
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
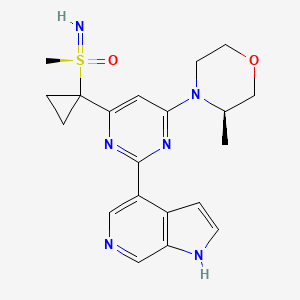
AZD-6738, Ceralasertib
- Molecular Formula C20H24N6O2S
- Average mass 412.509 Da

- 4-[4-[1-[[S(R)]-S-Methylsulfonimidoyl]cyclopropyl]-6-[(3R)-3-methyl-4-morpholinyl]-2-pyrimidinyl]-1H-pyrrolo[2,3-b]pyridine
- AZD 6738
- Ceralasertib
- Originator AstraZeneca; University of Pennsylvania
- Class Antineoplastics; Morpholines; Pyrimidines; Small molecules
- Mechanism of Action ATR protein inhibitors
- Phase II Breast cancer; Gastric cancer; Non-small cell lung cancer; Ovarian cancer
- Phase I/II Chronic lymphocytic leukaemia; Solid tumours
- Phase I Non-Hodgkin’s lymphoma
- Preclinical Diffuse large B cell lymphoma
- No development reported B-cell lymphoma; Lymphoid leukaemia
- 26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
- 18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
- 25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
- Kevin M. Foote


Patent
WO 2011154737
Example 1.01
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[((R)-S-methylsulfonimidoyl)methyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
(1 ml) to afford the title compound (34.6 mg, 49%); 1HNMR (400 MHz, CDCl3) 1.40 (3H, d), 3.17 (3H, s), 3.39 (1H, tt), 3.62 (1H, td), 3.77 (1H, dd), 3.85 (1H, d), 4.08 (1H, dd), 4.18 (1H, d), 4.37 – 4.48 (2H, q), 4.51 (1H, s), 6.59 (1H, s), 7.35 (1H, t), 7.46 (1H, d), 8.06 (1H, d), 8.42 (1H, d), 10.16 (1H, s); m/z: (ES+) MH+, 387.19.
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
3.29 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 3.99 (1H, dd), 4.02 (1H, s), 4.31 (1H, s), 6.41 (1H, s). Chiral HPLC: (HP1100 System 5, 20μm Chiralpak AD-H (250 mm × 4.6 mm) column eluting with Hexane/EtOH/TEA 50/50/0.1) Rf, 12.192 98.2%.
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
CDCI3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.23 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, d), 8.42 (1H, d); m/z: (ES+) MH+, 399.40.
Example 2.01 and example 2.02
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-blpyridine, and
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-blpyridine
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).


Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
(h) IBDA, trifluoroacetamide, MgO, DCM, Rh2(OAc)4 82%;
(i) 1,2-dibromoethane, sodium hydroxide, TOAB, 2-MeTHF, 47%;
(j) TsCl, tetrabutylammonium hydrogen sulfate, sodium hydroxide, DCM, 92%;
(k) bis(pinacolato)diboron, potassium acetate, 1,1′-bis(diphenylphosphino)ferrocene dichloro palladium(II), DMF, 62%;
(l) Pd(II)Cl2(PPh3)2, Na2CO3, DME, water, 80%;
(m) 2 N NaOH, DME, water, 92%.
Foote, K. M. N.; Johannes, W. M.; Turner, P.. Morpholino Pyrimidines and their use in therapy. WO 2011/154737 A1, 15 December 2011.
PAPER
Development and Scale-up of a Route to ATR Inhibitor AZD6738
- William R. F. Goundry et al




REFERENCES
1: Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN, Conrads TP, O’Connor MJ, Ferris RL, Tran PT, Delgoffe GM, Bakkenist CJ. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest. 2018 Jun 28. pii: 96519. doi: 10.1172/JCI96519. [Epub ahead of print] PubMed PMID: 29952768.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.



| Molecular Weight | 412.51 |
|---|---|
| Formula | C20H24N6O2S |
- Originator AstraZeneca; University of Pennsylvania
- Developer Acerta Pharma; AstraZeneca; Dana-Farber Cancer Institute; Gustave Roussy; National Cancer Institute (France); Samsung Medical Center; University of California at San Francisco; University of Pennsylvania
- Class Antineoplastics; Cyclopropanes; Imines; Ketones; Morpholines; Organic sulfur compounds; Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action ATR protein inhibitors
- Phase III Non-small cell lung cancer
- Phase IICholangiocarcinoma; Gynaecological cancer; Malignant melanoma; Osteosarcoma; Ovarian cancer; Pancreatic cancer; Prostate cancer; Small cell lung cancer; Solid tumours; Triple negative breast cancer
- Phase I/IIChronic lymphocytic leukaemia
- Phase IChronic myelomonocytic leukaemia; Myelodysplastic syndromes
- PreclinicalDiffuse large B cell lymphoma; Type 1 diabetes mellitus
- DiscontinuedHaematological malignancies; Non-Hodgkin’s lymphoma; Squamous cell cancer
- 22 Apr 2025AstraZeneca plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) (PO) in April 2025(NCT06929260)
- 23 Dec 2024AstraZeneca plans a phase I trial for Non-small Cell Lung Cancer, Ovarian Cancer, or Endometrial Cancer in United Kingdom(PO) In January 2025 (NCT06754761)
- 07 Dec 2024Updated efficacy and adverse event data from a phase I trial in Myelodysplastic syndrome presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)



Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry
- January 2022
- Organics 3(1):1-21

References
- ^ “Ceralasertib – AstraZeneca/University of Pennsylvania”. AdisInsight. Springer Nature Switzerland AG.
- ^ Mavroeidi D, Georganta A, Panagiotou E, Syrigos K, Souliotis VL (February 2024). “Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes”. International Journal of Molecular Sciences. 25 (5): 2767. doi:10.3390/ijms25052767. PMC 10932434. PMID 38474014.
- [1]. Vendetti FP, et al. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of CDDP to resolve ATM-deficient non-small cell lung cancer in vivo. [Content Brief][2]. Kim HJ, et al. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. [Content Brief]
| Clinical data | |
|---|---|
| Other names | AZD-6738 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1352226-88-0 |
| PubChem CID | 54761306 |
| IUPHAR/BPS | 9390 |
| DrugBank | DB14917 |
| ChemSpider | 58828171 |
| UNII | 85RE35306Z |
| KEGG | D11787 |
| ChEMBL | ChEMBL4285417 |
| PDB ligand | VJM (PDBe, RCSB PDB) |
| ECHA InfoCard | 100.232.607 |
| Chemical and physical data | |
| Formula | C20H24N6O2S |
| Molar mass | 412.51 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |


Dear Dr Castro,
I appreciate your reports on new drugs. I’m doring a survey of anthraquinone derivatives in the pharmaceutical space. Are you aware of any APIs based on anthrquinone chemistry?
Thanking you for any help.
Dr. G. Michael Laidlaw President Pinnachem llc
LikeLike