the like.
The example-222 of the US ‘794 discloses the process for the preparation of teneligliptin as trihydrochloride salt U.S. Patent No. 8,003,790 B2 (the US ‘790) discloses salts of proline derivative, solvate thereof and production method thereof. In particular, the US ‘790 discloses 2.0 hydrochloride or 2.5 hydrochloride; 2.0 hydrobromide or 2.5 hydrobromide, and hydrates thereof teneligliptin.
The US ‘790 B2 further discloses different salts 15 of teneligliptin which are incorporated herein as reference in their entirety U.S. PG-Pub. No. 2011/0282058 A1 discloses salts of 3-{((2S,4S)-4-(4-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-ylcarbonyl}thiazolidine with mono-, di- and tri-basic acids or a solvate thereof. 20 International (PCT) publication No. WO 2012/165547 A1 discloses a process for preparation of teneligliptin and pharmaceutically acceptable salts thereof.
International (PCT) publication No. WO 2007/127635 A2 (the WO ‘635 A2) discloses a process for the preparation of diketo-piperazine and piperidine 25 derivatives. In particular, the WO ‘635 A2 discloses the process for preparation of 4-oxo-2-(thiazolidine-3-carbonyl)-pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (III)] by reacting piperazine with aryl halide.
International (PCT) publication No. WO 2012/099915 A1 (the WO ‘915 A1) 5 discloses the process for the preparation of deuterated thiazolidine derivatives. The WO ‘915 A1 also discloses the process for the preparation of 1-(3-methyl-1- phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by condensation of 5- chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by deprotection of Boc-protected 1-(3-methyl-1-phenyl-1Hpyrazol-5-yl)piperazine with triflouroacetic acid.
U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine by condensation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine in presence of n-BuLi in tetrahydrofuran. Bioorganic & Medicinal Chemistry, 14(11), 3662-3671 (2006),
Bioorganic & Medicinal Chemistry, 20(16), 5033-5041 (2012) and U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate by reacting (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid with 25 thiazolidine in presence of HOBT and EDC.HCl in dimethylformamide solvent.
Bioorganic & Medicinal Chemistry, 15(2), 641-655 (2007) discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3- carbonyl)pyrrolidine-1-carboxylate by treating (2S,4S)-tert-butyl 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate with tetrabutylammonium fluoride in tetrahydrofuran.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the 5 process for the preparation of herein compound (II) after by reacting 1-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) with (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate in presence of sodium triacetoxyborohydride. There is provided different alternative processes for the preparation of teneligliptin and intermediates thereof.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) also discloses the process for the preparation of 4-[4-(5-methyl-2-phenyl-2H-pyrazol-3-yl)-piperazin- 1-yl]-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (II)] after by reacting 1-(3-methyl-1-phenyl-1H-pyrazol-5- 15 yl)piperazine [herein compound (V)] with (2S,4S)-tert-butyl 4-[[(1,1- dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate in presence of trifluoromethylsulfonic anhydride and diisopropylethylamine. 3 – [[(2S, 4S) -4- [4- (3- methyl-1-phenyl–1H- pyrazol-5-yl) -1-piperazinyl ] -2-pyrrolidinyl] carbamoyl] thiazolidine, having the formula below, is a very novel DPP-4 inhibitor potential.

World Patent Application No. W02012099915 for Ge Lieting discloses a process for the preparation route is as follows:

Journal B10rganic & Medicinal Chemistry, 2012, 20, 5705-5719 also discloses a preparation method for Ge Lieting, the route is as follows:

[0009] 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine, was prepared for the Ge Lieting key intermediate. Journals B10rganic & Medicinal Chemistry, 2012,20,5705-5719 reported the preparation of the intermediates prepared route is as follows:

[0011] The preparative route after the N-Boc-N- acetoacetyl piperazine phenylhydrazine and methanesulfonic acid in an ethanol solution of the reaction at room temperature 14h, concentrated under reduced pressure after addition of pyridine.Was added phosphorus oxychloride in pyridine, 20h post treatment reaction at room temperature the reaction system. The compound obtained above was then added trifluoroacetic acid was dissolved in methylene chloride after, after treatment at room temperature for 1.5h to give 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine.
The reaction process requires mesylate mesylate flammable, easy-absorbent deliquescence, and has a strong corrosive and irritating, easy to cause the body burns; phosphorus oxychloride, a highly toxic substance, water violent hair in the air smoke, hydrolyzed into phosphoric acid and hydrogen chloride, is very unstable, to operate a lot of trouble; trifluoroacetic acid is highly corrosive and irritant, can cause the body burns; low yield of the reaction (10%). Seeking a simple operation, high reaction yield, low cost and suitable for industrial production production process 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine has a very important role in the field of medicine.
…………………………………….


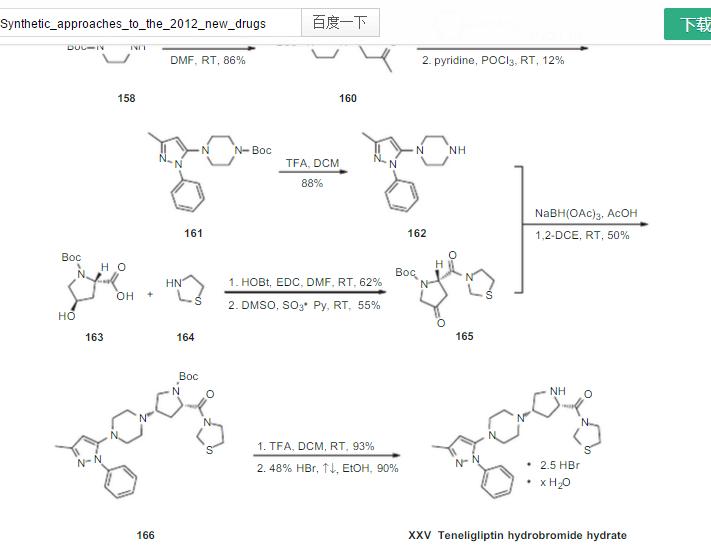
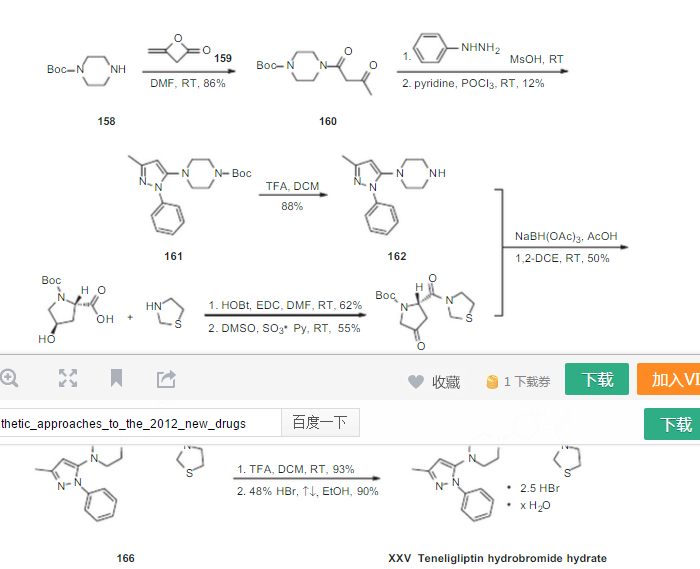
since the capture is staggered, compd 165 is not clear in above pic see below
…………
if above section iis not clear see at ……..http://www.allfordrugs.com/2015/07/03/teneligliptin/
…………………….
reaction scheme in http://www.google.com/patents/CN104177295A?cl=en

Description: LR as Lawesson reagent (Lawesson Reagent), is a sulfur oxygen exchange reagent. The present invention provides a method for preparing key intermediates Ge Lieting method, comprising the steps of: (I) N-Boc-N- acetoacetyl piperazine Lawesson’s reagent in the presence of an organic solvent, with a phenylhydrazine of the formula occurs ⑴ reaction shown:

(2) the step (1) The product was dissolved in an organic solvent, the following formula (II) in concentrated hydrochloric acid to deprotected shown:

Volume 20, Issue 19, 1 October 2012, Pages 5705–5719


………………………..
http://www.google.co.in/patents/WO2015019238A1?cl=en
Example 5: Preparation of {(2^,.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -vHpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
Activated carbon (10 g) was added to a solution of the residue (obtained in Example 4) in isopropyl alcohol (1000 mL) at 30°C to 35°C. The reaction mixture was filtered through a Hyflo® bed. The filtrate was heated to a temperature of 70°C to 75°C. Hydrobromic acid (48%; 168 g) was slowly added to the filtrate at 70°C to 75°C over a period of 10 minutes to 15 minutes. The reaction mixture was stirred for 2.5 hours at 70°C to 77°C. The progress of the reaction was monitored by HPLC. After completion of the reaction, the reaction mixture was cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes. The reaction mixture was filtered to obtain a solid. The solid obtained was washed with isopropyl alcohol (2 x 200 mL), and dried at 50°C under reduced pressure for 15 hours to obtain crude {(25*,45)-4-[4-(3-methyl-l-phenyl-lH- pyrazol-5 -yl)piperazin- 1 -yl]pyrrolidin-2-yl} ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate.
Yield: 90%
Example 6: Purification of {(2^’.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -yllpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
A reaction mixture containing {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5- yl)piperazin- 1 -yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate (100 g; prepared according to the process of Example 5) in ethanol (700 mL) was heated at 70°C to 75°C to obtain a solution. The solution was filtered at the same temperature. The filtrate was allowed to cool to a temperature of 65 °C to 68°C, and deionized water (10 mL) was added at the same temperature. The solution was cooled to a temperature of 55°C to 60°C, and stirred at the same temperature for 2 hours. The solution was further cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes to obtain a solid. The solid was filtered, washed with ethanol (100 mL), and dried at 45°C to 50°C under reduced pressure for 18 hours to 20 hours to obtain pure {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin-l- yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone hemipentahydrobromide hydrate .
Yield: 90%
HPLC Purity: 99.93%
| WO2012099915A1 * | 18 Jan 2012 | 26 Jul 2012 | Hongwen Zhu | Thiazolidine derivatives and their therapeutic use |
| WO2012165547A1 * | 31 May 2012 | 6 Dec 2012 | Mitsubishi Tanabe Pharma Corporation | Method for manufacturing pyrazole derivative |
| WO2014041560A2 * | 28 Aug 2013 | 20 Mar 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of teneligliptin |
| US7074794 | 10 Aug 2001 | 11 Jul 2006 | Mitsubishi Pharma Corporation | Proline derivatives and the use thereof as drugs |
| US8003790 | 17 Feb 2006 | 23 Aug 2011 | Mitsubishi Tanabe Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| US20050256310 * | 12 May 2005 | 17 Nov 2005 | Pfizer Inc | Therapeutic compounds |
| EP1854795A1 * | 17 Feb 2006 | 14 Nov 2007 | Mitsubishi Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| EP1894567A1 * | 2 Jun 2006 | 5 Mar 2008 | Mitsubishi Tanabe Pharma Corporation | Concomitant pharmaceutical agents and use thereof |
| US20040106655 * | 10 Aug 2001 | 3 Jun 2004 | Hiroshi Kitajima | Proline derivatives and the use thereof as drugs |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015019238A1 * | 28 Jul 2014 | 12 Feb 2015 | Ranbaxy Laboratories Limited | Process for the preparation of n-protected (5s)-5-(1,3-thiazolidin-3-ylcarbonyl)pyrrolidin-3-one |
| Patent | Submitted | Granted |
|---|---|---|
| Proline derivatives and use thereof as drugs [US7060722] | 2005-11-03 | 2006-06-13 |
| Proline derivatives and the use thereof as drugs [US7074794] | 2004-06-03 | 2006-07-11 |
| Proline derivatives and use thereof as drugs [US2006173056] | 2006-08-03 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US8003790] | 2009-08-27 | 2011-08-23 |
| METHOD OF TREATING ABNORMAL LIPID METABOLISM [US2010305139] | 2010-12-02 | |
| COMBINED USE OF DIPEPTIDYL PEPTIDASE 4 INHIBITOR AND SWEETENER [US2010113382] | 2010-05-06 | |
| CONCOMITANT PHARMACEUTICAL AGENTS AND USE THEREOF [US2009082256] | 2009-03-26 | |
| PROPHYLACTIC/THERAPEUTIC AGENT FOR ABNORMALITIES OF SUGAR/LIPID METABOLISM [US2009088442] | 2009-04-02 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US2011282058] | 2011-11-17 |
- Joanne Bronson, Amelia Black, T. G. Murali Dhar, Bruce A. Ellsworth, and J. Robert Merritt. “Teneligliptin (Antidiabetic)”. Annual Reports in Medicinal Chemistry 48: 523–524. doi:10.1016/b978-0-12-417150-3.00028-4.
- Kishimoto, M (2013). “Teneligliptin: A DPP-4 inhibitor for the treatment of type 2 diabetes”. Diabetes, metabolic syndrome and obesity : targets and therapy 6: 187–95. doi:10.2147/DMSO.S35682. PMC 3650886. PMID 23671395.
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
ANTHONY CRASTO VENTURES INTO CHINA…..MY KAIXIN BLOG 开心网 ON MEDICINAL CHEMISTRY
KAIXIN

MY EASTERN VENTURE TO PROPAGATE CHEMISTRY……………http://www.kaixin001.com/home/?_profileuid=159073878

 CHINA
CHINA
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY……………http://www.kaixin001.com/home/?_profileuid=159073878
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY……………http://www.kaixin001.com/home/?_profileuid=159073878
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY……………http://www.kaixin001.com/home/?_profileuid=159073878\


DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com

 Location in Madhya Pradesh
Location in Madhya Pradesh
-
Khajuraho Group of Monuments – Wikipedia, the free …
en.wikipedia.org/wiki/Khajuraho_Group_of_MonumentsThe Khajuraho Group of Monuments are a group of Hindu and Jain temples in Madhya Pradesh, India. About 620 kilometres (385 mi) southeast of New Delhi, …








Hotel Chandela – A Taj Leisure Hotel





Anthony crasto’s blog New drug approvals touches 3 lakh views…….Helping millions

link is https://newdrugapprovals.org/
All about Drugs, live, by DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Helping millions, 7 million hits on google, pushing boundaries, one lakh plus connections worldwide, 3 lakh plus VIEWS on this blog in 193 countries

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Matthew A. J. Duncton† *
Renovis, Inc. (a wholly-owned subsidiary of Evotec AG), Two Corporate Drive, South San Francisco, CA 94080, United States. E-mail: mattduncton@yahoo.com; Tel: +1 917-345-3183
First published on the web 22nd August 2011
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
The addition of a radical to a heteroaromatic base is commonly referred to as a Minsici reaction. Such reactions constitute a broad-set of selective CH-functionalization processes. This review describes some of the major applications of Minisci reactions and related processes to medicinal or biological chemistry, and highlights some potential developments within this area.
Introduction
The aim of this review is to summarize the use of Minisci reactions within medicinal chemistry, and to highlight some future opportunities to continue progression of this chemistry. As such, it is not an aim that detailed mechanistic information, or a comprehensive list of examples be described. For this, the reader is directed to excellent articles from Minisci, Harrowven and Bowman.1–3 Rather, the review is written to show that Minisci reactions are extremely valuable CH-functionalization processes within medicinal chemistry. However, their use has been somewhat under-utilized when compared with other well-known selective transformations (e.g. palladium-catalysed cross-couplings). Therefore, it is hoped that in the future, Minisci chemistry will continue to develop, such that the reactions become a staple-set of methods for medicinal and biological chemists alike.
To aid discussion, the review is divided in to several sections. First, some historical perspective is given. This is followed by a discussion of scope and limitations. The main-body of the review describes some specific examples of Minisci reactions and related processes, with a focus on their use within medicinal, or biological chemistry. Finally, brief mention is given to potential future applications, some of which may be beneficial in providing ‘high-content’ diverse libraries for screening.
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
…………………….
WIKI
The Minisci reaction is a named reaction in organic chemistry. It is a radical substitution to an aromatic compound, in particular to a heteroaromatic base, that introduces an alkyl group. The reaction was published about in 1971 by F. Minisci.[1] The aromatic compound is generally electron-deficient and with N-aromatic compounds the nitrogen atom is protonated.[2] A typical reaction is that between pyridine and pivalic acid to 2-tert-butylpyridine with silver nitrate, sulfuric acid and ammonium persulfate. The reaction resembles Friedel-Crafts alkylation but with opposite reactivity and selectivity.[3]
The Minisci reaction proceeds regioselectively and enables the introduction of a wide range of alkyl groups.[4] A side-reaction is acylation.[5] The ratio between alkylation and acylation depends on the substrate and the reaction conditions. Due to the simple raw materials and the simple reaction conditions the reaction has many applications in heterocyclic chemistry.[6][7]

Mechanism
A free radical is formed from the carboxylic acid in an oxidative decarboxylation with silver salts and an oxidizing agent. The oxidizing agent reoxidizes the silver salt. The radical then reacts with the aromatic compound. The ultimate product is formed by rearomatisation. The acylated product is formed from the acyl radical.[4][5]

References
- F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo: Nucleophilic character of alkyl radicals—VI : A new convenient selective alkylation of heteroaromatic bases, in: Tetrahedron 1971, 27, 3575–3579.
- Minisci reaction Jie Jack Li in Name Reactions 2009, 361-362, doi:10.1007/978-3-642-01053-8_163
- Strategic applications of named reactions in organic synthesis: background and detailed mechanisms László Kürti, Barbara Czakó 2005
- F. Fontana, F. Minisci, M. C. N. Barbosa, E. Vismara: Homolytic acylation of protonated pyridines and pyrazines with α-keto acids: the problem of monoacylation, in: J. Org. Chem. 1991, 56, 2866–2869; doi:10.1021/jo00008a050.
- M.-L. Bennasar, T. Roca, R. Griera, J. Bosch: Generation and Intermolecular Reactions of 2-Indolylacyl Radicals, in: Org. Lett. 2001, 3, 1697–1700; doi:10.1021/ol0100576.
- P. B. Palde, B. R. McNaughton, N. T. Ross, P. C. Gareiss, C. R. Mace, R. C. Spitale, B. L. Miller: Single-Step Synthesis of Functional Organic Receptors via a Tridirectional Minisci Reaction, in: Synthesis 2007, 15, 2287–2290; doi:10.1055/s-2007-983792.
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.
Nemonoxacin….TaiGen’s pneumonia antibiotic Taigexyn 奈诺沙星 gets marketing approval in Taiwan

Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
-
C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ — TaiGen Biotechnology Company, Limited (“TaiGen”) today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Nemonoxacin malate anhydrous
951163-60-3 CAS NO, MW: 505.5209
Nemonoxacin malate hemihydrate
951313-26-1, MW: 1029.0566
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


……………………..
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1…….DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1′) were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9′): Compound 9′ was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck’s Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9′) was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w – “ MsCI1TEA . „ _. – – _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1′)
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50″-65″C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1′ was synthesized from Compound 9′ as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
…………………
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck’s Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
………………..
REF
A. ARJONA ET AL: “Nemonoxacin“, DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: “TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia“, INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: “TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)“, 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: “TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic“, 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
|
7-4-2012
|
TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION
|
|
|
4-18-2012
|
Coupling Process For Preparing Quinolone Intermediates
|
|
|
10-19-2011
|
Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
|
|
|
6-18-2010
|
STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES
|
|
|
2-19-2010
|
PNEUMONIA TREATMENT
|
|
|
5-6-2009
|
Hydride reduction process for preparing quinolone intermediates
|
|
|
2-13-2009
|
ANTIMICROBIAL PARENTERAL FORMULATION
|
|
|
11-26-2008
|
Coupling process for preparing quinolone intermediates
|
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

Rapamycin (Sirolimus) For the prophylaxis of organ rejection in patients receiving renal transplants.

Rapamycin (Sirolimus)
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25, 26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone
Wyeth Pharmaceuticals (Originator)
M.Wt:914.18
Formula:C51H79NO13
53123-88-9 cas no
Antifungal and immunosuppressant. Specific inhibitor of mTOR (mammalian target of Rapamycin). Complexes with FKBP-12 and binds mTOR inhibiting its activity. Inhibits interleukin-2-induced phosphorylation and activation of p70 S6 kinase. Induces autophagy in yeast and mammalian cell lines.
Rapamycin is a triene macrolide antibiotic, which demonstrates anti-fungal, anti-inflammatory, anti-tumor and immunosuppressive properties. Rapamycin has been shown to block T-cell activation and proliferation, as well as, the activation of p70 S6 kinase and exhibits strong binding to FK-506 binding proteins. Rapamycin also inhibits the activity of the protein, mTOR, (mammalian target of rapamycin) which functions in a signaling pathway to promote tumor growth. Rapamycin binds to a receptor protein (FKBP12) and the rapamycin/FKB12 complex then binds to mTOR and prevents interaction of mTOR with target proteins in this signaling pathway. Rapamycin name is derived from the native word for Easter Island, Rapi Nui.
- (-)-Rapamycin
- Antibiotic AY 22989
- AY 22989
- AY-22989
- CCRIS 9024
- HSDB 7284
- NSC 226080
- Rapammune
- Rapamune
- Rapamycin
- SILA 9268A
- Sirolimus
- UNII-W36ZG6FT64
- WY-090217
- A 8167
A macrolide compound obtained from Streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to IMMUNOPHILINS. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties.
Sirolimus (INN/USAN), also known as rapamycin, is an immunosuppressant drug used to prevent rejection in organ transplantation; it is especially useful in kidney transplants. It prevents activation of T cells and B cells by inhibiting their response to interleukin-2 (IL-2). Sirolimus is also used as a coronary stent coating. Sirolimus works, in part, by eliminating old and abnormal white blood cells.[citation needed] Sirolimus is effective in mice with autoimmunity and in children with a rare condition called autoimmune lymphoproliferative syndrome (ALPS).

A macrolide, sirolimus was discovered by Brazilian researchers as a product of the bacterium Streptomyces hygroscopicus in a soil sample fromEaster Island[1] — an island also known as Rapa Nui.[2] It was approved by the FDA in September 1999 and is marketed under the trade nameRapamune by Pfizer (formerly by Wyeth).
Sirolimus was originally developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties. It has since been shown to prolong the life of mice and might also be useful in the treatment of certain cancers.
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response tointerleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12(FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits themammalian target of rapamycin (mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
 rapamycin
rapamycin
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response to interleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mammalian target of rapamycin(mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.

Rapamycin and its preparation are described in US Patent No. 3,929,992, issued December 30, 1975. Alternatively, rapamycin may be purchased commercially [Rapamune®, Wyeth].
Rapamycin (Sirolimus) is a 31-member natural macrocyclic lactone [C51H79N1O13; MWt=914.2] produced by Streptomyces hygroscopicus and found in the 1970s (U.S. Pat. No. 3,929,992; 3,993,749). Rapamycin (structure shown below) was approved by the Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection in 1999.

Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP-12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., IL2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S. Pat. No. 5,286,730], bowel disorders [U.S. Pat. No. 5,286,731], smooth muscle cell proliferation and intimal thickening following vascular injury [U.S. Pat. Nos. 5,288,711 and 5,516,781], adult T-cell leukemia/lymphoma [European Patent Application 525,960 A1], ocular inflammation [U.S. Pat. No. 5,387,589], malignant carcinomas [U.S. Pat. No. 5,206,018], cardiac inflammatory disease [U.S. Pat. No. 5,496,832], anemia [U.S. Pat. No. 5,561,138] and increase neurite outgrowth [Parker, E. M. et al, Neuropharmacology 39, 1913-1919, 2000].
Although rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803). Some of the analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No. 5,362,718), as well as alkyl, aryl, alkenyl, and alkynyl analogues (U.S. Pat. Nos. 5,665,772; 5,258,389; 6,384,046; WO 97/35575).
………………………………………..
Synthesis
ref are independent of body…see below for this clip
Several total synthese of rapamycin have been reported3,4as well as many fragments and part-syntheses. Rapamycin is a complicated molecule comprising a 31-membered ring including a pipecolinyl group and pyranose ring, a conjugated triene system and a tri-carbonyl region. It also has 15 chiral centres, meaning the number of possible stereoisomers is enormous. The synthesis of rapamycin therefore presents a huge challenge to synthetic chemists.
In the following synthesis, published in three separate papers5,6,7two fragments of C10-C21 and C22-C42 are prepared separately, before being combined to give the total synthesis of rapamycin. Only the main outline of the synthesis will be shown as it is too long and complicated to show in great detail. For the full experimental details of the synthesis see the literature (ref. nos. given above).

In the retro-synthesis shown the molecule is disconnected at the ester group next to carbon 1 and the C21-C22 double bond of the triene to give the synthetic precursors 2 and 3. Further disconnections of 3 will be shown later. First the C10-C21 fragment is synthesised.
Synthesis of C10-C21 fragment
The synthesis uses (R)-methyl 3-hydroxy-2-methylpropionate (8) as a starting material.

The starting material 8 is converted to an alcohol by a four-step process; protection of the alcohol as aTHP ether followed by reduction, ether formation and deprotection steps. Substitution of the hydroxyl group in the product for a bromine leads to the formation of the bromide 9. Reaction of 9 with methyl acetoacetate gave ester 10.

Catalytic reduction of 10 using the conditions of Noyori produced ester 11, which was then converted to its Weinreb amide 12. Overall, compound 12 was produced in 54% yield from an inexpensive starting material. Vinyl bromide 13 was metalated with t-BuLi and the resulting vinyllithium was combined with 12 and the PMB-protecting group was removed to give 14. The remaining carbonyl group in 14 was selectively reduced to a hyrdoxy group. In order to differentiate the 1,3-diol a lactol was formed, where one hydroxy group ended up in the ring. To acheive this an oxidation was performed using RuCl2(PPh3)3 resulting in formation of a lactol. The two remaining alcohol groups could then be methylated using MeI forming 15.

The lactol ring opening was achieved using TiCl4 and thiol HS(CH2)2SH to form a dithiolane. The freed alcohol was then protected as its TBS ether and the same protecting group selectively removed from the primary alcohol to form 16. To avoid removing the dithiolane group at a later stage in the synthesis the thio-acetal was converted to the dimethyl acetal 17 using PhI(OCOCF3)2 and methanol.

The next stage in the synthesis was to extend 17 for the building of the triene region. The terminal alcohol was oxidised to its aldehyde using BaMnO4 , then a Wittig reaction was carried out using Ph3P=CHCO2Et and CH2Cl2 to form the second double bond. Reduction of the ester group to an alcohol was carried out using DIBAL-H, then treatment with PPh3 and exposure to the air gave rapamycin fragment 2.
Synthesis of C22-C42 fragment
Here the retro-synthesis of 3 is shown, giving the three synthetic precursors 5, 6 and 7

It was thought 4 could be obtained by alkylative coupling of a vinyllithium species generated from 7 to the Weinreb amide 6. The nucleophilic opening of epoxide 5 by the lithiated sulfone from phenyl sulfone 4 would then produce the desired fragment.
The ester 18 was used as a starting material to make fragment 6.

A Wittig reaction followed by reduction and protection steps produced 19. This was hydrogenated using a rhodium catalyst to give syn-dimethyl product 20. The minor anti diastereomer was successfully separated off. 20 was oxidised then underwent an aldol condensation to give adduct 21.

Transamination of 21 and protection of the alcohol with PMB resulted in amide 6, corresponding to the C22-C28 segment of rapamycin.
The vinyl bromide 7 was prepared using ester 22 as a starting material.

Reduction of 22 followed by dibromoolefination resulted in product 23. Acetylene 24 was prepared using n-BuLi, THF and MeI, then sulfenylation with Ph2S2 and bromination gave fragment 7.

Iodination and alkylation of starting material 25 with the lithiated allylic sulfide shown followed by a number of further steps resulted in its conversion to fragment 5.

Fragments 7 was first converted to its vinyllithium using t-BuLi then combined with 6 forming an enone in 78% yield. Stereoselective reduction of the carbonyl group using Zn(BH4)2 gave an alcohol which was protected with DEIPS giving 28. The phenyl sulfide was oxidised to a sulfone using m-CPBA in excess pyridine.

Lithiation and addition of the epoxide 5 resulted in the hydroxy sulfone in a 4:1 ratio of two diastereomers which were separated by HPLC. Metalation using n-BuLi followed by oxidation formed the total C22-C42 fragment.
Total synthesis of rapamycin through the combination of C10-C21 and C22-C42 fragments.
Fragment 3 (C22-C42) was treated with (S)-Boc-pipecolinal, followed by a Swern oxidation resulted in the aldehyde 29.

Condensation with the lithium salt of phosphine oxide 2 (C10-C21) produced the triene shown below.

The triene was hydrolysed with pyridinium p-toluenesulfonic acid and an aldol reaction was performed. Treatment with triethylsilyl triflate produced an amino acid which was subjected to Mukaiyama macrocyclization conditions to form the 31-membered ring. Finally, deprotection steps were performed to give synthetic rapamyin (1). This was judged to be identical to natural rapamycin by comparison of physical properties, 1H-NMR, 13C-NMR, IR and UV spectral data.
3. K. C. Nicolaou, T. K. Chakraborty, A. D. Piscopio, N. Minowa, P. Bertinato; J. Am. Chem. Soc.; 115; 1993; 4419
4. C. M. Hayward, D. Yohannes, S. J. Danishefsky; J. Am. Chem. Soc.; 115; 1993; 9345
5. S. D. Meyer, T. Miwa, M. Nakatsuka, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5058-5060
6. D. Romo, D. D. Johnson, L. Plamondon, T. Miwa, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5060-5063
7. S. D. Meyer, D. Romo, D. D. Johnson, S. L. Schreiber; J. Am. Chem. Soc.; 115; 1993; 7906-7907
………………………………………….
Synthesis
PREPARATION
CUT PASTE FROM TEXT
In one embodiment of this invention rapamycin is prepared in the followingmanner: 4
A suitable fermenter is charged with production meis reached in the fermentation mixture after 2-8 days,
usually after about 5 days, as determined by the cup plate method and Candida albicans as the test organism. The mycelium is harvested by filtration with diatomaceous earth. Rapamycin is then extracted from the mycelium with a water-miscible solvent, for example a lower alkanol, preferably methanol or ethanol. The latter extract is then concentrated, preferably under reduced pressure, and the resulting aqueous phase is extracted with a water-immiscible solvent. A preferred water-immiscible solvent for this purpose is methylene dichloride although chloroform, carbon tetrachloride, benzene, n-butanol and the like may also be used. The latter extract is concentrated, preferably under reduced pressure, to afford the crude product as an oil.
The product may be purified further by a variety of methods. Among the preferred methods of purification is to dissolve the crude product in a substantially nonpolar, first solvent, for example petroleum ether or hexane, and to treat the resulting solution with a suit able absorbent, for example charcoal or silica gel, so that the antibiotic becomes absorbed on the absorbant. The absorbant is then separated and washed or eluted with a second solvent more polar than the first solvent, for example ethyl acetate, methylene dichloride, or a mixture of methylene dichloride and ether (preferred). Thereafter, concentration of the wash solution or eluate affords substantially pure rapamycin. Further purification is obtained by partial precipitation with a nonpolar solvent, for example, petroleum ether, hexane, pentane and the like, from a solution of the rapamycin in a more polar solvent, for example, ether, ethyl acetate, benzene and the like. Still-further purification is obtained by column chromatography, preferably employing silica gel, and by crystallization of the rapamycin from ether.
In another preferred embodiment of this invention a first stage inoculum of S treptomyces hygroscopicus NRRL 5491 is prepared in small batches in a medium containing soybean flour, glucose, ammonium sulfate, and calcium carbonate incubated at about 25C at pH 7.l-7.3 for 24 hrs. with agitation, preferably on a gyrotary shaker. The growth thus obtained is used to inoculate a number of somewhat larger batches of the same medium as described above which are incubated at about 25C and pH 7.1-7.3 for 18 hrs. with agitation, preferably on a reciprocating’shaker, to obtain a sec- “ond stagc inoculum which is used to inoculate the production stage fermenters.
6 5.86′.2.-The fermenters are inoculated with the second stage inoculum described above and incubated at about 25C with’ agitationand aeration while controlling and ‘mai’ntaining the mixture at approximately pH 6.0 by
addition offa base, for example, sodium hydroxide, potassium hydroxide or preferably ammonium hydroxide, as required from time to time. Addition of a source -of assimilable carbon, preferably glucose, is started when theconcentrationof the latter in the broth has dropped to about 0.5% wt/vol, normally about 48 hrs after. the start of fermentation, and is maintained until the end ofthe particular run. In this manner a fermentation broth containing about 60 ug/ml of rapamycin as determined by the assay method described above is obtained in 45 days, when fermentation is stopped.
‘ Filtration of the’mycelium, mixing the latter with a watef-miscible ‘lower’ alkanol, preferably methanol, followed by extraction with a halogenated aliphatic hydrocarbon, preferably trichloroethane, and evaporation of the solvents yields a first oily residue. This first oily residue is dissolved in a lower aliphatic ketone, preferably acetone, filtered from insoluble impurities, the filtrate evaporated to yield a second oily residue which is extractedjwith a water-miscible lower alkanol,
preferably methanol, and the latter extract is evaporated to yield crude rapamycin as a third oily residue. This third oily residue is dissolved in a mixture of a lower aliphatic ketone and a lower aliphatic hydrocarbon, preferably acetone-hexane, an absorbent such as charcoal or preferably silica gel is added to adsorb the rapamycin, the latter is eluted from the adsorbate with a similar but more polar solvent mixture, for example a mixture as above but containing a higher proportion of the aliphatic ketone, the eluates are evaporated and the residue is crystallized from diethyl ether, to yield pure crystalline rapamycin. In this manner a total of 45-5 8% of the rapamycin initially present in the fermentation mixture is recovered as pure crystalline rapamycin.
CHARACTERIZATION solvent systems; for example, ether-hexane 40:60 (Rf 0.42), ‘isopropyl alcoholvbenzene 15:85 (Rf= 0.5) and ethanol-benzene 20:80 (Rf f 0.43);
d. rapamycin obtained from four successive fermentation batchesgave the following values on repeated The production stage fermenters are equipped with 7 devices for controlling and maintaining pH at a predetermined level and for continuous metered addition of elemental analyses:
AVER- e. rapamycin exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum ethanol):
f. the infrared absorption spectrum of rapamycin in chloroform is reproduced in FIG. 1 and shows characteristic absorption bands at 3560, 3430, 1730, 1705 and 1630-1610 cm;
Further infrared absorption bands are characterized by the following data given in reciprocal centimeters with (s) denoting a strong, (m) denoting a medium, and ![]() denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak
denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak ![]() if its peak is less than one-third of the background in the same region.
if its peak is less than one-third of the background in the same region.
2990 cm (m) 1158 cm” (m) 2955 cm (s) 1129 cm (s) 2919 cm (s) 1080 cm (s) 2858 cm (s) 1060 cm (s) 2815 cm (m) 1040 cm (m) 1440 cm (s) 1020 crn’ (m) 1365 cm (m) 978 cm” (s) 1316 cm (in) 905 cm (m) 1272 cm (m) 888 cm” ![]() 1178 cm (s) 866 cm-
1178 cm (s) 866 cm- ![]() g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
CLAIMS
l. Rapamycin, an antibiotic which a. is a colourless, crystalline compound with a melting point of 183 to l8SC, after recrystallization from ether;
b. is soluble in ether, chloroform, acetone, methanol and dimethylformamide, very sparingly soluble in hexane and petroleum ether and substantially insoluble in water;
c. shows a uniform spot on thin layer plates of silica gel”,
d. has a characteristic elemental analysis of about C,
e. exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum (95% ff has ‘a characteristic infrared absorption spectrum shown in accompanying FIG. 1; SEE PATENT
……………………………………………..
Rapamycin synthetic studies. 1. Construction of the C(27)-C(42) subunit. Tetrahedron Lett 1994, 35, 28, 4907

A partial synthesis of rapamycin has been reported: The condensation of sulfone (I) with epoxide (II) by means of butyllithium followed by desulfonation with Na/Hg gives the partially protected diol (III), which is treated with methanesulfonyl chloride and NaH to afford the epoxide (IV). Ring opening of epoxide (IV) with LiI and BF3.Et2O followed by protection of the resulting alcohol with PMBOC(NH)CCl3 yields the primary iodo compound (V). The condensation of (V) with the fully protected dihydroxyaldehyde (VI) (see later) by means of butyllithium in THF/HMPT gives the fully protected trihydroxyketone (VII), which is hydrolyzed with camphorsulfonic acid (CSA) to the corresponding gemdiol and reprotected with pivaloyl chloride (the primary alcohol) and tert-butyldimethylsilyl trifluoromethanesulfonate (the secondary alcohol), yielding a new fully protected trihydroxyketone (VIII). Elimination of the pivaloyl group with DIBAL and the dithiane group with MeI/CaCO3 affords the hydroxyketone (IX), which is finally oxidized with oxalyl chloride to the ketoaldehyde (X), the C(27)-C(42) fragment [the C(12)-C(15) fragment with the C(12)-substituent based on the IUPAC nomenclature recommendations]. The fully protected dihydroxyaldehyde (VI) is obtained as follows: The reaction of methyl 3-hydroxy-2(R)-methylpropionate (XI) with BPSCl followed by reduction with LiBH4 to the corresponding alcohol and oxidation with oxalyl chloride gives the aldehyde (XII), which is protected with propane-1,3-dithiol and BF3.Et2O to afford the dithiane compound (XIII). Elimination of the silyl group with TBAF followed by esterification with tosyl chloride, reaction with NaI and, finally, with sodium phenylsulfinate gives the sulfone (XIV), which is condensed with the partially protected dihydroxyaldehyde (XV), oxidized with oxalyl chloride and desulfonated with Al/Hg to afford the dithianyl ketone (XVI). The reaction of (XVI) with lithium hexamethyldisilylazane gives the corresponding enolate, which is treated with dimethyllithium cuprate to yield the fully protected unsaturated dihydroxyaldehyde (VI).
……………………………………………
……………………………
The Ley Synthesis of RapamycinRapamycin (3) is used clinically as an immunosuppressive agent. The synthesis of 3 (Angew. Chem. Int. Ed. 2007, 46, 591. DOI: 10.1002/anie.200604053) by Steven V. Ley of the University of Cambridge was based on the assembly and subsequent coupling of the iododiene 1 and the stannyl alkene 2.
The lactone of 1 was prepared by Fe-mediated cyclocarbonylation of the alkenyl epoxide 5, following the protocol developed in the Ley group.
The cyclohexane of 2 was constructed by SnCl4-mediated cyclization of the allyl stannane 9, again employing a procedure developed in the Ley group. Hydroboration delivered the aldehyde 11, which was crotylated with 12, following the H. C. Brown method. The alcohol so produced (not illustrated) was used to direct the diastereoselectivity of epoxidation, then removed, to give 13. Coupling with 14 then led to 2.
Combination of 1 with 2 led to 15, which was condensed with catechol to give the macrocycle 16. Exposure of 16 to base effected Dieckmann cyclization, to deliver the ring-contracted macrolactone 17, which was carried on to (-)-rapamycin (3).
|




……………………………….
Total Synthesis of Rapamycin
Angewandte Chemie International Edition
Volume 46, Issue 4, pages 591–597, January 15, 2007

PREVIEW THIS ARTICLE WITH READCUBE
![]()
……………………..

Ley, Maddess, Tackett, Watanabe, Brennan, Spilling, Scott and Osborn. ACIEE, 2006, EarlyView. DOI:10.1002/anie.200604053.
It’s been in the works for quite a while, but Steve Ley’s synthesis of Rapamycin has just been published. This complex beast has a multitude of biological activities, including an interesting immunosuppressive profile, resulting in clinical usage following organ transplantation. So, unsurprisingly, it’s been the target of many projects, with complete total syntheses published by Smith, Danishefsky, Schreiber and KCN.
So what makes this one different? Well, it does have one of the most interesting macrocyclisations I’ve seen since Jamison’s paper, and a very nice demonstration of the BDA-aldol methodology. The overall strategy is also impressive, so on with the retro:

First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).

The rest of the fragment synthesis was completed in a succinct and competent manner, but using relatively well known chemistry. However, I was especially impressed with the macrocyclisation I mentioned:

Tethering the free ends of the linear precursor with a simple etherification/esterification onto catechol gave then a macrocycle holding the desired reaction centres together. Treatment of this with base then induces a Dieckmann-condensation type cyclisation to deliver the desired macrocycle. Of course, at this stage, only a few more steps were required to complete the molecule, and end an era of the Wiffen Lab.
………………………………
Drugs Fut 1999, 24(1): 22
DOI: 10.1358/dof.1999.024.01.474036

In CDCl3 rapamycin exists as a mixture of conformers in a 3:1 ratio, which complicates the NMR spectrum. In the table below the chemical shifts of the carbons and hydrogens of the major isomer only are given.
| Carbon No. | Carbon Type | Major carbon | Major proton | Carbon No. | Carbon Type | Major carbon | Major proton |
|
1
|
C=O | 169.2 |
–
|
28
|
CH-OH | 77.3 | 4.17 |
|
2
|
CH | 51.3 | 5.29 |
29
|
C=C | 136.1 |
–
|
|
3
|
CH2 | 27.0 | 2.34, 1.76 |
30
|
CH=C | 126.8 | 5.42 |
|
4
|
CH2 | 20.6 | 1.78, 1.47 |
31
|
CH | 46.6 | 3.33 |
|
5
|
CH2 | 25.3 | 1.75, 1.48 |
32
|
C=O | 208.2 |
–
|
|
6
|
CH2 | 44.2 | 3.59, 3.44 |
33
|
CH2 | 40.7 | 2.74, 2.60 |
|
8
|
C=O | 166.8 |
–
|
34
|
CH-OCO | 75.7 | 5.17 |
|
9
|
C=O | 192.5 |
–
|
35
|
CH | 33.1 | 1.98 |
|
10
|
O-C-OH | 98.5 |
–
|
36
|
CH2 | 38.4 | 1.22, 1.12 |
|
11
|
CH | 33.7 | 1.98 |
37
|
CH | 33.2 | 1.39 |
|
12
|
CH2 | 27.3 | 1.60, 1.60 |
38
|
CH2 | 34.2 | 2.10, 0.68 |
|
13
|
CH2 | 31.3 | 1.62, 1.33 |
39
|
CH-OCH3 | 84.4 | 2.93 |
|
14
|
67.2 | 3.86 |
40
|
CH-OH | 73.9 | 3.37 | |
|
15
|
CH2 | 38.8 | 1.85, 1.52 |
41
|
CH2 | 31.3 | 1.99, 1.33 |
|
16
|
CH-OCH3 | 84.4 | 3.67 |
42
|
CH2 | 31.7 | 1.70, 1.00 |
|
17
|
C=C | 135.5 |
–
|
43
|
11-CH3 | 16.2 | 0.95 |
|
18
|
CH=C | 129.6 | 5.97 |
44
|
17-CH3 | 10.2 | 1.65 |
|
19
|
CH=C | 126.4 | 6.39 |
45
|
23-CH3 | 21.5 | 1.05 |
|
20
|
CH=C | 133.6 | 6.32 |
46
|
25-CH3 | 13.8 | 1.00 |
|
21
|
CH=C | 130.1 | 6.15 |
47
|
29-CH3 | 13.0 | 1.74 |
|
22
|
CH=C | 140.2 | 5.54 |
48
|
31-CH3 | 16.0 | 1.11 |
|
23
|
CH | 35.2 | 2.32 |
49
|
35-CH3 | 15.9 | 0.92 |
|
24
|
CH2 | 40.2 | 1.50, 1.20 |
50
|
16-OCH3 | 55.8 | 3.13 |
|
25
|
CH | 41.4 | 2.74 |
51
|
27-OCH3 | 59.5 | 3.34 |
|
26
|
C=O | 215.6 |
–
|
52
|
39-OCH3 | 56.5 | 3.41 |
|
27
|
CH-OCH3 | 84.9 | 3.71 |
REFERENCES
- Vézina C, Kudelski A, Sehgal SN (October 1975). “Rapamycin (AY-22,989), a new antifungal antibiotic”. J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Pritchard DI (2005). “Sourcing a chemical succession for cyclosporin from parasites and human pathogens”. Drug Discovery Today 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
Wu X, Wang L, Han Y, Regan N, Li PK, Villalona MA, Hu X, Briesewitz R, Pei D.
ACS Comb Sci. 2011 Sep 12;13(5):486-95. doi: 10.1021/co200057n. Epub 2011 Jul 28.
Gibbons JJ, Abraham RT, Yu K.
Semin Oncol. 2009 Dec;36 Suppl 3:S3-S17. doi: 10.1053/j.seminoncol.2009.10.011. Review.
Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
6. Fluorescent probes to characterise FK506-binding proteins.
Kozany C, März A, Kress C, Hausch F.
Chembiochem. 2009 May 25;10(8):1402-10. doi: 10.1002/cbic.200800806.
7. Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs.
Graziani EI.
Nat Prod Rep. 2009 May;26(5):602-9. doi: 10.1039/b804602f. Epub 2009 Mar 5. Review.
8 Total synthesis of rapamycin.
Ley SV, Tackett MN, Maddess ML, Anderson JC, Brennan PE, Cappi MW, Heer JP, Helgen C, Kori M, Kouklovsky C, Marsden SP, Norman J, Osborn DP, Palomero MA, Pavey JB, Pinel C, Robinson LA, Schnaubelt J, Scott JS, Spilling CD, Watanabe H, Wesson KE, Willis MC.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
Evans AC, Longbottom DA, Matsuoka M, Davies JE, Turner R, Franckevicius V, Ley SV.
Org Biomol Chem. 2009 Feb 21;7(4):747-60. doi: 10.1039/b813494d. Epub 2009 Jan 6.
Maddess ML, Tackett MN, Ley SV.
Prog Drug Res. 2008;66:13, 15-186. Review.
Zhang J, Rodila R, Watson P, Ji Q, El-Shourbagy TA.
Biomed Chromatogr. 2007 Oct;21(10):1036-44.
Sormani R, Yao L, Menand B, Ennar N, Lecampion C, Meyer C, Robaglia C.
BMC Plant Biol. 2007 Jun 1;7:26.
13 Total synthesis of rapamycin.
Maddess ML, Tackett MN, Watanabe H, Brennan PE, Spilling CD, Scott JS, Osborn DP, Ley SV.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
14 Drug evaluation: AP-23573–an mTOR inhibitor for the treatment of cancer.
Elit L.
IDrugs. 2006 Sep;9(9):636-44.
15 lipase-catalyzed regioselective esterification of rapamycin: synthesis of temsirolimus (CCI-779).
Gu J, Ruppen ME, Cai P.
Org Lett. 2005 Sep 1;7(18):3945-8.
Elit L.
Curr Opin Investig Drugs. 2002 Aug;3(8):1249-53. Review.
Dumont FJ.
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
 SIROLIMUS
SIROLIMUS
FEMALE FERTILITY
PATENTS
| Canada | 2293793 | APPROVED2006-07-11 | EXP 2018-06-11 |
| Canada | 2103571 | 2003-04-29 | 2012-02-21 |
| United States | 5989591 | 1998-09-11 | 2018-09-11 |
| United States | 5212155 | 1993-05-18 | 2010-05-18 |
| WO1998054308A2 * | May 28, 1998 | Dec 3, 1998 | Biotica Tech Ltd | Polyketides and their synthesis and use |
| EP0589703A1 * | Sep 23, 1993 | Mar 30, 1994 | American Home Products Corporation | Proline derivative of rapamycin, production and application thereof |
| US20010039338 * | Jun 7, 2001 | Nov 8, 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO2007067560A2 * | Dec 6, 2006 | Jun 14, 2007 | Clifford William Coughlin | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| WO2012131019A1 | Mar 30, 2012 | Oct 4, 2012 | Sandoz Ag | Regioselective acylation of rapamycin at the c-42 position |
| US7622578 | Dec 6, 2006 | Nov 24, 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US3929992 | Apr 12, 1974 | Dec 30, 1975 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US5646160 | May 26, 1995 | Jul 8, 1997 | American Home Products Corporation | Method of treating hyperproliferative vascular disease with rapamycin and mycophenolic acid |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5728710 | Jul 16, 1993 | Mar 17, 1998 | Smithkline Beecham Corporation | Rapamycin derivatives |
| US5957975 | Dec 15, 1997 | Sep 28, 1999 | The Centre National De La Recherche Scientifique | Stent having a programmed pattern of in vivo degradation |
| US5985890 | Jun 5, 1996 | Nov 16, 1999 | Novartis Ag | Rapamycin derivatives |
| US6001998 | Oct 13, 1995 | Dec 14, 1999 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6015815 | Sep 24, 1998 | Jan 18, 2000 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| US6187568 | Aug 20, 1999 | Feb 13, 2001 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6273913 | Apr 16, 1998 | Aug 14, 2001 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US6585764 | Jun 4, 2001 | Jul 1, 2003 | Cordis Corporation | Stent with therapeutically active dosage of rapamycin coated thereon |
| US6641611 | Nov 26, 2001 | Nov 4, 2003 | Swaminathan Jayaraman | Therapeutic coating for an intravascular implant |
| US6805703 | Sep 18, 2001 | Oct 19, 2004 | Scimed Life Systems, Inc. | Protective membrane for reconfiguring a workpiece |
| US7025734 | Sep 28, 2001 | Apr 11, 2006 | Advanced Cardiovascular Systmes, Inc. | Guidewire with chemical sensing capabilities |
| US7056942 | Jan 16, 2004 | Jun 6, 2006 | Teva Pharmaceutical Industries Ltd. | Carvedilol |
| US7820812 * | Jul 23, 2007 | Oct 26, 2010 | Abbott Laboratories | Methods of manufacturing crystalline forms of rapamycin analogs |
| US20010027340 | Jun 4, 2001 | Oct 4, 2001 | Carol Wright | Stent with therapeutically active dosage of rapamycin coated thereon |
| US20010029351 | May 7, 2001 | Oct 11, 2001 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020005206 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Antiproliferative drug and delivery device |
| US20020007213 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US20020082680 | Sep 7, 2001 | Jun 27, 2002 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US20020123505 | Sep 10, 2001 | Sep 5, 2002 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20030129215 | Sep 6, 2002 | Jul 10, 2003 | T-Ram, Inc. | Medical devices containing rapamycin analogs |
| US20040072857 | Jul 2, 2003 | Apr 15, 2004 | Jacob Waugh | Polymerized and modified rapamycins and their use in coating medical prostheses |
| US20050033417 | Jul 1, 2004 | Feb 10, 2005 | John Borges | Coating for controlled release of a therapeutic agent |
| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050152842 | Dec 22, 2004 | Jul 14, 2005 | Chun Li | Poly (L-glutamic acid) paramagnetic material complex and use as a biodegradable MRI contrast agent |
| US20050175660 | Oct 29, 2004 | Aug 11, 2005 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20050208095 | Nov 22, 2004 | Sep 22, 2005 | Angiotech International Ag | Polymer compositions and methods for their use |
| US20050209244 | Feb 27, 2003 | Sep 22, 2005 | Prescott Margaret F | N{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine coated stents |
| US20050239178 | Apr 25, 2005 | Oct 27, 2005 | Wyeth | Labeling of rapamycin using rapamycin-specific methylases |
| US20060094744 | Sep 28, 2005 | May 4, 2006 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US20060229711 | Apr 4, 2006 | Oct 12, 2006 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697 | Nov 1, 2005 | Jan 18, 2007 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
| US20070059336 | Feb 27, 2006 | Mar 15, 2007 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US20070207186 | Mar 3, 2007 | Sep 6, 2007 | Scanlon John J | Tear and abrasion resistant expanded material and reinforcement |
| US20080086198 | May 24, 2007 | Apr 10, 2008 | Gary Owens | Nanoporous stents with enhanced cellular adhesion and reduced neointimal formation |
| EP1236478A1 | Feb 27, 2002 | Sep 4, 2002 | Medtronic Ave, Inc. | Peroxisome proliferator-activated receptor gamma ligand eluting medical device |
| EP1588727A1 | Apr 20, 2005 | Oct 26, 2005 | Cordis Corporation | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO1993016189A1 | Feb 11, 1993 | Aug 19, 1993 | Pfizer | Novel macrocyclic lactones and a productive strain thereof |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO1996041807A1 | Jun 5, 1996 | Dec 27, 1996 | Sylvain Cottens | Rapamycin derivatives |
| WO1998007415A2 | Aug 18, 1997 | Feb 26, 1998 | Ciba Geigy Ag | Methods for prevention of cellular proliferation and restenosis |
| WO2001087263A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087342A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087372A1 | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| WO2001087373A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087374A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087375A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087376A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO2002056790A2 | Dec 18, 2001 | Jul 25, 2002 | Avantec Vascular Corp | Delivery of therapeutic capable agents |
| WO2002065947A2 | Feb 18, 2002 | Aug 29, 2002 | Jomed Gmbh | Implants with fk506 for prophylaxis and treatment of restonoses |
| WO2003064383A2 | Feb 3, 2003 | Aug 7, 2003 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| WO2006116716A2 | Apr 27, 2006 | Nov 2, 2006 | William A Dunn | Materials and methods for enhanced degradation of mutant proteins associated with human disease |

![]()
A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
mTOR inhibitor
temsirolimus (CCI-779), everolimus (RAD001), deforolimus (AP23573), AP21967, biolimus, AP23102, zotarolimus (ABT 578), sirolimus (Rapamune), and tacrolimus (Prograf).\
SIROLIMUS
1H NMR

13 C NMR

HPLC


{kind=link}
{kind=link}