PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Selpercatinib is a tyrosine kinase inhibitor with antineoplastic properties.
A phase I/II trial is also under way in pediatric patients and young adults with activating RET alterations and advanced solid or primary CNS tumors.
Loxo Oncology (a wholly-owned subsidiary of Eli Lilly ), under license from Array , is developing selpercatinib, a lead from a program of RET kinase inhibitors, for treating cancer, including non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma and soft tissue sarcoma
In 2018, the compound was granted orphan drug designation in the U.S. for the treatment of pancreatic cancer and in the E.U. for the treatment of medullary thyroid carcinoma.
Trk is a high affinity receptor tyrosine kinase activated by a group of soluble growth factors called neurotrophic factor (NT). The Trk receptor family has three members, namely TrkA, TrkB and TrkC. Among the neurotrophic factors are (1) nerve growth factor (NGF) which activates TrkA, (2) brain-derived neurotrophic factor (BDNF) and NT4/5 which activate TrkB, and (3) NT3 which activates TrkC. Trk is widely expressed in neuronal tissues and is involved in the maintenance, signaling and survival of neuronal cells.
The literature also shows that Trk overexpression, activation, amplification and/or mutations are associated with many cancers including neuroblastoma, ovarian cancer, breast cancer, prostate cancer, pancreatic cancer, multiple myeloma, astrocytoma. And medulloblastoma, glioma, melanoma, thyroid cancer, pancreatic cancer, large cell neuroendocrine tumor and colorectal cancer. In addition, inhibitors of the Trk/neurotrophin pathway have been shown to be effective in a variety of preclinical animal models for the treatment of pain and inflammatory diseases.
The neurotrophin/Trk pathway, particularly the BDNF/TrkB pathway, has also been implicated in the pathogenesis of neurodegenerative diseases, including multiple sclerosis, Parkinson’s disease, and Alzheimer’s disease. The modulating neurotrophic factor/Trk pathway can be used to treat these and related diseases.
It is believed that the TrkA receptor is critical for the disease process in the parasitic infection of Trypanosoma cruzi (Chagas disease) in human hosts. Therefore, TrkA inhibitors can be used to treat Chagas disease and related protozoal infections.
Trk inhibitors can also be used to treat diseases associated with imbalances in bone remodeling, such as osteoporosis, rheumatoid arthritis, and bone metastasis. Bone metastases are a common complication of cancer, up to 70% in patients with advanced breast or prostate cancer and about 15 in patients with lung, colon, stomach, bladder, uterine, rectal, thyroid or kidney cancer Up to 30%. Osteolytic metastases can cause severe pain, pathological fractures, life-threatening hypercalcemia, spinal cord compression, and other neurostress syndromes. For these reasons, bone metastases are a serious cancer complication that is costly. Therefore, an agent that can induce apoptosis of proliferating bone cells is very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, almost all osteoblast apoptosis agents are very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, localization of NGF was observed in almost all osteoblasts. Recently, it was demonstrated that pan-Trk inhibitors in human hFOB osteoblasts inhibit tyrosine signaling activated by neurotrophic factors that bind to all three Trk receptors. This data supports the theory of using Trk inhibitors to treat bone remodeling diseases, such as bone metastases in cancer patients.
Developed by Loxo Oncology, Larotrectinib (LOXO-101) is a broad-spectrum antineoplastic agent for all tumor patients expressing Trk, rather than tumors at an anatomical location. LOXO-101 chemical name is (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)pyrazolo[1,5-a] Pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide, the structural formula is as follows. LOXO-101 began treatment of the first patient in March 2015; on July 13, 2016, the FDA granted a breakthrough drug qualification for the inoperable removal or metastatic solid tumor of adults and children with positive Trk fusion gene mutations; Key entry was completed in February 2017; in November 2018, the FDA approved the listing under the trade name Vitrakvi.
Poor absorption, distribution, metabolism, and/or excretion (ADME) properties are known to be the primary cause of clinical trial failure in many drug candidates. Many of the drugs currently on the market also limit their range of applications due to poor ADME properties. The rapid metabolism of drugs can lead to the inability of many drugs that could be effectively treated to treat diseases because they are too quickly removed from the body. Frequent or high-dose medications may solve the problem of rapid drug clearance, but this approach can lead to problems such as poor patient compliance, side effects caused by high-dose medications, and increased treatment costs. In addition, rapidly metabolizing drugs may also expose patients to undesirable toxic or reactive metabolites.
Although LOXO-101 is effective as a Trk inhibitor in the treatment of a variety of cancers and the like, it has been found that a novel compound having a good oral bioavailability and a drug-forming property for treating a cancer or the like is a challenging task. Thus, there remains a need in the art to develop compounds having selective inhibitory activity or better pharmacodynamics/pharmacokinetics for Trk kinase mediated diseases useful as therapeutic agents, and the present invention provides such compounds.
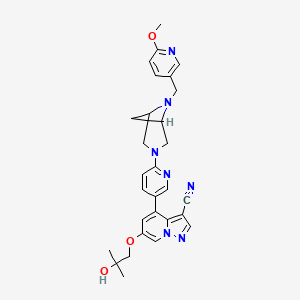
Compounds of Formula I-IV, 4-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula I); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula II); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-(6-methoxynicotinoyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula III); and 6-(2-hydroxy-2-methylpropoxy)-4-(6-(4-hydroxy-4-(pyridin-2-ylmethyl)piperidin-1-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula IV) are inhibitors of RET kinase, and are useful for treating diseases such as proliferative diseases, including cancers.
[0007] Accordingly, provided herein is a compound of Formula I-IV:
and pharmaceutically acceptable salts, amorphous, and polymorph forms thereof.
PATENT
WO 2019075114
PATENT
WO-2019120194
Novel deuterated analogs of pyrazolo[1,5-a]pyrimidine compounds, particularly selpercatinib , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, inflammation, cancer and certain infectious diseases.
Example 2(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl-2,3,3-d 3)-pyrazolo[ 1,5-a] pyrimidin-3-yl) -3-hydroxypyrazole prepared pyrrolidine-1-carboxamide (compound L-2) a.
No development reported B-cell lymphoma; Lymphoid leukaemia
26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
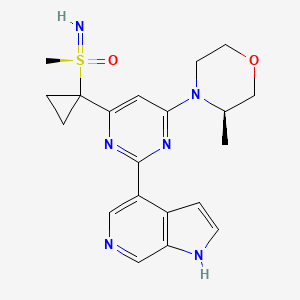
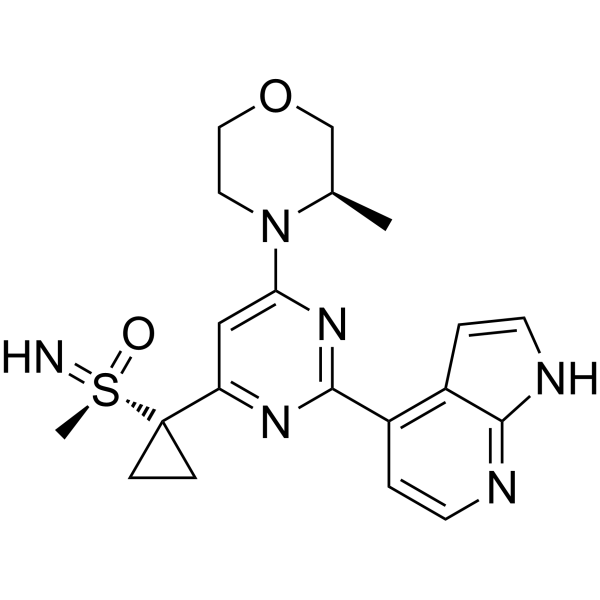
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
The kinase ataxia telangiectasia mutated and rad3 related (ATR) is a key regulator of the DNA-damage response and the apical kinase which orchestrates the cellular processes that repair stalled replication forks (replication stress) and associated DNA double-strand breaks. Inhibition of repair pathways mediated by ATR in a context where alternative pathways are less active is expected to aid clinical response by increasing replication stress. Here we describe the development of the clinical candidate 2(AZD6738), a potent and selective sulfoximine morpholinopyrimidine ATR inhibitor with excellent preclinical physicochemical and pharmacokinetic (PK) characteristics. Compound 2 was developed improving aqueous solubility and eliminating CYP3A4 time-dependent inhibition starting from the earlier described inhibitor 1 (AZ20). The clinical candidate 2 has favorable human PK suitable for once or twice daily dosing and achieves biologically effective exposure at moderate doses. Compound 2 is currently being tested in multiple phase I/II trials as an anticancer agent.
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).
Scheme 1. Medicinal Chemistry Route to AZD6738
Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
AZD6738 is currently being tested in multiple phase I/II trials for the treatment of cancer. Its structure, comprising a pyrimidine core decorated with a chiral morpholine, a cyclopropyl sulfoximine, and an azaindole, make it a challenging molecule to synthesize on a large scale. We describe the evolution of the chemical processes, following the manufacture of AZD6738 from the initial scale-up through to multikilos on plant scale. During this evolution, we developed a biocatalytic process to install the sulfoxide with high enantioselectivity, followed by introduction of the cyclopropyl group first in batch, then in a continuous flow plate reactor, and finally through a series of continuous stirred tank reactors. The final plant scale process to form AZD6738 was operated on 46 kg scale with an overall yield of 18%. We discuss the impurities formed throughout the process and highlight the limitations of this route for further scale-up.
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) (30.0 g) were added at 75 °C, and the reaction mixture was held for 2 h. The mixture was cooled to 20 °C, and n-heptane (141.9 kg) was added at the rate of 40 kg/h. The solid was collected by filtration, washed with a mixture of 1-butanol and n-heptane (9.3 and 22.4 kg respectively), and then given a further wash with n-heptane (32.2 kg). The solid was dried at 40 °C to give imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) as a whit solid (41.4 kg, 92% yield): Assay (HPLC) 99.9%; Assay (NMR) 99% wt/wt.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.
//////AZD6738, AZD-6738, AZD 6738, AstraZeneca, University of Pennsylvania, Phase II, Breast cancer, Gastric cancer, Non-small cell lung cancer, Ovarian cancer, Ceralasertib
Ceralasertib
CAS 1352226-88-0
(R)-imino(methyl)(1-{6-[(3R)-3-methylmorpholin-4-yl]-2-{1H-pyrrolo[2,3-c]pyridin-4-yl}pyrimidin-4-yl}cyclopropyl)-lambda6-sulfanone
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane
UNII 85RE35306Z
Molecular Weight
412.51
Formula
C20H24N6O2S
AZD 6738, ATR KINASE INHIBITOR AZD6738, AZD-6738, AZD6738, WHO 10842
Ceralasertib (AZD6738) is an orally active and bioavailable inhibitor of ATR kinase with an IC50 of 1 nM.
Ceralasertib is an investigational new drug that is being evaluated for the treatment of cancer.[1] It is an ATR kinase inhibitor.[2]
Originator AstraZeneca; University of Pennsylvania
Developer Acerta Pharma; AstraZeneca; Dana-Farber Cancer Institute; Gustave Roussy; National Cancer Institute (France); Samsung Medical Center; University of California at San Francisco; University of Pennsylvania
Class Antineoplastics; Cyclopropanes; Imines; Ketones; Morpholines; Organic sulfur compounds; Pyridines; Pyrimidines; Pyrroles; Small molecules
Mechanism of Action ATR protein inhibitors
Phase III Non-small cell lung cancer
Phase IICholangiocarcinoma; Gynaecological cancer; Malignant melanoma; Osteosarcoma; Ovarian cancer; Pancreatic cancer; Prostate cancer; Small cell lung cancer; Solid tumours; Triple negative breast cancer
PreclinicalDiffuse large B cell lymphoma; Type 1 diabetes mellitus
DiscontinuedHaematological malignancies; Non-Hodgkin’s lymphoma; Squamous cell cancer
22 Apr 2025AstraZeneca plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) (PO) in April 2025(NCT06929260)
23 Dec 2024AstraZeneca plans a phase I trial for Non-small Cell Lung Cancer, Ovarian Cancer, or Endometrial Cancer in United Kingdom(PO) In January 2025 (NCT06754761)
07 Dec 2024Updated efficacy and adverse event data from a phase I trial in Myelodysplastic syndrome presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
SCHEME
PATENT
WO2011154737
CN112939966
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00482
4-{4-[(3R)-3-methyl-4-morpholinyl]-6-[1-(S-methylsulfonimidoyl)cyclopropyl]-2-
pyrimidinyl}-1H-pyrrolo[2,3-b]pyridine (1).
Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry
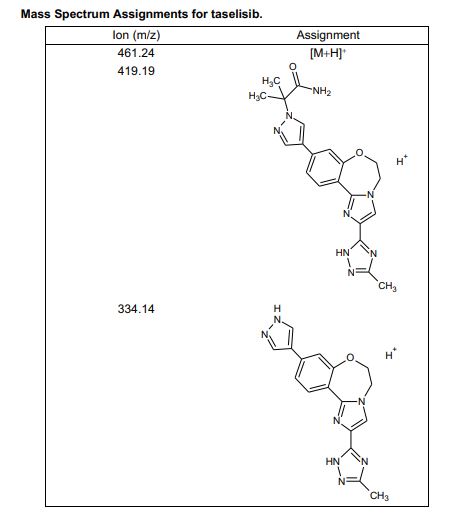
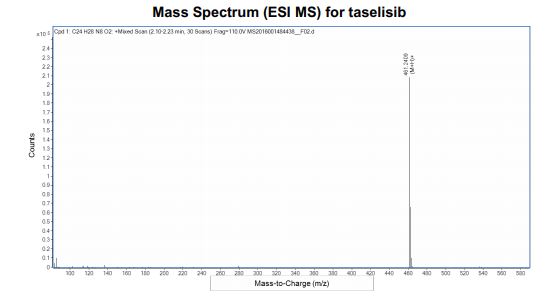
Taselisib (GDC-0032) is an experimental cancer drug in development by Roche. Molecule is a complex heterocycle with no chiral centres, hazardous materials are used in synthesis, preparation of impurities is a challenge. Taselisib is in phase III with Roche , clinical trials for treatment of metastatic breast cancer and non-small cell lung cancer
Taselisib (GDC-0032) is an experimental cancer drug in development by Roche. It is a small molecule inhibitor targeting phosphoinositide 3-kinase subtype PIK3CA.[1]
Taselisib is in phase III with Roche , clinical trials for treatment of metastatic breast cancer and non-small cell lung cancer.[2]
Taselisib is a phosphatidylinositol 3-kinase (PI3Kalpha) inhibitor in phase III clinical studies at Roche for the treatment of postmenopausal women with histologically or cytologically confirmed locally advanced or metastatic estrogen-receptor positive (ER+) breast cancer.
Taselisib is an orally bioavailable inhibitor of the class I phosphatidylinositol 3-kinase (PI3K) alpha isoform (PIK3CA), with potential antineoplastic activity. Taselisib selectively inhibits PIK3CA and its mutant forms in the PI3K/Akt/mTOR pathway, which may result in tumor cell apoptosis and growth inhibition in PIK3CA-expressing tumor cells. By specifically targeting class I PI3K alpha, this agent may be more efficacious and less toxic than pan PI3K inhibitors. Dysregulation of the PI3K/Akt/mTOR pathway is frequently found in solid tumors and causes increased tumor cell growth, survival, and resistance to both chemotherapy and radiotherapy. PIK3CA, which encodes the p110-alpha catalytic subunit of the class I PI3K, is mutated in a variety of cancer cell types and plays a key role in cancer cell growth and invasion.
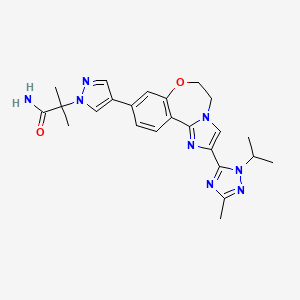
Discovery of 2-(3-(2-(1-Isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo(f)imidazo(1,2-d)(1,4)oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide (GDC-0032): A -sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity
J Med Chem 2013, 56(11): 4597
Condensation of 4-bromo-2-hydroxybenzaldehyde with glyoxal in the presence of NH3 in MeOH gives 5-bromo-2-(1H-imidazol-2-yl)phenol
Which upon annulation with 1,2-dibromoethane in the presence of Cs2CO3 in DMF at 90 °C yields 9-bromo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine .
Iodination of oxazepine with NIS in DMF provides 9-bromo-2,3-diiodo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine,
Which upon mono-deiodination by means of EtMgBr in THF at -15 °C affords 9-bromo-2-iodo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine .
Amidation of iodide with CO in the presence of PdCl2(PPh3)2 and HMDS in DMF at 70 °C produces the intermediate,
Which upon reaction with N,N-dimethylacetamide dimethyl acetal in the presence of DME at 65 °C furnishes intermediate . Intramolecular cyclization of this compound with isopropylamine hydrochloride in AcOH generates triazole derivative,
Which upon Suzuki coupling with dioxaborolane derivative in the presence of Pd(PPh3)4 and KOAc in CH3CN/H2O at 120 °C yields the target compound Taselisib.
Taselisib has been used in trials studying the treatment and basic science of LYMPHOMA, Breast Cancer, Ovarian Cancer, Solid Neoplasm, and HER2/Neu Negative, among others.
Taselisib (GDC 0032) is a potent, next-generation β isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with Ki of 0.29 nM/0.12 nM/0.97nM, >10 fold selective over PI3Kβ.
GDC-0032 is an orally bioavailable, potent, and selective inhibitor of Class I PI3Kα, δ, and γ isoforms, with 30 fold less inhibition of the PI3K β isoform relative to the PI3Kα isoform. Preclinical data show that GDC-0032 has increased activity against PI3Kα isoform (PIK3CA) mutant and HER2-amplified cancer cell lines. GDC-0032 inhibits MCF7-neo/HER2 cells proliferation with IC50 of 2.5 nM. [1]
Cell Data
Cell Lines
Assay Type
Concentration
Incubation Time
Formulation
Activity Description
PMID
human MOLM16 cells
Proliferation assay
72 h
Antiproliferative activity against human MOLM16 cells after 72 hrs by Cell Titer-Blue assay
22727640
In vivo
GDC-0032 pharmacokinetics is approximately dose proportional and time independent with a mean t1/2 of 40 hours. The combination of GDC-0032 enhances activity of fulvestrant resulting in tumor regressions and tumor growth delay (91% tumor growth inhibition (TGI)). In addition, the combination of GDC-0032 with tamoxifen enhances the efficacy of tamoxifen in vivo (102%TGI for GDC-0032). [1]
The invention relates to methods of making the PI3K inhibitor I (GDC-0032), named as 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanamide, having the structure:
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof.
Another aspect of the invention includes novel intermediates useful for preparing GDC- 0032 and having the structures:
The following Schemes 1-15 illustrate the chemical reactions, processes, methodology for the synthesis of GDC-0032, Formula I, and certain intermediates and reagents. Scheme 1:
Scheme 1 shows the synthesis of intermediate isopropylhydrazine hydrochloride 4 from Boc-hydrazine 1. Condensation of 1 with acetone and magnesium sulfate gave Boc-hydrazone, tert-butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 (Example 1). Palladium-catalyzed hydrogenation of 2 in acetic acid and methanol gave Boc-isopropyl-hydrazine 3 (Example 2) which was treated in situ with hydrogen chloride gas to give 4 (Example 3).
Alternatively, the double bond of 2 can be reduced with a hydride reagent such as sodium cyanoborohydride (Example 2).
Scheme 2:
Scheme 2 shows the synthesis of l-isopropyl-3-methyl-lH-l,2,4-triazole 7 from methyl acetimidate hydrochloride 5 and isopropylhydrazine hydrochloride 4. Reaction of 5 and 4 in triethylamine and methanol followed by cyclization of condensation product, N’- isopropylacetohydrazonamide 6 (Example 4) with triethyl orthoformate (triethoxymethane) gave 7 (Example 5). Alternatively, 4 and acetamidine can be reacted to give 6.
Or, 4 can be reacted with acetonitrile and an acid to form the corresponding salt of 6. Scheme 3:
0 K2C03, H20, MTBE w
Scheme 3 shows the synthesis of intermediate, 2-chloro-N-methoxy-N-methylacetamide 10. Reaction of 2-chloroacetyl chloride 8 and Ν,Ο-dimethylhydroxylamine hydrochloride 9 in aqueous potassium carbonate and methyl, tert-butyl ether (MTBE) gave 10 (Example 6).
Scheme 4:
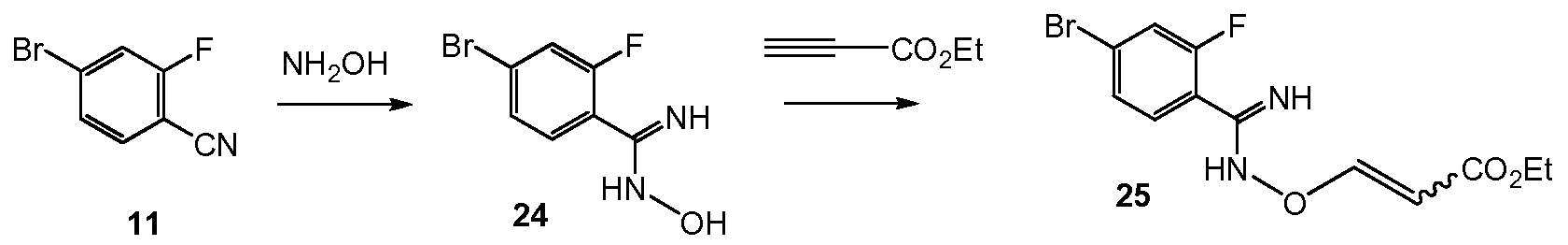
Scheme 4 shows the synthesis of intermediate 4-bromo-2-fluorobenzimidamide hydrochloride 12 formed by reaction of 4-bromo-2-fluorobenzonitrile 11 with lithium hexamethyldisilazide (LiHMDS) in tetrahydrofuran (Example 7). Alternatively, 11 is treated with hydrogen chloride in an alcohol, such as ethanol, to form the imidate, ethyl 4-bromo-2- fluorobenzimidate hydrochloride, followed by ammonia in an alcohol, such as ethanol, to form 12 (Example 7).
Scheme 5:
Scheme 5 shows the synthesis of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole V from l-isopropyl-3-methyl-lH-l,2,4-triazole 7.
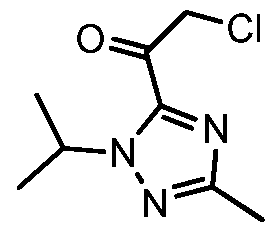
Deprotonation of 7 with n-butyllithium and acylation with 2-chloro-N-methoxy-N- methylacetamide 10 gave intermediate 2-chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)ethanone 13 (Example 8). Cyclization of 13 with 4-bromo-2-fluorobenzimidamide hydrochloride 12 and potassium hydrogen carbonate in water and THF (tetrahydrofuran) formed the imidazole V (Example 9).
Scheme 6:
Scheme 6 shows the synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from V. Alkylation of the imidazole nitrogen of V with a 2-hydroxyethylation reagent such as, l,3-dioxolan-2-one, gave 2-(2-(4- bromo-2-fluorophenyl)-4-( 1 -isopropyl-3-methyl- 1 H- 1 ,2,4-triazol-5 -yl)- 1 H-imidazol- 1 – yl)ethanol 14 (Example 10). Cyclization of 14 with an aqueous basic reagent, such as methyltributylammonium chloride in aqueous potassium hydroxide, gave III, which can be cystallized from ethanol and water (Example 11). Scheme 7:
IV
Scheme 7 shows the synthesis of ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate IV starting from 2-bromo-2-methylpropanoic acid 15. Alkylation of pyrazole with 15 gave 2- methyl-2-(lH-pyrazol-l-yl)propanoic acid 16 (Example 12). Esterification of 16 with sulfuric acid in ethanol gave ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 (Example 13).
Regiospecific bromination of 17 with N-bromosuccinimide (NBS) gave IV (Example 14). Alternatively, 16 was treated in situ with a brominating reagent such as l,3-dibromo-5,5- dimethylhydantoin (DBDMH) to give 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoic acid which was esterified to give IV, where R is ethyl. Other esters can also be prepared, such as methyl, iso-propyl, or any alkyl, benzyl or aryl ester.
Scheme 8:
Scheme 8 shows an alternative synthesis of ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2- methylpropanoate IV starting from ethyl 2-bromo-2-methylpropanoate 18. Alkylation of pyrazole with 18 in the presence of a base such as sodium tert-butyloxide or cesium carbonate gave a mixture of ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 and ethyl 2-methyl-3-(lH- pyrazol-l-yl)propanoate 19. Bromination of the mixture with l,3-dibromo-5,5- dimethylimidazolidine-2,4-dione (DBDMH) gave a mixture containing IV, ethyl 3-(4-bromo- lH-pyrazol-l-yl)-2-methylpropanoate 20, and 4-bromo-lH-pyrazole 21 which was treated with a strong base under anhydrous conditions, such as lithium hexamethyldisilazide in tetrahydrofuran. Acidification with hydrochloric acid gave IV.
Scheme 9:
Pd(O) catalyst
KOAc, EtOH
Scheme 9 shows the synthesis of 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)- 5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanamide, GDC-0032, 1 from ethyl 2-(4-bromo- 1 H-pyrazol- 1 -yl)-2-methylpropanoate IV (CAS Registry Number: 1040377-17-0, WO 2008/088881) and 9-bromo-2-(l-isopropyl-3-methyl-lH- 1,2,4- triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III (CAS Registry Number: 1282514-63-9, US 2012/0245144, US 8242104). Other esters besides ethyl can also be used which can be hydrolyzed with aqueous base, such as methyl, iso-propyl, or any alkyl, benzyl or aryl ester. In a one-pot Miyaura Borylation /Suzuki, Buchwald system, ethyl 2-(4-bromo-lH- pyrazol-l-yl)-2-methylpropanoate IV is reacted with 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l,3,2- dioxaborolane), CAS Reg. No. 73183-34-3, also referred to as B2Pin2, and a palladium catalyst such as XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, CAS Reg. No. 564483- 18-7), with a salt such as potassium acetate, in a solvent such as ethanol, at about 75 °C to form the intermediate ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol- l-yl)propanoate 22 (Example 15, CAS Registry Number: 1201657-32-0, US 8242104, US 8263633, WO 2009/150240).
XPhos ligandIntermediate 22 can be isolated or reacted in situ (one pot) with III to form 23.
A variety of low valent, Pd(II) and Pd(0) palladium catalysts can be used during the Suzuki coupling step to form 23 (Example 16) from 22 and III, including PdCl2(PPh3)2, Pd(t- Bu)3, PdCl2 dppf CH2C12, Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Pd[(Pet3)]2, Pd(DIPHOS)2, Cl2Pd(Bipy), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[P(o-tol)3]2, Pd2(dba)3/P(o-tol)3, Pd2(dba)/P(furyl)3,
The ester group of 23 is saponified with an aqueous basic reagent such as lithium hydroxide, to give 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoic acid II (Example 17). Intermediate 23 can be isolated or further reacted in situ with the aqueous basic reagent to form II. The carboxylic acid group of II is activated with an acyl activating reagent such as di(lH-imidazol-l-yl)methanone (carbonyl diimidazole, CDI) or Ν,Ν,Ν’,Ν’-tetramethyl- 0-(7-azabenzotriazol-l-yl)uronium hexafluorophosphate (HATU), and then reacted with an alcoholic ammonia reagent, such as ammonia dissolved in methanol, ethanol, or isopropanol, aqueous ammonium hydroxide, aqueous ammonium chloride, or ammonia dissolved in THF, to give I (Example 18).
A variety of solid adsorbent palladium scavengers can be used to remove palladium after the Suzuki coupling step to form compound I. Exemplary embodiments of palladium scavengers include FLORISIL®, SILIABOND®Thiol, and SILIABOND® Thiourea. Other palladium scavengers include silica gel, controlled-pore glass (TosoHaas), and derivatized low crosslinked polystyrene QUADRAPURE™ AEA, QUADRAPURE™ IMDAZ, QUADRAPURE™ MPA, QUADRAPURE™ TU (Reaxa Ltd., Sigma-Aldrich Chemical Co.).
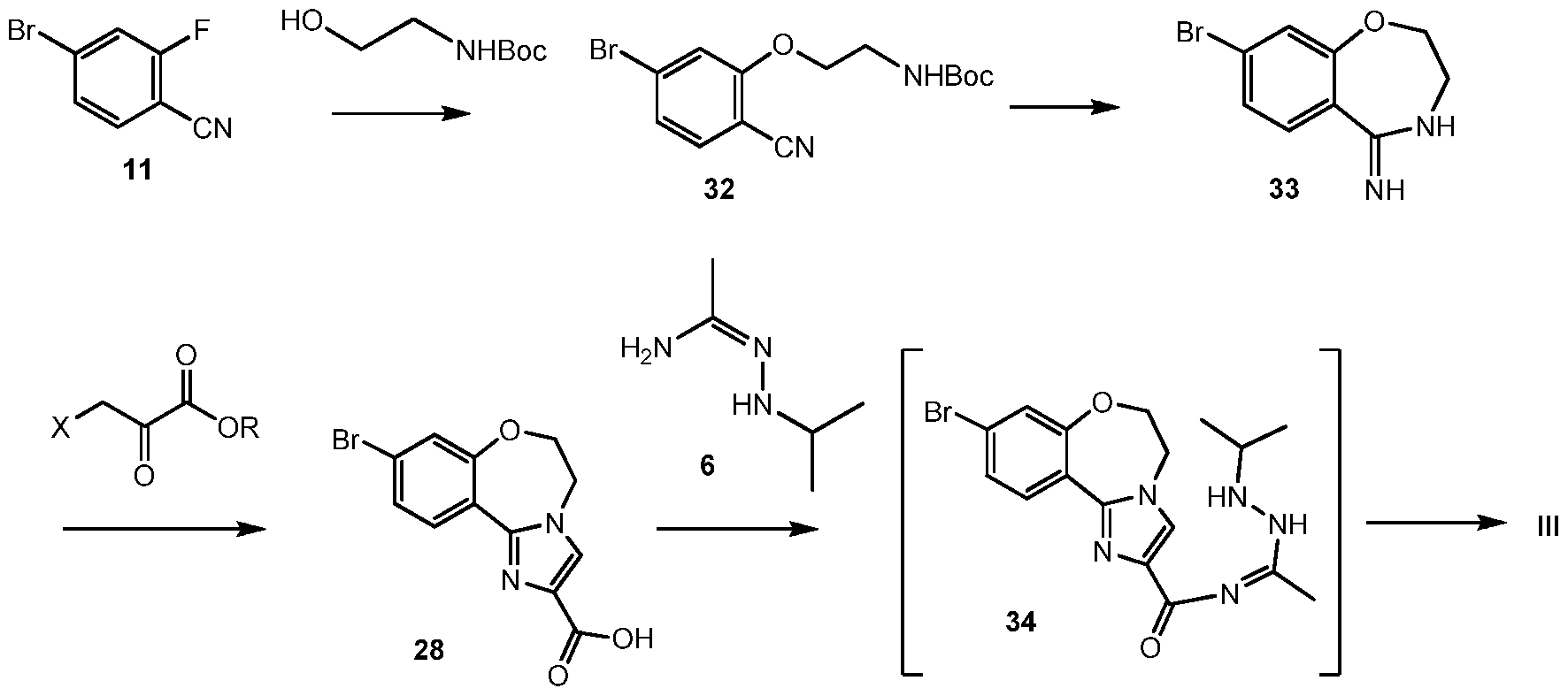
Scheme 10 shows the synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from 4-bromo-2-fluorobenzonitrile 11. Addition of hydroxylamine to the nitrile of 11 gave 4-bromo-2-fluoro-N-hydroxybenzimidamide 24. Michael addition of 24 to ethyl propiolate gave ethyl 3-(4-bromo-2- fluorobenzimidamidooxy)acrylate 25. Heating 25 in a high-boiling solvent such as toluene, xylene, ethylbenzene, or diphenyl oxide gave cyclized imidazole, ethyl 2-(4-bromo-2- fluorophenyl)-lH-imidazole-4-carboxylate 26, along with by-product pyrimidine, 2-(4-bromo-2- fluorophenyl)pyrimidin-4-ol. Alternatively, 25 can be cyclized to 26 with catalytic Lewis acids such as Cu(I) or Cu(II) salts. Alkylation of 26 with a 2-hydroxyethylation reagent, such as 1,3- dioxolan-2-one, in a base, such as N-methylimidazole or cesium carbonate, gave ethyl 2-(4- bromo-2-fluorophenyl)-l-(2-hydroxyethyl)-lH-imidazole-4-carboxylate 27. Ring-cyclization of 27 with an aqueous basic reagent, such as potassium hydroxide, lithium hydroxide, and methyl tributylammonium hydrochloride, gave 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepine-2-carboxylic acid 28. Addition of acetamidine to 28 with triphenylphosphine gave 9-bromo-N-(l-iminoethyl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2- carboxamide 29. Ring-cyclization of 29 with isopropylhydrazine hydrochloride 4 in acetic acid gave 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepine III.
Alternatively, 28 can be reacted with N’-isopropylacetohydrazonamide 6 to give III (Scheme 12).
Scheme 11 :
Scheme 11 shows the synthesis of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l ,2,4-triazole V from 4-bromo-2-fluorobenzimidamide hydrochloride 12. 3-Chloro-2-oxopropanoic acid and 12 are reacted with base to give 2-(4-bromo-2-fluorophenyl)- lH-imidazole-4-carboxylic acid 30. Alternatively, 3-bromo-2-oxopropanoic acid can be reacted with 12 to give 30. Reaction of 30 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU (N,N,N’,N’-tetramethyl-0-(lH-benzotriazol-l-yl)uronium hexafluorophosphate, O- (Benzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate, CAS Ref. No. 94790- 37-1) in DMF gives intermediate, 2-(4-bromo-2-fluorophenyl)-N-(l-(2- isopropylhydrazinyl)ethylidene)-lH-imidazole-4-carboxamide 31 which need not be isolated and cyclizes upon heating to give V.
Alternatively, 5-(2-(4-chloro-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl- lH-l,2,4-triazole 44, the chloro version of V, can be prepared from 4-chloro-2-fluorobenzonitrile 38 (Scheme 15) Scheme 12:
Scheme 12 shows an alternative synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4- triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from 4-bromo-2- fluorobenzonitrile 11. Alkylation of 11 with tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2-cyanophenoxy)ethylcarbamate 32. Cyclization of 32 under acidic conditions, such as hydrochloric acid in ethanol, gives 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33. It will be noted that 33 has an alternative tautomeric form where the double bond is inside the oxazepine ring. Formation of the imidazole ring occurs by reaction of 3-bromo-2- oxopropanoic acid (X = Br, R = OH), or other 3-halo-2-oxopropanoic acid or ester (R = alkyl), and 33 to give 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28. Coupling of 28 with N’-isopropylacetohydrazonamide 6 and a coupling reagent such as HBTU, HATU or CDI in DMF gives intermediate, 9-bromo-N-(l-(2-isopropylhydrazinyl)ethylidene)- 5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxamide 34, which need not be isolated and forms 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III upon heating.
Alternatively, N’-isopropylacetohydrazonamide 6 is used as monohydrochloride salt, which has to be set free under the reaction conditions with an appropriate base, such as K2CO3. Scheme 13:
Scheme 13 shows an alternative synthesis of 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin- 5(2H)-imine 33 from 4-bromo-2-fluorobenzonitrile 11. Reaction of 11 with sodium methoxide in methanol gives methyl 4-bromo-2-fluorobenzimidate 35. Alkylation of 35 with 2- aminoethanol gives 4-bromo-2-fluoro-N-(2-hydroxyethyl)benzimidamide 36, followed by cyclization to 33.
Scheme 14:
37
11
Scheme 14 shows another alternative synthesis of 8-bromo-3,4- dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 from 4-bromo-2-fluorobenzonitrile 11. Reaction of 11 with 2-aminoethanol and potassium tert-butoxide displaces fluorine to give 2-(2- aminoethoxy)-4-bromobenzonitrile hydrochloride 37. Ring closure of 37 with
trimethylaluminum gave 33. Alternatively, other trialkylaluminum reagents can be used, or magnesium alkoxide reagents such as magnesium ethoxide (magnesium bisethoxide, CAS Reg. No. 2414-98-4) to cyclize 37 to 33.
Scheme 15 shows the synthesis of 5-(2-(4-chloro-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole 44 from 4-chloro-2-fluorobenzonitrile 38. Addition of hydroxylamine to the nitrile of 38 gave 4-chloro-2-fluoro-N-hydroxybenzimidamide 39.
Michael addition of 39 to ethyl propiolate gave ethyl 3-(4-chloro-2- fluorobenzimidamidooxy)acrylate 40. Heating 40 in diphenyl oxide gave cyclized imidazole, ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41. Saponification of the ester of 41 with aqueous sodium hydroxide in tetrahydrofuran gave 2-(4-chloro-2-fluorophenyl)-lH- imidazole-4-carboxylic acid 42. Reaction of 42 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in DMF gives intermediate, 2-(4-chloro-2-fluorophenyl)-N-(l-(2- isopropylhydrazinyl)ethylidene)-lH-imidazole-4-carboxamide 43 which cyclizes upon heating to give 44.
EXAMPLES
Example 1 tert-butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2
To a solution of tert-butyl hydrazinecarboxylate 1 (CAS Reg. No. 870-46-2) (25.1 g, 0.190 mol) in acetone (185 mL) was added the magnesium sulfate (6 g) and 12 drops acetic acid (Wu et al (2012) Jour. Med. Chem. 55(6):2724-2736; WO 2007/056170; Zawadzki et al (2003) Polish Jour. Chem. 77(3):315-319). The mixture was heated to reflux for 2.5 h and cooled to rt and filtered. The filtrate was concentrated to give tert-butyl 2-(propan-2- ylidene)hydrazinecarboxylate 2 (CAS Reg. No. 16689-34-2) as an off-white solid (32 g, 98%) (used in the next step without further purification). LC-MS [M+H]+ = 172.9, RT = 2.11 min. 1H NMR 300 MHz (CDC13) d 7.35 (br s, 1H, NH), 2.04 (s, 3H), 1.82 (s, 3H), 1.54 (s, 9H); 13C NMR 300 MHz (CDC13) d 152.9, 149.7, 80.7, 28.1, 25.3, 15.9. Example 2 tert-butyl 2-isopropylhydrazinecarboxylate 3
tert-Butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 was reduced with palladium catalyst on carbon with hydrogen gas in acetic acid and methanol to give tert-butyl 2- isopropylhydrazinecarboxylate 3 (CAS Reg. No. 16689-35-3).
Alternatively, tert-Butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 (0.51 g, 3.0 mmol) was dissolved in 20 mL of THF, treated with NaB¾CN (0.19 g, 3.0 mmol) and a few mg of bromocresol green, followed by a solution of p-toluenesulfonic acid (0.57 g, 3.0 mmol) in 1.5 mL of THF which was added dropwise over approximately 1 h to maintain the reaction pH between 3.5-5.0. After stirring at room temperature for an additional hour, the solvent was removed by rotary evaporation, and the residue was partitioned between EtOAc (30 mL) and brine. The organic phase was extracted with sat. NaHCC>3, 20 mL and brine, evaporated to a residue and dissolved in 10 mL of ethanol. The ethanolic solution was treated with 3.6 mL of 1M NaOH solution (3.6 mmol) and left to stir at rt for 30 min. The solvent was removed by rotary evaporation and the residue was taken up into ethyl acetate and extracted with water. The organic layer was evaporated under reduced pressure and the residue was purified by column chromatography using 5 % MeOH in DCM as eluent to collect tert-butyl 2- isopropylhydrazinecarboxylate 3 (0.4 g, 77 % yield): mp = 47-49 °C; Rf = 0.44 (5 % MeOH in DCM); IH NMR 300 MHz (CDC13) d 6.03 (s, N-H, IH), 3.92 (s, N-H, IH), 3.14 (m, IH), 1.46 (s, 9H), 1.02 (d, 6H, J = 6 Hz); 13C NMR 300 MHz (CDC13) d 157.2, 80.8, 51.2, 28.7, 21.0.
Example 3 isopropylhydrazine hydrochloride 4
tert-butyl 2-isopropylhydrazinecarboxylate 3 was treated with hydrochloric acid to remove the Boc protecting group and give 4 (CAS Reg. No. 16726-41-3).
Example 4 N’-isopropylacetohydrazonamide 6
Methyl acetimidate hydrochloride 5 (CAS Reg. No. 14777-27-6), isopropylhydrazine hydrochloride 4, and triethylamine were reacted in methanol to give 6 (CAS Reg. No. 73479-06- 8).
Example 5 l-isopropyl-3 -methyl- lH-l,2,4-triazole 7
N’-isopropylacetohydrazonamide 6 was treated with triethylorthoformate in ethanol, followed by triethylamine and tetrahydrofuran to give 7 (CAS Reg. No. 1401305-30-3). Example 6 2-chloro-N-methoxy-N-methylacetamide 10
To a solution of 21.2 kg potassium carbonate K2CO3 (153.7 mol, 3.0 eq) in 30 L H20 was added, Ν,Ο-dimethylhydroxylamine 9 (CAS Reg. No. 1117-97-1) (5.0 kg, 51.3 mol, 1.0 eq) at 15-20 °C. The reaction was stirred at rt for 30min and 30 L methyl tert-butyl ether (TBME) was added. After stirred for 30min, the mixture was cooled to 5°C, and 11.6 kg of 2-Chloroacetyl chloride 8 (CAS Reg. No. 79-04-9 (102.7 mol, 2.0 eq) were added slowly. The reaction was stirred at rt overnight. Organics were separated from aqueous, and aqueous was extracted with TBME (30 L). The combined organics were washed with H20 (50 L), brine (50 L) and dried over Na2S04. Filtered and concentrated under vacuum afforded 5.1 kg of 2-chloro-N-methoxy- N-methylacetamide 10 (CAS Reg. No. 67442-07-3) as a white solid.
Example 7 4-bromo-2-fluorobenzimidamide hydrochloride 12
To 35.0 L of lithium hexamethyldisilazide LiHMDS (35.0 mol, 1.4 eq, 1.0 M in THF) under N2 was added a THF solution of 4-Bromo-2-fluorobenzonitrile 11 (CAS Reg. No. 105942- 08-3) (5.0 kg in 10 L THF) at 10 °C, the mixture was stirred at rt for 3h. Cooled to -20°C and 8.3 L of HCl-EtOH (6.6 M) were added. The mixture was stirred at -10 °C for additional lh, filtered. The wet cake was washed with EA (10 L) and H20 (6 L). Drying in vacuo yielded 5.8 kg 4- bromo-2-fluorobenzimidamide hydrochloride 12 (CAS Reg. No. 1187927-25-8) as an off-white solid.
Alternatively, to a 200-L vessel was charged 4-bromo-2-fluorobenzonitrile 11 (10 kg, 50.00 mol, 1.00 equiv) and ethanol (100 L) followed by purging 40 kg Hydrogen chloride (g) at – 10 °C with stirring (Scheme 4). The resulting solution was allowed to react for an additional 36 h at 10 °C. The reaction progress was monitored by TLC until 11 was consumed completely. The resulting mixture was concentrated under vacuum while maintaining the temperature below 60 °C. The volume was concentrated to 10-15 L before 60 L MTBE was added to precipitate the product. The precipitates were collected by filtration to afford in 12 k g of ethyl 4-bromo-2- fluorobenzimidate hydrochloride 12 as a white solid. (Yield: 85%). 1H NMR δ 7.88-7.67 (m), 4.89 (br s), 4.68 (q), 3.33 (m), 1.61 (t). MS M+l: 245.9, 248.0.
To a 200L vessel, was charged ethyl 4-bromo-2-fluorobenzimidate hydrochloride (12.5 k g, 44mol, 1.00 equiv, 99%) and ethanol (125 L) followed by purging NH3 (g) at -5 °C for 12 h. The resulting solution was stirred at 30 °C for an additional 24 h. The reaction progress was monitored by TLC until SM was consumed completely. The precipitates were filtered and the filtrate was concentrated under vacuum. The product was precipitated and collected by filtration to afford 6.1 kg (54.5%) of 4-bromo-2-fluorobenzamidine hydrochloride 12 as a white solid. 1H NMR δ 9.60 (br), 7.91-7.64 (m), 3.40 (s), 2.50 (m). MS M+l: 216.9, 219.9.
Example 8 2-chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)ethanone 13
To a 10L four necked flask was charged l-Isopropyl-3-methyl-lH-l,2,4-triazole 7 (400 g) in THF (2.5 L). The resulting solution was cooled to -40 °C and 2.5 M n-butyllithium BuLi in n- hexanes (1.41 L) was added while keeping the internal temp, below -20°C. The resulting yellow suspension was stirred at -40°C for 1 hour before being transferred. To a 20L flask was charged 2-chloro-N-methoxy-N-methylacetamide 10 (485 g) in THF (4 L). The resulting solution was cooled to -40 °C at which point a white suspension was obtained, and to this was added the solution of lithiated triazole 7 keeping the internal temp, below -20°C. At this point a yellow orange solution was obtained which was stirred at – 30°C for lhour. Propionic acid (520 mL) was added keeping the internal temp, below -20°C. The resulting off-white to yellowish suspension was warmed to -5 °C over 30 minutes. Citric acid (200 g) in water (0.8 L) was added and after stirring for 5 minutes a clear biphasic mixture was obtained. At this point stirring was stopped and the bottom aqueous layer was removed. The organic phase was washed with 20w% K3PO4 solution (1 L), 20w% K2HP04 solution (2 L), and 20w% NaCl solution (1 L). The organics was reduced to ca 4L via distillation under vacuum to afford 2-chloro-l-(l-isopropyl-3- methyl-lH-l,2,4-triazol-5-yl)ethanone 13 as a dark amber liquid which was used “as is” in the next step.
Example 9 5-(2-(4-bromo-2-fluorophenyl)- lH-imidazol-4-yl)-l -isopropyl-3-methyl- lH-l,2,4-triazole V
To a 10 L four- neck flask were charged with THF (5.6 L), 4-bromo-2- fluorobenzimidamide hydrochloride 12 (567 g), KHCO3 (567 g) and water (1.15 L). The resulting white suspension was heated to 60°C over 2 hours. At this point a hazy solution was obtained to which was added a solution of 2-Chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)ethanone 13 in THF (2 L). This solution was stirred at 60-65 °C for 24 hours. Then the aqueous bottom layer was removed. The organic layer was concentrated under vacuum. The residue was slurried in a mixture of MIBK (1.25 L) and toluene (0.7 L), and the precipitated product was filtered giving 552 g of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole V (98.0% purity, 254 nm) as a brown solid Example 10 2-(2-(4-bromo-2-fluorophenyl)-4-(l-isopropyl-3-methyl-lH-l,2,4-triazol- 5-yl)- 1 H-imidazol- 1 -yl)ethanol 14
5-(2-(4-Bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl-lH- 1,2,4- triazole V (2.75 kg, 7.55 mol) was added to a solution of 3-dioxolan-2-one (ethylene carbonate, 3.99 kg, 45.3 mol) inN-methylimidazole (12 L) at 50 °C. The suspension was heated at 80°C for 7 h until the reaction was judged complete by HPLC. The solution of 14 was cooled to 35 °C and used directly in the subsequent cyclization.
Example 11 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine III
To a solution of 2-(2-(4-Bromo-2-fluorophenyl)-4-(l-isopropyl-3-methyl-lH-l,2,4- triazol-5-yl)-l H-imidazol- l-yl)ethanol (7.55 mmol) 14 inN-methylimidazole(12 L) at 35 °C was added methyl tributylammonium chloride (115 g, 0.453 mol), toluene (27.5 L) and 35% potassium hydroxide solution (10.6 kg, 25 mol in 22 L of water). The biphasic solution was stirred vigorously at 65 °C for 18 h when it was judged complete by HPLC. Stirring was stopped but heating was continued and the bottom aqueous layer was removed. Added isopropyl acetate (13.8 L) and the organic phase was washed twice with water (13.8 L and 27.5 L). The solvent was removed via vacuum distillation and after 30 L had been removed, isopropanol (67.6 L) was added. Vacuum distillation was resumed until an additional 30 L of solvent had been removed. Added additional isopropanol (28.8 L) and continued vacuum distillation until the volume was reduced by 42 L. Added isopropanol (4L) and the temperature was increased to >50 °C. Added water (28 L) such that the internal temperature was maintained above 50 °C, then heated to 75 °C to obtain a clear solution. The mixture was allowed to cool slowly and the product crystallized out of solution. The resulting suspension was cooled to 0 °C, held for 1 h then filtered and the cake was washed with water (5.5 L). The cake was dried at 45 °C under a nitrogen sweep to give III as a tan solid (3.30 kg, 71.6 wt %, 80.6% yield).
Example 12 2-methyl-2-(lH-pyrazol-l-yl)propanoic acid 16
2-Bromo-2-methylpropanoic acid 15 and pyrazole were reacted in triethylamine and 2- methyltetrahydrofuran to give 16.
Example 13 ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17
2-Methyl-2-(lH-pyrazol-l-yl)propanoic acid 16 was treated with sulfuric acid in ethanol to give 17. Alternatively, pyrazole (10 g, 147 mmol, 1.0 eq.) was dissolved in DMF (500 ml) at room temperature (Scheme 8). 2-Bromoisobutyrate 18 (22 ml, 147 mmol, 1.0 eq.), cesium carbonate CS2CO3 (53 g, 162 mmol, 1.1 eq) and catalytic sodium iodide Nal (2.2 g, 15 mmol, 0.1, eq) were added to the mixture that was then heated to 60 °C for 24 hr. Reaction was followed by 1H NMR and pyrazole was not detected after 24 hr. The reaction mixture was quenched with a saturated solution of NaHCC>3 (200 ml) and ethyl acetate EtOAc (150 ml) was added and organics were separated from aqueous. Organics were dried over Na2S04, filtered and concentrated under vacuum to afford an oil which was purified by flash chromatography to give 17.
Example 14 Ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate IV
Method A: Ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 was reacted with N- bromosuccinimide (NBS) in 2-methyltetrahydrofuran to give IV (CAS Reg. No. 1040377-17-0).
Method B: Ethyl 2-bromo-2-methylpropanoate 18 and pyrazole were reacted with sodium tert-butoxide in dimethylformamide (DMF) to give a mixture of ethyl 2-methyl-2-(lH- pyrazol-l-yl)propanoate 17 and ethyl 2-methyl-3-(lH-pyrazol-l-yl)propanoate 19 which was treated with l,3-dibromo-5,5-dimethylimidazolidine-2,4-dione to give a mixture of IV, ethyl 3- (4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate 20, and 4-bromo-lH-pyrazole 21. The mixture was treated with a catalytic amount of lithium hexamethyldisilazide in tetrahydrofuran followed by acidification with hydrochloric acid to give IV.
Example 15 ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazol-l-yl)propanoate 22
To a 50 L glass reactor was charged ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2- methylpropanoate IV (1.00 kg, 3.85 mol, 1.00 equiv), potassium acetate, KOAc (0.47 kg, 4.79 mol 1.25 equiv), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane),
bis(pinacolato)diboron, B2Pin2 (1.22 kg, 4.79 mol, 1.25 equiv) and ethanol (10 L, 10 vol) and the mixture was stirred until a clear solution was obtained. The solution was vacuum/degassed 3x with nitrogen. To this mixture was charged XPhos ligand (0.023 kg, 0.048 mol, 1.0 mol ) and the Pd precatalyst (0.018 kg, 0.022 mol, 0.5 mol ) resulting in a homogeneous orange solution. The solution was vacuum/degassed once with nitrogen. The internal temperature of the reaction was set to 75 °C and the reaction was sampled every 30 min once the set temperature was reached and was monitored by LC (IPC method: XTerra MS Boronic). After 5 h, conversion to 22 (CAS Reg. No. 1201657-32-0) was almost complete, with 1.3% IV remaining. Example 16 ethyl 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoate 23
Ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propanoate 22 and 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III were reacted under Suzuki conditions with palladium catalyst, in isopropanol and aqueous phosphate buffer to give 23.
A 1M solution of K3PO4 (1.60 kg in 7.6 L of water, 7.54 mol, 2.00 equiv) was charged to the above reaction mixture from Example 15, followed by the addition of a solution of III in THF (1.33 kg in 5.0 L, 3.43 mol, 0.90 equiv) over 2 min. The reaction mixture was warmed to 75 °C (internal temperature) over 45 min and stirred for 13 h at 75 °C, then analyzed by HPLC (III not detected) showing the formation of 23.
Ethyl 2-(4-(2-(l -isopropyl-3 -methyl- 1 H- 1 ,2,4-triazol-5-yl)-5 ,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanoate 23 was treated with aqueous lithium hydroxide to give II.
The ester saponification reaction was initiated with the addition of 3.5 M aqueous LiOH (0.74 kg in 5.0 L, 17.64 mol, 5 equiv) to the reaction mixture from Example 16 and allowed to warm to 75 °C. The mixture was sampled every 30 min (IPC method: XTerra MS Boronic) and the saponification was complete after 4.5 h (with less than 0.3% 23 remaining). The reaction mixture was concentrated via distillation to approximately half volume (starting vol = 37 L; final vol = 19 L) to remove EtOH and THF, resulting in tan-brown slurry. Water (5 L, 5 vol) was charged to the mixture and then distilled (starting vol = 25 L; final vol = 21 L). The temperature was set at 60 °C (jacket control) and then charged with isopropyl acetate, IP Ac (4 L, 4 vol). The biphasic mixture was stirred a minimum of 5 min and then the layers allowed to separate for a minimum of 5 min. The bottom aqueous layer was removed into a clean carboy and the organics were collected into a second carboy. The extraction process was repeated a total of four times, until the organic layer was visibly clear. The aqueous mixture was transferred back to the reactor and then cooled to 15 °C. A 6 M solution of HC1 (6.4 L, 38.40 mol, 10 equiv) was charged slowly until a final pH = 1 was obtained. The heterogeneous mixture was then filtered. The resulting solids were washed twice with 5 L (2 x 5 vol) of water. The filter was then heated to 80 °C and the vacuum set to -10 Psi (with nitrogen bleed) and the solids were dried for 24 h (KF = 2.0 % H20) to give 1.54 kg (95% corrected yield) of II as a white solid; 98% wt, 97.3 % pure.
2-(4-(2-(l-Isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanoic acid II was treated with di(lH- imidazol-l-yl)methanone (carbonyldiimidazole, CDI) in tetrahydrofuran followed by methanolic ammonia to give crude I.
Solid II (1.44 kg, 3.12 mol, 1.00 equiv) was transferred into a 20 L bottle and then THF
(10 L, 7 vol) was charged. The slurry was transferred under reduced pressure into a second 50 L reactor and additional THF (5 L, 3 vol) was added for the rinse. The internal temperature of the slurry was set to 22 °C and Γ1 -carbonyldiimidazole, CDI (0.76 Kg, 5.12 mol, 1.50 equiv) was charged to the mixture and a clear solution was observed after 5 min. The reaction mixture was sampled every 30 min and analyzed by HPLC (IPC: XTerra MS Boronic method) which showed almost complete conversion to the acyl-imidazole intermediate and 1.2% remaining II after 30 min. An additional portion of CDI (0.07 kg, 0.15 mol, 0.14 equiv) was added, and the reaction mixture was stirred for 1 h and then analyzed by HPLC (IPC: XTerra MS Boronic method) which showed 0.8% remaining II.
Into a second 50-L reactor, was added NH3/MeOH (1.5 L, 10.5 mol, 3.37 equiv) and THF
(5 L, 3 vol). The acyl-imidazole intermediate was transferred to a second reactor under reduced pressure (transfer time -10 min). The internal temperature was then set to 45 °C and the volume of solvent was distilled down from 35 L to 12 L. Water (6 L, 4 vol) was then added to the mixture that was further distilled from 18 L to 11 L. Finally, another portion of water (6 L, 4 vol) was added and the solvents were distilled one last time from 17 L to 14 L, until no more THF was coming out. The reaction was then cooled down to 10 °C (internal temperature). The white slurry was filtered and the filter cake was washed with water (2 x 6 L, 2 x 4 vol). The solids were then dried at 80 °C (jacket temp) in the Aurora filter for 24 h (KF = 1.5 % H20) under vacuum to give 1.25 kg crude I, GDC-0032 (84% corrected yield, 96% wt, 97.3 % pure by HPLC) as a white solid.
A slurry of crude I (1.15 kg, 2.50 moles) in MeOH (6 L, 5 vol) was prepared and then charged to a 50 L glass reactor. Additional MeOH (24 L, 21 vol) was added to the mixture, which was then heated to 65 °C. A homogenous mixture was obtained. Si-thiol (Silicycle, Inc., 0.23 kg, 20% wt) was added to the solution via the addition port and the mixture was stirred for 3 hours. It was then filtered warm via the Aurora filter (jacket temperature = 60 °C, polish filtered and transferred directly into a second 50 L reactor with reduced pressure. The solution was then heated back to 65 °C internal temperature (IT). The homogeneous solution was cooled down to 54 °C and I seeds (12 g, 1 % wt) in MeOH (50 mL) were added with reduced pressure applied to the reactor. The mixture was then cooled down to 20 °C over 16 hours. The solids were then filtered via the Aurora filter and dried at 80 °C for 72 hours to give 921 g, 80% yield of I as a methanoate solvate (form A by XRPD,) and transferred to a pre-weighed charge-point bag.
In an isolator, the solids were slurred in IP Ac (8 L, 7 vol) and transferred to a clean 10 L reactor. The mixture was stirred for 1 h at 60 °C (IT). The solids were then filtered via the Aurora system and dried at 80 °C (jacket) for 96 h. A sample of I was removed and analyzed by GC (IP Ac = 1 %). To attempt more efficient drying, the API was transferred to two glass trays in an isolator and sealed with a drying bag before being dried in a vacuum oven set at 100 °C for 16 h. GC (IPC: Q12690V2) showed 1 % solvent was still present. The process afforded 760 g (68% corrected yield, 68% wt, 99.9 % purity by LC) of a white solid (form B by XRPD).
Crude I (340.7 g) was charged to a 2-1 . 1 1 DPI · bottle and slurried with 0.81 ,
isoamylalcohol (I A A). The slurry was transferred to a 20 L reactor and diluted with 6.7 L round- bottom flask (22 vol total). The white slurry was heated until a solution was observed (internal temperature rose to 118 °C and then cooled to 109 °C). The solution was polish filtered (0.2 μ .Μ filter). A flask was equipped with overhead stirring and the filtrate was slurried in isoamyl alcohol (344 ml ., 21 vol). The mixture was warmed to 95 °C (internal) until the solids dissolved. A slurry of charcoal (10 wt%, 0.16g) and silicycle thiol (10 wt%, 0.16g) in isoamyl alcohol (1 vol, 1 6 ml . ) was charged and the mixture was stirred at 90-95 °C for 1 h and then filtered (over Celite® pad). The clear amber colored solution was cooled to 73 °C (seeding temp range = 70 ±5 °C) and a GDC-0032 I seed (10 wt%, 0.16g) was added. The temperature of the heating mantle was turned off and the mixture was allowed to cool to room temperature overnight with stirrin (200 rpni). After 17 hr, the white solids were filtered starting with slow gravity filtration and then vacuum was applied. The solids were suction dried for 20 min with mixing until a free flowing powder was obtained. ( “rude weight prior to oven drying = 16 g. The solids were oven- dried at 100 °C for 24 h and then sampled for testing. Drying continued at 100 °C for another 24 hr. I l l NMR (DMSO d6) δ 8.38 (t), 8.01 (s), 7.87 (s), 7.44, 7.46 (d), 7.36 (s), 7.18 (br s), 6.81
Purified 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- lH-pyrazol- 1 -yl)-2-methylpropanamide I (GDC-0032) was dry granulation formulated in tablet form by the roller compaction method (He et al (2007) Jour, of Pharm. Sci., 96(5):1342-1355) with excipients including lactose, microcrystalline cellulose (AVICEL® PH 01, FMC BioPolymer, 50 μΜ particle),
croscarmellose sodium (Ac-Di-Sol®, FMC BioPolymer), and magnesium stearate.
Example 19 4-bromo-2-fluoro-N-hydroxybenzimidamide 24
To a solution of 4-Bromo-2-fluorobenzonitrile 11 (800 g, 4 mol, 1 eq), hydroxylamine hydrochloride (695 g, 10 mol, 2.5 eq) in MeOH (2 L, 2.5 vol) was added Et3N (485 g, 4.8 mol, 1.2 eq), then the mixture was stirred at 60 °C for 40 min and checked by HPLC (no nitrile remaining). Reaction was then quenched by H20 (30 L), and lots of off-white solid was separated out, and then filtered, the filter cake was washed with water (10 L x 2) and 1350 g wet 4-bromo-2-fluoro-N-hydroxybenzimidamide 24 was obtained with 96% purity.
Example 20 ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25
To a solution of 4-Bromo-2-fluoro-N-hydroxybenzimidamide 24 (800 g, 3.43 mol, 1 eq) and Amberlyst® A21 (20 wt%, 160 g) in PhMe (12 L, 15 vol) was added ethyl propiolate (471 g, 4.8 mol, 1.4 eq) at 10 °C. The reaction was stirred at 50 °C overnight and checked by LC-MS (ca 14A% of starting material 24 was left). Reaction was then filtered and the filtrate was concentrated under vacuum, and 1015 g ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25 was obtained as a yellow oil with 84.9% LC purity (yield: 89%).
Example 21 ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26
A solution of ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25 (300 g, 0.91 mol, 1 eq) in diphenyl oxide (900 mL, 3 vol) was stirred at 190 °C under N2 for 1 h and checked by LC-MS (no 25 remaining). Cooled the mixture to rt and TBME (600 mL, 2 vol of 25) was added, and then PE (1.8 L, 6 vol of 25) was dropwise added to separate out solids. The mixture was stirred at rt for 20 min, and filtered to give 160 g wet cake. The wet cake was washed with PE (1 L) and dried to afford 120 g ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26 with 92% LC purity as brown solids. Example 22 ethyl 2-(4-bromo-2-fluorophenyl)- 1 -(2-hydroxyethyl)- 1 H-imidazole-4- carboxylate 27
Ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26 and l,3-dioxolan-2- one and N-methylimidazole were reacted to give 27.
Example 23 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28
Ethyl 2-(4-bromo-2-fluorophenyl)-l -(2-hydroxyethyl)- lH-imidazole-4-carboxylate 27, potassium hydroxide and methyl tributylammonium hydrochloride were reacted at 65 °C, cooled, and concentrated. The mixture was dissolved in ethanol and water to crystallize 28.
Example 24 9-bromo-N-(l-iminoethyl)-5,6-dihydrobenzo[f]imidazo[l,2- d] [ 1 ,4]oxazepine-2-carboxamide 29
9-Bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28, triphenylphosphine, and acetamidine were reacted to give 29.
Example 25 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III
9-Bromo-N-( 1 -iminoethyl)-5 ,6-dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine-2- carboxamide 29 was reacted with isopropylhydrazine hydrochloride 4 in acetic acid to give III.
Example 26 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 30
3-Chloro-2-oxopropanoic acid and 4-bromo-2-fluorobenzimidamide hydrochloride 12 are reacted with base to give 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 30.
Alternatively, to a solution of ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4- carboxylate 26 (1350 g, 4.3 mol) in THF (8.1 L, 6 vol) and H20 (4 L, 3 vol) was added NaOH (520 g, 13 mol, 3 eq), and the reaction was stirred at 65 °C for 48 h till it completed (checked by LC-MS). Adjust the mixture with 2 M HC1 to pH = 5, and product was separated out as a yellow solid, filtered to give 2.2 kg wet cake, the wet cake was washed with H20 (1.5 L), DCM (1.5 L x 3), PE (1 L), and dried to afford 970 g pure 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4- carboxylic acid 30 (Scheme 10). Example 27 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl- lH-l,2,4-triazole V
Reaction of 30 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in DMF gives intermediate, 2-(4-bromo-2-fluorophenyl)-N-(l-(2-isopropylhydrazinyl)ethylidene)- lH-imidazole-4-carboxamide 31 which cyclizes upon heating to give V.
Example 28 tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2- cyanophenoxy)ethylcarbamate 32
Alkylation of 4-bromo-2-fluorobenzonitrile 11 with tert-butyl 2-hydroxyethylcarbamate gives 32.
Example 29 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33
Cyclization of tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2- cyanophenoxy)ethylcarbamate 32 under acidic conditions, such as hydrochloric acid in ethanol, gives 33.
Example 30 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28
Reaction of 3-bromo-2-oxopropanoic acid and 8-bromo-3,4- dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 gives 28 (CAS Reg. No. 1282516-74-8).
Example 31 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine III
Coupling of 28 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in
DMF gives intermediate, 9-bromo-N-(l-(2-isopropylhydrazinyl)ethylidene)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxamide 34, which forms III upon heating.
Example 32 methyl 4-bromo-2-fluorobenzimidate 35
Reaction of 4-bromo-2-fluorobenzonitrile 11 with sodium methoxide in methanol gives 35.
Example 33 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33
Alkylation of methyl 4-bromo-2-fluorobenzimidate 35 with 2-aminoethanol gives 4- bromo-2-fluoro-N-(2-hydroxyethyl)benzimidamide 36, followed by cyclization to 33 (Scheme 13). Alternatively, reaction of 11 with 2- aminoethanol and potassium tert-butoxide displaces fluorine to give 2-(2-aminoethoxy)-4-bromobenzonitrile hydrochloride 37. Ring closure of 37 with trimethylaluminum gave 33 (Scheme 14). A solution of 11 (10 g, 50 mmol) and 2- aminoethanol (3.1 mL, 50.8 mmol) in 2-methyltetrahydrofuran (80 mL) was cooled to 0 °C and a solution of 1M potassium tert-butoxide in tetrahydrofuran (55 mL, 55 mmol) was slowly added while maintaining the solution temperature below 5 °C. The reaction was stirred at 0 °C for 30 min until judged complete by HPLC at which point it was warmed to 25 °C. A solution of 0.5M HC1 in isopropanol (100 mL, 50 mmol) was added and the desired HC1 salt 3 crystallized directly from the solution. The solid was collected by filtration and dried under vacuum with a nitrogen bleed to give 2-(2-aminoethoxy)-4-bromobenzonitrile hydrochloride 37 as a white solid. (12.1 g, 87 % yield).
To a flask was charged 37 (9.00 g, 32.4 mmol) and toluene (90.0 ml). The suspension was cooled to 0 °C and was added trimethylaluminum (1.8 equiv., 58.4 mmol, 2M in toluene) drop-wise over 30 minutes. The suspension was then stirred at room temperature for 1 h and then warmed to 100 °C. After 5 h, the solution was cooled to 0 °C and quenched with aqueous NaOH (2N, 90.0 ml). The suspension was extracted with EtOAc (4 x 90 ml) and the combined extracts were dried over then filtered through Celite®. The solution was concentrated and the residue triturated with EtOAc to afford 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 (6.26 g, 26.0 mmol, 80% yield) as white crystalline solid.
Example 34 4-chloro-2-fluoro-N-hydroxybenzimidamide 39
To a solution of 4-chloro-2-fluorobenzonitrile 38 (400 g, 2.58 mol, 1.0 eq),
hydroxylamine hydrochloride (448 g, 6.45 mol, 2.5 eq) in MeOH (1 L, 2.5 vol) was added Et3N (313 g, 3.1 mol, 1.2 eq), then the mixture was stirred at 60 °C for 40 min and checked by HPLC (no nitrile remaining). Reaction was then quenched by H20 (10 L), and lots of off-white solid was separated out, and then filtered, the filter cake was washed with water (10 L x 2) and 378 g 4-chloro-2-fluoro-N-hydroxybenzimidamide 39 was obtained with 93% purity (Scheme 15).
Example 35 ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40
To a solution of 4-chloro-2-fluoro-N-hydroxybenzimidamide 39 (378 g, 2 mol, 1.0 eq) and Amberlyst® A21 (20 wt%, 75.6 g) in toluene PhMe (5.6 L, 15 vol) was added ethyl propiolate (275 g, 2.8 mol, 1.4 eq) at 30 °C. The reaction was stirred at 30 °C overnight and checked by LC-MS. Reaction was then filtered and the filtrate was concentrated under vacuum, and 550 g ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40 was obtained as a yellow oil with 83% LC purity (Scheme 15).
Example 36 ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41
A solution of ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40 (550 g, 1.9 mol, 1.0 eq, 83% LC purity) in diphenyl oxide (1.65 L, 3 vol) was stirred at 190 °C under N2 for 1 h and checked by LC-MS (no 40 remaining). Cooled the mixture to rt and PE (10 L) was added dropwise. The mixture was stirred at rt for 20 min, and filtered to give 400 g wet cake, after purified by chromatography on silica gel (PE / EA=1 / 5) to get 175 g pure ethyl 2-(4-chloro-2- fluorophenyl)-lH-imidazole-4-carboxylate 41 with 98% LC purity (Scheme 15).
Example 37 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 42
To a solution of ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41 (175 g, 4.3 mol) in THF (1 L, 6 vol) and H20 (500 mL, 3 vol) was added NaOH (78 g, 1.95 mol, 3.0 eq), and the reaction was stirred at 65 °C for 48 h till it completed (checked by LC-MS). Adjust the mixture with 2 N HC1 to pH = 5, and product was separated out as a yellow solid, filtered to give 210 g wet cake, the wet cake was washed with H20 (300 mL), DCM (3 x 300 mL), PE (500 mL), and dried to afford 110 g pure 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 42 (CAS Reg. No. 1260649-87-3) (Scheme 15 ). I l l NMR (DMSO d6) δ: 12.8 (br s), 8.0, 7.9 (br s), 7.46, 7.4 (m).
Discovery of 2-{3-[2-(1-Isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity
†Departments of Discovery Chemistry, ‡Drug Metabolism and Pharmacokinetics, §Translational Oncology, and ∥Biochemical Pharmacology, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
⊥ Argenta Discovery, 8-9 Spire Green Centre, Flex Meadow, Harlow, Essex, CM19 5TR, United Kingdom
Dysfunctional signaling through the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway leads to uncontrolled tumor proliferation. In the course of the discovery of novel benzoxepin PI3K inhibitors, we observed a strong dependency of in vivo antitumor activity on the free-drug exposure. By lowering the intrinsic clearance, we derived a set of imidazobenzoxazepin compounds that showed improved unbound drug exposure and effectively suppressed growth of tumors in a mouse xenograft model at low drug dose levels. One of these compounds, GDC-0032 (11l), was progressed to clinical trials and is currently under phase I evaluation as a potential treatment for human malignancies.
GDC-0032, also known as taselisib, RG7604, or the IUPAC name: 2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide, has potent PI3K activity (Ndubaku et al (2013) J. Med. Chem. 56(11):4597-4610; WO 2013/182668; WO 2011/036280; U.S. Pat. No. 8,242,104; U.S. Pat. No. 8,343,955) and is being studied in patients with locally advanced or metastatic solid tumors (Juric et al “GDC-0032, a beta isoform-sparing PI3K inhibitor: Results of a first-in-human phase Ia dose escalation study”, 2013 (Apr. 7) Abs LB-64 American Association for Cancer Research Annual Meeting).
the invention relates to polymorph forms of the PI3K inhibitor I (taselisib, GDC-0032, RG7604, CAS Reg. No. 1282512-48-4, Genentech, Inc.), named as 2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide, having the structure:
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof.
POLYMORPHS OF 2-(4-(2-(1-ISOPROPYL-3-METHYL-1H-1, 2, 4-TRIAZOL-5-YL)-5, 6-DIHYDROBENZO[F]IMIDAZO[1, 2-D][1, 4]OXAZEPIN-9-YL)-1H-PYRAZOL-1-YL)-2-METHYLPROPANAMIDE, METHODS OF PRODUCTION, AND PHARMACEUTICAL USES THEREOF
This compound and its uses are investigational and have not been approved by the US Food and Drug Administration. Efficacy and safety have not been established. The information presented should not be construed as a recommendation for use. The relevance of findings in preclinical studies to humans is currently being evaluated.
Taselisib, a PI3K inhibitor
Taselisib, an investigational PI3K inhibitor, is currently in clinical development based on its potential selectivity for the PI3Kα isoform.1,2 Preclinical data have shown that taselisib induced growth inhibition in PI3Kα-mutant cell lines.1 Taselisib continues to be investigated in ongoing clinical studies.
1Taselisib is an investigational PI3K inhibitor currently being studied for its potential to selectively inhibit the PI3Kα isoform.1,2
2Taselisib is designed to bind to the ATP-binding pocket of PI3Kα to potentially prevent subsequent downstream signaling.1
3In preclinical studies, taselisib induced growth inhibition in PI3Kα-mutant xenograft mouse models.1 Taselisib continues to be investigated in ongoing clinical studies.
References
Lopez S, Schwab CL, Cocco E, et al. Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol.2014;135:312-317. PMID: 25172762
Ndubaku CO, Heffron TP, Staben ST, et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597-4610. PMID: 23662903
A highly efficient and regioselective manufacturing route for the phosphoinositide 3-kinase β-sparing inhibitor taselisib was developed. Highlights of the synthesis include: (1) magnesium-mediated formation of a challenging cyclic amidine; (2) regioselective imidazole construction via alkylation/condensation with bromopyruvic acid; and (3) triazole formation with 2-isopropyl acetamidrazone to generate the key bromobenzoxazepine core intermediate. Subsequent highly efficient one-pot palladium-catalyzed Miyaura borylation/Suzuki cross-coupling/saponification, followed by a 1,1′-carbonyldiimidazole-mediated coupling with ammonia, led to the pentacyclic taselisib. This new synthetic approach provides a more efficient route to taselisib with a significant decrease in process mass intensity compared to the previous early development routes to the bromobenzoxazepine core. Finally, implementation of a controlled crystallization provided the active pharmaceutical ingredient with the desired polymorphic form.
Taselisib was obtained in 90% yield (16.2 kg) as a white to off-white solid (>99.9 wt %; >99.9 A%) as Form B crystal.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....