
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Ataluren (Translarna)
3-(5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl)benzoic acid
3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
CAS 775304-57-9
PTC Therapeutics (Originator)
- Molecular FormulaC15H9FN2O3
- Average mass284.242 Da
- EC-000.2051
NCGC00168759-02PTC-124, PTC124,UNII:K16AME9I3V
- EU 2014-07-31 APPROVED

Ataluren, formerly known as PTC124, is a pharmaceutical drug for the treatment of Duchenne muscular dystrophy and potentially other genetic disorders. It was designed by PTC Therapeutics and is sold under the trade name Translarna in the European Union.
Ataluren was approved by European Medicine Agency (EMA) on July 31, 2014. It was developed and marketed as Translarna® by PTC Therapeutics.

Ataluren was regulator of nonsense mutations indicated for the treatment of Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene, in ambulatory patients aged 5 years and older.
Translarna® is available as granules for oral use, containing 125 mg, 250 mg or 1000 mg of free Ataluren. The recommended dose is 10 mg/kg body weight in the morning, 10 mg/kg body weight at midday, and 20 mg/kg body weight in the evening.
Medical uses
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[1][2] such as some people with cystic fibrosis and Duchenne muscular dystrophy. It is approved for the use in Duchenne in the European Union.

Mechanism of action
Ataluren makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. Studies have demonstrated that PTC124 treatment increases expression of full-length dystrophin protein in human and mouse primary muscle cells containing the premature stop codon mutation for Duchenne muscular dystrophy and rescues striated muscle function.[3] Studies in mice with the premature stop codon mutation for cystic fibrosis demonstrated increased CFTR protein production and function.[4] The European Medicines Agency review on the approval of ataluren concluded that “the non-clinical data available were considered sufficient to support the proposed mechanism of action and to alleviate earlier concerns on the selectivity of ataluren for premature stop codons.” [5]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[6] Ataluren appears to be most effective for the stop codon ‘UGA’.[1]

History
Clinical trials
In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[7] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug.
Phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[8] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[9]
Approval
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[10]Translarna was first available in Germany, the first EU country to launch the new medicine.[11]
In August 2014, ataluren received market authorization from the European Commission to treat patients with nonsense mutation Duchenne muscular dystrophy. A confirmatory phase III clinical trial is ongoing.[11] The drug does not yet have approval by the US Food and Drug Administration.
In October 2015, NICE asked for further evidence of benefit to justify the “very high cost”.[12] NICE estimated that for a typical patient, treatment would cost £220,256 per year.
In February 2016, FDA declined to approve or even discuss PTC Therapeutics application for ataluren because it deemed the data presented by the developer “insufficient to warrant a review”.[13]

Ataluren Molecule
PAPER
http://www.pnas.org/content/106/9/3585.full
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental/Appendix_PDF.pdf
Samples were analyzed for purity on an Agilent 1200 series LC/MS equipped with a Luna® C18 reverse phase (3 micron, 3 x 75 mm) column having a flow rate of 0.8-1.0 mL/min. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and temperature was maintained at 50 °C. A gradient of 4% to 100% acetonitrile over 7 minutes was used during analytical analysis. Purity of final compounds was determined to be >95%, using a 5 μL injection with quantitation by AUC at 220 and 254 nM. High resolution mass spectra were obtained with an Agilent 6210 Time-of-Flight LC/MS with a 3.5 um Zorbax SB-C18 column (2.1 x 30 mm) (solvents are Water and ACN with 0.1% Formic Acid). A 3 minute gradient at 1 mL/min from 5% to 100% acetonitrile was used.
3-[5-(2-fluorophenyl)-[1,2,4]-oxadiazol-3-yl]-benzoic acid (1a, PTC124).
1 H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH = 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).
13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz). LC-
MS: rt (min) = 5.713; [M+H]+ 285.1;
HRMS: (CI+, m/z), calcd for C15H10FN2O3 (MH+ ), 285.06814; found, 285.06769.
CLIP
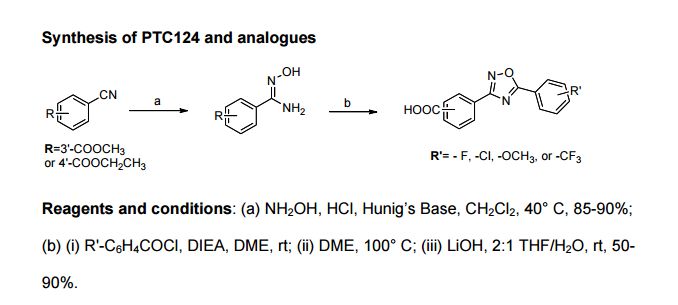
Ataluren (Translarna) Ataluren is a drug marketed under the trade name Translarna which was developed by PTC Therapeutics and approved by the European Union in May 2014 for the treatment of Duchenne’s muscular dystrophy (DMD) and potentially other genetic disorders.50
Ataluren renders ribosomes less sensitive to premature stop or ‘read-through’ codons, which are thought to be beneficial in diseases such as DMD and cystic fibrosis.51 Of the reported synthetic approaches to ataluren,52–55 the most likely process-scale approach consists of the sequence described in Scheme 7, which reportedly has been exemplified on kilogram scale.56
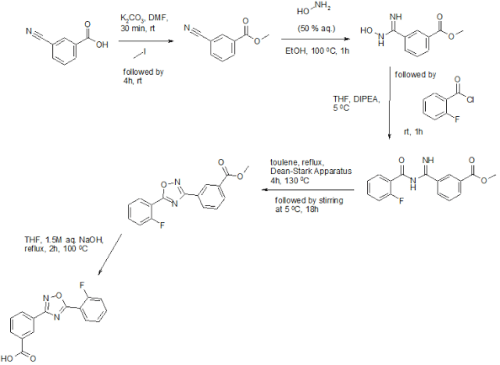
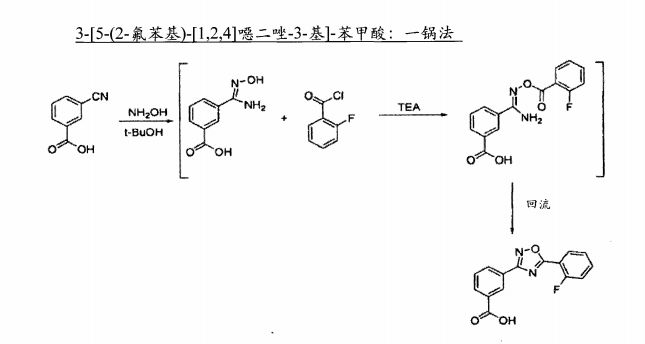
The sequence to construct ataluren, which was described by the authors at PTC Therapeutics, commenced with commercially available methyl 3-cyanobenzoate (38).56 This ester was exposed to hydroxylamine in aqueous tert-butanol and warmed gently until the reaction was deemed complete.
Then this mixture was treated with 2-fluorobenzoyl chloride dropwise and subsequently triethylamine dropwise. To minimize exotherm and undesired side products, careful control of the addition of reagents was achieved through slow dropwise addition of these liquid reagents.
Upon complete consumption of starting materials and formation of amidooxime 39, the aqueous reaction mixture was then heated to 85 C to facilitate 1,2,4-oxadiazole formation, resulting in the tricyclic ester 40 in excellent yield across the three steps.
Finally,saponification of ester 40 through the use of sodium hydroxide followed by acidic quench gave ataluren (V) in 96% over the two-step sequence.57
50. Welch, E. M.; Barton, E. R.; Zhuo, J.; Tomizawa, Y.; Friesen, W. J.; Trifillis, P.;Paushkin, S.; Patel, M.; Trotta, C. R.; Hwang, S.; Wilde, R. G.; Karp, G.; Takasugi,J.; Chen, G.; Jones, S.; Ren, H.; Moon, Y. C.; Corson, D.; Turpoff, A. A.; Campbell,J. A.; Conn, M. M.; Khan, A.; Almstead, N. G.; Hedrick, J.; Mollin, A.; Risher, N.;Weetall, M.; Yeh, S.; Branstrom, A. A.; Colacino, J. M.; Babiak, J.; Ju, W. D.;Hirawat, S.; Northcutt, V. J.; Miller, L. L.; Spatrick, P.; He, F.; Kawana, M.; Feng,H.; Jacobson, A.; Peltz, S. W.; Sweeney, H. L. Nature 2007, 447, 87.
51. Hirawat, S.; Welch, E. M.; Elfring, G. L.; Northcutt, V. J.; Paushkin, S.; Hwang,S.; Leonard, E. M.; Almstead, N. G.; Ju, W.; Peltz, S. W.; Miller, L. L. J. Clin.Pharmacol. 2007, 47, 430.
52Karp, G. M.; Hwang, S.; Chen, G.; Almstead, N. G. US Patent 2004204461A1,2004.
53. Andersen, T. L.; Caneschi, W.; Ayoub, A.; Lindhardt, A. T.; Couri, M. R. C.;Skrydstrup, T. Adv. Synth. Catal. 2014, 356, 3074.
54. Gupta, P. K.; Hussain, M. K.; Asad, M.; Kant, R.; Mahar, R.; Shukla, S. K.; Hajela,K. New J. Chem. 2014, 38, 3062.
55. Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; PalumboPiccionello, A.; Pibiri, I. Mol. Pharm. 2014, 11, 653.
56. Almstead, N. G.; Hwang, P. S.; Pines, S.; Moon, Y. -C.; Takasugi, J. J. WO Patent2008030570A1, 2008.
57. Almstead, N. G.; Chen, G.; Hirawat, S.; Hwang, S.; Karp, G. M.; Miller, L.; Moon,Y. C.; Ren, H.; Takasugi, J. J.; Welch, E. M.; Wilde, R. G. WO Patent2007117438A2, 2007.
CLIP

Ataluren trial success: trial aborted.
07 September 2011 – Pharma……..http://chem.vander-lingen.nl/info/item/September_2011/id/190/mid/140
Last week the newspaper NRC Handelsblad reported on a court case in which the parents of two young boys sued a pharmaceutical company over access to one of their developmental drugs. The drug in question wasAtaluren, the pharmaceutical companyPTC Therapeutics. The boys suffer from Duchenne muscular dystrophyand had taken part in a clinical trial. Whereas the results of this trial on the whole were inconclusive the boys did seriously benefit from the drug. Hardly any wonder the parents took action when the whole development program was canceled.
And the judge? He threw the case out arguing that doctors do not make the compound themselves and arguing that the compound is not commercially available. Are these arguments valid? and do the boys have options?
It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).
Except for the fluorine atom in it the compound is unremarkable. If you have to believe the Internet many Chinese companies produce and sell it. Ataluren is also still in the running as a potential treatment for some other diseases. So if need be the compound will be around for some time to come.
CLIP
Ataluren [3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid] is an orally available, small molecule compound that targets nonsense mutation. It is the first drug in its class and appears to allow cellular machinery to read through premature stop codons in mRNA, and thus enables the translation process to produce full-length, functional proteins.
Ataluren is developed and approved for the treatment of nonsense mutation Duchenne muscular dystrophy (nmDMD) by EU in July 2014 [1].
| Ataluren: 2D and 3D Structure |
Nonsense Mutations as Target for DMD
A single nucleotide change in the DNA sequence that introduces a premature stop codon is known as a nonsense mutation, a subset of a major class of premature termination codon (PTC) mutations. Nonsense mutations cause premature termination of translation resulting in the production of truncated polypeptides, which in turn halts the ribosomal translation process at an earlier site than normal, producing a truncated, non-functional protein [1].
Nonsense mutations are implicated in 5-70 % of individual cases of most inherited diseases, including Duchenne muscular dystrophy (DMD) and cystic fibrosis. Ataluren appears to allow cellular machinery to read through premature stop codons in mRNA, enabling the translation process to produce full length, functional proteins.
Ataluren Synthesis
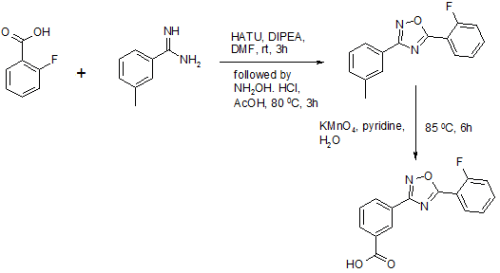
New J Chem 2014, 38, 3062-3070: The text reports one pot synthesis of Ataluren with an overall yield of 40%. It also reports few interesting and potent derivatives too.
WO 2007117438A2: It appears to be the industrial process. The patent also reports various pharmaceutically relevant assay and their results wrt Ataluren.
Identifications:
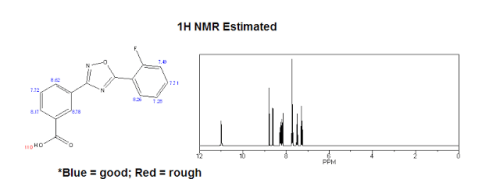
| 1H NMR (Estimated) for Ataluren |
Experimental: 1H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH= 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).
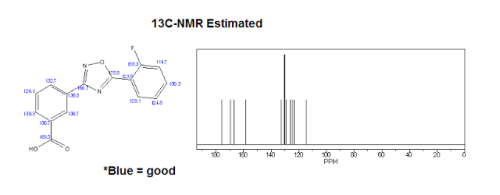
| 13C-NMR (Estimated) for Ataluren |
Experimental: 13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz)……https://ayurajan.blogspot.in/2016/05/ataluren-treatment-for-duchenne.html
CLIP


Reference:1. WO2004091502A2 / US6992096B2.
2. WO2008045566A1 / US2008114039A1.
3. WO2008030570A1 / US2008139818A1.
Reference:1. New. J. Chem. 2014, 38, 3062-3070.

Reference:1. Adv. Synth. Catal. 2014, 356, 3074-3082.
CLIP
Carcinogenicity
Carcinogenicity bioassays in transgenic mice (26 weeks) and in rats (24 months):
● For Tg.rasH2 mouse: Ataluren did not increase the incidence of tumors up to the HDs in males (600 mg/kg/day) and in females (300 mg/kg/day). The non-neoplastic findings included endometrial hyperplasia and nephropathy in females.
● For rats: Urinary bladder tumors (benign urothelial cell papilloma [2 rats] and malignant urothelial cell carcinoma [1 rat]) were observed in 3/60 female rats dosed at 300 mg/kg/day. In addition, one case of malignant hibernoma was observed in 1/60 male rats at the dose of 300 mg/kg/day. The non-neoplastic toxicity consisted of a decrease of body weight.
Example 1 (prepared by known ataluren)
Method ataluren according to Patent Document 2 is described in Example W02004091502A2 prepared.
Specific methods of preparation:
To a solution of 0.6 l of DMF was 44. 14g3- cyano acid 62.19 g of potassium carbonate was added, followed by stirring at room temperature for 30 minutes. 20 minutes To the suspension was added 28 ml of methyl iodide (450mmol), and the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was poured into 1.2 l of ice water, stirred for 30 minutes, the precipitate was filtered out thereof. The white cake was dissolved in 70 ml of methanol, and then reprecipitated in cold water. To give 79% yield of 3-cyano-benzoic acid methyl ester.
50 g of 3-cyano-benzoic acid methyl ester was dissolved in 500 ml of ethanol, to which was added 41 ml of 50% aqueous hydroxylamine (620mmol). 100 ° C and the reaction mixture was stirred for 1 hour, the solvent was removed under reduced pressure. So that the oily residue is dissolved in 100 ml of 20/80 ethanol / toluene, concentrated again. To give 61 g 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester.
60 g of 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester was dissolved in 200 ml of anhydrous tetrahydrofuran, followed by adding thereto 75 ml of diisopropylethylamine (434 mmol), and then 20 minutes this mixture was added 48.1 ml 2- fluorobenzoyl chloride (403mmol). The reaction mixture was stirred at room temperature for 1 hour. The precipitate was filtered off, the filtrate was concentrated under reduced pressure. The residue was dissolved in 400 ml of ethyl acetate, washed with 400 ml of water and then twice. The solvent was removed under reduced pressure, containing 60% ethyl acetate in hexane to give the desired product, generating 81 g 3- (N-2- amidino-fluorobenzoyl) – benzoate.
at 130 ° C with a Dean-Stark apparatus was dissolved in 500 ml of toluene was heated under reflux in 44 g of 3- (N-2- fluorobenzoyl) -1,2,3,4-_ benzoate 4 hours. 5 ° C and the reaction mixture was stirred for 18 hours. The white precipitate was filtered off, the filtrate was concentrated, recrystallized in toluene. To give 38 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester.
33 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester was dissolved in 400 ml of tetrahydrofuran, to which was added 100 ml of 1. 5M aqueous sodium hydroxide solution. At 100 ° C and the reaction mixture was heated at reflux for 2 hours. The solvent was removed under reduced pressure at 5 ° C the solution was stirred for 2 hours. The organic solvent was removed, washed with 50 mL of water. The aqueous solution was then acidified with hydrochloric acid to pH 1. The white precipitate was filtered off, the filter cake washed with cold water, then dried with a freeze dryer. To give 3.0 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] benzoic acid. 1H-NMR (500MHz, d6-DMS0): 8. 31 (1H), 8 18 (2H), 8 08 (1H), 7 88 (2H), 7 51 (2H)….. Display: ataluren- Sample Preparation Example 1 prepared in Preparation Example 2 and TO2004091502A2 induced.
Each prepared in Example 2 (prepared according to known Form A)
Method [0084] A known polymorph according to Patent Document W02008039431A2 Example 5. 1. 1.1 prepared as described. Specifically: ataluren be prepared 1 100 mg Preparation Example, 60 ° C add 16.2 ml of isopropanol ultrasound clear solution, the solution by 2 square micron filter and the filtrate was kept covered with aluminum foil having a small hole. vial, 60 ° C and evaporated. The solid formed was isolated to give ataluren the A polymorph.
as needles.
its XRPD shown in Figure 1, the display ataluren polymorph A disclosed in Patent Document W02008039431A2 consistent.
novel crystalline forms of 3-[5-(2-fluorophenyl)-
[l,2,4]oxadiazol-3-yl]-benzoic acid, which has the following chemical structure (I):

(I)
In particular, crystalline forms of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are useful for the treatment, prevention or management of diseases ameliorated by modulation of premature translation termination or nonsense-mediated mRNA decay, as described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, which is incorporated herein by reference in its entirety. In addition, the present provides a crystalline form of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]-benzoic acid which is substantially pure, i.e., its purity greater than about 90%.
Processes for the preparation of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, and U.S. patent application no. 1 1/899,813, filed September 9, 2007, both of which are incorporated by reference in their entirety.
PATENT
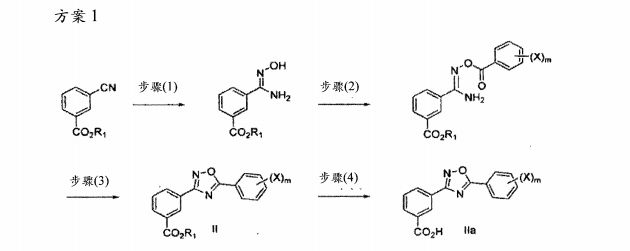
Example
3- ‘5- (2-fluorophenyl) – “1,2,41 oxadiazol-3-yl benzoate 1- Batch 1
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 2 hours 48 minutes, 50. /. Aqueous hydroxylamine (43L, 47.4 kg) was added to a clear solution of 3-cyano benzoic acid methyl ester in a molten in t-butanol. The addition of a 50% aqueous solution of hydroxylamine period, the maximum temperature batch of about 43 ° C. 50% aqueous solution of hydroxylamine addition rate of from about 9L / h when the changes start adding to about 30L / hr. To maintain the temperature of the batch by varying the reactor jacket set point. In particular, the set value is about 40.5 ° C, with the addition of a rate increase at the beginning join, change the setting to about 29.6 ° C. After about 40-45t stirred for about 4 hours, the reaction was deemed complete (i.e., less than about 0.5% ester).
The batch was transferred to a drying reactor, additional (chased through) approximately 10L molten tert-butanol. Jacket setpoint from about 33 when the batch was received when dried reactor. C is reduced to about 27 after the completion of the transfer. C. Batch crystallization was observed part, which does not adversely affect stirring. The batch was cooled to about 34.4 ° C, triethylamine (72.6 kg, IOOL) added to the reactor. The jacket temperature set value from about 20.4. C is increased to about 31.0 ° C, in order to maintain the batch temperature in the range of about 30-35t. With molten tert-butanol (IO L) was washed with a linear (line rinse) After the batch was added to the 2-fluorobenzoyl chloride (113.7 kg, 86.0L).Charge is added to the first third of the rate of about 25L / hr. In the meantime, the jacket inlet temperature was lowered to about 15 ° C, the batch temperature is maintained at about 34.6 ° C. In about 5.5 hours after the addition was complete.During the addition, the maximum temperature of the batch was about 38.8 ° C. Near the end of the addition, the addition rate slowed to about 11L / hr was added last 27 liters of 2-fluoro-benzoyl chloride. 30-35. C After stirring for about 2 hours, that the reaction was complete (i.e., less than about 0.5% of methyl 3-amidinophenoxy). Then, after about 1 hour 42 minutes, the batch was heated to reflux temperature (about 82 ° C), and then stirred for about 18 hours. During the stirring, a number of product partially crystallized to form a slurry. The slurry was cooled to about 40. C thus sampled, during which complete crystallization occurs. The batch was then heated to reflux temperature and stirred for about 1 hour 50 minutes.Then, after about two hours, the batch was cooled to about 69 ° C, and after about four hours and 15 minutes, slowly added 630L of pure water, while maintaining the batch temperature at about 66-69 ° C. After about 3 hours 14 minutes, the slurry was cooled to about 22.4 ° C, and transferred to 2x200L ceramic filter, the ceramic filter equipped 25-30n polypropylene mesh filter cloth. In about 55 minutes after the completion of material from the container to the filter transfer. With 50n /. The tert-butanol solution (210L) was washed cake was washed for about 10 minutes so that the cleaning liquid can penetrate into each cake. Then, the cake was dried in a vacuum for about 5-10 minutes. The purified water as a second washing (158L / cake) applied to the filter cake to remove residual t-butanol and triethylammonium chloride salt. Dried in a vacuum for about 5 minutes, the solution was removed. In vacuo and then the cake was dried for about 2 hours, and then sampled using liquid chromatography. The filter cake was measured by liquid chromatography purity of about 99.6%.
The filter cake was dried in vacuo for about 8 hours 25 minutes later, the wet cake (207.4kg) is transferred to an air oven. At about 50-55. C, the oven dried in air for about 52 hours. The product was isolated in a total yield of about 89.9% (174.65kg), in the calculation of cost of materials sampling, you can adjust the overall yield of about 90.7%.
Batch 2
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 3 hours 29 minutes, 50% aqueous solution of hydroxylamine (47.85 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. At about 40-45. C After stirring for about 3 hours 16 minutes, that the reaction was complete (i.e., less than about 0.5% ester). As for the drying reactor, the batch was transferred to one of the batch in. The batch was cooled
To about 34.4 ° C, and triethylamine (72.6 kg, 100 L). During about 45 minutes was added, while maintaining the batch temperature between about 30-35 ° C. During the addition, the jacket inlet temperature of from about 31.4. C increased to about 32.6. C. After the molten tert-butanol linear washed, was added to the batch 2- fluorobenzoyl chloride (l 13.7 kg, 86.0 L). After about 3 hours, 27 minutes, add the acid chloride. 35. C under stirring for about 8 hours, that the reaction is not complete (i.e., more than about 0.5% residual 3-amidino-benzoyl ester). Then, 1.5% by weight of the original charge of triethylamine and 2-fluorobenzoyl chloride was added to the batch. Linear washed with tert-butanol (IO L) associated with each additional charge. During the addition of the acid chloride, no additional cooling. The batch was maintained at a temperature of about 30-35 ° C, the jacket inlet temperature range was maintained at about 30.3. C to about 33.0 ° C. After stirring for about 2 hours at 30-35t, that reaction was complete (i.e., less than 0.5% of methyl 3-amidinophenoxy).
After about 1 hour and 44 minutes, the batch was heated to reflux temperature (about 83 ° C), and stirred for about 18 hours.The same batch 1, during cooling the sample, the solid was completely crystallized. The batch was then heated to reflux temperature and stirred for about 1 hour and 2 minutes. Then, after about 2 hours and 20 minutes, the batch was cooled to about 69.2 ° C, and after about four hours and 30 minutes, slowly added 630 L of pure water, while the temperature of the batch was maintained at about 65.6-69.2 ° C. After about 3 hours and 30 minutes, the slurry was cooled to about 23.4 ° C, and, as for, the contents were transferred to one of the double batch of the ceramic filter. About 5 hours and 6 minutes, to complete the transfer of the material. With about 50% of t-butanol (2 volumes / cake) was washed filter cake was washed with 10 minutes to allow the cleaning liquid to penetrate into each cake, then dried in vacuo. About 1 hour and 40 minutes, the filter is completed. The purified water was added to a final wash the filter cake. The liquid was removed by drying under vacuum for about 10 minutes. In vacuo and then the cake was dried for about 2 hours and 5 minutes, and then sampled using liquid chromatography. The cake purity liquid chromatography were about 99.5% and 99.6%. After the cake was then dried in vacuo for about 2 hours and 5 minutes, the wet cake (191.5 kg) is transferred to an air oven. At about 50-55. C under dry in an air oven for about 48 hours. The product was isolated in a total yield of about 92.5% (179.7 kg).
Lot 3
The 3-cyano-benzoic acid methyl ester (52.5 kg) and molten tert-butanol (228 kg) added to the reaction vessel. The vessel was sealed, the batch temperature set of about 40-45 ° C, and the stirrer is started. Under an inert atmosphere, after 2 hours 40 minutes, 50% of the shoes amine solution (24 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. In about 42. Under C, then further stirred for about 5 hours to complete the reaction.
The batch was cooled to 30-35 ° C, and after 15 minutes, was added triethylamine (36 kg). After about 2 hours 44 minutes, was added 2-fluorobenzoyl chloride (57 kg). During the addition, batch temperature was maintained at about 30-35 ° C.Under the 32t, the batch was stirred for 2 hours 10 minutes to complete the reaction.
After about 50 minutes, the batch was heated to reflux temperature (about 83-86 ° C), at about 8rc, stirred for about 18 hours. Then, over about two hours, the batch was cooled to about 65-70 ° C, and after about 6 hours 25 minutes, slowly added to purified water (315 L), while the batch temperature was maintained at about 65- 70 ° C. After about 2 hours and 15 minutes, the slurry was cooled to about 22 ° C, and the contents were transferred to a centrifuge filter (2 batches). About 1 hour and 40 minutes, the filter is completed. After about 20 minutes, with about 50% aqueous solution of tert-butyl alcohol (90 kg / cake), dried cake. The purified water (79 kg / cake) as the last added to the filter cake washed. At about 900 rpm drying the cake for about 1 hour and 5 minutes, then filled cylinder. Liquid chromatography wet cake (91.5 kg, LOD = 5% w / w) of a purity of about 99.75% area.
3- ‘5- (2-fluorophenyl) -fl, 2,41 oxadiazol-3-yl l- acid batch 1
3- [5- (2-fluorophenyl) – [l, 2,4] oxadiazol-3-yl] – benzoic acid methyl ester (74.0kg) added to the reaction vessel, the vessel is sealed, evacuated and purification. Jacket set value of about 35. C, start the stirrer in the container. Molten tert-butanol (222 L, 3 volumes) and purified water (355 L, 4.8 vol) was added to the vessel. After the addition was added 25.1% w / w aqueous sodium hydroxide solution (43.5 kg, 1.1 molar equivalents), and with additional purified water (100L, 1.35 mol) was washed linear. During the addition, the batch temperature from about 39.0t reduced to about 38.8 ° C. After about 1 hour and 54 minutes, the batch temperature to about 63-67. C, and then, after about 30 minutes, which was adjusted to about 68-72.C. About 68-72t, stirring the mixture for about 3 hours. Then, after about five hours 11 minutes, the solution was cooled to about 40-45 ° C. Then, after the above process, after about three hours 33 minutes, the solution was then heated to about 68-72. C.
Jacket temperature of the reaction vessel was set to about 60 ° C, the stirrer started, and at about 70 ° C, a slightly positive pressure of nitrogen (1.5 to 5.6 psig), the heat transfer liquid through a micron filter . During the transfer, the product temperature is reduced to about 64.3 ° C, the transfer is completed in about 45 minutes. Was added to the purified water container (61 L, 0.82 vol) and the contents were heated to about 68-72. C.
The batch temperature was adjusted to about 69.4 ° C, and after about four hours and 18 minutes, with 13.9% w / w sulfuric acid (100.7 kg, 1.15 mol equiv.). During the addition, batch temperature was maintained at about 68.0-70.8 ° C. After the addition of the acid, with purified water (50 L, 0.68 vol) line wash at about 68-72 ° C, the stirring was continued for 31 minutes.
After about 4 hours and 10 minutes, the batch in a linear fashion from about 69.2t cooled to about 41.2 ° C. The stirrer Rosenmund filter / dryer was elevated to the highest position and jacket set value is set at about 40 ° C. The slurry was transferred to the two portions of the filter / drier. Applying a constant nitrogen pressure to the first portion (less than about 15 psig). During the transfer, a pressure of about 23.9 to about 28.8 psi, the transfer is complete in about 1 hour and 5 minutes. The second part of the slurry was transferred onto the filter cake, and the composite was stirred briefly to homogenize the batch. Use about 26.1 to about 29.1 psi nitrogen pressure filters the second part, after about three hours, squeeze the cake so that it does not contain liquid. With about 38-42 ° C hot tert-butanol solution (352 kg, 5 volumes) and about 65-70 ° C in 3x hot purified water (370 L, 5 volumes) and the filter 々.
Said filter / dryer jacket temperature was set to about 43 ° C, the product was dried under vacuum for about 26 hours while stirring periodically. Determination of purity of about 99.7%. The product was isolated in a total yield of about 74.4% (52.45 kg).
Batch 2
Was added to the reactor vessel 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (47 kg, wet cake) and melt-hyun tert-butyl alcohol (111.4 kg). A sealed container, and the batch temperature was set at 30-40t, and start the stirrer. The purified water (51.6 kg) was added to the vessel. After the addition was added 3.47% w / w aqueous sodium hydroxide solution (202.4 kg). After about l hour, the batch temperature to about 67-73. C, then, at about 7 (under TC, stirred for about three hours.
Under a slight positive pressure of nitrogen, with a 1 micron polypropylene bag filter the batch, and then transferred to the new reactor. Was added to the vessel pure water (146 kg), and heating the batch to about 68-72. C.
After about four hours, the 10.7% aqueous hydrochloric acid was added to the batch. During the addition, batch temperature was maintained at about 68-72 ° C. PH was measured by using the batch pH of about 2.2, and then stirring was continued at about 7 (under TC about 1 hour.
After about two hours, the batch in a linear fashion from about 70. C is cooled to about 60 ° C. After about two hours, about 60. C of the batch in a linear fashion from about 6 (TC was cooled to about 40 ° C. In 40t, the batch was stirred for 2 hours, and the slurry was transferred to a centrifuge filter. After about 30 minutes, filtered completion . After about 30 minutes, with about 42Mw / w in t-butanol solution (165kg) cake was washed. The purified water (118kg, 4 (TC) as the last added to the filter cake was washed. The filter cake was dried at about 900rpm about 1 hour, then filled cylinder.
The wet cake was transferred to a paddle dryer (a double cone drier also suitable for this step), the jacket temperature was set to about 70. C. At about 70. C, the product was dried under vacuum for about 48 hours while stirring periodically.Determination of purity of about 99.8%. The product was isolated overall yield of about 74% (68.5 kg).
Lot 3
To the reaction vessel was added 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (10 g) and t-butanol fused (128mL ). The batch temperature was set to 30-40 ° C, and the stirrer is started. After about 30 minutes, the aqueous sodium hydroxide solution 4.48% w / w of (32.5 g) was added to the vessel. The batch was maintained at a temperature of about 40-50 ° C. After about l hour, the batch temperature is raised to about 78-82 ° C, and then, at about 78-82t, and then stirred for about one hour. Under positive pressure of nitrogen, a polyethylene bag with a 5 micron filter the batch, and then transferred to a new reaction vessel. The batch was maintained at a temperature of about 78-82 ° C.
It was added to a new vessel 37% aqueous hydrochloric acid (4 mL) and tert-butanol molten (8 mL). The temperature was maintained at about 30-40. Under C, and stirring the mixture for about 30 minutes.
After about four hours, using a metering pump was added to the batch of hydrochloric acid in tert-butanol. After about SO-SO minutes before adding half filled. The stirrer speed is set at about 200rpm. After about 3.5 hours, add the remaining charge. The stirrer speed is set at about 100 rpm. During the addition, batch temperature was maintained at about 78-82 ° C.PH meter with a final batch pH was adjusted to about 1.2, at about 78-82t, then continue stirring for about l hour. After about one hour, the batch in a linear fashion from about 78-82. C is cooled to about 70 ° C. After about four hours, about 7 (TC batches in a linear fashion from 70.C cooled to about 50 ° C, and the stirrer speed was set at about 80 rpm. After about four hours, about 50 ° C Batch linearly cooled from 50 ° C to about 40t, and stirrer speed was set at approximately 60rpm. In 40.C, the batch was stirred for a further 4 hours.
The temperature of the filter is set to about 40-45 ° C. The slurry was transferred to the filter. After about one minute to complete filtration. After about two minutes, with tert-butanol (50 mL, 50.C) washing the filter cake. The pure water (IOO mLx2, 60.C) as the last wash was added to the cake. Under vacuum at about 60-70 ° C the cake was dried for about 12 hours, and then loaded into the container.
Determination of HPLC purity of about 99.9% of the area. The yield of isolated product was about 94% (9.0g).
3- “5- (2-fluorophenyl) -” 1,2,41-oxadiazol-3-yl 1- acid: One-pot
The methyl 3-cyanophenyl Yue (7.35 g) and tert-butanol molten (100 mL) added to the reactor vessel. Sealed containers, the batch temperature was set to 60 ° C, and the stirrer is started. The suspension was stirred for 1 hour and then the batch temperature was set to 40. C. Under an inert atmosphere, after three hours, 50% aqueous solution of hydroxylamine (3.63 g) was added to the reactor. During the addition, batch temperature was maintained at 38-41 ° C. 40. C After stirring for 18 hours, to complete the reaction.
The batch was cooled to 27 ° C, and after two minutes, triethylamine (5.56 g). After 3 hours, was added 2-fluorobenzoyl chloride (7.82 g). During the addition, batch temperature was maintained at 24-27 ° C. 40. C, the batch was stirred for a further 4 hours.
After 30 minutes, the batch was heated to 79 ° C, and at about 79. C was stirred for 16 hours. After 3 hours, the white suspension was added to the water (IOO mL), while the batch temperature was maintained at 70 ° C. After 20 minutes, a 37% aqueous hydrochloric acid were added to the batch. PH was measured by using the batch pH of about 2.2, stirring was continued at about 70t for about 1 hour.
After three hours, the batch in a linear manner from 7 (TC cooled to 30 ° C, and the slurry is transferred to the filter. After 5 minutes, the filtering is done. After five minutes, with tert-butanol (50mL, 40 .C) filter cake was washed. The purified water (IOO mL, 60.C) is added to a final wash the filter cake. In 70.C of the filter cake was dried in a vacuum oven for 18 hours and then removed. Determination of purity approximately 98.68%. The total yield of isolated product of about 76% (10.8g).
PICS

References
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91. Bibcode:2007Natur.447…87W.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444.doi:10.1177/0091270006297140. PMID 17389552.
- Nature. 2007 May 3;447(7140):87-91.
- Proc Natl Acad Sci U S A. 2008 Feb 12;105(6):2064-9.
- Neuromuscul Disord. 2015 Jan;25(1):5-13.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery Medicine 15 (81): 127–133. PMID 23449115.
- “PTC Therapeutics and Genzyme Corporation announce preliminary results from the phase 2b clinical trial of ataluren for nonsense mutation Duchenne/Becker muscular dystrophy (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271.Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272. doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved2013-11-28.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
- “PTC Therapeutics Announces Launch of Translarna™ (ataluren) in Germany”.marketwatch.com. 3 Dec 2014. Retrieved 27 Dec 2014.
- “NICE asks for further evidence for the benefits of a new treatment for Duchenne muscular dystrophy to justify its very high cost”.
- http://uk.reuters.com/article/us-ptc-therapeutics-fda-idUKKCN0VW1FG
External links
References:
1. Ryan, N. J. Ataluren: first global approval. Drugs 2014, 74(14), 1709-14. (FMO only)
2. Gupta, P. K.; et. al. A metal-free tandem approach to prepare structurally diverse N-heterocycles: synthesis of 1,2,4-oxadiazoles and pyrimidinones. New J Chem 2014, 38, 3062-3070 (FMO only)
3. Almstead, N. G.; et. al. Methods for the production of functional protein from dna having a nonsense mutation and the treatment of disorders associated therewith. WO2007117438A2
| WO2004091502A2 | Apr 9, 2004 | Oct 28, 2004 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8486982 | Jun 22, 2012 | Jul 16, 2013 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acids |
| US8796322 | Jun 19, 2013 | Aug 5, 2014 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-oxadiazole benzoic acid compounds |
| US8975287 | Jun 18, 2014 | Mar 10, 2015 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-Oxadiazole benzoic acid compounds |
| US9205088 | Jan 28, 2015 | Dec 8, 2015 | Ptc Therapeutics, Inc. | Compositions of 1,2,4-oxadiazol benzoic acid compounds and methods for their use |
| US9289398 | Mar 29, 2007 | Mar 22, 2016 | Ptc Therapeutics, Inc. | Methods for the production of functional protein from DNA having a nonsense mutation and the treatment of disorders associated therewith |
| Preparation | CN101535284A | CN101535284B | ||||
| 10 | Crystal | CN101541770A | ||||
| 11 | Crystal | CN104341371A | ||||
| 12 | Crystal | CN102382075A |
| Formula | CN1802360A | CN1802360B | ||||
| 2 | Combination | CN104056278A | ||||
| 3 | Indication | CN101076703A | ||||
| 4 | Indication | CN101076332A | ||||
| 5 | Indication | CN101076337A | ||||
| 6 | Indication | CN101193632A | ||||
| 7 | Formulation | CN103720688A | ||||
| 8 | Indication | CN101505739A |

 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
|
|
| Other names
PTC124
|
|
| Identifiers | |
| 775304-57-9 |
|
| ChEMBL | ChEMBL256997 |
| ChemSpider | 9394889 |
| 7341 | |
| Jmol 3D model | Interactive image |
| KEGG | D09323 |
| PubChem | 11219835 |
| UNII | K16AME9I3V |
| Properties | |
| C15H9FN2O3 | |
| Molar mass | 284.24 g/mol |
| Pharmacology | |
| M09AX03 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////ORPHAN DRUG, Ataluren, Translarna, Duchenne Muscular Dystrophy, EU, 775304-57-9, PTC Therapeutics, PTC 124
O=C(O)c1cccc(c1)c2nc(on2)c3ccccc3F

