
Home » Posts tagged 'Duchenne muscular dystrophy'
Tag Archives: Duchenne muscular dystrophy
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
IDEBENONE
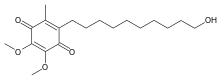
IDEBENONE
2-(10-Hydroxydecyl)-5,6-dimethoxy-3-methyl-1,4-benzoquinone
- Molecular FormulaC19H30O5
- Average mass338.439 Da
- 58186-27-9
- Idebenona, Idebenonum, CV 2619
IdesolKS-5193NemocebralSNT-MC17идебенонإيديبينون艾地苯醌
Puldysa (idebenone), for the treatment of Duchenne muscular dystrophyTitle: Idebenone
CAS Registry Number: 58186-27-9
CAS Name: 2-(10-Hydroxydecyl)-5,6-dimethoxy-3-methyl-2,5-cyclohexadiene-1,4-dione
Additional Names: 6-(10-hydroxydecyl)-2,3-dimethoxy-5-methyl-1,4-benzoquinone; 2,3-dimethoxy-5-methyl-6-(10¢-hydroxydecyl)-1,4-benzoquinone; 6-(10-hydroxydecyl)ubiquinone
Manufacturers’ Codes: CV-2619
Trademarks: Avan (Takeda); Daruma (Takeda); Lucebanol (Hormona); Mnesis (Takeda)
Molecular Formula: C19H30O5Molecular Weight: 338.44
Percent Composition: C 67.43%, H 8.93%, O 23.64%
Literature References: Ubiquinone derivative with protective effects against cerebral ischemia. Prepn: H. Morimoto et al.,DE2519730; eidem,US4271083 (1975, 1981 both to Takeda); K. Okamoto et al.,Chem. Pharm. Bull.30, 2797 (1982); C.-A. Yu, L. Yu, Biochemistry21, 4096 (1982). Effect on ischemia-induced amnesia in rats: N. Yamazaki et al.,Jpn. J. Pharmacol.36, 349 (1984). Metabolism in animals: T. Kobayashi et al.,J. Pharmacobio-Dyn.8, 448 (1985). Disposition: H. Torii et al.,ibid. 457. Pharmacokinetics and tolerance in humans: M. F. Barkworth et al.,Arzneim.-Forsch.35, 1704 (1985). Series of articles on pharmacology and clinical studies: Arch. Gerontol. Geriatr.8, 193-366 (1989). Review of chemistry, toxicology and pharmacology: I. Zs-Nagy, Arch. Gerontol. Geriatr.11, 177-186 (1990).Properties: Orange needles from ligroin, mp 46-50° (Morimoto); also reported as crystals from hexane + ethyl acetate, mp 52-53° (Okamoto). Sol in organic solvents. Practically insol in water.Melting point: mp 46-50° (Morimoto); mp 52-53° (Okamoto)Therap-Cat: Nootropic.Keywords: Nootropic.
Idebenone is a member of the class of 1,4-benzoquinones which is substituted by methoxy groups at positions 2 and 3, by a methyl group at positions 5, and by a 10-hydroxydecyl group at positions 6. Initially developed for the treatment of Alzheimer’s disease, benefits were modest; it was subsequently found to be of benefit for the symptomatic treatment of Friedreich’s ataxia. It has a role as an antioxidant. It is a primary alcohol and a member of 1,4-benzoquinones.
Idebenone (pronounced eye-deb-eh-known, trade names Catena, Raxone, Sovrima, among others) is a drug that was initially developed by Takeda Pharmaceutical Company for the treatment of Alzheimer’s disease and other cognitive defects.[1] This has been met with limited success. The Swiss company Santhera Pharmaceuticals has started to investigate it for the treatment of neuromuscular diseases. In 2010, early clinical trials for the treatment of Friedreich’s ataxia[2] and Duchenne muscular dystrophy[3] have been completed. As of December 2013 the drug is not approved for these indications in North America or Europe. It is approved by the European Medicines Agency (EMA) for use in Leber’s hereditary optic neuropathy (LHON) and was designated an orphan drug in 2007.[4]
Chemically, idebenone is an organic compound of the quinone family. It is also promoted commercially as a synthetic analog of coenzyme Q10 (CoQ10).
Uses
Indications that are or were approved in some territories
Nootropic effects and Alzheimer’s disease
Idebenone improved learning and memory in experiments with mice.[5] In humans, evaluation of Surrogate endpoints like electroretinography, auditory evoked potentials and visual analogue scales also suggested positive nootropic effects,[6] but larger studies with hard endpoints are missing.
Research on idebenone as a potential therapy of Alzheimer’s disease have been inconsistent, but there may be a trend for a slight benefit.[7][8] In May 1998, the approval for this indication was cancelled in Japan due to the lack of proven effects. In some European countries, the drug is available for the treatment of individual patients in special cases.[1]
Friedreich’s ataxia (Sovrima)
Preliminary testing has been done in humans and found idebenone to be a safe treatment for Friedreich’s ataxia (FA), exhibiting a positive effect on cardiac hypertrophy and neurological function.[9] The latter was only significantly improved in young patients.[10] In a different experiment, a one-year test on eight patients, idebenone reduced the rate of deterioration of cardiac function, but without halting the progression of ataxia.[11]
The drug was approved for FA in Canada in 2008 under conditions including proof of efficacy in further clinical trials.[12] However, on February 27, 2013, Health Canada announced that idebenone would be voluntarily recalled as of April 30, 2013 by its Canadian manufacturer, Santhera Pharmaceuticals, due to the failure of the drug to show efficacy in the further clinical trials that were conducted.[13] In 2008, the European Medicines Agency (EMA) refused a marketing authorisation for this indication.[1] As of 2013 the drug was not approved for FA in Europe[14] nor in the US, where there is no approved treatment.[15]
Leber’s hereditary optic neuropathy (Raxone)
Leber’s hereditary optic neuropathy (LHON) is a mitochondrially inherited (mother to all offspring) degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; this affects predominantly young adult males. Santhera completed a Phase III clinical trial in this indication in Europe with positive results,[16] and submitted an application to market the drug to European regulators in July 2011.[17] It is approved by EMA for this indication and was designated an orphan drug in 2007.[4]
Indications being explored
Duchenne muscular dystrophy (Catena)
After experiments in mice[18] and preliminary studies in humans, idebenone has entered Phase II clinical trials in 2005[3] and Phase III trials in 2009.[19]
Other neuromuscular diseases
Phase I and II clinical trials for the treatment of MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes)[20] and primary progressive multiple sclerosis[21] are ongoing as of December 2013.
Life style
Idebenone is claimed to have properties similar to CoQ10 in its antioxidant properties, and has therefore been used in anti-aging on the basis of free-radical theory. Clinical evidence for this use is missing. It has been used in topical applications to treat wrinkles.[22]
Pharmacology
In cellular and tissue models, idebenone acts as a transporter in the electron transport chain of mitochondria and thus increases the production of adenosine triphosphate (ATP) which is the main energy source for cells, and also inhibits lipoperoxide formation. Positive effects on the energy household of mitochondria has also been observed in animal models.[1][23] Clinical relevance of these findings has not been established.
Pharmacokinetics
Idebenone is well absorbed from the gut but undergoes excessive first pass metabolism in the liver, so that less than 1% reach the circulation. This rate can be improved with special formulations (suspensions) of idebenone and by administering it together with fat food; but even taking these measures bioavailability still seems to be considerably less than 14% in humans. More than 99% of the circulating drug are bound to plasma proteins. Idebenone metabolites include glucuronides and sulfates, which are mainly (~80%) excreted via the urine.[1]
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0040402014014306


SYN
The palladium-catalyzed olefination of a sp2 or benzylic carbon attached to a (pseudo)halogen is known as the Heck reaction.2,63 It is a powerful tool, mainly used for the synthesis of vinylarenes, and it has also been employed for the construction of conjugated double bonds. The widespread application of this reaction can be illustrated by numerous examples in both academia small-scale64 and industrial syntheses.5 As an example, in 2011, a idebenone (124) total synthesis based on a Heck reaction was described (Scheme 35).65 This compound, initially designed for the treatment of Alzheimer’s and Parkinson’s diseases, presented a plethora of other interesting activities, such as free radical scavenging and action against some muscular illnesses. The key step in the synthesis was the coupling of 2-bromo-3,4,5-trimethoxy-1-methylbenzene (125) with dec-9-en-1-ol affording products 126. Under non-optimized conditions (Pd(OAc)2, PPh3, Et3N, 120 ºC), a mixture composed of 60% linear olefins 126 and 15% of the undesired branched product 127 was obtained after three days of reaction. Therefore, the conditions were optimized, allowing the preparation of 126 in 67% yield with no detection of 127 after only 30 min of reaction employing DMF, Pd(PPh3)4, iPr2NEt under microwave heating. To conclude the synthesis, the Heck adducts were submitted to hydroxyl protection/deprotection, hydrogenation, and ring oxidation. After these reactions, idebenone was obtained with 20% overall yield over 6 steps.

Scheme 35 Synthesis of idebenone (124) based on Heck reaction of 2-bromo-3,4,5-trimethoxy-1-methylbenzene with dec-9-en-1-ol under microwave irradiation.
Syn
- Duveau, Damien Y.; Bioorganic & Medicinal Chemistry 2010, V18(17), P6429-6441
- Okada, Taiiti; EP 289223 A1 1988
- Watanabe, Masazumi; EP 58057 A1 1982
- Okamoto, Kayoko; Chemical & Pharmaceutical Bulletin 1982, V30(8), P2797-819
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
Paper
Tsoukala, Anna; Organic Process Research & Development 2011, V15(3), P673-680
https://pubs.acs.org/doi/10.1021/op200051v
An environmentally benign, convenient, high yielding, and cost-effective synthesis leading to idebenone is disclosed. The synthesis includes a bromination process for the preparation of 2-bromo-3,4,5-trimethoxy-1-methylbenzene, a protocol for the Heck cross-coupling reaction using either thermal or microwave heating, olefin reduction by palladium catalyzed hydrogenation, and a green oxidation protocol with hydrogen peroxide as oxidant to achieve the benzoquinone framework. The total synthesis is composed of six steps that provide an overall yield of 20% that corresponds to a step yield of 76%.





PAPER
Bioorganic & Medicinal Chemistry 2010, V18(17), P6429-6441
https://www.sciencedirect.com/science/article/abs/pii/S0968089610006322

Analogues of mitoQ and idebenone were synthesized to define the structural elements that support oxygen consumption in the mitochondrial respiratory chain. Eight analogues were prepared and fully characterized, then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain. While oxygen consumption was strongly inhibited by mitoQ analogues 2–4 in a chain length-dependent manner, modification of idebenone by replacement of the quinone methoxy groups by methyl groups (analogues 6–8) reduced, but did not eliminate, oxygen consumption. Idebenone analogues 6–8 also displayed significant cytoprotective properties toward cultured mammalian cells in which glutathione had been depleted by treatment with diethyl maleate.
Idebenone (5)18 To a stirred solution containing 200 mg (0.467 mmol) of 2,3- dimethoxy-6-methyl-5-benzyloxydecyl-p-benzoquinone (38) in 5 mL of anhydrous methanol at 23 C was added 15 mg of 10 % Pd/C in one portion. The reaction mixture was stirred at 23 C under an atmosphere of hydrogen for 24 h. Air was then bubbled through the reaction mixture at 23 C for 24 h. The suspension was filtered through Celite and the filtrate was concentrated under diminished pressure to afford idebenone (5) as an orange solid: yield 130 mg (82%); mp: 46–47 C; 1 H NMR (400 MHz, CDCl3) d 1.34 (m, 14H), 1.60 (quint, 2H, J = 7.6 Hz), 2.04 (s, 3H), 2.44 (t, 2H, J = 8.0 Hz), 3.63 (t, 2H, J = 6.8 Hz), and 3.99 (s, 6H); 13C NMR (100 MHz, CDCl3) d 11.9, 25.7, 26.4, 28.7, 29.3, 29.3, 29.4, 29.5, 29.8, 32.7
References
- ^ Jump up to:a b c d e “CHMP Assessment Report for Sovrima” (PDF). European Medicines Agency. 20 November 2008: 6, 9–11, 67f.
- ^ Clinical trial number NCT00229632 for “Idebenone to Treat Friedreich’s Ataxia” at ClinicalTrials.gov
- ^ Jump up to:a b Clinical trial number NCT00654784 for “Efficacy and Tolerability of Idebenone in Boys With Cardiac Dysfunction Associated With Duchenne Muscular Dystrophy (DELPHI)” at ClinicalTrials.gov
- ^ Jump up to:a b “Raxone”. http://www.ema.europa.eu. Retrieved 12 July 2019.
- ^ Liu, XJ; Wu, WT (1999). “Effects of ligustrazine, tanshinone II A, ubiquinone, and idebenone on mouse water maze performance”. Zhongguo Yao Li Xue Bao. 20 (11): 987–90. PMID 11270979.
- ^ Schaffler, K; Hadler, D; Stark, M (1998). “Dose-effect relationship of idebenone in an experimental cerebral deficit model. Pilot study in healthy young volunteers with piracetam as reference drug”. Arzneimittel-Forschung. 48 (7): 720–6. PMID 9706371.
- ^ Gutzmann, H; Kühl, KP; Hadler, D; Rapp, MA (2002). “Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: results of a randomized, double-blind, parallel-group multicenter study”. Pharmacopsychiatry. 35 (1): 12–8. doi:10.1055/s-2002-19833. PMID 11819153.
- ^ Parnetti, L; Senin, U; Mecocci, P (1997). “Cognitive enhancement therapy for Alzheimer’s disease. The way forward”. Drugs. 53 (5): 752–68. doi:10.2165/00003495-199753050-00003. PMID 9129864. S2CID 46987059.
- ^ Di Prospero NA, Baker A, Jeffries N, Fischbeck KH (October 2007). “Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial”. Lancet Neurol. 6 (10): 878–86. doi:10.1016/S1474-4422(07)70220-X. PMID 17826341. S2CID 24749816.
- ^ Tonon C, Lodi R (September 2008). “Idebenone in Friedreich’s ataxia”. Expert Opin Pharmacother. 9 (13): 2327–37. doi:10.1517/14656566.9.13.2327. PMID 18710357. S2CID 73285881.
- ^ Buyse G, Mertens L, Di Salvo G, et al. (May 2003). “Idebenone treatment in Friedreich’s ataxia: neurological, cardiac, and biochemical monitoring”. Neurology. 60 (10): 1679–81. doi:10.1212/01.wnl.0000068549.52812.0f. PMID 12771265. S2CID 36556782.
- ^ “Heath Canada Fact Sheet – Catena”. Archived from the original on 19 June 2014.
- ^ Voluntary Withdrawal of Catena from the Canadian Market
- ^ Margaret Wahl for Quest Magazine, MAY 28, 2010. FA Research: Idebenone Strikes Out Again
- ^ NINDS Fact Sheet
- ^ Klopstock, T; et al. (2011). “A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy”. Brain. 134 (9): 2677–86. doi:10.1093/brain/awr170. PMC 3170530. PMID 21788663.
- ^ Staff (26 July 2011). “Santhera publishes pivotal trial results of idebenone and goes for EU approval”. European Biotechnology News. Archived from the original on 2013-02-17.
- ^ Buyse, GM; Van Der Mieren, G; Erb, M; D’hooge, J; Herijgers, P; Verbeken, E; Jara, A; Van Den Bergh, A; et al. (2009). “Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance”. European Heart Journal. 30 (1): 116–24. doi:10.1093/eurheartj/ehn406. PMC 2639086. PMID 18784063.
- ^ Clinical trial number NCT01027884 for “Phase III Study of Idebenone in Duchenne Muscular Dystrophy (DMD) (DELOS)” at ClinicalTrials.gov
- ^ Clinical trial number NCT00887562 for “Study of Idebenone in the Treatment of Mitochondrial Encephalopathy Lactic Acidosis & Stroke-like Episodes (MELAS)” at ClinicalTrials.gov
- ^ Clinical trial number NCT00950248 for “Double Blind Placebo-Controlled Phase I/II Clinical Trial of Idebenone in Patients With Primary Progressive Multiple Sclerosis (IPPoMS)” at ClinicalTrials.gov
- ^ McDaniel D, Neudecker B, Dinardo J, Lewis J, Maibach H (September 2005). “Clinical efficacy assessment in photodamaged skin of 0.5% and 1.0% idebenone”. J Cosmet Dermatol. 4 (3): 167–73. doi:10.1111/j.1473-2165.2005.00305.x. PMID 17129261. S2CID 2394666.
- ^ Suno M, Nagaoka A (May 1988). “[Effect of idebenone and various nootropic drugs on lipid peroxidation in rat brain homogenate in the presence of succinate]”. Nippon Yakurigaku Zasshi (in Japanese). 91 (5): 295–9. doi:10.1254/fpj.91.295. PMID 3410376.
| Clinical data | |
|---|---|
| Trade names | Catena, Raxone, Sovrima |
| AHFS/Drugs.com | International Drug Names |
| License data | EU EMA: by INN |
| ATC code | N06BX13 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | <1% (high first pass effect) |
| Protein binding | >99% |
| Elimination half-life | 18 hours |
| Excretion | Urine (80%) and feces |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 58186-27-9 |
| PubChem CID | 3686 |
| ChemSpider | 3558 |
| UNII | HB6PN45W4J |
| KEGG | D01750 |
| ChEMBL | ChEMBL252556 |
| CompTox Dashboard (EPA) | DTXSID0040678 |
| Chemical and physical data | |
| Formula | C19H30O5 |
| Molar mass | 338.444 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=C1/C(=C(\C(=O)C(\OC)=C1\OC)C)CCCCCCCCCCO | |
| InChI[hide]InChI=1S/C19H30O5/c1-14-15(12-10-8-6-4-5-7-9-11-13-20)17(22)19(24-3)18(23-2)16(14)21/h20H,4-13H2,1-3H3 Key:JGPMMRGNQUBGND-UHFFFAOYSA-N |
////////////IDEBENONE, Puldysa, Duchenne muscular dystrophy, Idesol, KS 5193, Nemocebral, SNT MC17, идебенон, إيديبينون , 艾地苯醌 , CV 2619
CC1=C(C(=O)C(=C(C1=O)OC)OC)CCCCCCCCCCO
Golodirsen, ゴロジルセン;

Golodirsen
- RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(G-m5U-m5U-G-C-C-m5U-C-C-G-G-m5U-m5U-C-m5U-G-A-A-G-G-m5U-G-m5U-m5U-C), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N-dimethylphosphonamidate]
- Nucleic Acid Sequence
- Sequence Length: 25
| Formula |
C305H481N138O112P25
|
|---|---|
| CAS |
1422959-91-8
|
| Mol weight |
8647.2841
|
- Exon 53: NG-12-0163
- Golodirsen
- SRP 4053
Nucleic Acid Sequence
Sequence Length: 252 a 6 c 8 g 9 umodified
FDA APPROVED, Vyondys 53, 019/12/12
Antisense oligonucleotide
|
ゴロジルセン;
|
Duchenne muscular dystrophy (DMD variant amenable to exon 53 skipping)
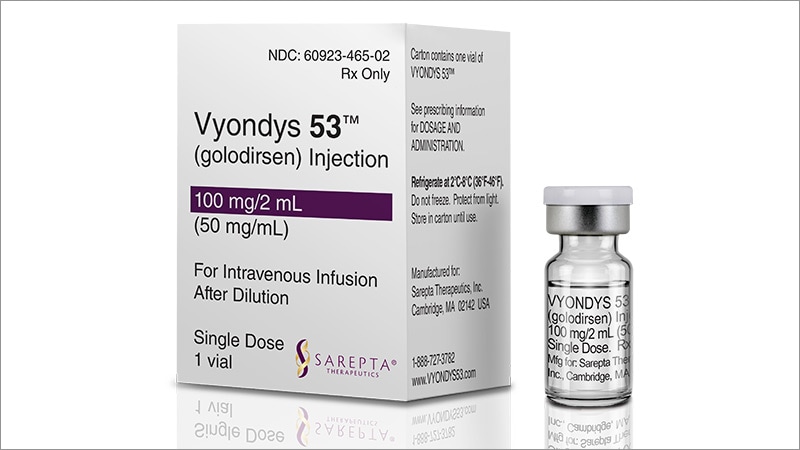
VYONDYS 53 (golodirsen) injection is a sterile, aqueous, preservative-free, concentrated solution for dilution prior to intravenous administration. VYONDYS 53 is a clear to slightly opalescent, colorless liquid. VYONDYS 53 is supplied in single-dose vials containing 100 mg golodirsen (50 mg/mL). VYONDYS 53 is formulated as an isotonic phosphate buffered saline solution with an osmolality of 260 to 320 mOSM and a pH of 7.5. Each milliliter of VYONDYS 53 contains: 50 mg golodirsen; 0.2 mg potassium chloride; 0.2 mg potassium phosphate monobasic; 8 mg sodium chloride; and 1.14 mg sodium phosphate dibasic, anhydrous, in water for injection. The product may contain hydrochloric acid or sodium hydroxide to adjust pH.
Golodirsen is an antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass. PMOs are synthetic molecules in which the five-membered ribofuranosyl rings found in natural DNA and RNA are replaced by a six-membered morpholino ring. Each morpholino ring is linked through an uncharged phosphorodiamidate moiety rather than the negatively charged phosphate linkage that is present in natural DNA and RNA. Each phosphorodiamidate morpholino subunit contains one of the heterocyclic bases found in DNA (adenine, cytosine, guanine, or thymine). Golodirsen contains 25 linked subunits. The sequence of bases from the 5′ end to 3′ end is GTTGCCTCCGGTTCTGAAGGTGTTC. The molecular formula of golodirsen is C305H481N138O112P25 and the molecular weight is 8647.28 daltons. The structure of golodirsen is:
|
SIDE EFFECTS
- Hypersensitivity Reactions [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the VYONDYS 53 clinical development program, 58 patients received at least one intravenous dose of VYONDYS 53, ranging between 4 mg/kg (0.13 times the recommended dosage) and 30 mg/kg (the recommended dosage). All patients were male and had genetically confirmed Duchenne muscular dystrophy. Age at study entry was 6 to 13 years. Most (86%) patients were Caucasian.
VYONDYS 53 was studied in 2 double-blind, placebo-controlled studies.
In Study 1 Part 1, patients were randomized to receive once-weekly intravenous infusions of VYONDYS 53 (n=8) in four increasing dose levels from 4 mg/kg to 30 mg/kg or placebo (n=4), for at least 2 weeks at each level. All patients who participated in Study 1 Part 1 (n=12) were continued into Study 1 Part 2, an open-label extension, during which they received VYONDYS 53 at a dose of 30 mg/kg IV once weekly [see Clinical Studies].
In Study 2, patients received VYONDYS 53 (n=33) 30 mg/kg or placebo (n=17) IV once weekly for up to 96 weeks, after which all patients received VYONDYS 53 at a dose of 30 mg/kg.
Adverse reactions observed in at least 20% of treated patients in the placebo-controlled sections of Studies 1 and 2 are shown in Table 1.
Table 1: Adverse Reactions That Occurred in At Least 20% of VYONDYS 53-Treated Patients and at a Rate Greaterthan Placebo in Studies 1 and 2
| Adverse Reaction | VYONDYS 53 (N = 41) % |
Placebo (N = 21) % |
| Headache | 41 | 10 |
| Pyrexia | 41 | 14 |
| Fall | 29 | 19 |
| Abdominal pain | 27 | 10 |
| Nasopharyngitis | 27 | 14 |
| Cough | 27 | 19 |
| Vomiting | 27 | 19 |
| Nausea | 20 | 10 |
Other adverse reactions that occurred at a frequency greater than 5% of VYONDYS 53-treated patients and at a greater frequency than placebo were: administration site pain, back pain, pain, diarrhea, dizziness, ligament sprain, contusion, influenza, oropharyngeal pain, rhinitis, skin abrasion, ear infection, seasonal allergy, tachycardia, catheter site related reaction, constipation, and fracture.
Hypersensitivity reactions have occurred in patients treated with VYONDYS 53 [see WARNINGS AND PRECAUTIONS].
Antisense technology provides a means for modulating the expression of one or more specific gene products, including alternative splice products, and is uniquely useful in a number of therapeutic, diagnostic, and research applications. The principle behind antisense technology is that an antisense compound, e.g., an oligonucleotide, which hybridizes to a target nucleic acid, modulates gene expression activities such as transcription, splicing or translation through any one of a number of antisense mechanisms. The sequence specificity of antisense compounds makes them attractive as tools for target validation and gene functionalization, as well as therapeutics to selectively modulate the expression of genes involved in disease.
Duchenne muscular dystrophy (DMD) is caused by a defect in the expression of the protein dystrophin. The gene encoding the protein contains 79 exons spread out over more than 2 million nucleotides of DNA. Any exonic mutation that changes the reading frame of the exon, or introduces a stop codon, or is characterized by removal of an entire out of frame exon or exons, or duplications of one or more exons, has the potential to disrupt production of functional dystrophin, resulting in DMD.
Recent clinical trials testing the safety and efficacy of splice switching
oligonucleotides (SSOs) for the treatment of DMD are based on SSO technology to induce alternative splicing of pre-mRNAs by steric blockade of the spliceosome (Cirak et al., 2Q\ \; Goemans et al., 2011; Kinali et al., 2009; van Deutekom et al., 2007). However, despite these successes, the pharmacological options available for treating DMD are limited. Golodirsen is a phosphorodiamidate morpholino oligomer (PMO) designed to skip exon 53 of the human dystrophin gene in patients with DMD who are amendable to exon 53 skipping to restore the read frame and produce a functional shorter form of the dystrophin protein.
Although significant progress has been made in the field of antisense technology, there remains a need in the art for methods of preparing phosphorodiamidate morpholino oligomers with improved antisense or antigene performance.
PATENT
https://patents.google.com/patent/WO2017205880A1/en
Provided herein are processes for preparing phosphorodiamidate morpholino oligomers (PMOs). The synthetic processes described herein allow for a scaled-up PMO synthesis while maintaining overall yield and purity of a synthesized PMO.
Accordingly, in one aspect, provided herein is a process for preparing an oligomeric compound of Formula A):

(A).
In certain embodiments, provided herein is a process for preparing an oligomeric compound of Formula (G):

In yet another embodiment, the oligomeric compound of the disclosure including, for example, some embodiments of an oligomeric compound of Formula (G), is an oligomeric compound of Formula (XII):

(XII).
For clarity, the structural formulas including, for example, oligomeric compound of Formula (C) and Golodirsen depicted by Formula (XII), are a continuous structural formula from 5′ to 3′, and, for the convenience of depicting the entire formula in a compact form in the above structural formulas, Applicants have included various illustration breaks labeled “BREAK A” and “BREAK B.” As would be understood by the skilled artisan, for example, each indication of “BREAK A” shows a continuation of the illustration of the structural formula at these points. The skilled artisan understands that the same is true for each instance of “BREAK B” in the structural formulas above including Golodirsen. None of the illustration breaks, however, are intended to indicate, nor would the skilled artisan understand them to mean, an actual discontinuation of the structural formulas above including
Example 1: NCP2 Anchor Synthesis
1. Preparation of Meth l 4-Fluoro-3-Nitrobenzoate (1)

To a 100L flask was charged 12.7kg of 4-fluoro-3-nitrobenzoic acid was added 40kg of methanol and 2.82kg concentrated sulfuric acid. The mixture was stirred at reflux (65° C) for 36 hours. The reaction mixture was cooled to 0° C. Crystals formed at 38° C. The mixture was held at 0° C for 4 hrs then filtered under nitrogen. The 100L flask was washed and filter cake was washed with 10kg of methanol that had been cooled to 0° C. The solid filter cake was dried on the funnel for 1 hour, transferred to trays, and dried in a vacuum oven at room temperature to a constant weight of 13.695kg methyl 4-fluoro-3-nitrobenzoate (100% yield; HPLC 99%).
2. Preparation of 3-Nitro-4-(2-oxopropyl)benzoic Acid
A. (Z)-Methyl 4-(3 -Hydroxy- l-Methoxy-l-Oxobut-2-en-2-yl)-3-Nitrobenzoate (2)

To a 100L flask was charged 3.98kg of methyl 4-fluoro-3-nitrobenzoate (1) from the previous step 9.8kg DMF, 2.81kg methyl acetoacetate. The mixture was stirred and cooled to 0° C. To this was added 3.66kg DBU over about 4 hours while the temperature was maintained at or below 5° C. The mixture was stirred an additional 1 hour. To the reaction flask was added a solution of 8.15kg of citric acid in 37.5kg of purified water while the reaction temperature was maintained at or below 15° C. After the addition, the reaction mixture was stirred an addition 30 minutes then filtered under nitrogen. The wet filter cake was returned to the 100L flask along with 14.8kg of purified water. The slurry was stirred for 10 minutes then filtered. The wet cake was again returned to the 100L flask, slurried with 14.8kg of purified water for 10 minutes, and filtered to crude (Z)-methyl 4-(3 -hydroxy- 1 – methoxy-l-oxobut-2-en-2-yl)-3-nitrobenzoate.
B. 3-Nitro-4-(2-oxopropyl)benzoic Acid

2 3
The crude (Z)-m ethyl 4-(3 -hydroxy- 1-methoxy-l -ox obut-2-en-2-yl)-3-nitrobenzoate was charged to a 100L reaction flask under nitrogen. To this was added 14.2kg 1,4-dioxane and the stirred. To the mixture was added a solution of 16.655kg concentrated HC1 and 13.33kg purified water (6M HC1) over 2 hours while the temperature of the reaction mixture was maintained below 15° C. When the addition was complete, the reaction mixture was heated at reflux (80° C) for 24 hours, cooled to room temperature, and filtered under nitrogen. The solid filter cake was triturated with 14.8kg of purified water, filtered, triturated again with 14.8kg of purified water, and filtered. The solid was returned to the 100L flask with 39.9kg of DCM and refluxed with stirring for 1 hour. 1.5kg of purified water was added to dissolve the remaining solids. The bottom organic layer was split to a pre-warmed 72L flask, then returned to a clean dry 100L flask. The solution was cooled to 0° C, held for 1 hour, then filtered. The solid filter cake was washed twice each with a solution of 9.8kg DCM and 5kg heptane, then dried on the funnel. The solid was transferred to trays and dried to a constant weight of 1.855kg 3-Nitro-4-(2-oxopropyl)benzoic Acid. Overall yield 42% from compound 1. HPLC 99.45%.
3. Preparation of N-Tritylpiperazine Succinate (NTP)

To a 72L jacketed flask was charged under nitrogen 1.805kg triphenylmethyl chloride and 8.3kg of toluene (TPC solution). The mixture was stirred until the solids dissolved. To a 100L jacketed reaction flask was added under nitrogen 5.61kg piperazine, 19.9kg toluene, and 3.72kg methanol. The mixture was stirred and cooled to 0° C. To this was slowly added in portions the TPC solution over 4 hours while the reaction temperature was maintained at or below 10° C. The mixture was stirred for 1.5 hours at 10° C, then allowed to warm to 14° C. 32.6kg of purified water was charged to the 72L flask, then transferred to the 100L flask while the internal batch temperature was maintained at 20+/-50 C. The layers were allowed to split and the bottom aqueous layer was separated and stored. The organic layer was extracted three times with 32kg of purified water each, and the aqueous layers were separated and combined with the stored aqueous solution.
The remaining organic layer was cooled to 18° C and a solution of 847g of succinic acid in 10.87kg of purified water was added slowly in portions to the organic layer. The mixture was stirred for 1.75 hours at 20+/-50 C. The mixture was filtered, and the solids were washed with 2kg TBME and 2kg of acetone then dried on the funnel. The filter cake was triturated twice with 5.7kg each of acetone and filtered and washed with 1kg of acetone between triturations. The solid was dried on the funnel, then transferred to trays and dried in a vacuum oven at room temperature to a constant weight of 2.32kg of NTP. Yield 80%. 4. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l-yl)Methanone A. Preparation of l-(2-Nitro-4(4-Tritylpiperazine-l-Carbonyl)Phenyl)Propan-2-one

3 4
To a 100L jacketed flask was charged under nitrogen 2kg of 3-Nitro-4-(2- oxopropyl)benzoic Acid (3), 18.3 kg DCM, 1.845kg N-(3-dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDC.HC1). The solution was stirred until a homogenous mixture was formed. 3.048kg of NTP was added over 30 minutes at room temperature and stirred for 8 hours. 5.44kg of purified water was added to the reaction mixture and stirred for 30 minutes. The layers were allowed to separate and the bottom organic layer containing the product was drained and stored. The aqueous layer was extracted twice with 5.65kg of DCM. The combined organic layers were washed with a solution of 1.08kg sodium chloride in 4.08kg purified water. The organic layers were dried over 1.068kg of sodium sulfate and filtered. The sodium sulfate was washed with 1.3kg of DCM. The combined organic layers were slurried with 252g of silica gel and filtered through a filter funnel containing a bed of 252g of silica gel. The silica gel bed was washed with 2kg of DCM. The combined organic layers were evaporated on a rotovap. 4.8kg of THF was added to the residue and then evaporated on the rotovap until 2.5 volumes of the crude l-(2-nitro-4(4-tritylpiperazine-l- carbonyl)phenyl)propan-2-one in THF was reached.
B. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l- yl)Methano

To a 100L jacketed flask was charged under nitrogen 3600g of 4 from the previous step and 9800g THF. The stirred solution was cooled to <5° C. The solution was diluted with 11525g ethanol and 194g of sodium borohydride was added over about 2 hours at <5° C. The reaction mixture was stirred an additional 2 hours at <5° C. The reaction was quenched with a solution of about 1.1kg ammonium chloride in about 3kg of water by slow addition to maintain the temperature at <10° C. The reaction mixture was stirred an additional 30 minutes, filtered to remove inorganics, and recharged to a 100L jacketed flask and extracted with 23kg of DCM. The organic layer was separated and the aqueous was twice more extracted with 4.7kg of DCM each. The combined organic layers were washed with a solution of about 800g of sodium chloride in about 3kg of water, then dried over 2.7kg of sodium sulfate. The suspension was filtered and the filter cake was washed with 2kg of DCM. The combined filtrates were concentrated to 2.0 volumes, diluted with about 360g of ethyl acetate, and evaporated. The crude product was loaded onto a silica gel column of 4kg of silica packed with DCM under nitrogen and eluted with 2.3kg ethyl acetate in 7.2kg of DCM. The combined fractions were evaporated and the residue was taken up in 11.7kg of toluene. The toluene solution was filtered and the filter cake was washed twice with 2kg of toluene each. The filter cake was dried to a constant weight of 2.275kg of compound 5 (46% yield from compound 3) HPLC 96.99%. 5. Preparation of 2,5-Dioxopyrrolidin-l-yl(l-(2-Nitro-4-(4-triphenylmethylpiperazine-l Carbon l)Phenyl)Propan-2-yl) Carbonate (NCP2 Anchor)

3 NCP2 Anchor
To a 100L jacketed flask was charged under nitrogen 4.3kg of compound 5 (weight adjusted based on residual toluene by 1H MR; all reagents here after were scaled accordingly) and 12.7kg pyridine. To this was charged 3.160 kg of DSC (78.91 weight % by 1H NMR) while the internal temperature was maintained at <35° C. The reaction mixture was aged for about 22 hours at ambience then filtered. The filter cake was washed with 200g of pyridine. In two batches each comprising ½ the filtrate volume, filtrate wash charged slowly to a 100L jacketed flask containing a solution of about 11kg of citric acid in about 50 kg of water and stirred for 30 minutes to allow for solid precipitation. The solid was collected with a filter funnel, washed twice with 4.3kg of water per wash, and dried on the filter funnel under vacuum.
The combined solids were charged to a 100L jacketed flask and dissolved in 28kg of DCM and washed with a solution of 900g of potassium carbonate in 4.3kg of water. After 1 hour, the layers were allowed to separate and the aqueous layer was removed. The organic layer was washed with 10kg of water, separated, and dried over 3.5kg of sodium sulfate. The DCM was filtered, evaporated, and dried under vacuum to 6.16kg of NCP2 Anchor (114% yield).
Example 2: Anchor Loaded Resin Synthesis
To a 75L solid phase synthesis reactor was charged about 52L of NMP and 2600g of aminomethyl polystyrene resin. The resin was stirred in the NMP to swell for about 2 hours then drained. The resin was washed twice with about 39L DCM per wash, then twice with 39L Neutralization Solution per wash, then twice with 39L of DCM per wash. The NCP2 Anchor Solution was slowly added to the stirring resin solution, stirred for 24 hours at room temperature, and drained. The resin was washed four times with 39L of NMP per wash, and six times with 39L of DCM per wash. The resin was treated and stirred with ½ the DEDC Capping Solution for 30 minutes, drained, and was treated and stirred with the 2nd ½ of the DEDC Capping Solution for 30 minutes and drained. The resin was washed six times with 39L of DCM per wash then dried in an oven to constant weight of 3573.71g of Anchor Loaded Resin.
Example 3: Preparation of Activated EG3 Tail
1. Preparation of Trityl Piperazine Phenyl Carbamate 35

To a cooled suspension of NTP in dichloromethane (6 mL/g NTP) was added a solution of potassium carbonate (3.2 eq) in water (4 mL/g potassium carbonate). To this two- phase mixture was slowly added a solution of phenyl chloroformate (1.03 eq) in
dichloromethane (2 g/g phenyl chloroformate). The reaction mixture was warmed to 20° C. Upon reaction completion (1-2 hr), the layers were separated. The organic layer was washed with water, and dried over anhydrous potassium carbonate. The product 35 was isolated by crystallization from acetonitrile. Yield=80%
2. Preparation of Carbamate Alcohol (36)

Sodium hydride (1.2 eq) was suspended in l-methyl-2-pyrrolidinone (32 mL/g sodium hydride). To this suspension were added triethylene glycol (10.0 eq) and compound 35 (1.0 eq). The resulting slurry was heated to 95° C. Upon reaction completion (1-2 hr), the mixture was cooled to 20° C. To this mixture was added 30% dichloromethane/methyl tert- butyl ether (v:v) and water. The product-containing organic layer was washed successively with aqueous NaOH, aqueous succinic acid, and saturated aqueous sodium chloride. The product 36 was isolated by crystallization from dichloromethane/methyl tert-butyl ether/heptane. Yield=90%.
3. Preparation of EG3 Tail Acid (37)

To a solution of compound 36 in tetrahydrofuran (7 mL/g 36) was added succinic anhydride (2.0 eq) and DMAP (0.5 eq). The mixture was heated to 50° C. Upon reaction completion (5 hr), the mixture was cooled to 20° C and adjusted to pH 8.5 with aqueous NaHC03. Methyl tert-butyl ether was added, and the product was extracted into the aqueous layer. Dichloromethane was added, and the mixture was adjusted to pH 3 with aqueous citric acid. The product-containing organic layer was washed with a mixture of pH=3 citrate buffer and saturated aqueous sodium chloride. This dichloromethane solution of 37 was used without isolation in the preparation of compound 38. 4. Preparation of Activated EG3 Tail (38)

To the solution of compound 37 was added N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (HONB) (1.02 eq), 4-dimethylaminopyridine (DMAP) (0.34 eq), and then l-(3- dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) (1.1 eq). The mixture was heated to 55° C. Upon reaction completion (4-5 hr), the mixture was cooled to 20° C and washed successively with 1 : 1 0.2 M citric acid/brine and brine. The dichloromethane solution underwent solvent exchange to acetone and then to Ν,Ν-dimethylformamide, and the product was isolated by precipitation from acetone/N,N-dimethylformamide into saturated aqueous sodium chloride. The crude product was reslurried several times in water to remove residual Ν,Ν-dimethylformamide and salts. Yield=70% of Activated EG3 Tail 38 from compound 36.
Example 4: 50L Solid-phase Synthesis of
Golodirsen [Oligomeric Compound (XII)] Crude Drug Substance
1. Materials
Table 2: Starting Materials

Activated Phosphoramidochloridic acid, 1155373-31-1 C37H37CIN5O5P 698.2 C Subunit N,N-dimethyl-,[6-[4-
(benzoylamino)-2-oxo-l(2H)- pyrimidinyl]-4-
(triphenylmethyl)-2- morpholinyljmethyl ester
Activated Propanoic Acid, 2,2-dimethyl- 1155309-89-9 C5iH53ClN707P 942.2
DPG ,4-[[[9-[6-
Subunit [[[chloro(dimethylamino)phosp
hinyl]oxy]methyl]-4-
(triphenylmethyl)-2- morpholinyl]-2-[(2- phenylacetyl)amino]-9H-purin-
6-yl]oxy]methyl]phenyl ester
Activated Phosphoramidochloridic acid, 1155373-34-4 C3iH34ClN405P 609.1 T Subunit N,N-dimethyl-,[6-(3,4-dihydro- 5-methyl-2,4-dioxo- 1 (2H)- pyrimidinyl)]-4- (triphenylmethyl)-2- morpholinyljmethyl ester
Activated Butanedioic acid, 1- 1380600-06-5 C43H47N3Oio 765.9 EG3 Tail [3aR,4S,7R,7aS)-l,3,3a,4,7,7a- hexahydro- 1 ,3 -dioxo-4,7- methano-2H-isoindol-2-yl] 4- [2-[2-[2-[[[4-(triphenylmethyl)- 1- piperazinyl ] carb onyl ] oxy] ethox
y]ethoxy] ethyl] ester
Golodirsen.
Example 5: Purification of Golodirsen Crude Drug Substance
The deprotection solution from Example 4, part E, containing the Golodirsen crude drug substance was loaded onto a column of ToyoPearl Super-Q 650S anion exchange resin (Tosoh Bioscience) and eluted with a gradient of 0-35% B over 17 column volume (Buffer A: 10 mM sodium hydroxide; Buffer B: 1 M sodium chloride in 10 mM sodium hydroxide) and fractions of acceptable purity (CI 8 and SCX HPLC) were pooled to a purified drug product solution. HPLC: 93.571% (C18; Fig. 3) 88.270% (SCX; Fig. 4).
The purified drug substance solution was desalted and lyophilized to 1450.72 g purified Golodirsen drug substance. Yield 54.56 %; HPLC: 93.531% (Fig. 5; C18) 88.354% (Fig. 6; SCX).
PATENT
WO 2019067979
Duchenne Muscular Dystrophy (DMD) is a serious, progressively debilitating, and ultimately fatal inherited X-linked neuromuscular disease. DMD is caused by mutations in the dystrophin gene characterized by the absence, or near absence, of functional dystrophin protein that disrupt the mRNA reading frame, resulting in a lack of dystrophin, a critically important part of the protein complex that connects the cytoskeletal actin of a muscle fiber to the extracellular matrix. In the absence of dystrophin, patients with DMD follow a predictable disease course. Affected patients, typically boys, develop muscle weakness in the first few years of life, lose the ability to walk during childhood, and usually require respiratory support by their late teens. Loss of functional abilities leads to loss of independence and increasing caregiver burden. Once lost, these abilities cannot be recovered. Despite improvements in the standard of care, such as the use of glucocorticoids, DMD remains an ultimately fatal disease, with patients usually dying of respiratory or cardiac failure in their mid to late 20s.
Progressive loss of muscle tissue and function in DMD is caused by the absence or near absence of functional dystrophin; a protein that plays a vital role in the structure and function of muscle cells. A potential therapeutic approach to the treatment of DMD is suggested by Becker muscular dystrophy (BMD), a milder dystrophinopathy. Both dystrophinopathies are caused by mutations in the DMD gene. In DMD, mutations that disrupt the pre-mRNA reading frame,
referred to as “out-of-frame” mutations, prevent the production of functional dystrophin. In BMD, “in-frame” mutations do not disrupt the reading frame and result in the production of internally shortened, functional dystrophin protein.
An important approach for restoring these “out-of-frame” mutations is to utilize an antisense oligonucleotide to exclude or skip the molecular mutation of the DMD gene
(dystrophin gene). The DMD or dystrophin gene is one of the largest genes in the human body and consists of 79 exons. Antisense oligonucleotides (AONs) have been specifically designed to target specific regions of the pre-mRNA, typically exons to induce the skipping of a mutation of the DMD gene thereby restoring these out-of-frame mutations in-frame to enable the production of internally shortened, yet functional dystrophin protein.
The skipping of exon 53 in the dystrophin gene has been an area of interest for certain research groups due to it being the most prevalent set of mutations in this disease area, representing 8% of all DMD mutations. A prominent AON being developed by Sarepta
Therapeutics, Inc., for DMD patients that are amenable to exon 53 skipping is golodirsen.
Golodirsen is a phosphorodiamidate morpholino oligomer, or PMO. Another AON being developed by Nippon Shinyaku CO., LTD., for DMD patients that are amenable to exon 53 skipping is viltolarsen (NS-065 which is a PMO.
Exondys 51 ® (eteplirsen), is another PMO that was approved in 2016 by the United States Food and Drug Administration (FDA) for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. However, the current standard of care guidelines for the treatment of DMD in patients that are not amenable to exon 51 skipping include the administration of glucocorticoids in conjunction with palliative interventions. While glucocorticoids may delay the loss of ambulation, they do not sufficiently ameliorate symptoms, modify the underlying genetic defect or address the absence of functional dystrophin characteristic of DMD.
Previous studies have tested the efficacy of an antisense oligonucleotides (AON) for exon skipping to generate at least partially functional dystrophin in combination with a steroid for reducing inflammation in a DMD patient (see WO 2009/054725 and van Deutekom, et al., N. Engl. J. Med. 2007; 357:2677-86, the contents of which are hereby incorporated herein by reference for all purposes). However, treatment with steroids can result in serious complications, including compromise of the immune system, reduction in bone strength, and growth
suppression. Notably, none of the previous studies suggest administering an antisense
oligonucleotide for exon skipping with a non-steroidal anti-inflammatory compound to a patient for the treatment of DMD.
Thus, there remains a need for improved methods for treating muscular dystrophy, such as DMD and BMD in patients.
EXAMPLE 1
CAT- 1004 in Combination with M23D PMO Reduces Inflammation and Fibrosis in Mdx Mice.
To assess the effectiveness of a combination treatment of an exon skipping antisense oligonucleotide and an F-Kb inhibitor in Duchenne muscular dystrophy, M23D PMO and
CAT-1004 were utilized in the Mdx mouse model. The effect on inflammation and fibrosis was determined by analyzing samples of muscle taken from the quadriceps, of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT-1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT-1004. The tissue sections were analyzed for fibrosis by picrosirius red staining and for inflammation and fibrosis by Hematoxylin and Eosin (H&E) staining, as described in the Materials and Methods section above.
Treatment of Mdx mice with either M23D PMO or CAT-1004 as monotherapies resulted in a reduction of inflammation and fibrosis as compared to Mdx mice treated with saline.
Surprisingly, treatment of Mdx mice with the M23D PMO in combination with CAT-1004 resulted in reduced inflammation and fibrosis as compared with mice treated with CAT-1004
alone or M23D alone (Fig. 9). These results indicate the combination treatment enhances muscle fiber integrity.
EXAMPLE 2
Exon Skipping and Dystrophin Production in Mdx Mice Treated with the M23D
PMO and the M23D PMO in Combination with CAT- 1004
To analyze the extent of exon skipping and dystrophin production in mice treated with the M23D PMO in combination with CAT- 1004, samples of muscle were taken from the quadriceps, diaphragm, and heart of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT- 1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT- 1004. RT-PCR analysis for exon 23 skipping was performed as well as Western blot analysis to determine dystrophin protein levels.
Exon skipping was observed in the muscle of the quadriceps, diaphragm, and heart of the Mdx mice treated with the M23D PMO as well as mice treated with the M23D PMO in combination with CAT-1004 (Fig. 10). Surprisingly, enhanced dystrophin production was observed in the muscle of the quadriceps, diaphragm, and heart of the mice treated with the M23D PMO in combination with CAT-1004 as compared to treatment with M23D PMO monotherapy (Fig. 11). These results indicated the increase in dystrophin levels extended to the heart, a tissue known to have low efficiency of dystrophin upregulation by these agents when used alone. Notably, neither exon skipping nor dystrophin production were observed in mdx mice treated with CAT-1004 monotherapy (Figs. 10 and 11).
PATENT
WO 2019046755
PAPER
Methods in Molecular Biology (New York, NY, United States) (2018), 1828(Exon Skipping and Inclusion Therapies), 31-55.
PAPER
Human Molecular Genetics (2018), 27(R2), R163-R172.
///////////Golodirsen, ゴロジルセン , FDA 2019, ANTISENSE, Exon 53: NG-12-0163, SRP 4053, OLIGONUCLEOTIDE, Duchenne Muscular Dystrophy
SYN 01

SYN-01, SYN-510
Synthena AG
Preclinical
Synthena , presumed to be under license from University of Bern , is investigating (presumably SYN-01 ), a lead from the tricyclo(tc)-DNA based antisense oligonucleotides (AON) developed using its proprietary tricyclo-DNA technology platform, for the treatment of Duchenne muscular dystrophy. In January 2017, the drug was listed as being in preclinical development.
Patent
WO-2019142135
Process for preparing tricyclo-deoxyribonucleic acid (tc-DNA) which may be used as building blocks for tc-DNA containing antisense oligonucleotide-based therapies.
Antisense technology is an effective means for reducing the expression of specific gene products and can therefore be useful in therapeutic, diagnostic, and research applications.
Generally, the principle behind antisense technology is that an antisense oligomeric compound (a sequence of nucleotides or analogues thereof) hybridizes to a target nucleic acid and modulates gene expression activities or function, such as transcription and/or translation.
[003] Antisense oligomeric compounds may be prepared from chemically-modified antisense oligonucleotides, which may include a variety of different structural variations depending upon the therapeutic strategy. For example, tricyclo-deoxyribonucleic acids (tc-DNA) are conformationally constrained DNA analogs.
[004] There is a need in the field for processes that allow for the bulk preparation of tc-DNA nucleoside precursors that may be used as building blocks for tc-DNA containing antisense oligonucleotide-based therapies.
Example 4 – Cvclopropanation of Compound 17 with Carbenoid Prepared from CH2I2 and Et2Zn in the Absence of Additives
[00127] According to the following scheme, compound 17 was converted to tc-DNA Nucleoside Precursor 18 using the cyclopropanation conditions set forth in Examples 4 to 7 :
[00128] 1.07 g purified a-anomer (3.736 mmol) 17 was dissolved in 37 ml of dry CH2C12 and cooled to 0 °C (ice). Subsequently, 22.3 ml (22.3 mmol, 6 eq.) Et2Zn 1.0 M in hexane (Aldrich) were added dropwise and stirred under Ar for 30 min at 0 °C. Then, 3.02 ml (37.2 mmol, 10 eq.) of CH2I2 were added dropwise over 15 min at the same temperature and stirred for further 2 h at 0 °C. Afterwards the cooling bath was removed and the mixture was stirred for additional 21 h at ambient temperature. TLC showed substantial amount of unreacted a- 17. It was diluted by addition of EtOAc and quenched with 50 mL of sat. aqueous NH4Cl. Extractive work-up provided 1.79 g of crude which was purified by chromatography on silica-gel giving 0.43 g (39%) of 18 and 0.49 g of mixture of compound 17 and 18 (approximately 20:80).
PATENT
WO2018193428
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018193428
claiming a composition comprising an oligomeric compound having tricyclo-deoxyribonucleic acid (tc-DNA) nucleosides and a lipid moiety.
EXAMPLE 1
Inventive compositions for the treatment of Duchenne muscular dystrophy
Evaluation of efficacy
[00464] Adult mdx mice were treated weekly over 4 weeks with intravenous injections of different 13-mer AONs targeting the donor splice site of exon 23 of the dystrophin pre-mRNA (M23D: +2-11), namely with either SY-0308, SY-0210 and the inventive SY-0299, SY-0343, SY-0442 and SY-0455. SY-0308 (also named “tcDNA-PO M23D” interchangeably herein) corresponds to p-CCTCGGCTTACCT-OH of SEQ ID NO: l, with all nucleotides being tc-DNAs and all internucleosidic linkage groups being phosphorodiester linkage groups, and p being a phosphate moiety at the 5′ end. SY-0210 (also named “tcDNA-PS M23D” interchangeably herein) corresponds to p-CCTCGGCTTACCT-OH of SEQ ID NO: 1, with all nucleotides being tc-DNAs and all internucleosidic linkage groups being phosphorothioate linkage groups, and p being a phosphate moiety at the 5′ end. The inventive composition SY-0343 is herein interchangeably referred to as “Palm-2PS-tcDNA-PO M23D” which is depicted in the following:
[00465] The inventive composition SY-0442 is herein interchangeably referred to as “Palm-lPS-tcDNA-PO M23D” which is depicted in the following:
[00466] The inventive composition SY-0299 is herein interchangeably referred to as “Palm-2PO-tcDNA-PO M23D” which is depicted in the following:
//////////////////SYN-01, SYN 01, SYN01, preclinical , Duchenne muscular dystrophy, University of Bern,
FDA approves drug to treat Duchenne muscular dystrophy
FDA approves drug to treat Duchenne muscular dystrophy
Feb. 9, 2017
The U.S. Food and Drug Administration today approved Emflaza (deflazacort) tablets and oral suspension to treat patients age 5 years and older with Duchenne muscular dystrophy (DMD), a rare genetic disorder that causes progressive muscle deterioration and weakness. Emflaza is a corticosteroid that works by decreasing inflammation and reducing the activity of the immune system.

FDA grants accelerated approval to first drug for Duchenne muscular dystrophy


Structure credit http://lgmpharma.com/eteplirsen-still-proves-efficacious-duchenne-drug/
FDA grants accelerated approval to first drug for Duchenne muscular dystrophy
New therapy addresses unmet medical need
The U.S. Food and Drug Administration today approved Exondys 51 (eteplirsen) injection, the first drug approved to treat patients with Duchenne muscular dystrophy (DMD). Exondys 51 is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13 percent of the population with DMD.
FDA grants accelerated approval to first drug for Duchenne muscular dystrophy
September 19, 2016
Release
The U.S. Food and Drug Administration today approved Exondys 51 (eteplirsen) injection, the first drug approved to treat patients with Duchenne muscular dystrophy (DMD). Exondys 51 is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13 percent of the population with DMD.
“Patients with a particular type of Duchenne muscular dystrophy will now have access to an approved treatment for this rare and devastating disease,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “In rare diseases, new drug development is especially challenging due to the small numbers of people affected by each disease and the lack of medical understanding of many disorders. Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial that the company must conduct after approval.”
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness. It is the most common type of muscular dystrophy. DMD is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between three and five years of age, and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about one out of every 3,600 male infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary.
Exondys 51 was approved under the accelerated approval pathway, which provides for the approval of drugs that treat serious or life-threatening diseases and generally provide a meaningful advantage over existing treatments. Approval under this pathway can be based on adequate and well-controlled studies showing the drug has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit to patients (how a patient feels or functions or whether they survive). This pathway provides earlier patient access to promising new drugs while the company conducts clinical trials to verify the predicted clinical benefit.
The accelerated approval of Exondys 51 is based on the surrogate endpoint of dystrophin increase in skeletal muscle observed in some Exondys 51-treated patients. The FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in some patients with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping. A clinical benefit of Exondys 51, including improved motor function, has not been established. In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease for these children and the lack of available therapy.
Under the accelerated approval provisions, the FDA is requiring Sarepta Therapeutics to conduct a clinical trial to confirm the drug’s clinical benefit. The required study is designed to assess whether Exondys 51 improves motor function of DMD patients with a confirmed mutation of the dystrophin gene amenable to exon 51 skipping. If the trial fails to verify clinical benefit, the FDA may initiate proceedings to withdraw approval of the drug.
The most common side effects reported by participants taking Exondys 51 in the clinical trials were balance disorder and vomiting.
The FDA granted Exondys 51 fast track designation, which is a designation to facilitate the development and expedite the review of drugs that are intended to treat serious conditions and that demonstrate the potential to address an unmet medical need. It was also granted priority review and orphan drug designation.Priority review status is granted to applications for drugs that, if approved, would be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan drug designation provides incentives such as clinical trial tax credits, user fee waiver and eligibility for orphan drug exclusivity to assist and encourage the development of drugs for rare diseases.
The manufacturer received a rare pediatric disease priority review voucher, which comes from a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. This is the seventh rare pediatric disease priority review voucher issued by the FDA since the program began.
Exondys 51 is made by Sarepta Therapeutics of Cambridge, Massachusetts.

| Systematic (IUPAC) name | |
|---|---|
|
(P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(C-m5U-C-C-A-A-C-A-m5U-C-A-A-G-G-A-A-G-A-m5U-G-G-C-A-m5U-m5U-m5U-C-m5U-A-G),5′-(P-(4-((2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)carbonyl)-1-piperazinyl)-N,N-dimethylphosphonamidate) RNA
|
|
| Clinical data | |
| Routes of administration |
Intravenous infusion |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1173755-55-9 |
| ATC code | None |
| ChemSpider | 34983391 |
| UNII | AIW6036FAS |
| Chemical data | |
| Formula | C364H569N177O122P30 |
| Molar mass | 10305.738 |
///////////Exondys 51, Sarepta Therapeutics, Cambridge, Massachusetts, eteplirsen, Orphan drug designation, Priority review, fast track designation, Duchenne muscular dystrophy, этеплирсен , إيتيبليرسان ,
Ataluren (Translarna) drug for Duchenne Muscular Dystrophy

Ataluren (Translarna)
3-(5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl)benzoic acid
3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
CAS 775304-57-9
PTC Therapeutics (Originator)
- Molecular FormulaC15H9FN2O3
- Average mass284.242 Da
- EC-000.2051
NCGC00168759-02PTC-124, PTC124,UNII:K16AME9I3V
- EU 2014-07-31 APPROVED

Ataluren, formerly known as PTC124, is a pharmaceutical drug for the treatment of Duchenne muscular dystrophy and potentially other genetic disorders. It was designed by PTC Therapeutics and is sold under the trade name Translarna in the European Union.
Ataluren was approved by European Medicine Agency (EMA) on July 31, 2014. It was developed and marketed as Translarna® by PTC Therapeutics.

Ataluren was regulator of nonsense mutations indicated for the treatment of Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene, in ambulatory patients aged 5 years and older.
Translarna® is available as granules for oral use, containing 125 mg, 250 mg or 1000 mg of free Ataluren. The recommended dose is 10 mg/kg body weight in the morning, 10 mg/kg body weight at midday, and 20 mg/kg body weight in the evening.
Medical uses
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[1][2] such as some people with cystic fibrosis and Duchenne muscular dystrophy. It is approved for the use in Duchenne in the European Union.

Mechanism of action
Ataluren makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. Studies have demonstrated that PTC124 treatment increases expression of full-length dystrophin protein in human and mouse primary muscle cells containing the premature stop codon mutation for Duchenne muscular dystrophy and rescues striated muscle function.[3] Studies in mice with the premature stop codon mutation for cystic fibrosis demonstrated increased CFTR protein production and function.[4] The European Medicines Agency review on the approval of ataluren concluded that “the non-clinical data available were considered sufficient to support the proposed mechanism of action and to alleviate earlier concerns on the selectivity of ataluren for premature stop codons.” [5]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[6] Ataluren appears to be most effective for the stop codon ‘UGA’.[1]

History
Clinical trials
In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[7] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug.
Phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[8] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[9]
Approval
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[10]Translarna was first available in Germany, the first EU country to launch the new medicine.[11]
In August 2014, ataluren received market authorization from the European Commission to treat patients with nonsense mutation Duchenne muscular dystrophy. A confirmatory phase III clinical trial is ongoing.[11] The drug does not yet have approval by the US Food and Drug Administration.
In October 2015, NICE asked for further evidence of benefit to justify the “very high cost”.[12] NICE estimated that for a typical patient, treatment would cost £220,256 per year.
In February 2016, FDA declined to approve or even discuss PTC Therapeutics application for ataluren because it deemed the data presented by the developer “insufficient to warrant a review”.[13]

Ataluren Molecule
PAPER
http://www.pnas.org/content/106/9/3585.full
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental/Appendix_PDF.pdf
Samples were analyzed for purity on an Agilent 1200 series LC/MS equipped with a Luna® C18 reverse phase (3 micron, 3 x 75 mm) column having a flow rate of 0.8-1.0 mL/min. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and temperature was maintained at 50 °C. A gradient of 4% to 100% acetonitrile over 7 minutes was used during analytical analysis. Purity of final compounds was determined to be >95%, using a 5 μL injection with quantitation by AUC at 220 and 254 nM. High resolution mass spectra were obtained with an Agilent 6210 Time-of-Flight LC/MS with a 3.5 um Zorbax SB-C18 column (2.1 x 30 mm) (solvents are Water and ACN with 0.1% Formic Acid). A 3 minute gradient at 1 mL/min from 5% to 100% acetonitrile was used.
3-[5-(2-fluorophenyl)-[1,2,4]-oxadiazol-3-yl]-benzoic acid (1a, PTC124).
1 H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH = 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).
13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz). LC-
MS: rt (min) = 5.713; [M+H]+ 285.1;
HRMS: (CI+, m/z), calcd for C15H10FN2O3 (MH+ ), 285.06814; found, 285.06769.
CLIP
Ataluren (Translarna) Ataluren is a drug marketed under the trade name Translarna which was developed by PTC Therapeutics and approved by the European Union in May 2014 for the treatment of Duchenne’s muscular dystrophy (DMD) and potentially other genetic disorders.50
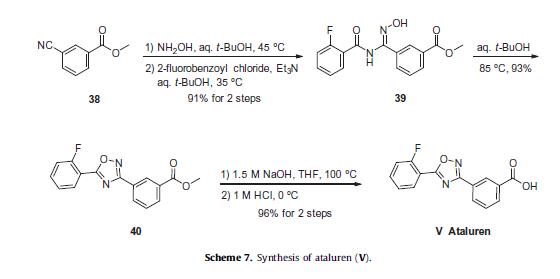
Ataluren renders ribosomes less sensitive to premature stop or ‘read-through’ codons, which are thought to be beneficial in diseases such as DMD and cystic fibrosis.51 Of the reported synthetic approaches to ataluren,52–55 the most likely process-scale approach consists of the sequence described in Scheme 7, which reportedly has been exemplified on kilogram scale.56
The sequence to construct ataluren, which was described by the authors at PTC Therapeutics, commenced with commercially available methyl 3-cyanobenzoate (38).56 This ester was exposed to hydroxylamine in aqueous tert-butanol and warmed gently until the reaction was deemed complete.
Then this mixture was treated with 2-fluorobenzoyl chloride dropwise and subsequently triethylamine dropwise. To minimize exotherm and undesired side products, careful control of the addition of reagents was achieved through slow dropwise addition of these liquid reagents.
Upon complete consumption of starting materials and formation of amidooxime 39, the aqueous reaction mixture was then heated to 85 C to facilitate 1,2,4-oxadiazole formation, resulting in the tricyclic ester 40 in excellent yield across the three steps.
Finally,saponification of ester 40 through the use of sodium hydroxide followed by acidic quench gave ataluren (V) in 96% over the two-step sequence.57
50. Welch, E. M.; Barton, E. R.; Zhuo, J.; Tomizawa, Y.; Friesen, W. J.; Trifillis, P.;Paushkin, S.; Patel, M.; Trotta, C. R.; Hwang, S.; Wilde, R. G.; Karp, G.; Takasugi,J.; Chen, G.; Jones, S.; Ren, H.; Moon, Y. C.; Corson, D.; Turpoff, A. A.; Campbell,J. A.; Conn, M. M.; Khan, A.; Almstead, N. G.; Hedrick, J.; Mollin, A.; Risher, N.;Weetall, M.; Yeh, S.; Branstrom, A. A.; Colacino, J. M.; Babiak, J.; Ju, W. D.;Hirawat, S.; Northcutt, V. J.; Miller, L. L.; Spatrick, P.; He, F.; Kawana, M.; Feng,H.; Jacobson, A.; Peltz, S. W.; Sweeney, H. L. Nature 2007, 447, 87.
51. Hirawat, S.; Welch, E. M.; Elfring, G. L.; Northcutt, V. J.; Paushkin, S.; Hwang,S.; Leonard, E. M.; Almstead, N. G.; Ju, W.; Peltz, S. W.; Miller, L. L. J. Clin.Pharmacol. 2007, 47, 430.
52Karp, G. M.; Hwang, S.; Chen, G.; Almstead, N. G. US Patent 2004204461A1,2004.
53. Andersen, T. L.; Caneschi, W.; Ayoub, A.; Lindhardt, A. T.; Couri, M. R. C.;Skrydstrup, T. Adv. Synth. Catal. 2014, 356, 3074.
54. Gupta, P. K.; Hussain, M. K.; Asad, M.; Kant, R.; Mahar, R.; Shukla, S. K.; Hajela,K. New J. Chem. 2014, 38, 3062.
55. Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; PalumboPiccionello, A.; Pibiri, I. Mol. Pharm. 2014, 11, 653.
56. Almstead, N. G.; Hwang, P. S.; Pines, S.; Moon, Y. -C.; Takasugi, J. J. WO Patent2008030570A1, 2008.
57. Almstead, N. G.; Chen, G.; Hirawat, S.; Hwang, S.; Karp, G. M.; Miller, L.; Moon,Y. C.; Ren, H.; Takasugi, J. J.; Welch, E. M.; Wilde, R. G. WO Patent2007117438A2, 2007.
CLIP

Ataluren trial success: trial aborted.
07 September 2011 – Pharma……..http://chem.vander-lingen.nl/info/item/September_2011/id/190/mid/140
Last week the newspaper NRC Handelsblad reported on a court case in which the parents of two young boys sued a pharmaceutical company over access to one of their developmental drugs. The drug in question wasAtaluren, the pharmaceutical companyPTC Therapeutics. The boys suffer from Duchenne muscular dystrophyand had taken part in a clinical trial. Whereas the results of this trial on the whole were inconclusive the boys did seriously benefit from the drug. Hardly any wonder the parents took action when the whole development program was canceled.
And the judge? He threw the case out arguing that doctors do not make the compound themselves and arguing that the compound is not commercially available. Are these arguments valid? and do the boys have options?
It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).
Except for the fluorine atom in it the compound is unremarkable. If you have to believe the Internet many Chinese companies produce and sell it. Ataluren is also still in the running as a potential treatment for some other diseases. So if need be the compound will be around for some time to come.
CLIP
Ataluren [3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid] is an orally available, small molecule compound that targets nonsense mutation. It is the first drug in its class and appears to allow cellular machinery to read through premature stop codons in mRNA, and thus enables the translation process to produce full-length, functional proteins.
Ataluren is developed and approved for the treatment of nonsense mutation Duchenne muscular dystrophy (nmDMD) by EU in July 2014 [1].

| Ataluren: 2D and 3D Structure |
Nonsense Mutations as Target for DMD
A single nucleotide change in the DNA sequence that introduces a premature stop codon is known as a nonsense mutation, a subset of a major class of premature termination codon (PTC) mutations. Nonsense mutations cause premature termination of translation resulting in the production of truncated polypeptides, which in turn halts the ribosomal translation process at an earlier site than normal, producing a truncated, non-functional protein [1].
Nonsense mutations are implicated in 5-70 % of individual cases of most inherited diseases, including Duchenne muscular dystrophy (DMD) and cystic fibrosis. Ataluren appears to allow cellular machinery to read through premature stop codons in mRNA, enabling the translation process to produce full length, functional proteins.
Ataluren Synthesis

New J Chem 2014, 38, 3062-3070: The text reports one pot synthesis of Ataluren with an overall yield of 40%. It also reports few interesting and potent derivatives too.

WO 2007117438A2: It appears to be the industrial process. The patent also reports various pharmaceutically relevant assay and their results wrt Ataluren.
Identifications:

| 1H NMR (Estimated) for Ataluren |
Experimental: 1H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH= 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).
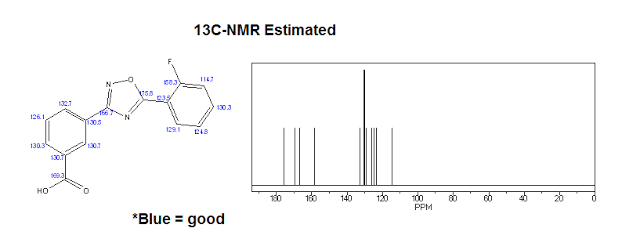
| 13C-NMR (Estimated) for Ataluren |
Experimental: 13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz)……https://ayurajan.blogspot.in/2016/05/ataluren-treatment-for-duchenne.html
CLIP


Reference:1. WO2004091502A2 / US6992096B2.
2. WO2008045566A1 / US2008114039A1.
3. WO2008030570A1 / US2008139818A1.
Reference:1. New. J. Chem. 2014, 38, 3062-3070.

Reference:1. Adv. Synth. Catal. 2014, 356, 3074-3082.
CLIP
Carcinogenicity
Carcinogenicity bioassays in transgenic mice (26 weeks) and in rats (24 months):
● For Tg.rasH2 mouse: Ataluren did not increase the incidence of tumors up to the HDs in males (600 mg/kg/day) and in females (300 mg/kg/day). The non-neoplastic findings included endometrial hyperplasia and nephropathy in females.
● For rats: Urinary bladder tumors (benign urothelial cell papilloma [2 rats] and malignant urothelial cell carcinoma [1 rat]) were observed in 3/60 female rats dosed at 300 mg/kg/day. In addition, one case of malignant hibernoma was observed in 1/60 male rats at the dose of 300 mg/kg/day. The non-neoplastic toxicity consisted of a decrease of body weight.
Example 1 (prepared by known ataluren)
Method ataluren according to Patent Document 2 is described in Example W02004091502A2 prepared.
Specific methods of preparation:
To a solution of 0.6 l of DMF was 44. 14g3- cyano acid 62.19 g of potassium carbonate was added, followed by stirring at room temperature for 30 minutes. 20 minutes To the suspension was added 28 ml of methyl iodide (450mmol), and the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was poured into 1.2 l of ice water, stirred for 30 minutes, the precipitate was filtered out thereof. The white cake was dissolved in 70 ml of methanol, and then reprecipitated in cold water. To give 79% yield of 3-cyano-benzoic acid methyl ester.
50 g of 3-cyano-benzoic acid methyl ester was dissolved in 500 ml of ethanol, to which was added 41 ml of 50% aqueous hydroxylamine (620mmol). 100 ° C and the reaction mixture was stirred for 1 hour, the solvent was removed under reduced pressure. So that the oily residue is dissolved in 100 ml of 20/80 ethanol / toluene, concentrated again. To give 61 g 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester.
60 g of 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester was dissolved in 200 ml of anhydrous tetrahydrofuran, followed by adding thereto 75 ml of diisopropylethylamine (434 mmol), and then 20 minutes this mixture was added 48.1 ml 2- fluorobenzoyl chloride (403mmol). The reaction mixture was stirred at room temperature for 1 hour. The precipitate was filtered off, the filtrate was concentrated under reduced pressure. The residue was dissolved in 400 ml of ethyl acetate, washed with 400 ml of water and then twice. The solvent was removed under reduced pressure, containing 60% ethyl acetate in hexane to give the desired product, generating 81 g 3- (N-2- amidino-fluorobenzoyl) – benzoate.
at 130 ° C with a Dean-Stark apparatus was dissolved in 500 ml of toluene was heated under reflux in 44 g of 3- (N-2- fluorobenzoyl) -1,2,3,4-_ benzoate 4 hours. 5 ° C and the reaction mixture was stirred for 18 hours. The white precipitate was filtered off, the filtrate was concentrated, recrystallized in toluene. To give 38 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester.
33 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester was dissolved in 400 ml of tetrahydrofuran, to which was added 100 ml of 1. 5M aqueous sodium hydroxide solution. At 100 ° C and the reaction mixture was heated at reflux for 2 hours. The solvent was removed under reduced pressure at 5 ° C the solution was stirred for 2 hours. The organic solvent was removed, washed with 50 mL of water. The aqueous solution was then acidified with hydrochloric acid to pH 1. The white precipitate was filtered off, the filter cake washed with cold water, then dried with a freeze dryer. To give 3.0 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] benzoic acid. 1H-NMR (500MHz, d6-DMS0): 8. 31 (1H), 8 18 (2H), 8 08 (1H), 7 88 (2H), 7 51 (2H)….. Display: ataluren- Sample Preparation Example 1 prepared in Preparation Example 2 and TO2004091502A2 induced.
Each prepared in Example 2 (prepared according to known Form A)
Method [0084] A known polymorph according to Patent Document W02008039431A2 Example 5. 1. 1.1 prepared as described. Specifically: ataluren be prepared 1 100 mg Preparation Example, 60 ° C add 16.2 ml of isopropanol ultrasound clear solution, the solution by 2 square micron filter and the filtrate was kept covered with aluminum foil having a small hole. vial, 60 ° C and evaporated. The solid formed was isolated to give ataluren the A polymorph.
as needles.
its XRPD shown in Figure 1, the display ataluren polymorph A disclosed in Patent Document W02008039431A2 consistent.
novel crystalline forms of 3-[5-(2-fluorophenyl)-
[l,2,4]oxadiazol-3-yl]-benzoic acid, which has the following chemical structure (I):

(I)
In particular, crystalline forms of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are useful for the treatment, prevention or management of diseases ameliorated by modulation of premature translation termination or nonsense-mediated mRNA decay, as described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, which is incorporated herein by reference in its entirety. In addition, the present provides a crystalline form of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]-benzoic acid which is substantially pure, i.e., its purity greater than about 90%.
Processes for the preparation of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, and U.S. patent application no. 1 1/899,813, filed September 9, 2007, both of which are incorporated by reference in their entirety.
PATENT
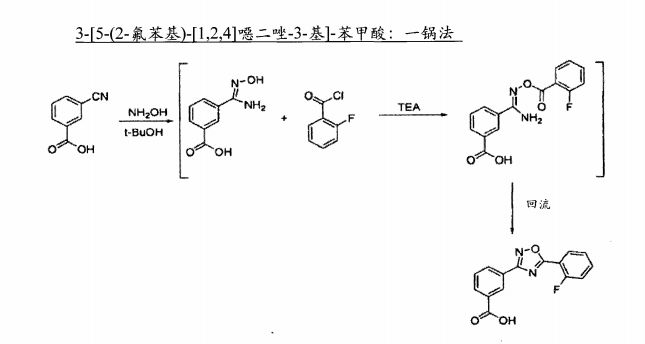
Example
3- ‘5- (2-fluorophenyl) – “1,2,41 oxadiazol-3-yl benzoate 1- Batch 1
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 2 hours 48 minutes, 50. /. Aqueous hydroxylamine (43L, 47.4 kg) was added to a clear solution of 3-cyano benzoic acid methyl ester in a molten in t-butanol. The addition of a 50% aqueous solution of hydroxylamine period, the maximum temperature batch of about 43 ° C. 50% aqueous solution of hydroxylamine addition rate of from about 9L / h when the changes start adding to about 30L / hr. To maintain the temperature of the batch by varying the reactor jacket set point. In particular, the set value is about 40.5 ° C, with the addition of a rate increase at the beginning join, change the setting to about 29.6 ° C. After about 40-45t stirred for about 4 hours, the reaction was deemed complete (i.e., less than about 0.5% ester).
The batch was transferred to a drying reactor, additional (chased through) approximately 10L molten tert-butanol. Jacket setpoint from about 33 when the batch was received when dried reactor. C is reduced to about 27 after the completion of the transfer. C. Batch crystallization was observed part, which does not adversely affect stirring. The batch was cooled to about 34.4 ° C, triethylamine (72.6 kg, IOOL) added to the reactor. The jacket temperature set value from about 20.4. C is increased to about 31.0 ° C, in order to maintain the batch temperature in the range of about 30-35t. With molten tert-butanol (IO L) was washed with a linear (line rinse) After the batch was added to the 2-fluorobenzoyl chloride (113.7 kg, 86.0L).Charge is added to the first third of the rate of about 25L / hr. In the meantime, the jacket inlet temperature was lowered to about 15 ° C, the batch temperature is maintained at about 34.6 ° C. In about 5.5 hours after the addition was complete.During the addition, the maximum temperature of the batch was about 38.8 ° C. Near the end of the addition, the addition rate slowed to about 11L / hr was added last 27 liters of 2-fluoro-benzoyl chloride. 30-35. C After stirring for about 2 hours, that the reaction was complete (i.e., less than about 0.5% of methyl 3-amidinophenoxy). Then, after about 1 hour 42 minutes, the batch was heated to reflux temperature (about 82 ° C), and then stirred for about 18 hours. During the stirring, a number of product partially crystallized to form a slurry. The slurry was cooled to about 40. C thus sampled, during which complete crystallization occurs. The batch was then heated to reflux temperature and stirred for about 1 hour 50 minutes.Then, after about two hours, the batch was cooled to about 69 ° C, and after about four hours and 15 minutes, slowly added 630L of pure water, while maintaining the batch temperature at about 66-69 ° C. After about 3 hours 14 minutes, the slurry was cooled to about 22.4 ° C, and transferred to 2x200L ceramic filter, the ceramic filter equipped 25-30n polypropylene mesh filter cloth. In about 55 minutes after the completion of material from the container to the filter transfer. With 50n /. The tert-butanol solution (210L) was washed cake was washed for about 10 minutes so that the cleaning liquid can penetrate into each cake. Then, the cake was dried in a vacuum for about 5-10 minutes. The purified water as a second washing (158L / cake) applied to the filter cake to remove residual t-butanol and triethylammonium chloride salt. Dried in a vacuum for about 5 minutes, the solution was removed. In vacuo and then the cake was dried for about 2 hours, and then sampled using liquid chromatography. The filter cake was measured by liquid chromatography purity of about 99.6%.
The filter cake was dried in vacuo for about 8 hours 25 minutes later, the wet cake (207.4kg) is transferred to an air oven. At about 50-55. C, the oven dried in air for about 52 hours. The product was isolated in a total yield of about 89.9% (174.65kg), in the calculation of cost of materials sampling, you can adjust the overall yield of about 90.7%.
Batch 2
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 3 hours 29 minutes, 50% aqueous solution of hydroxylamine (47.85 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. At about 40-45. C After stirring for about 3 hours 16 minutes, that the reaction was complete (i.e., less than about 0.5% ester). As for the drying reactor, the batch was transferred to one of the batch in. The batch was cooled
To about 34.4 ° C, and triethylamine (72.6 kg, 100 L). During about 45 minutes was added, while maintaining the batch temperature between about 30-35 ° C. During the addition, the jacket inlet temperature of from about 31.4. C increased to about 32.6. C. After the molten tert-butanol linear washed, was added to the batch 2- fluorobenzoyl chloride (l 13.7 kg, 86.0 L). After about 3 hours, 27 minutes, add the acid chloride. 35. C under stirring for about 8 hours, that the reaction is not complete (i.e., more than about 0.5% residual 3-amidino-benzoyl ester). Then, 1.5% by weight of the original charge of triethylamine and 2-fluorobenzoyl chloride was added to the batch. Linear washed with tert-butanol (IO L) associated with each additional charge. During the addition of the acid chloride, no additional cooling. The batch was maintained at a temperature of about 30-35 ° C, the jacket inlet temperature range was maintained at about 30.3. C to about 33.0 ° C. After stirring for about 2 hours at 30-35t, that reaction was complete (i.e., less than 0.5% of methyl 3-amidinophenoxy).
After about 1 hour and 44 minutes, the batch was heated to reflux temperature (about 83 ° C), and stirred for about 18 hours.The same batch 1, during cooling the sample, the solid was completely crystallized. The batch was then heated to reflux temperature and stirred for about 1 hour and 2 minutes. Then, after about 2 hours and 20 minutes, the batch was cooled to about 69.2 ° C, and after about four hours and 30 minutes, slowly added 630 L of pure water, while the temperature of the batch was maintained at about 65.6-69.2 ° C. After about 3 hours and 30 minutes, the slurry was cooled to about 23.4 ° C, and, as for, the contents were transferred to one of the double batch of the ceramic filter. About 5 hours and 6 minutes, to complete the transfer of the material. With about 50% of t-butanol (2 volumes / cake) was washed filter cake was washed with 10 minutes to allow the cleaning liquid to penetrate into each cake, then dried in vacuo. About 1 hour and 40 minutes, the filter is completed. The purified water was added to a final wash the filter cake. The liquid was removed by drying under vacuum for about 10 minutes. In vacuo and then the cake was dried for about 2 hours and 5 minutes, and then sampled using liquid chromatography. The cake purity liquid chromatography were about 99.5% and 99.6%. After the cake was then dried in vacuo for about 2 hours and 5 minutes, the wet cake (191.5 kg) is transferred to an air oven. At about 50-55. C under dry in an air oven for about 48 hours. The product was isolated in a total yield of about 92.5% (179.7 kg).
Lot 3
The 3-cyano-benzoic acid methyl ester (52.5 kg) and molten tert-butanol (228 kg) added to the reaction vessel. The vessel was sealed, the batch temperature set of about 40-45 ° C, and the stirrer is started. Under an inert atmosphere, after 2 hours 40 minutes, 50% of the shoes amine solution (24 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. In about 42. Under C, then further stirred for about 5 hours to complete the reaction.
The batch was cooled to 30-35 ° C, and after 15 minutes, was added triethylamine (36 kg). After about 2 hours 44 minutes, was added 2-fluorobenzoyl chloride (57 kg). During the addition, batch temperature was maintained at about 30-35 ° C.Under the 32t, the batch was stirred for 2 hours 10 minutes to complete the reaction.
After about 50 minutes, the batch was heated to reflux temperature (about 83-86 ° C), at about 8rc, stirred for about 18 hours. Then, over about two hours, the batch was cooled to about 65-70 ° C, and after about 6 hours 25 minutes, slowly added to purified water (315 L), while the batch temperature was maintained at about 65- 70 ° C. After about 2 hours and 15 minutes, the slurry was cooled to about 22 ° C, and the contents were transferred to a centrifuge filter (2 batches). About 1 hour and 40 minutes, the filter is completed. After about 20 minutes, with about 50% aqueous solution of tert-butyl alcohol (90 kg / cake), dried cake. The purified water (79 kg / cake) as the last added to the filter cake washed. At about 900 rpm drying the cake for about 1 hour and 5 minutes, then filled cylinder. Liquid chromatography wet cake (91.5 kg, LOD = 5% w / w) of a purity of about 99.75% area.
3- ‘5- (2-fluorophenyl) -fl, 2,41 oxadiazol-3-yl l- acid batch 1
3- [5- (2-fluorophenyl) – [l, 2,4] oxadiazol-3-yl] – benzoic acid methyl ester (74.0kg) added to the reaction vessel, the vessel is sealed, evacuated and purification. Jacket set value of about 35. C, start the stirrer in the container. Molten tert-butanol (222 L, 3 volumes) and purified water (355 L, 4.8 vol) was added to the vessel. After the addition was added 25.1% w / w aqueous sodium hydroxide solution (43.5 kg, 1.1 molar equivalents), and with additional purified water (100L, 1.35 mol) was washed linear. During the addition, the batch temperature from about 39.0t reduced to about 38.8 ° C. After about 1 hour and 54 minutes, the batch temperature to about 63-67. C, and then, after about 30 minutes, which was adjusted to about 68-72.C. About 68-72t, stirring the mixture for about 3 hours. Then, after about five hours 11 minutes, the solution was cooled to about 40-45 ° C. Then, after the above process, after about three hours 33 minutes, the solution was then heated to about 68-72. C.
Jacket temperature of the reaction vessel was set to about 60 ° C, the stirrer started, and at about 70 ° C, a slightly positive pressure of nitrogen (1.5 to 5.6 psig), the heat transfer liquid through a micron filter . During the transfer, the product temperature is reduced to about 64.3 ° C, the transfer is completed in about 45 minutes. Was added to the purified water container (61 L, 0.82 vol) and the contents were heated to about 68-72. C.
The batch temperature was adjusted to about 69.4 ° C, and after about four hours and 18 minutes, with 13.9% w / w sulfuric acid (100.7 kg, 1.15 mol equiv.). During the addition, batch temperature was maintained at about 68.0-70.8 ° C. After the addition of the acid, with purified water (50 L, 0.68 vol) line wash at about 68-72 ° C, the stirring was continued for 31 minutes.
After about 4 hours and 10 minutes, the batch in a linear fashion from about 69.2t cooled to about 41.2 ° C. The stirrer Rosenmund filter / dryer was elevated to the highest position and jacket set value is set at about 40 ° C. The slurry was transferred to the two portions of the filter / drier. Applying a constant nitrogen pressure to the first portion (less than about 15 psig). During the transfer, a pressure of about 23.9 to about 28.8 psi, the transfer is complete in about 1 hour and 5 minutes. The second part of the slurry was transferred onto the filter cake, and the composite was stirred briefly to homogenize the batch. Use about 26.1 to about 29.1 psi nitrogen pressure filters the second part, after about three hours, squeeze the cake so that it does not contain liquid. With about 38-42 ° C hot tert-butanol solution (352 kg, 5 volumes) and about 65-70 ° C in 3x hot purified water (370 L, 5 volumes) and the filter 々.
Said filter / dryer jacket temperature was set to about 43 ° C, the product was dried under vacuum for about 26 hours while stirring periodically. Determination of purity of about 99.7%. The product was isolated in a total yield of about 74.4% (52.45 kg).
Batch 2
Was added to the reactor vessel 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (47 kg, wet cake) and melt-hyun tert-butyl alcohol (111.4 kg). A sealed container, and the batch temperature was set at 30-40t, and start the stirrer. The purified water (51.6 kg) was added to the vessel. After the addition was added 3.47% w / w aqueous sodium hydroxide solution (202.4 kg). After about l hour, the batch temperature to about 67-73. C, then, at about 7 (under TC, stirred for about three hours.
Under a slight positive pressure of nitrogen, with a 1 micron polypropylene bag filter the batch, and then transferred to the new reactor. Was added to the vessel pure water (146 kg), and heating the batch to about 68-72. C.
After about four hours, the 10.7% aqueous hydrochloric acid was added to the batch. During the addition, batch temperature was maintained at about 68-72 ° C. PH was measured by using the batch pH of about 2.2, and then stirring was continued at about 7 (under TC about 1 hour.
After about two hours, the batch in a linear fashion from about 70. C is cooled to about 60 ° C. After about two hours, about 60. C of the batch in a linear fashion from about 6 (TC was cooled to about 40 ° C. In 40t, the batch was stirred for 2 hours, and the slurry was transferred to a centrifuge filter. After about 30 minutes, filtered completion . After about 30 minutes, with about 42Mw / w in t-butanol solution (165kg) cake was washed. The purified water (118kg, 4 (TC) as the last added to the filter cake was washed. The filter cake was dried at about 900rpm about 1 hour, then filled cylinder.
The wet cake was transferred to a paddle dryer (a double cone drier also suitable for this step), the jacket temperature was set to about 70. C. At about 70. C, the product was dried under vacuum for about 48 hours while stirring periodically.Determination of purity of about 99.8%. The product was isolated overall yield of about 74% (68.5 kg).
Lot 3
To the reaction vessel was added 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (10 g) and t-butanol fused (128mL ). The batch temperature was set to 30-40 ° C, and the stirrer is started. After about 30 minutes, the aqueous sodium hydroxide solution 4.48% w / w of (32.5 g) was added to the vessel. The batch was maintained at a temperature of about 40-50 ° C. After about l hour, the batch temperature is raised to about 78-82 ° C, and then, at about 78-82t, and then stirred for about one hour. Under positive pressure of nitrogen, a polyethylene bag with a 5 micron filter the batch, and then transferred to a new reaction vessel. The batch was maintained at a temperature of about 78-82 ° C.
It was added to a new vessel 37% aqueous hydrochloric acid (4 mL) and tert-butanol molten (8 mL). The temperature was maintained at about 30-40. Under C, and stirring the mixture for about 30 minutes.
After about four hours, using a metering pump was added to the batch of hydrochloric acid in tert-butanol. After about SO-SO minutes before adding half filled. The stirrer speed is set at about 200rpm. After about 3.5 hours, add the remaining charge. The stirrer speed is set at about 100 rpm. During the addition, batch temperature was maintained at about 78-82 ° C.PH meter with a final batch pH was adjusted to about 1.2, at about 78-82t, then continue stirring for about l hour. After about one hour, the batch in a linear fashion from about 78-82. C is cooled to about 70 ° C. After about four hours, about 7 (TC batches in a linear fashion from 70.C cooled to about 50 ° C, and the stirrer speed was set at about 80 rpm. After about four hours, about 50 ° C Batch linearly cooled from 50 ° C to about 40t, and stirrer speed was set at approximately 60rpm. In 40.C, the batch was stirred for a further 4 hours.
The temperature of the filter is set to about 40-45 ° C. The slurry was transferred to the filter. After about one minute to complete filtration. After about two minutes, with tert-butanol (50 mL, 50.C) washing the filter cake. The pure water (IOO mLx2, 60.C) as the last wash was added to the cake. Under vacuum at about 60-70 ° C the cake was dried for about 12 hours, and then loaded into the container.
Determination of HPLC purity of about 99.9% of the area. The yield of isolated product was about 94% (9.0g).
3- “5- (2-fluorophenyl) -” 1,2,41-oxadiazol-3-yl 1- acid: One-pot
The methyl 3-cyanophenyl Yue (7.35 g) and tert-butanol molten (100 mL) added to the reactor vessel. Sealed containers, the batch temperature was set to 60 ° C, and the stirrer is started. The suspension was stirred for 1 hour and then the batch temperature was set to 40. C. Under an inert atmosphere, after three hours, 50% aqueous solution of hydroxylamine (3.63 g) was added to the reactor. During the addition, batch temperature was maintained at 38-41 ° C. 40. C After stirring for 18 hours, to complete the reaction.
The batch was cooled to 27 ° C, and after two minutes, triethylamine (5.56 g). After 3 hours, was added 2-fluorobenzoyl chloride (7.82 g). During the addition, batch temperature was maintained at 24-27 ° C. 40. C, the batch was stirred for a further 4 hours.
After 30 minutes, the batch was heated to 79 ° C, and at about 79. C was stirred for 16 hours. After 3 hours, the white suspension was added to the water (IOO mL), while the batch temperature was maintained at 70 ° C. After 20 minutes, a 37% aqueous hydrochloric acid were added to the batch. PH was measured by using the batch pH of about 2.2, stirring was continued at about 70t for about 1 hour.
After three hours, the batch in a linear manner from 7 (TC cooled to 30 ° C, and the slurry is transferred to the filter. After 5 minutes, the filtering is done. After five minutes, with tert-butanol (50mL, 40 .C) filter cake was washed. The purified water (IOO mL, 60.C) is added to a final wash the filter cake. In 70.C of the filter cake was dried in a vacuum oven for 18 hours and then removed. Determination of purity approximately 98.68%. The total yield of isolated product of about 76% (10.8g).
PICS

References
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91. Bibcode:2007Natur.447…87W.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444.doi:10.1177/0091270006297140. PMID 17389552.
- Nature. 2007 May 3;447(7140):87-91.
- Proc Natl Acad Sci U S A. 2008 Feb 12;105(6):2064-9.
- Neuromuscul Disord. 2015 Jan;25(1):5-13.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery Medicine 15 (81): 127–133. PMID 23449115.
- “PTC Therapeutics and Genzyme Corporation announce preliminary results from the phase 2b clinical trial of ataluren for nonsense mutation Duchenne/Becker muscular dystrophy (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271.Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272. doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved2013-11-28.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
- “PTC Therapeutics Announces Launch of Translarna™ (ataluren) in Germany”.marketwatch.com. 3 Dec 2014. Retrieved 27 Dec 2014.
- “NICE asks for further evidence for the benefits of a new treatment for Duchenne muscular dystrophy to justify its very high cost”.
- http://uk.reuters.com/article/us-ptc-therapeutics-fda-idUKKCN0VW1FG
External links
References:
1. Ryan, N. J. Ataluren: first global approval. Drugs 2014, 74(14), 1709-14. (FMO only)
2. Gupta, P. K.; et. al. A metal-free tandem approach to prepare structurally diverse N-heterocycles: synthesis of 1,2,4-oxadiazoles and pyrimidinones. New J Chem 2014, 38, 3062-3070 (FMO only)
3. Almstead, N. G.; et. al. Methods for the production of functional protein from dna having a nonsense mutation and the treatment of disorders associated therewith. WO2007117438A2
| WO2004091502A2 | Apr 9, 2004 | Oct 28, 2004 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8486982 | Jun 22, 2012 | Jul 16, 2013 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acids |
| US8796322 | Jun 19, 2013 | Aug 5, 2014 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-oxadiazole benzoic acid compounds |
| US8975287 | Jun 18, 2014 | Mar 10, 2015 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-Oxadiazole benzoic acid compounds |
| US9205088 | Jan 28, 2015 | Dec 8, 2015 | Ptc Therapeutics, Inc. | Compositions of 1,2,4-oxadiazol benzoic acid compounds and methods for their use |
| US9289398 | Mar 29, 2007 | Mar 22, 2016 | Ptc Therapeutics, Inc. | Methods for the production of functional protein from DNA having a nonsense mutation and the treatment of disorders associated therewith |
| Preparation | CN101535284A | CN101535284B | ||||
| 10 | Crystal | CN101541770A | ||||
| 11 | Crystal | CN104341371A | ||||
| 12 | Crystal | CN102382075A |
| Formula | CN1802360A | CN1802360B | ||||
| 2 | Combination | CN104056278A | ||||
| 3 | Indication | CN101076703A | ||||
| 4 | Indication | CN101076332A | ||||
| 5 | Indication | CN101076337A | ||||
| 6 | Indication | CN101193632A | ||||
| 7 | Formulation | CN103720688A | ||||
| 8 | Indication | CN101505739A |

 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
|
|
| Other names
PTC124
|
|
| Identifiers | |
| 775304-57-9 |
|
| ChEMBL | ChEMBL256997 |
| ChemSpider | 9394889 |
| 7341 | |
| Jmol 3D model | Interactive image |
| KEGG | D09323 |
| PubChem | 11219835 |
| UNII | K16AME9I3V |
| Properties | |
| C15H9FN2O3 | |
| Molar mass | 284.24 g/mol |
| Pharmacology | |
| M09AX03 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////ORPHAN DRUG, Ataluren, Translarna, Duchenne Muscular Dystrophy, EU, 775304-57-9, PTC Therapeutics, PTC 124
O=C(O)c1cccc(c1)c2nc(on2)c3ccccc3F
PTC Therapeutics’ large-scale multinational trial of ataluren for nonsense-mutation Duchenne or Becker MD has opened its first site in Cincinnati, Ohio

ATALUREN
A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
Ataluren, formerly known as PTC124, is a novel small-molecular agent designed to makeribosomes become less sensitive to, or possibly ignore premature stop codons. This may be particularly beneficial in genetic disorders where the mRNA contains a mutation causing premature stop codon or nonsense codon. However, it is not equally effective with every stop codon, working best on the sequence ‘UGA’.
PTC124 has been tested on healthy humans and humans carrying genetic disorderscaused by nonsense mutations,such as some people with cystic fibrosis andDuchenne muscular dystrophy. Clinical trials are proceeding for several genetic disorders, in the subset of affected people who have nonsense mutations (typically <10% of those with the disorder). PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.However, phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.
PTC124 has been developed by PTC Therapeutics.
Phase 2 , Sarepta, shows eteplirsen for Duchenne muscular dystrophy
ETEPLIRSEN
27 MAR 2013
The clock is ticking for Sarepta Therapeutics , a multitude of biotech investors and the boys who suffer from Duchenne muscular dystrophy.
Armed with promising Phase IIb data from a small study of eteplirsen involving only 12 patients with the lethal disease, Sarepta CEO Chris Garabedian is completing one of the most closely-watched high wire acts in the industry. At a time most companies would be focused solely on organizing a pivotal late-stage study, there’s intense speculation that the biotech will shoot for an accelerated approval with the data in hand. And it all comes down to their sit-down with the FDA to review mid-stage data.
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
The drug is a Morpholino antisense oligomer which triggers excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.

Phase 2, Sarepta Therapeutics, Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
s excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Clinical studies
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
References
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods.. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303.. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.

{kind=link}