
Home » Posts tagged 'organic synthesis'
Tag Archives: organic synthesis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Simple and effective method for two-step synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile

Simple and effective method for two-step synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile (75% overall yield) and molecular modeling calculation of the mechanism by B3LYP and the 6-311++G(2df,2p) basis set.
http://dx.doi.org/10.5935/0100-4042.20140308
Método alternativo para a síntese e mecanismo de 2-(1,3-ditiano-2-ilideno)-acetonitrila
Marcelle S. Ferreira; José D. Figueroa-Villar*
Quim. Nova, Vol. 38, No. 2, 233-236, 2015
Artigo http://dx.doi.org/10.5935/0100-4042.20140308
*e-mail: jdfv2009@gmail.com
MÉTODO ALTERNATIVO PARA A SÍNTESE E MECANISMO DE 2-(1,3-DITIANO-2-ILIDENO)-ACETONITRILA
Marcelle S. Ferreira e José D. Figueroa-Villar* Departamento de Química, Instituto Militar de Engenharia, Praça General Tiburcio 80, 22290-270
Rio de Janeiro – RJ, Brasil
Recebido em 18/08/2014; aceito em 15/10/2014; publicado na web em 12/12/2014
ALTERNATIVE METHOD FOR SYNTHESIS AND MECHANISM OF 2-(1,3-DITHIAN-2-YLIDENE)-ACETONITRILE. We report an alternative method for the synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile using 3-(4-chlorophenyl)-3-oxopropanenitrile and carbon disulfide as starting materials. The methanolysis of the intermediate 3-(4-chlorophenyl)-2-(1,3-dithian-2-ylidene)-3- oxopropanenitrile occurs via three possible intermediates, leading to the formation of the product at a 75% overall yield. Molecular modeling simulation of the reaction pathway using B3LYP 6-311G++(2df,2p) justified the proposed reaction mechanism. Keywords: 2-(1,3-dithian-2-ylidene)-acetonitrile; reaction mechanism; methanolysis; molecular modeling.
3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3): Cristal amarelo. Rendimento: 95%, 2,80 g, pf 158-160 °C, lit.21 159-160 °C;
IV (KBr, cm-1): 2198 (CN), 1612 (C=O), 1585, 1560 (aromático), 678 cm -1 (C-S);
1H RMN (300 MHz, CDCl3) δ 2,38 (m, J 6,9, 2H, CH2); 3,01 (t, J 6,6, 2H, SCH2); 3,17 (t, J 7,2 , 2H, SCH2); 7,43 (d, J 8,5, 2H); 7,83 (d, J 8,5, 2H);
13C RMN (75 MHz, CDCl3) δ 23,9 (CH2), 30,4 (SCH2), 104,2 (CCO), 117,5 (CN), 128,9, 130,5, 135,6, 139,2 (aromático), 185,2 (C=CS), 185,4 (CO).
21…….Rudorf, W. D.; Augustin, M.; Phosphorus Sulfur Relat. Elem. 1981, 9, 329.
…………………………………….
Síntese da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) Em um balão de fundo redondo de 100 mL foram adicionados 0,400 g (1,4 mmol) de 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3- -oxopropanonitrila (2) dissolvidos em 15 mL de THF seco, 0,140 g (20 mmol) de sódio e 15 mL de metanol seco sob atmosfera de nitrogênio. A mistura reacional foi mantida sob agitação à 25 °C por 48 h. Em seguida, a mistura reacional foi dissolvida em 30 mL de água destilada e extraída com acetato de etila (3 x 20 mL). A fase orgânica foi seca em sulfato de sódio anidro, filtrada e concentrada a vácuo para se obter o produto bruto, que foi purificado por cromatografia em coluna (silica gel e hexano:acetato de etila 7:3).
2-(1,3-ditiano-2-ilideno)-acetonitrila (1): Cristal branco. Rendimento: 75%, 165 mg, pf. 60-63 °C, lit1 60-62 °C;
1 H RMN (300 MHz, CDCl3) δ 2,23 (m, J 6,8, 2H, CH2); 3,01 (t, J 7,5, 2H, SCH2); 3,06 (t, J 6,9, 2H, SCH2), 5,39 (s, 1H, CH);
13C RMN (75 MHz, CDCl3) δ 22,9 (CH2), 28,7 (SCH2), 28,8 (SCH2), 90,4 (CHCN), 116,3 (CN), 163,8 (C=CS).
1………Yin, Y.; Zangh, Q.; Liu, Q.; Liu, Y.; Sun, S.; Synth. Commun. 2007, 37, 703.

CAS 113998-04-2
- C6 H7 N S2
- Acetonitrile, 2-(1,3-dithian-2-ylidene)-
- 157.26
| Melting Point | 60-62 °C |
1H NMR predict
2-(1,3-dithian-2-ylidene)-acetonitrile
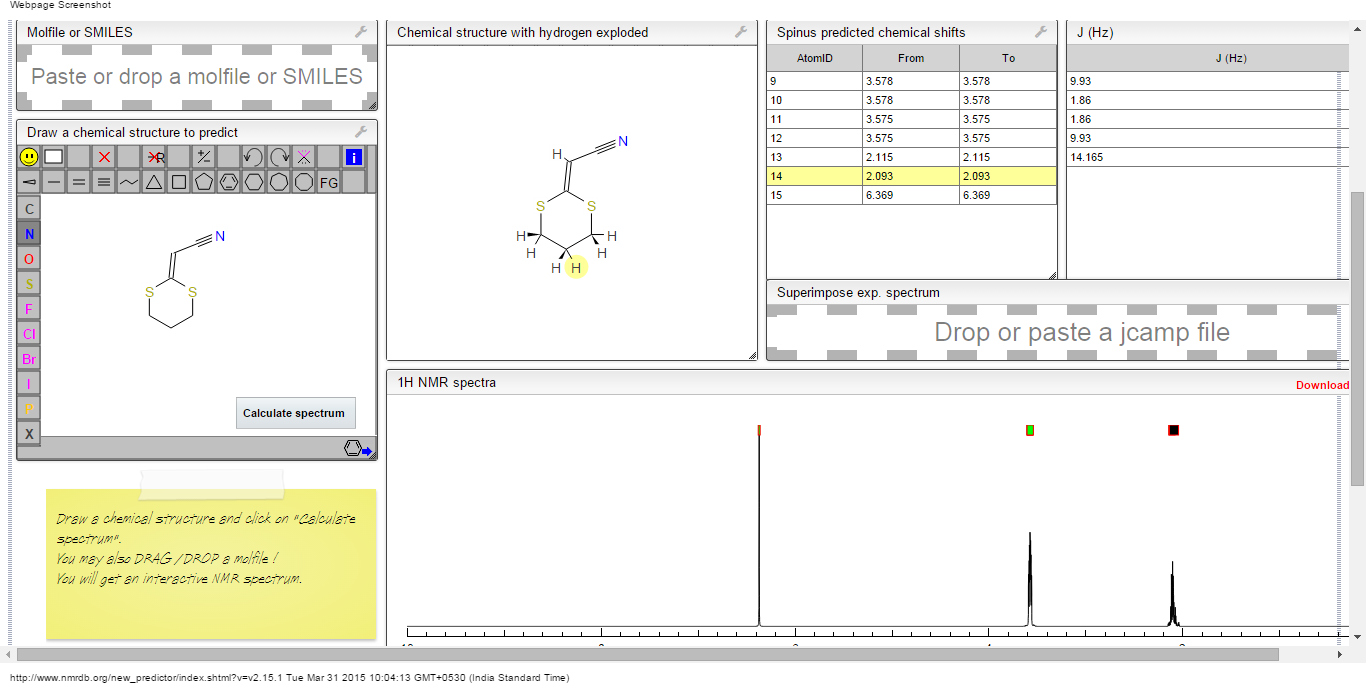

ACTUAL 1H NMR VALUES
1 H RMN (300 MHz, CDCl3)
δ 2,23 (m, J 6,8, 2H, CH2);
3,01 (t, J 7,5, 2H, SCH2);
3,06 (t, J 6,9, 2H, SCH2),
5,39 (s, 1H, CH);
……………………..
13C NMR PREDICT
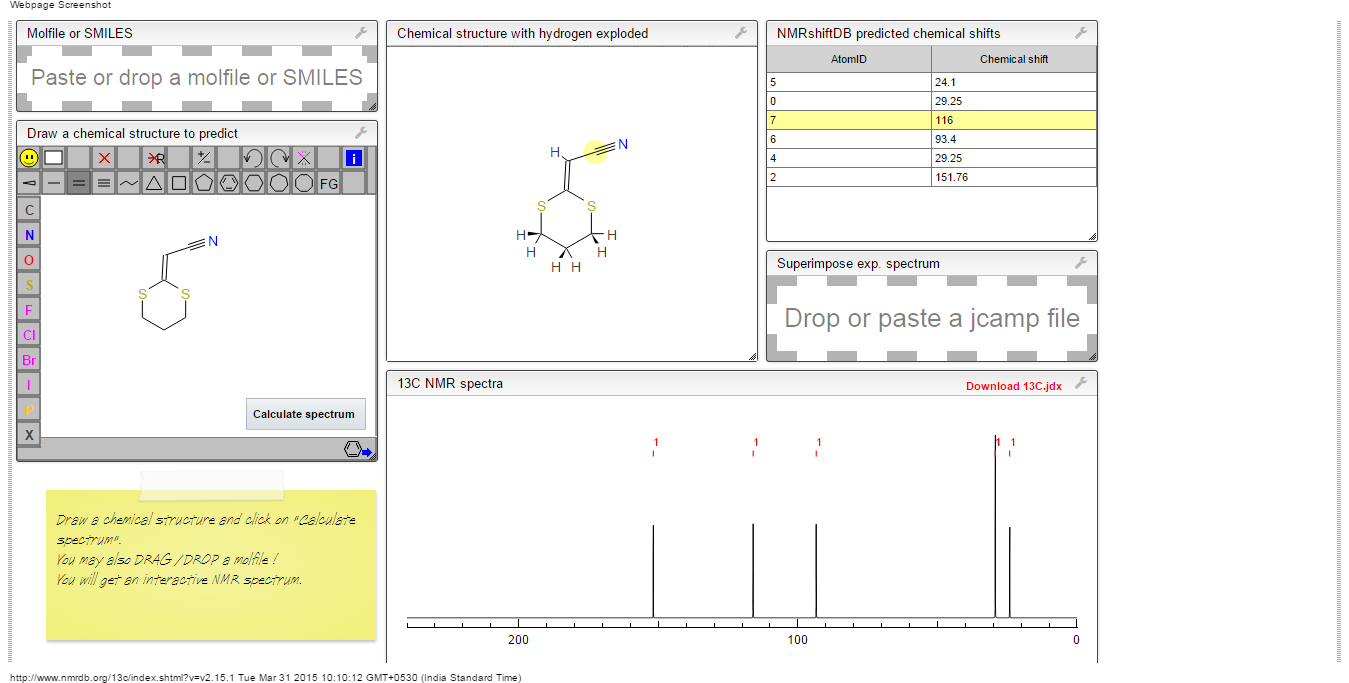

ACTUAL 13C NMR VALUE
13C RMN (75 MHz, CDCl3)
δ 22,9 (CH2),
28,7 (SCH2),
28,8 (SCH2),
90,4 (CHCN),
116,3 (CN),
163,8 (C=CS)
COSY NMR PREDICT

SYNTHESIS
2-(1,3-ditiano-2-ilideno)-acetonitrila (1): Cristal branco. Rendimento: 75%, 165 mg, pf. 60-63 °C, lit1 60-62 °C;
1 H RMN (300 MHz, CDCl3) δ 2,23 (m, J 6,8, 2H, CH2); 3,01 (t, J 7,5, 2H, SCH2); 3,06 (t, J 6,9, 2H, SCH2), 5,39 (s, 1H, CH);
13C RMN (75 MHz, CDCl3) δ 22,9 (CH2), 28,7 (SCH2), 28,8 (SCH2), 90,4 (CHCN), 116,3 (CN), 163,8 (C=CS).
WILL BE UPDATED WATCH OUT…………………
Departamento de Química, Instituto Militar de Engenharia, Praça General Tiburcio

Instituto Militar de Engenharia, Rio de Janeiro. BELOW


Entrada do antigo Instituto de Química da UFRGS, um prédio histórico

Equipe – Os módulos foram fabricados na Unisanta sob a supervisão do professor Luiz Renato Lia, coordenador do Curso de Engenharia Química, …

Instituto de Florestas da Universidade Federal Rural do Rio de Janeiro

Praça General Tibúrcio

Praça General Tibúrcio com o Morro da Urca ao fundo
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

A Microcapillary Flow Disc (MFD) Reactor for Organic Synthesis

A Microcapillary Flow Disc (MFD) Reactor for Organic Synthesis,
C.H. Hornung, M.R. Mackley, I.R. Baxendale and S.V. Ley and, Org. Proc. Res. Dev., 2007, 11, 399-405.
http://pubs.acs.org/doi/abs/10.1021/op700015f
This paper reports proof of concept, development, and trials for a novel plastic microcapillary flow disc (MFD) reactor. The MFD was constructed from a flexible, plastic microcapillary film (MCF), comprising parallel capillary channels with diameters in the range of 80−250 μm. MCFs were wound into spirals and heat treated to form solid discs, which were then capable of carrying out continuous flow reactions at elevated temperatures and pressures and with a controlled residence time. Three reaction schemes were conducted in the system, namely the synthesis of oxazoles, the formation of an allyl-ether, and a Diels−Alder reaction. Reaction scales of up to four kilograms per day could be achieved. The potential benefits of the MFD technology are compared against those of other reactor geometries including both conventional lab-scale and other microscale devices.
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Matthew A. J. Duncton† *
Renovis, Inc. (a wholly-owned subsidiary of Evotec AG), Two Corporate Drive, South San Francisco, CA 94080, United States. E-mail: mattduncton@yahoo.com; Tel: +1 917-345-3183
First published on the web 22nd August 2011
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
The addition of a radical to a heteroaromatic base is commonly referred to as a Minsici reaction. Such reactions constitute a broad-set of selective CH-functionalization processes. This review describes some of the major applications of Minisci reactions and related processes to medicinal or biological chemistry, and highlights some potential developments within this area.
Introduction
The aim of this review is to summarize the use of Minisci reactions within medicinal chemistry, and to highlight some future opportunities to continue progression of this chemistry. As such, it is not an aim that detailed mechanistic information, or a comprehensive list of examples be described. For this, the reader is directed to excellent articles from Minisci, Harrowven and Bowman.1–3 Rather, the review is written to show that Minisci reactions are extremely valuable CH-functionalization processes within medicinal chemistry. However, their use has been somewhat under-utilized when compared with other well-known selective transformations (e.g. palladium-catalysed cross-couplings). Therefore, it is hoped that in the future, Minisci chemistry will continue to develop, such that the reactions become a staple-set of methods for medicinal and biological chemists alike.
To aid discussion, the review is divided in to several sections. First, some historical perspective is given. This is followed by a discussion of scope and limitations. The main-body of the review describes some specific examples of Minisci reactions and related processes, with a focus on their use within medicinal, or biological chemistry. Finally, brief mention is given to potential future applications, some of which may be beneficial in providing ‘high-content’ diverse libraries for screening.
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
…………………….
WIKI
The Minisci reaction is a named reaction in organic chemistry. It is a radical substitution to an aromatic compound, in particular to a heteroaromatic base, that introduces an alkyl group. The reaction was published about in 1971 by F. Minisci.[1] The aromatic compound is generally electron-deficient and with N-aromatic compounds the nitrogen atom is protonated.[2] A typical reaction is that between pyridine and pivalic acid to 2-tert-butylpyridine with silver nitrate, sulfuric acid and ammonium persulfate. The reaction resembles Friedel-Crafts alkylation but with opposite reactivity and selectivity.[3]
The Minisci reaction proceeds regioselectively and enables the introduction of a wide range of alkyl groups.[4] A side-reaction is acylation.[5] The ratio between alkylation and acylation depends on the substrate and the reaction conditions. Due to the simple raw materials and the simple reaction conditions the reaction has many applications in heterocyclic chemistry.[6][7]

Mechanism
A free radical is formed from the carboxylic acid in an oxidative decarboxylation with silver salts and an oxidizing agent. The oxidizing agent reoxidizes the silver salt. The radical then reacts with the aromatic compound. The ultimate product is formed by rearomatisation. The acylated product is formed from the acyl radical.[4][5]

References
- F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo: Nucleophilic character of alkyl radicals—VI : A new convenient selective alkylation of heteroaromatic bases, in: Tetrahedron 1971, 27, 3575–3579.
- Minisci reaction Jie Jack Li in Name Reactions 2009, 361-362, doi:10.1007/978-3-642-01053-8_163
- Strategic applications of named reactions in organic synthesis: background and detailed mechanisms László Kürti, Barbara Czakó 2005
- F. Fontana, F. Minisci, M. C. N. Barbosa, E. Vismara: Homolytic acylation of protonated pyridines and pyrazines with α-keto acids: the problem of monoacylation, in: J. Org. Chem. 1991, 56, 2866–2869; doi:10.1021/jo00008a050.
- M.-L. Bennasar, T. Roca, R. Griera, J. Bosch: Generation and Intermolecular Reactions of 2-Indolylacyl Radicals, in: Org. Lett. 2001, 3, 1697–1700; doi:10.1021/ol0100576.
- P. B. Palde, B. R. McNaughton, N. T. Ross, P. C. Gareiss, C. R. Mace, R. C. Spitale, B. L. Miller: Single-Step Synthesis of Functional Organic Receptors via a Tridirectional Minisci Reaction, in: Synthesis 2007, 15, 2287–2290; doi:10.1055/s-2007-983792.
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.
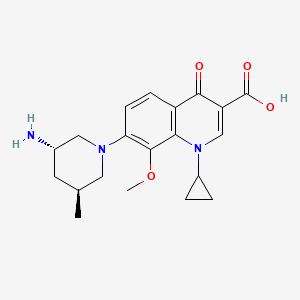
Nemonoxacin….TaiGen’s pneumonia antibiotic Taigexyn 奈诺沙星 gets marketing approval in Taiwan

Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
-
C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ — TaiGen Biotechnology Company, Limited (“TaiGen”) today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Nemonoxacin malate anhydrous
951163-60-3 CAS NO, MW: 505.5209
Nemonoxacin malate hemihydrate
951313-26-1, MW: 1029.0566
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


……………………..
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1…….DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1′) were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9′): Compound 9′ was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck’s Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9′) was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w – “ MsCI1TEA . „ _. – – _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1′)
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50″-65″C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1′ was synthesized from Compound 9′ as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
…………………
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck’s Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
………………..
REF
A. ARJONA ET AL: “Nemonoxacin“, DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: “TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia“, INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: “TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)“, 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: “TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic“, 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
|
7-4-2012
|
TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION
|
|
|
4-18-2012
|
Coupling Process For Preparing Quinolone Intermediates
|
|
|
10-19-2011
|
Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
|
|
|
6-18-2010
|
STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES
|
|
|
2-19-2010
|
PNEUMONIA TREATMENT
|
|
|
5-6-2009
|
Hydride reduction process for preparing quinolone intermediates
|
|
|
2-13-2009
|
ANTIMICROBIAL PARENTERAL FORMULATION
|
|
|
11-26-2008
|
Coupling process for preparing quinolone intermediates
|
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

TEDIGLUTIDE ..Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.


TEDUGLUTIDE
Glucagon-like peptide 2 (GLP-2) analog; protects small intestinal stem cells from radiation damage.
Gattex (teduglutide) is a recombinant analog of human glucagon-like peptide 2 for the treatment of adults with short bowel syndrome.
- (Gly2)GLP-2
- ALX 0600
- ALX-0600
- Gattex
- Gly(2)-GLP-2
- Teduglutide
- UNII-7M19191IKG
[Gly2]hGLP-2, [Gly2]-hGLP-2, ALX-0600,
Gattex, Revestive
| CAS number | 197922-42-2 |
|---|
L-histidylglycyl-L-α-aspartylglycyl-L-seryl-L-phenylalanyl-L-seryl-L-α-aspartyl-L-α-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-α-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-α-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid
| Formula | C164H252N44O55S |
|---|---|
| Mol. mass | 3752.082 g/mol |
Gattex, ALX-0600, (Gly2)GLP-2, Gly(2)-GLP-2, ALX 0600, [Gly2]GLP-2, Glucagon-like peptide II (2-glycine) (human), UNII-7M19191IKG
LAUNCHED 2013, NPS Pharmaceuticals
APPROVAL FDA
Company: NPS Pharmaceuticals, Inc.
Date of Approval: December 21, 2012 FDA
NDA 203441
POWDER; SUBCUTANEOUS GATTEX
U-1320=TREATMENT OF ADULT PATIENTS WITH SHORT BOWEL SYNDROME WHO ARE DEPENDENT ON PARENTERAL SUPPORT
| Patent No | Patent Expiry Date | Patent use code |
|---|---|---|
| 5789379 | Apr 14, 2015 | U-1320 |
| 7056886 | Sep 18, 2022 | U-1320 |
| 7847061 | Nov 1, 2025 | U-1320 |
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ORPHAN DRUG EXCLUSIVITY | Dec 21, 2019 |
| NEW CHEMICAL ENTITY | Dec 21, 2017 |
SEE FDA
http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203441Orig1s000lbl.pdf
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=Teduglutide+OR+ALX-0600
The active ingredient in GATTEX (teduglutide [rDNA origin]) for injection is teduglutide (rDNA origin), which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified byrecombinant DNA technology. The chemical name of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is:
Figure 1: Structural formula of teduglutide

Teduglutide has a molecular weight of 3752 Daltons. Teduglutide drug substance is a clear, colorless to light-straw–colored liquid.
Each single-use vial of GATTEX contains 5 mg of teduglutide as a white lyophilized powder for solution for subcutaneous injection. In addition to the active pharmaceutical ingredient (teduglutide), each vial of GATTEX contains 3.88 mg L-histidine, 15 mg mannitol, 0.644 mg monobasic sodium phosphate monohydrate, 3.434 mg dibasic sodium phosphate heptahydrate as excipients. No preservatives are present.
At the time of administration the lyophilized powder is reconstituted with 0.5 mL of Sterile Water for Injection, which is provided in a prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution. Up to 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn for subcutaneous injection upon reconstitution.
Teduglutide (brand names Gattex and Revestive) is a 36-membered polypeptide andglucagon-like peptide-2 analog that is used for the treatment of short bowel syndrome. It works by promoting mucosal growth and possibly restoring gastric emptying and secretion.[1] In Europe it is marketed under the brand Revestive by Nycomed. It was approved by the United States under the name Gattex on December 21, 2012.
Teduglutide is a proprietary analogue of glucagon-like peptide 2 (GLP-2) which was approved in the U.S. in December 2012 for the once-daily treatment of short-bowel syndrome in adults who are dependent on parenteral support. Commercial launch took place in 2013.The product was filed for approval in the E.U. in 2011 by Nycomed for this indication. In June 2012, a positive opinion was received in the E.U. and final approval was assigned in September 2012.
At NPS Pharmaceuticals, the compound is in phase III clinical development for this indication in pediatric patients and in phase II clinical studies for the treatment of Crohn’s disease. Preclinical studies are also ongoing at the company for the treatment of chemotherapy-induced enterocolitis and for the prevention and treatment of necrotizing enterocolitis (NEC) in preterm infants.
Teduglutide has been found to induce intestinal hyperplasia, reduce apoptosis and inflammation and improve cell barrier integrity in animal models. In 2001, orphan drug designation was assigned to teduglutide for the treatment of short-bowel syndrome.
In 2007, the compound was licensed to Nycomed for development and commercialization outside the U.S., Canada and Mexico for the treatment of gastrointestinal disorders. In 2012, the product was licensed to Neopharm by NPS Pharmaceuticals in Israel for development and commercialization for the treatment of gastrointestinal disorders.
The estimated prevalence of short bowel syndrome (SBS) patients with non-malignant disease requiring home parenteral nutrition (HPN) is at least 40 per million of the U.S. population. SBS usually results from surgical resection of some or most of the small intestine for conditions such as Crohn’s disease, mesenteric infarction, volvulus, trauma, congenital anomalies, and multiple strictures due to adhesions or radiation. Surgical resection may also include resection of all or part of the colon. SBS patients suffer from malabsorption that may lead to malnutrition, dehydration and weight loss. Some patients can maintain their protein and energy balance through hyperphagia; more rarely they can sustain fluid and electrolyte requirements to become independent from parenteral fluid.
Although long-term parenteral nutrition (PN) is life saving in patients with intestinal failure, it is expensive, impairs quality of life and is associated with serious complications such as catheter sepsis, venous occlusions and liver failure. Treatments that amplify absolute intestinal absorption, and eliminate or minimize the need for PN have great potential significance to SBS patients.
The endogenous meal-stimulated hormone, glucagon-like peptide-2 (GLP-2), raises considerable interest for SBS patients. GLP-2 functions to slow gastric emptying, reduce gastric secretions, increase intestinal blood-flow and stimulate growth of the small and large intestine. In animal studies, GLP-2 administration induces mucosal epithelial proliferation in the stomach and small and large intestine by stimulation of crypt cell proliferation and inhibition of enterocyte apoptosis.
SBS patients with end-jejunostomy and no colon have low basal GLP-2 levels and limited meal-stimulated GLP-2 secretion due to removal of GLP-2 secreting L-cells, which are located primarily in the terminal ileum and colon. This GLP-2 deficiency results in a minimal adaptive response following resection and could explain the gastric hypersecretion, rapid intestinal transit and lack of intestinal adaptation observed in these SBS patients.
Jeppesen et al. (Gastroenterology 2001; 120:806-815) have described positive benefit in an open-label study using pharmacologic doses of native GLP-2 in SBS jejunostomy patients. There was significant improvement in intestinal wet weight absorption and a more modest improvement in energy absorption that led to an increase in body weight, lean body mass and a rise in urinary creatinine excretion.
In contrast, SBS patients with colon-in-continuity have elevated basal endogenous GLP-2 levels resulting in an adaptive response to resection characterized by improved wet weight gain and energy absorption. The potential for added benefit of pharmacologic doses of GLP-2 receptor agonists in these patients is not obvious and has not been studied.

TEDUGLUTIDE
- Jeppesen PB (May 2012). “Teduglutide, a novel glucagon-like peptide 2 analog, in the treatment of patients with short bowel syndrome”. Therap Adv Gastroenterol 5 (3): 159–71. doi:10.1177/1756283X11436318. PMC 3342570. PMID 22570676.
- US 2013157954
- WO 2006050244
- WO 2005021022
- US 6586399
- WO 2002066062
- US 6297214
- US 2001021767
- WO 2001041779
- WO 1999058144
- WO 1998052600
Gattex Approved By FDA For Short Bowel Syndrome
 Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
Gattex (teduglutide) has been approved by the U.S. Food and Drug Administration to be used in patients that have short bowel syndrome and require parenteral nutrition.
The drug, once it is in the market, will compete against two others that have been approved by the FDA for this type of patient population. Those two medications are Nutrestore (glutamine) and Zorbtive (Somatropin).
Short bowel syndrome comes on following the removal surgically of part of the large or small intestine or part of both. Patients who are affected must have parenteral nutrition due to the poor absorption they have of nutrients and fluids. Teduglutide is injected one time each day and improves the absorption making it less important to have nutrition assistance.
The advisory committee for the FDA voted unanimously in October to recommend the drug’s approval after seeing the results from a pair of clinical trials that showed the advantage teduglutide had over just a placebo in at least a reduction of 20% in the amount of parenteral nutrition at 6 months.
During the first clinical trial, 46% of the patients that took the drug saw a level of reduction, which was compared to only 6% who had taken only a placebo. In the other study, the figure increased to 63%, while the placebo rated was up to 30%
The side effects most common found in those who use teduglutide during the trials included nausea, reactions around the injection site, abdominal pain abdominal distension and headaches.
………..
| US5789379 | Jun 28, 1996 | Aug 4, 1998 | 1149336 Ontario Inc. | Glucagon-like peptide-2 analogs |
| US6077949 | Apr 24, 1997 | Jun 20, 2000 | Allelix Biopharmaceuticals, Inc. | Cloned glucagon-like peptide 2 receptors |
| US6184201 * | Apr 8, 1997 | Feb 6, 2001 | Nps Allelix Corp. | Intestinotrophic glucagon-like peptide-2 analogs |
| US7411039 | Oct 14, 2003 | Aug 12, 2008 | Novo Nordisk A/S | GLP-2 compounds, formulations, and uses thereof |
| EP1231219A1 | Apr 11, 1997 | Aug 14, 2002 | 1149336 Ontario Inc. | GLucagon-like peptide-2 analogs |
| WO1997039031A1 | Apr 11, 1997 | Oct 23, 1997 | Allelix Biopharma | Glucagon-like peptide-2 analogs |
| WO1997039091A1 | Apr 16, 1997 | Oct 23, 1997 | Burckett St Laurent James Char | Mid-chain branched surfactants |
| WO2002066511A2 | Feb 15, 2002 | Aug 29, 2002 | Conjuchem Inc | Long lasting glucagon-like peptide 2 (glp-2) for the treatment of gastrointestinal diseases and disorders |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
TEDIZOLID (torezolid)
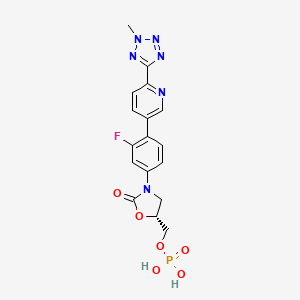
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
……………………………..
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
…………………………………………………….
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,

These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
…………………………………………………………..
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
…………………………………………..
imp patents
|
12-3-2010
|
OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE
|
|
|
10-20-2010
|
Oxazolidinone derivatives
|
|
|
7-31-2009
|
NOVEL OXAZOLIDINONE DERIVATIVES
|
…………………………………………..
TEDIZOLID disodium salt
59 nos in
http://www.google.com/patents/US20130102523

 38 nos
38 nos
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
……………………………………
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
………………………………………………………………………………………………………………………..
SYNTHESIS

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

………………………………………………………………………………………………………………………….
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
………………………………………………………………………………………………………………………………………………………….
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
…………………………………………………………………………………………………………………………………………..
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

………………………………………………………………………………………………………………………………………………………
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
………………………………………………………
IMPURITIES
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |
 |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |
 |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |
  |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |
 |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |
 |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |
 |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |
 |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |
 |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |
 |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
……………………………………………..
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
……………………………………………………………………………………….

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
EPROSARTAN MESYLATE

TEVETEN® (eprosartan mesylate) is a non-biphenyl non-tetrazole angiotensin II receptor (AT1) antagonist. A selective non-peptide molecule, TEVETEN® is chemically described as the monomethanesulfonate of (E)-2-butyl-1 -(p-carboxybenzyl)-α-2-thienylmethylimid-azole-5 -acrylic acid.
Its empirical formula is C23H24N2O4S•CH4O3S and molecular weight is 520.625. Its structural formula is:
 |
EPROSARTAN MESYLATE
tevetenEprosartan mesilate, SK&F-108566-J(?, SK&F-108566, Teveten SB, Navixen, Regulaten, Tevetenz, Teveten
| US 5656650 | exp Aug 12, 2014 |
CAS EPROSARTAN
144143-96-4
133040-01-4
| Chemical Name: | Eprosartan mesylate |
| Synonyms: | EPROSARTAN MESYLATE;Eprosartan Methanesulfonate;4-[[2-butyl-5-(2-carboxy-3-thiophen-2-yl-prop-1-enyl)-imidazol-1-yl]methyl]benzoic acid mesylate;4-({2-butyl-5-[(1E)-2-carboxy-2-(thiophen-2-ylMethyl)eth-1-en-1-yl]-1H-iMidazol-1-yl}Methyl)benzoic acid;(E)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid Methanesulfonate;(αE)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid MonoMethanesulfonate |
| CBNumber: | CB4842192 |
| Molecular Formula: | C24H28N2O7S2 |
| Formula Weight: | 520.61832 |
Eprosartan is an angiotensin II receptor antagonist used for the treatment of high blood pressure. It is marketed as Teveten byAbbott Laboratories in the United States.It is marketed as Eprozar by INTAS Pharmaceuticals in India and by Abbott Laboratorieselsewhere. It is sometimes paired with hydrochlorothiazide, marketed in the US as Teveten HCT and elsewhere as TevetenPlus.
The drug acts on the renin-angiotensin system in two ways to decrease total peripheral resistance. First, it blocks the binding ofangiotensin II to AT1 receptors in vascular smooth muscle, causing vascular dilatation. Second, it inhibits sympatheticnorepinephrine production, further reducing blood pressure.
As with other angiotensin II receptor antagonists, eprosartan is generally better tolerated than enalapril (an ACE inhibitor), especially among the elderly.[1]
- Ruilope L, Jäger B, Prichard B (2001). “Eprosartan versus enalapril in elderly patients with hypertension: a double-blind, randomized trial”. Blood Press. 10 (4): 223–9. doi:10.1080/08037050152669747. PMID 11800061.
PAT APR EXP
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| United States | 5185351 | 1993-02-09 | 2010-02-09 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
J Med Chem1991,34,(4):1514-7
J Med Chem1993,36,(13):1880-92
Synth Commun1993,23,(22):3231-48
AU 9056901, EP 403159, JP 91115278, US 5185351.
Drugs Fut1997,22,(10):1079

Eprosartan mesylate was developed successfully by SmithKline Beecham Corporation in 1997, and marketed in Germany in 1998 under the trade-name Teveten and in the United States later in 1999. Eprosartan mesylate, as an angiotensin II receptor blocker, is an antihypertensive drug of the latest generation. Eprosartan mesylate is potent to lower systolic and diastolic pressures in mild, moderate and severe hypertensive patients, and is safe and tolerable. Eprosartan mesylate is rapidly absorbed when administrated orally, with a bioavailability of 13% and a protein binding rate of 98%. The blood peak concentration and AUC (Area Under Curve) can be elevated by about 50% in patients with liver and kidney dysfunction, or fullness after administration, and can be elevated by 2 to 3 folds in elderly patients. Eprosartan mesylate has a structure shown as follows:

U.S. Pat. No. 5,185,351 discloses a method for preparing eprosartan mesylate using Eprosartan and methanesulfonic acid in isopropanol (U.S. Pat. No. 5,185,351, Example 41 (ii)). However, it is found when following this method for preparing eprosartan mesylate in industry, an esterification reaction can occur between eprosartan and isopropanol and the following two impurities can be generated:

In addition to the above two esterification impurities, the salifying method provided by the above patent is prone to produce isopropyl mesylate. Considering currently known potential risk of gene toxicity of methylsulfonic acid ester on human as well as the stringent requirements of methylsulfonic acid ester from the Europe and the America authorities, it is important to produce eprosartan mesylate in a non-alcohol solvent during the process of producing eprosartan mesylate, since it avoids the formation of methylsulfonic acid ester and the residue thereof in the final product. Since the dosage of eprosartan mesylate is high, it is particularly important to strictly control methylsulfonic acid ester in eprosartan mesylate.
In addition, for the above salifying method, solid eprosartan is suspended in propanol at a low temperature, then methanesulfonic acid is added, about ten seconds later a great deal of eprosartan mesylate precipitate is obtained. Therefore, solid eprosartan may be embedded by the precipitated eprosartan mesylate. Since isopropyl alcohol has a high viscosity at low temperature, a heavy filtering operation burden is needed to obtain solid from isopropanol, and the obtained solid contains quite an amount of isopropanol.

Eprosartan has been obtained by several different ways: 1) The iodination of 2-butylimidazole (I) with I2 and Na2CO3 in dioxane/water gives 2-butyl-4,5-diiodoimidazole (II), which is treated with benzyl chloromethyl ether (III) and K2CO3 in DMF yielding the imidazole derivative (IV). The condensation of (IV) with N-methyl-N-(2-pyridyl)formamide (V) by means of butyllithium in THF affords 1-(benzyloxymethyl)-2-butyl-4-iodoimidazole-5-carbaldehyde (VI), which is deprotected with concentrated HCl ethanol to give 2-butyl-4-iodoimidazole-5-carbaldehyde (VII). The acylation of (VII) with methyl 4-(bromomethyl)benzoate (VIII) by means of K2CO3 in hot DMF yields 4-(2-butyl-5-formyl-4-iodoimidazol-1 ylmethyl)benzoic acid methyl ester (IX), which is deiodinated by hydrogenation with H2 over Pd/C in methanol affording compound (X). The condensation of (X) with methyl 3-(2-thienyl)propionate (XI) by means of lithium diisopropylamide (LDA) in THF gives (XII), which is acylated with acetic anhydride and dimethylaminopyridine (DMAP) in dichloromethane yielding the corresponding acetate (XIII). Elimination of acetic acid from (XIII) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in hot toluene affords the expected propenoic ester (XIV), which is finally saponified with NaOH or KOH in ethanol/water.
…………………………………………………………………………………………………….
WO 1998035962 A1

…………………………………………………………………………………………..



Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder

cyclic pyranopterin monophosphate (cPMP, ALXN1101)
Alexion Pharma International Sàrl has received a breakthrough therapy designation from the US Food and Drug Administration (FDA) for its cyclic pyranopterin monophosphate (cPMP, ALXN1101), an enzyme co-factor replacement therapy to treat patients with molybdenum cofactor deficiency (MoCD) type A.
Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder
read all at
Cyclic pyranopterin monophosphate (cPMP) is an experimental treatment formolybdenum cofactor deficiency type A, which was developed by José Santamaría-Araujo and Schwarz at the German universities TU Braunschweig and the University of Cologne.[1][2]
cPMP is a precursor to molybdenum cofactor, which is required for the enyzme activity ofsulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
- Guenter Schwarz Laboratory, Institute for Biochemistry – University of Cologne (English, German)
- Günter Schwarz, José Angel Santamaria-Araujo, Stefan Wolf, Heon-Jin Lee, Ibrahim M. Adham, Hermann-Josef Gröne, Herbert Schwegler, Jörn Oliver Sass, Tanja Otte, Petra Hänzelmann, Ralf R. Mendel, Wolfgang Engel and Jochen Reiss (2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli“. Human Molecular Genetics 13 (12): 1249–1255. doi:10.1093/hmg/ddh136.PMID 15115759.
- Doctors risk untried drug to stop baby’s brain dissolving, TimesOnline, November 5, 2009
- José Angel Santamaria-Araujo, Berthold Fischer, Tanja Otte, Manfred Nimtz, Ralf R. Mendel, Victor Wray and Günter Schwarz (2004). “The Tetrahydropyranopterin Structure of the Sulfur-free and Metal-free Molybdenum Cofactor Precursor”. The Journal of Biological Chemistry 279 (16): 15994–15999.doi:10.1074/jbc.M311815200. PMID 14761975.
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclicpyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, unbeatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis {see U.S. Patent No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
WO 2012112922 A1
In this synthesis, the deprotection may involve, for example, either sequential or one-pot deprotection of certain amino and hydroxyl protecting groups on a compound of formula (VII) to furnish the compound of formula (I). Suitable reagents and conditions for the deprotection of a compound of formula (VII) can be readily determined by those of ordinary skill in the art. For example, compound (I) may be formed upon treatment of a compound of formula (VII) under conditions so that hydroxyl protecting groups, such as acetate, isopropylidine, and benzylidine protecting groups, are removed from the formula (VII) structure. The acetate group can be cleaved, for example, under Zemplen conditions using catalytic NaOMe as a base in methanol. The benzylidene and isopropylidene groups can be cleaved by hydrogenation or using acidic hydrolysis as reported by R.M. Harm et ah, J. Am. Chem. Soc, 72, 561 (1950). In yet another example, the deprotection can be performed so that amino protecting groups, such as 9- fluorenylmethyl carbamate (Fmoc), t-butyl carbamate (Boc), and carboxybenzyl carbamate (cbz) protecting groups are cleaved from the compound of formula (VII). 9-fluorenylmethyl carbamate (Fmoc) can be removed under mild conditions with an amine base (e.g. , piperidine) to afford the free amine and dibenzofulvene, as described by E. Atherton et al, “The
Fluorenylmethoxycarbonyl Amino Protecting Group,” in The Peptides, S. Udenfriend and J. Meienhofer, Academic Press, New York, 1987, p. 1. t-butyl carbamate (Boc) can be removed, as reported by G.L. Stahl et al., J. Org. Chem., 43, 2285 (1978), under acidic conditions (e.g., 3 M HC1 in EtOAc). Hydrogenation can be used to cleave the carboxybenzyl carbamate (cbz) protecting group as described by J. Meienhofer et al., Tetrahedron Lett., 29, 2983 (1988).
To prevent oxidation of formula (I) during the reaction, the deprotection may be performed under anaerobic conditions. The deprotection may also be performed at ambient temperature or at temperatures of from about 20 – 60 °C (e.g. , 25, 30, 35, 40, 45, 50, or 55 °C).
The compound of formula (I) may be isolated in the form of a pharmaceutically acceptable salt. For example, the compound of formula (I) may be crystallized in the presence of HC1 to form the HC1 salt form of the compound. In some embodiments, the compound of formula (I) may be crystallized as the HBr salt form of the compound. The compound of formula (I) may also be isolated, e.g., by precipitation as a sodium salt by treating with NaOH. The compound of formula (I) is labile under certain reaction and storage conditions. In some embodiments, the final solution comprising the compound of formula (I) may be acidified by methods known in the art. For example, the compound of formula (I), if stored in solution, can be stored in an acidic solution.
In some embodiments, the compound of formula (I) may be prepared, for example, by: reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV- A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-A):

wherein:
Rj is a protecting group, as defined above;
phosphorylating the compound of formula (V-A) to prepare a compound of formula (VI- A):

oxidizing the compound of formula (VI-A) to prepare a compound of formula (VII- A):

; and deprotecting the compound of formula (VII-A) to prepare the compound of formula (I). For example, a compound of formula (I) can be prepared as shown in Scheme 3.
Scheme 3.

5 R = Fraoc

In another embodiment, the compound of formula (I) is prepared by:
reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV-A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-B):

wherein:
each Ri is independently a protecting group, as defined above;
phosphorylating the compound of formula (V-B) to prepare a compound of formula (VI-B):

oxidizing the compound of formula (VI-B) to prepare a compound of formula (VII-B):

; and deprotecting the compound of formula (VII-B) to prepare the compound of formula (I), example, a compound of formula (I) can be prepared as shown in Scheme 4.
Scheme 4.

Alternatively, a compound of formula (I) can be formed as shown in Scheme 5. A diaminopyrimidinone compound of formula (II) can be coupled with a phosphorylated hexose sugar of formula (VIII), to give a compound of formula (IX). The piperizine ring nitrogen atoms can be protected to give a compound of formula (X) which can be oxidized to give a diol of formula (XI). The diol of formula (XI) can then be deprotected using appropriate conditions and converted to the compound of formula (I).
Scheme 5

In this embodiment, the phosphate may be introduced at the beginning of the synthesis to avoid undesirable equilibrium between the pyrano and furano isomers during subsequent steps of the synthesis. For example, a compound of formula (I) can be prepared as shown in Scheme 6.
Scheme 6.
ridine

A compound of formula (I) can also be formed as shown in Scheme 7. A diaminopyrimidinone compound of formula (II) can be coupled to a compound of formula (III) to afford the piperizine derivative of formula (IV). The piperizine ring nitrogen atoms of the compound of formula (IV) can be protected under standard conditions to give a derivative of formula (V). The formula (V) structure can be oxidized to afford compounds of formula (XII). Phosphorylation of a compound of formula (XII) gives a compound of formula (VII). Global deprotection of the compound of formula (VII) can afford the compound of formula (I).
Scheme 7
Piperizine ring protection
sphorylation
(VII)
For example, a compound of formula (I) can be prepared as shown in Scheme 8.
Scheme 8.

RABEPRAZOLE

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
-
US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.



Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
EFAVIRENZ – Huahai Pharma China-Approved to Produce AIDS Treatment

Efavirenz
DMP 266
- Sustiva (USA, Bristol-Myers Squibb)
- Stocrin (EU, MSD)
- Aspen Efavirenz (Sub-Saharan Africa, Aspen Pharmacare)
- E.F (McNeil & Argus)
- Efavir (Cipla)
- Efcure (Emcure Pharmaceuticals)
- Efferven (Ranbaxy Laboratories)
- Estiva (Hetero)
- Evirenz (Alkem Laboratories)
- Viranz (Aurobindo Pharma)
Zhejiang Huahai Pharma received CFDA approval to produce efavirenz, an oral non-nucleoside reverse transcriptase inhibitor (NNRTI) used to control the symptoms of AIDS. Huahai is the first China drugmaker approved to make the drug. Huahai produced efavirenz API for Merck, which marketed the drug under the name Stocrin
read at
http://www.sinocast.com/readbeatarticle.do?id=99634
Efavirenz (EFV), sold under the brand names Sustiva among others, is a non-nucleoside reverse transcriptase inhibitor (NNRTI). It is used as part of highly active antiretroviral therapy (HAART) for the treatment of a human immunodeficiency virus (HIV) type 1. For HIV infection that has not previously been treated, the United States Department of Health and Human Services Panel on Antiretroviral Guidelines currently recommends the use of efavirenz in combination with tenofovir/emtricitabine (Truvada) as one of the preferred NNRTI-based regimens in adults and adolescents.[1] Efavirenz is also used in combination with other antiretroviral agents as part of an expanded postexposure prophylaxis regimen to reduce the risk of HIV infection in people exposed to a significant risk (e.g. needlestick injuries, certain types of unprotected sex etc.).
It is usually taken on an empty stomach at bedtime to reduce neurological and psychiatric adverse effects.
Efavirenz was combined with the HIV medications tenofovir and emtricitabine, all of which are reverse transcriptase inhibitors. This combination of three medications under the brand name Atripla, provides HAART in a single tablet taken once a day.
Efavirenz was discovered at Merck Research Laboratories. It is on the WHO Model List of Essential Medicines, the most important medication needed in a basic health system.[2] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[3]
Efavirenz (EFV, brand names Sustiva, Stocrin, Efavir etc.) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) and is used as part of highly active antiretroviral therapy(HAART) for the treatment of a human immunodeficiency virus (HIV) type 1.
For HIV infection that has not previously been treated, the United States Department of Health and Human Services Panel on Antiretroviral Guidelines currently recommends the use of efavirenz in combination with tenofovir/emtricitabine (Truvada) as one of the preferred NNRTI-based regimens in adults and adolescents.
Efavirenz is also used in combination with other antiretroviral agents as part of an expanded postexposure prophylaxis regimen to reduce the risk of HIV infection in people exposed to a significant risk (e.g. needlestick injuries, certain types of unprotected sex etc.).
The usual adult dose is 600 mg once a day. It is usually taken on an empty stomach at bedtime to reduce neurological and psychiatric adverse effects.
Efavirenz was combined with the popular HIV medication Truvada, which consists oftenofovir and emtricitabine, all of which are reverse transcriptase inhibitors. This combination of three medications approved by the U.S. Food and Drug Administration(FDA) in July 2006 under the brand name Atripla, provides HAART in a single tablet taken once a day. It results in a simplified drug regimen for many patients.

doi:10.1016/0040-4039(95)01955-H

Merck synthesis of Efavirenz
History
Efavirenz was approved by the FDA on September 21, 1998, making it the 14th approved antiretroviral drug.
-
Efavirenz is a non-nucleoside reverse trancriptase inhibitor being studied clinically for use in the treatment of HIV infections and AIDS.
- Efavirenz chemically known as (-) 6-Chloro-4-cyclopropylethynyl-4-trifluoromethyl- 1 , 4- dihydro-2H-3, 1-benzoxa zin-2-one, is a highly potent non-nucleoside reverse transcriptase inhibitor (NNRTI).A number of compounds are effective in the treatment of the human immunodeficiency virus (HIV) which is the retrovirus that causes progressive destruction of the human immune system. Effective treatment through inhibition of HIV reverse transcriptase is known for non- nucleoside based inhibitors. Benzoxazinones have been found to be useful non-nucleoside based inhibitors of HIV reverse transcriptase.(-) β-chloro^-cyclopropylethynyM-trifluoromethyl-l ,4-dihydro-2H-3,l -ben zoxazin-2-one (Efavirenz) is efficacious against HIV reverse transcriptase resistance. Due to the importance of (-)6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-l,4-dihydro-2H-3,l-ben zoxazin-2- one, economical and efficient synthetic processes for its production needs to be developed.The product patent US5519021. discloses the preparation of Efavirenz, in Example-6, column-29, involving cyclisation of racemic mixture of 2-(2-amino-5-chlorophenyl)-4- cyclopropyl-l,l,l-trifluoro-3-butyn-2-ol using l ,l ‘-carbonyldiimidazole as carbonyl delivering agent to give racemic Efavirenz. Further, resolution of the racemic Efavirenz is carried out using (-) camphanic acid chloride to yield optically pure Efavirenz. However, research article published in the Drugs of the future, 1998, 23(2), 133-141 discloses process for manufacture of optically pure Efavirenz. The process involves cyclisation of racemic 2-(2-amino-5-chlorophenyl)-4-cyclopropyl-l, 1, l-trifluoro-3-butyn-2- ol using 1, 1-carbonyldiimidazole as carbonyl delivering agent to give racemic Efavirenz and further resolution by (-) camphanic acid chloride.Similarly research article published in Synthesis 2000, No. 4, 479-495 discloses stereoselective synthesis of Efavirenz (95%yield, 99.5%ee), as shown below

Even though many prior art processes report method for the preparation of Efavirenz, each process has some limitations with respect to yield, purity, plant feasibility etc. Hence in view of the commercial importance of Efavirenz there remains need for an improved process.
- US 6 028 237 discloses a process for the manufacture of optically pure Efavirenz.
-
The synthesis of efavirenz and structurally similar reverse transcriptase inhibitors are disclosed in US Patents 5,519,021, 5,663,169, 5,665,720 and the corresponding PCT International Patent Application WO 95/20389, which published on August 3, 1995. Additionally, the asymmetric synthesis of an enantiomeric benzoxazinone by a highly enantioselective acetylide addition and cyclization sequence has been described by Thompson, et al., Tetrahedron Letters 1995, 36, 8937-8940, as well as the PCT publication, WO 96/37457, which published on November 28, 1996.
-
Additionally, several applications have been filed which disclose various aspects of the synthesis of(-)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one including: 1) a process for making the chiral alcohol, U.S.S.N. 60/035,462, filed 14 January 1997; 2) the chiral additive, U.S.S.N. 60/034,926, filed 10 January 1997; 3) the cyclization reaction, U.S.S.N. 60/037,059, filed 12 February 1997; and the anti-solvent crystallization procedure, U.S.S.N. 60/037,385 filed 5 February 1997 and U.S.S.N. 60/042,807 filed 8 April 1997.




Syntheses of EFV API; different routes of manufacturingAPI, active pharmaceutical ingredient; EFV efavirenz. BELOW
Related substances and degradants (partial listing) in EFVAPI, active pharmaceutical ingredient; CPA, cyclopropylacetylene; EFV, efavirenz
Syntheses of EFV API; different routes of manufacturingAPI, active pharmaceutical ingredient; EFV efavirenz.
Chemical properties
Efavirenz is chemically described as (S)-6-chloro-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one. Its empirical formula is C14H9ClF3NO2. Efavirenz is a white to slightly pink crystalline powder with a molecular mass of 315.68 g/mol. It is practically insoluble in water (<10 µg/mL).
History
Efavirenz was approved by the FDA on September 21, 1998, making it the 14th approved antiretroviral drug.
Society and culture
Pricing information
A one-month supply of 600 mg tablets cost approximately $550 in April 2008.[16] Merck provides efavirenz in certain developing countries at cost, currently about $0.65 per day.[17] Some emerging countries have opted to purchase Indian generics[18] such as Efavir by Cipla Ltd.[19] In Thailand, one month supply of efavirenz + truvada, as of June 2012, costs THB 2900 ($90), there’s also a social program for poorer patients who can’t afford even this price. In South Africa, a license has been granted to generics giant Aspen Pharmacare to manufacture, and distribute to Sub-Saharan Africa, a cost-effective antiretroviral drug.[20]
PATENT
http://www.google.com/patents/WO1999061026A1?cl=en
EXAMPLE 1
Cl

1a

To a solution of trifluoroethanol and (IR, 2S)-N-pyrrolidinyl norephedrine in THF (9 L) under nitrogen is added a solution of diethylzinc in hexane at 0 °C slowly enough to keep the temperature below 30 °C. The mixture is stirred at room temperature for 0.5 ~ 1 h. In another dry flask a solution of chloromagnesium cyclopropyl acetylide is prepared as follows: To neat cyclopropyl acetylene at 0 °C is added a solution of rc-butylmagnesium chloride slowly enough to keep the internal temperature < 30 °C. The solution is stirred at 0 °C for ~ 40 min and transfered to the zinc reagent via cannula with 0.36 L of THF as a wash. The mixture is cooled to -10 °C and ketoaniline la is added. The mixture is stirred at -2 to -8 °C for 35 h, warmed to room temperature, stirred for 3 h, and quenched with 30% potassium carbonate over 1.5 h. The mixture is stirred for 4 h and the solid is removed by filtration and washed with THF (2 cake volume). The wet solid still contains -18 wt% of pyrrolidinyl norephedrine and is saved for further study. The filtrate and wash are combined and treated with 30% citric acid. The two layers are separated. The organic layer is washed with water (1.5 L). The combined aqueous layers are extracted with 2.5 L of toluene and saved for norephedrine recovery. The toluene extract is combined with the organic solution and is concentrated to ~ 2.5 L. Toluene is continuously feeded and distilled till THF is not detectable by GC. The final volume is controlled at 3.9 L. Heptane (5.2 L) is added over 1 h. The slurry is cooled to 0 °C, aged for 1 h, and filtered. The solid is washed with heptane (2 cake volume) and dried to give 1.234 Kg (95.2% yield) of amino alcohol 3 as a white crystalline. The material is 99.8 A% pure and 99.3% ee.
EXAMPLE 2


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3, MTBE (500 L), and aqueous KHCO3 (45 g in 654 mL H2O). Solid 4-nitrophenyl chloroformate was added, in 4 batches, at 25°C. During the addition the solution pH was monitored. The pH was maintained between 8.5 and 4 during the reaction and ended up at 8.0. The mixture was stirred at 20-25°C for two hours. Aqueous KOH (2N) was added over 20 minutes, until the pH of the aqueous layer reached 11.0.
The layers were separated and 500 mL brine was added to the MTBE layer. 0.1 N Acetic acid was added until the pH was 6-7. The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 3


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3a, toulene (500 mL), and aqueous KHCO3 (86.5 g in 500 L H2O). Phosgene solution in toulene was added at 25°C, and the mixture was stirred at 20-25°C for two hours.
The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 4


To a three necked round bottom flask, equipped with a mechanical stirrer, nitrogen line, and thermocouple, was charged the solid amino alcohol 3a, MTBE (500 mL), and aqueous KHCO3 (86.5 g in 500 mL H2O). Phosgene gas was slowly passed into the solution at 25°C, until the reaction was complete.
The layers were separated and the organic phase was washed with brine (500 mL). At this point the mixture was solvent switched to EtOH/IPA and crystallized as recited in Examples 5 and 6.
EXAMPLE 5
Crystallization of efavirenz from 30% 2-Propanol in Water using a ratio of 15 ml solvent per gram efavirenz Using Controlled Anti-Solvent Addition on a 400 g Scale.
400 g. of efavirenz starting material is dissolved in 1.8 L of 2- propanol. The solution is filtered to remove extraneous matter. 1.95 L of deionized (DI) water is added to the solution over 30 to 60 minutes. 10 g. to 20 g. of efavirenz seed (Form II wetcake) is added to the solution. The seed bed is aged for 1 hour. The use of Intermig agitators is preferred to mix the slurry. If required (by the presence of extremely long crystals or a thick slurry), the slurry is wet-milled for 15 – 60 seconds. 2.25 L of DI water is added to the slurry over 4 to 6 hours. If required (by the presence of extremely long crystals or a thick slurry), the slurry is wet- milled for 15 – 60 seconds during the addition. The slurry is aged for 2 to 16 hours until the product concentration in the supernatant remains constant. The slurry is filtered to isolate a crystalline wet cake. The wet cake is washed with 1 to 2 bed volumes of 30 % 2-propanol in water and then twice with 1 bed volume of DI water each. The washed wet cake is dried under vacuum at 50°C.
EXAMPLE 6
Crystallization of efavirenz from 30% 2-Propanol in Water using a ratio of 15 ml solvent per gram efavirenz Using a Semi-Continuous Process on a 400 g Scale.
400 g. of efavirenz starting material is dissolved in 1.8 L of 2- propanol. A heel slurry is produced by mixing 20 g. of Form II efavirenz in 0.3 L of 30 % (v/v) 2-propanol in water or retaining part of a slurry froma previous crystallization in the crystallizer. The dissolved batch and 4.2 L of DI water are simultaneously charged to the heel slurry at constant rates over 6 hours to maintain a constant solvent composition in the crystallizer. Use of Intermig agitators during the crystallization is preferred. During this addition the slurry is wet-milled when the crystal lengths become excessively long or the slurry becomes too thick. The slurry is aged for 2 to 16 hours until the product concentration in the supernatant remains constant. The slurry is filtered to isolate a crystalline wet cake. The wet cake is washed with 1 to 2 bed volumes of 30 % 2-propanol in water and then twice with 1 bed volume of DI water each. The washed wet cake is dried under vacuum at 50°C.
EXAMPLE 7 Preparation of Amino Alcohol 3 and ee Upgrading— Through Process

1a

A solution of diethyl zinc in hexane was added to a solution of trifluoroethanol (429.5 g, 4.29’mol) and (IR, 2S)-N-pyrrolidinyl norephedrine (1.35 kg, 6.58 mol) in THF (9 L), under nitrogen, at 0 °C. The resulting mixture was stirred at room temperature for approx. 30 min. In another dry flask a solution of chloromagnesium- cyclopropylacetylide was prepared as follows. To a solution of n- butylmagnesium chloride in THF (2 M, 2.68 L, 5.37 mol) was added neat cyclopropylacetylene at 0 °C keeping the temperature < 25 °C. The solution was stirred at 0 °C for 1 ~ 2 h. The solution of chloromagnesiumcyclopropylacetylide was then warmed to room temperature and was transferred into the zinc reagent via cannula over 5 min followed by vessel rinse with 0.36 L of THF. The resulting mixture was aged at ~ 30 °C for 0.5 h and was then cooled to 20 °C. The ketoaniline 1 (1.00 kg, 4.47 mol) was added in one portion as a solid, and the resulting mixture was stirred at 20-28 °C for 3 h.
The reaction was quenched with 30% aq. potassium carbonate (1.2 L) and aged for 1 h. The solid waste was filtered and the cake was washed with THF (3 cake volumes). The filtrate and wash were combined and solvent switched to IP Ac.
The IPAc solution of product 3 and pyrrolidinyl norephedrine was washed with citric acid (3.5 L) and with water (1.5 L). The combined aqueous layers were extracted with IPAc (2 L) and saved for norephedrine recovery. To the combined organic layers was added
12N HC1 (405 mL, 4.88 mol), to form a thin slurry of the amino alcohol-
HC1 salt. The mixture was aged for 30 min at 25 °C and was then dried azeotropically. The slurry was aged at 25 °C for 30 min and filtered. The cake was washed with 2.5 L of IPAc and dried at 25 °C under vacuum/nitrogen for 24 h to give 1.76 kg of the wet HC1 salt.
The salt was dissolved in a mixture of MTBE (6 L) and aq Na2Cθ3 (1.18 kg in 6.25 L water). The layers were separated and the organic layer was washed with 1.25 L of water. The organic layer was then solvent switched into toluene.
Heptane (5 L) was added over 1 h at 25 °C. The slurry was cooled to 0 °C, aged for 1 h, and filtered. The solid was washed with heptane (2 cake volumes) and was dried to give 1.166 kg (90% overall yield) of amino alcohol 3 as a white crystalline solid. Norephedrine recovery
The aqueous solution was basified to pH13 using 50% aq NaOH, and extracted with heptane (2 L). The heptane solution was washed with water (1 L) and concentrated to remove residual IPAc and water. The final volume was adjusted to about 3 L. The heptane solution was cooled to -20 °C, aged for 2 h, and filtered. The solid was washed with cold heptane (1 cake volume) and dried to give 1.269 kg solid (94% recovery)




CLIPS
http://www.mdpi.com/1420-3049/21/2/221/htm



| WO2007013047A2 * | Jul 31, 2006 | Feb 1, 2007 | Ranbaxy Lab Ltd | Water-dispersible anti-retroviral pharmaceutical compositions |
| WO2007013047A3 * | Jul 31, 2006 | May 31, 2007 | Ranbaxy Lab Ltd | Water-dispersible anti-retroviral pharmaceutical compositions |
| WO2007052289A2 * | Jul 24, 2006 | May 10, 2007 | Rubicon Res Pvt Ltd | Novel dispersible tablet composition |
| WO2007052289A3 * | Jul 24, 2006 | Dec 27, 2007 | Rubicon Res Pvt Ltd | Novel dispersible tablet composition |
| WO2011131943A2 | Apr 20, 2011 | Oct 27, 2011 | Cipla Limited | Pharmaceutical compositions |
| WO2012048884A1 | Oct 14, 2011 | Apr 19, 2012 | Lonza Ltd | Process for the synthesis of cyclic carbamates |
| WO2012048886A1 | Oct 14, 2011 | Apr 19, 2012 | Lonza Ltd | Process for the synthesis of cyclic carbamates |
| WO2015059466A1 | Oct 22, 2014 | Apr 30, 2015 | Cipla Limited | Pharmaceutical compositions comprising efavirenz |
| EP1448170A2 * | Nov 26, 2002 | Aug 25, 2004 | Bristol-Myers Squibb Company | Efavirenz tablet formulation having unique biopharmaceutical characteristics |
| EP2441759A1 | Oct 14, 2010 | Apr 18, 2012 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| EP2447255A1 | Oct 14, 2010 | May 2, 2012 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US6238695 | Apr 6, 1999 | May 29, 2001 | Dupont Pharmaceuticals Company | Formulation of fast-dissolving efavirenz capsules or tablets using super-disintegrants |
| US6555133 | Apr 2, 2001 | Apr 29, 2003 | Bristol-Myers Squibb Company | Formulation of fast-dissolving efavirenz capsules or tablets using super-disintegrants |
| US8871271 | Jul 29, 2013 | Oct 28, 2014 | Gilead Sciences, Inc. | Method and composition for pharmaceutical product |
| US8957204 | Oct 14, 2011 | Feb 17, 2015 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US8969550 | Oct 14, 2011 | Mar 3, 2015 | Lonza Ltd. | Process for the synthesis of cyclic carbamates |
| US9018192 | Oct 10, 2013 | Apr 28, 2015 | Bristol-Myers Squibb & Gilead Sciences, Inc. | Unitary pharmaceutical dosage form |
| US9198862 | Jul 24, 2006 | Dec 1, 2015 | Rubicon Research Private Limited | Dispersible tablet composition |
| WO1995020389A1 * | Jan 24, 1995 | Aug 3, 1995 | Merck & Co Inc | Benzoxazinones as inhibitors of hiv reverse transcriptase |
| WO1996037457A1 * | May 21, 1996 | Nov 28, 1996 | Merck & Co Inc | Asymmetric synthesis of (-) 6-chloro-4-cyclopropyl-ethynyl-4-trifluoromethyl-1,4-dihydro-2h-3,1-benzoxazin-2-one |
| WO1998052570A1 * | May 14, 1998 | Nov 26, 1998 | David Walter Barry | Antiviral combinations containing the carbocyclic nucleoside 1592u89 |
References
- 1 “Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents”. Retrieved 10 May 2013.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 62. ISBN 9781284057560.
- 4
- Cespedes, MS; Aberg, JA (2006). “Neuropsychiatric complications of antiretroviral therapy.”. Drug safety : an international journal of medical toxicology and drug experience 29 (10): 865–74. doi:10.2165/00002018-200629100-00004. PMID 16970510.
- 5
- “www.accessdata.fda.gov” (PDF).
- 6
- DHHS panel. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents (October 10, 2006). (Available for download from AIDSInfo)
- 7
- Ford, N.; Mofenson, L.; Kranzer, K.; Medu, L.; Frigati, L.; Mills, E. J.; Calmy, A. (2010). “Safety of efavirenz in first-trimester of pregnancy: A systematic review and meta-analysis of outcomes from observational cohorts”. AIDS 24 (10): 1461–1470. doi:10.1097/QAD.0b013e32833a2a14. PMID 20479637.
- 8
- Rossi, S; Yaksh, T; Bentley, H; Van Den Brande, G; Grant, I; Ellis, R (2006). “Characterization of interference with 6 commercial delta9-tetrahydrocannabinol immunoassays by efavirenz (glucuronide) in urine”. Clinical Chemistry 52 (5): 896–7. doi:10.1373/clinchem.2006.067058. PMID 16638958.
- 9
- Röder, CS; Heinrich, T; Gehrig, AK; Mikus, G (2007). “Misleading results of screening for illicit drugs during efavirenz treatment”. AIDS (London, England) 21 (10): 1390–1. doi:10.1097/QAD.0b013e32814e6b3e. PMID 17545727.
- 10
- Ren J, Bird LE, Chamberlain PP; et al. (2002). “Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors”. Proc Natl Acad Sci USA 99 (22): 14410–15. doi:10.1073/pnas.222366699. PMC 137897. PMID 12386343.
- 11
- Sustiva (efavirenz) capsules and tablets. Product information (April 2005)
- 12
- Simen AA, Ma J, Svetnik V, Mayleben D, Maynard J, Roth A, Mixson L, Mogg R, Shera D, George L, Mast TC, Beals C, Stoch A, Struyk A, Shire N, Fraser I (2014). “Efavirenz modulation of sleep spindles and sleep spectral profile”. J Sleep Res 24: 66–73. doi:10.1111/jsr.12196. PMID 25113527.
- 13
- Gatch MB, Kozlenkov A, Huang RQ, Yang W, Nguyen JD, González-Maeso J, Rice KC, France CP, Dillon GH, Forster MJ, Schetz JA (2013). “The HIV antiretroviral drug efavirenz has LSD-like properties”. Neuropsychopharmacology 38 (12): 2373–84. doi:10.1038/npp.2013.135. PMC 3799056. PMID 23702798.
- 14
- Dabaghzadeh F, Ghaeli P, Khalili H, Alimadadi A, Jafari S, Akhondzadeh S, Khazaeipour Z (2013). “Cyproheptadine for prevention of neuropsychiatric adverse effects of efavirenz: a randomized clinical trial”. AIDS Patient Care STDS 27 (3): 146–54. doi:10.1089/apc.2012.0410. PMID 23442031.
- 15
- Dabaghzadeh F, Khalili H, Ghaeli P, Dashti-Khavidaki S (2012). “Potential benefits of cyproheptadine in HIV-positive patients under treatment with antiretroviral drugs including efavirenz”. Expert Opin Pharmacother 13 (18): 2613–24. doi:10.1517/14656566.2012.742887. PMID 23140169.
- 16
- Price listed on http://drugstore.com website, 4/20/2008
- 17
- “Merck & Co., Inc., Again Reduces Price of Stocrin (efavirenz) for Patients in Least Developed Countries and Countries Hardest Hit by Epidemic – Drugs.com MedNews”.
- 18
- IndiaDaily – A new trend in emerging nations – Brazil opts for Indian generic drug ignoring US pharmaceutical giant Merck’s patent on AIDS drug Efavirenz
- 19
- http://www.cipla.com
- 20
- Patrick Lumumba Osewe; Yvonne Korkoi Nkrumah; Emmanuel K. Sackey (15 June 2008). Improving Access to HIV/AIDS Medicines in Africa: Trade-Related Aspects of Intellectual Property Rights (TRIPS) Flexibilities Utilization. World Bank Publications. pp. 35–39. ISBN 978-0-8213-7544-0. Retrieved 30 June 2012.
- 21
- http://www.sustiva.com/
- 22
- http://www.medsafe.govt.nz/consumers/cmi/s/stocrin.pdf
- 23
- Drugsupdate.com generic brands list: http://www.drugsupdate.com/brand/generic/Efavirenz/87
- 24
- http://mcneilargusindia.com/
- 25
- http://www.alkemlabs.com/
- 26
- “Regast® (efavirenz) film-coated tablets.”. http://www.pharmasyntez.com (in Russian). Pharmasyntez, 2011. Retrieved 28 June 2015. External link in
|website=(help) - 27
- IOL: Thugs get high on stolen Aids drugs IOL News May 12, 2007
- 28
- Getting high on HIV drugs in S Africa. BBC News, 8 December 2008.
- 29
- ‘No Turning Back’: Teens Abuse HIV Drugs. ABC News, April 6, 2009.
- 30
- Getting High On HIV Medication Vice 7.04.2014.
- 31
- Gatch, M. B.; Kozlenkov, A.; Huang, R. Q.; Yang, W.; Nguyen, J. D.; González-Maeso, J.; Rice, K. C.; France, C. P.; Dillon, G. H.; Forster, M. J.; Schetz, J. A. (2013). “The HIV Antiretroviral Drug Efavirenz has LSD-Like Properties”. Neuropsychopharmacology 38 (12): 2373–84. doi:10.1038/npp.2013.135. PMC 3799056. PMID 23702798.
- Sütterlin, S.; Vögele, C.; Gauggel, S. (2010). “Neuropsychiatric complications of Efavirenz therapy: suggestions for a new research paradigm”. The Journal of Neuropsychiatry and Clinical Neurosciences 22 (4): 361–369. doi:10.1176/jnp.2010.22.4.361.
External links


|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(4S)-6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-2,4-dihydro-1H-3,1-benzoxazin-2-one
|
|
| Clinical data | |
| Trade names | Sustiva, Stocrin, others |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699004 |
| Pregnancy category |
|
| Routes of administration |
By mouth (capsules, tablets) |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 40–45% (under fasting conditions) |
| Protein binding | 99.5–99.75% |
| Metabolism | Hepatic (CYP2A6 and CYP2B6-mediated) |
| Onset of action | 3–5 hours |
| Biological half-life | 40–55 hours |
| Excretion | Urine (14–34%) and feces (16–61%) |
| Identifiers | |
| CAS Number | 154598-52-4 |
| ATC code | J05AG03 (WHO) |
| PubChem | CID 64139 |
| DrugBank | DB00625 |
| ChemSpider | 57715 |
| UNII | JE6H2O27P8 |
| KEGG | D00896 |
| ChEBI | CHEBI:119486 |
| ChEMBL | CHEMBL223228 |
| NIAID ChemDB | 032934 |
| PDB ligand ID | EFZ (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C14H9ClF3NO2 |
| Molar mass | 315.675 g/mol |



