
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

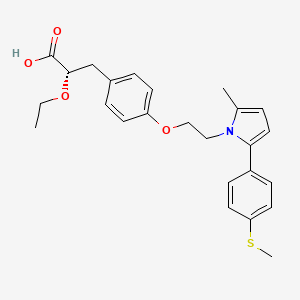
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
-
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |


 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid | |
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Mol. mass | 439.56 g/mol |
MORE DETAILS
Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable glycemic control by reducing the fasting plasma glucose and HBA1c in diabetes patients.
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched . The company is also developing the drug for the potential treatment of lipodystrophy. In May 2014, a phase III trial was initiated . In June 2012, the company was seeking to outlicense the drug for regional/global partnerships
By June 2012, an NDA filing had been made for dyslipidemia. In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
_MoA.png)
Clinical trials
The approval for saroglitazar was based on the results obtained from clinical studies, which were conducted for more than eight years.
The studies evaluated the efficacy, safety, pharmacokinetics and pharmacodynamics of the drug. Phase I clinical trials on saroglitazar were conducted in 2005. The highest dose of saroglitazar evaluated in a Phase I trial was 128 mg, several times the estimated therapeutic doses (1–4 mg). The pharmacokinetics of saroglitazar support a once daily dosage schedule. No serious adverse events were reported.[3] Phase II studies were completed in 2006.
The Phase III clinical trials were conducted between 2008 and 2011. The first Phase III clinical trials on saroglitazar compared saroglitazar 4 mg dose with pioglitazone 45 mg. The results of the study demonstrated that patients who were administered with saroglitazar 4 mg dose showed reduction in LDL cholesterol and triglycerides, and increase in HDL cholesterol. The study also showed that saroglitazar administered patients showed a reduction in fasting plasma glucose and glycosylated hemoglobin.
Saroglitazar 2 mg and 4 mg significantly reduced (P < 0.001) plasma triglycerides from baseline by 26.4% (absolute change ± SD: −78.2 ± 81.98 mg/dL) and 45% (absolute change ± SD −115.4 ± 68.11 mg/dL), respectively, as compared to pioglitazone -15.5% (absolute change ± SD: −33.3 ± 162.41 mg/dL) at week 24. Saroglitazar 4 mg treatment also demonstrated marked decrease in low-density lipoprotein (5%), very-low-density lipoprotein (45.5%), total cholesterol (7.7%), and apolipoprotein-B (10.9%).[4]
The second Phase III clinical trials on saroglitazar were conducted to evaluate the diabetic dyslipidemic patients insufficiently controlled with statin therapy. The second Phase III study results showed that patients treated with saroglitazar showed pronounced beneficial effect on both the lipid and glycaemic parameters.
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by -45.5±3.03% and -46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.[5]
Safety
Saroglitazar was found to be safe and well tolerated during the clinical program. In Phase III trials, There was no edema or weight gain reported in any of the study arms. During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no adverse events reported as far as cardiac safety is concerned.
After 12 weeks of treatment, there were a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, andγ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms.[6][7]
In Phase I clinical trials saroglitazar was used up to 128 mg and found well tolerated. No serious adverse events were reported. Adverse events were generally mild and moderate in nature and did not show any clinically relevant findings in clinical laboratory investigations, physical examinations, vital signs and electrocardiograms.[8]
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
PATENT
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
References
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
- “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
-
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- 7 “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- 8 “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O


at least one NCE from india
LikeLike
Reblogged this on Vijaya Shastry Ph.D Physical Chemistry.
LikeLike
Reblogged this on My Blog on Medicinal Chemistry.
LikeLike
Reblogged this on GREEN MED CHEMISTRY.
LikeLike
Pls inform whether it is available in market for patient use. Send me product monogram of Lipaglyn.
LikeLike
thanks to being first indian company ………..plz send product monograph
LikeLike
Reblogged this on New Drug Approvals.
LikeLike
It’s worth noting thazt other kinds of impairments like dementia
would not show that mch disparity, and is also only appliable on Alzheimer’s disease so
far. At Always Beest Care in Chapel Hill, we know that as
your spouse and childcren gett older, they’re going to
start to fasce new challenges that youu can not have predicted.
Alzheimer’s disease is a commmon type of dementia the place that the brain tissue degenerates.
LikeLike
[…] https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/ […]
LikeLike
Hello,
Is it FDA approved ?
LikeLike
[…] READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/ […]
LikeLike