
Home » Posts tagged 'breast cancer'
Tag Archives: breast cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BQ-788
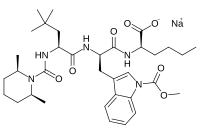
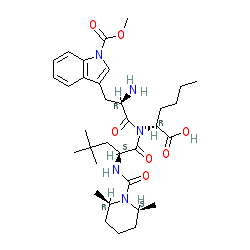
BQ-788
- Molecular FormulaC34H50N5NaO7
- Average mass663.780 Da
SP ROT +3.8 ° Conc: 1.032 g/100mL; methanol; Wavlenght: 589.3 nm, Development of an efficient strategy for the synthesis of the ETB receptor antagonist BQ-788 and some related analogues
Peptides (New York, NY, United States) (2005), 26, (8), 1441-1453., https://doi.org/10.1016/j.peptides.2005.03.022
FOR FREE FORM +19.6 °, Conc: 0.998 g/100mL; : N,N-dimethylformamide; 589.3 nm
BQ-788 is a selective ETB antagonist.[1]
presumed to be under license from Banyu , was investigating BQ-788, a selective endothelin receptor B (ETRB) antagonist, for treating metastatic melanoma. By December 2009, the drug was in validation.
Also claimed is their use as an ETBR antagonist and for treating cancers, such as brain cancer, pancreas cancer, colon cancer, breast cancer, ovary cancer, prostate cancer, glioblastoma, solid tumor, melanoma and squamous cell carcinoma. Represent a first filing from ENB Therapeutics Inc and the inventors on these deuterated forms of BQ-788. Melcure SarL ,

SYN
By Brosseau, Jean-Philippe et alFrom Peptides (New York, NY, United States), 26(8), 1441-1453; 2005
CONTD…………

PAPER
https://pubs.acs.org/doi/pdf/10.1021/jo00130a028

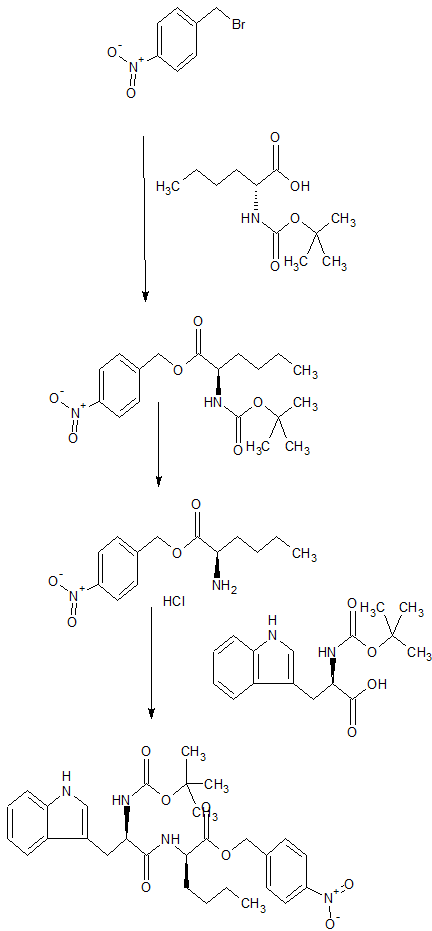
N-(cw-2,6-Dimethylpiperidinocarbonyl)-y-methylleucylD-l-(methoxycarbonyl)tryptophanyl-D-norleucine Sodium Salt (1, BQ-788). To a solution of 15 (3.5 g, 5.5 mmol) in methanol (50 mL) was slowly added 5% aqueous NaHCOs (300 mL) over a period of 30 min. The solution was stirred until clarity was achieved (30 min, 23 °C). The solution was diluted with water (200 mL), and the resulting solution was passed through a C18 (60 mL) cartridge preequilbrated in water. BQ-788 (1) was eluted with methanol (2 x 50 mL), concentrated under reduced pressure, resuspended in water (50 mL), and lyophilized to quantitatively yield compound 1 as a white powder:
HPLC £r = 16.4 (gradient A, > 99%);
NMR (400 MHz, DMSO-d6) ó 0.80 (s, 9H), 0.74-0.85 (m, 3H), 1.00 (d, 3H), 1.02 (d, 3H), 1.10-1.25 (m, 6H), 1.30-1.55 (m, 6H), 1.60-1.75 (m, 2H), 2.92 (dd, 1H), 3.12 (dd, 1H), 3.78 (m, 1H), 3.95 (s, 3H), 4.08 (m, 1H), 4.13 (m, 1H), 4.29 (m, 1H), 4.50 (m, 1H), 5.98 (d, 1H), 7.22 (t, 1H), 7.32 (t, 1H), 7.50 (s, 1H), 7.58 (br d, 1H), 7.65 (d, 1H), 8.05 (d, 1H), 8.15 (br d, 1H) ESMS m/z 640.6 (M).
PATENT
WO-2019140324
Novel deuterated analogs of a substituted heterocyclic compound, particularly BQ-788 , processes for their preparation and compositions and combinations comprising them are claimed.
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0196978105001415

PAPER
By He, John X.; Cody, Wayne L.; Doherty, Annette M., From Journal of Organic Chemistry (1995), 60(25), 8262-6
Journal of medicinal chemistry (1996), 39(12), 2313-30.
References
 |
|
| Names | |
|---|---|
| Systematic IUPAC name
Sodium N-{[(2R,6S)-2,6-dimethyl-1-piperidinyl]carbonyl}-4-methyl-L-leucyl-N-[(1R)-1-carboxylatopentyl]-1-(methoxycarbonyl)-D-tryptophanamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C34H50N5NaO7 | |
| Molar mass | 663.792 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////BQ-788, BQ 788, BQ788, ETBR antagonist, cancers, brain cancer, pancreas cancer, colon cancer, breast cancer, ovary cancer, prostate cancer, glioblastoma, solid tumor, melanoma, squamous cell carcinoma, PEPTIDE
CCCC[C@H](C(=O)O)NC(=O)[C@@H](Cc1cn(c2c1cccc2)C(=O)OC)NC(=O)[C@H](CC(C)(C)C)NC(=O)N3[C@@H](CCC[C@@H]3C)C
Picropodophyllin
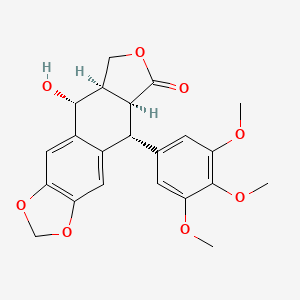
Picropodophyllin
Picropodophyllotoxin
CAS 477-47-4
AXL1717, NSC 36407, BRN 0099161
414.4 g/mol, C22H22O8
(5R,5aR,8aS,9R)-5-hydroxy-9-(3,4,5-trimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one
Furo(3′,4′:6,7)naphtho(2,3-d)-1,3-dioxol-6(5aH)-one, 5,8,8a,9-tetrahydro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-, (5R-(5-alpha,5a-alpha,8a-alpha,9-alpha))-
5-19-10-00665 (Beilstein Handbook Reference)
Axelar is developing picropodophyllin, a small-molecule IGF-1 receptor antagonist for the treatment of cancer including NSCLC and malignant astrocytoma. In February 2019, a phase Ia study was planned to initiate for solid tumor in March 2019.
Picropodophyllin is a cyclolignan alkaloid found in the mayapple plant family (Podophyllum peltatum), and a small molecule inhibitor of the insulin-like growth factor 1 receptor (IGF1R) with potential antineoplastic activity. Picropodophyllin specifically inhibits the activity and downregulates the cellular expression of IGF1R without interfering with activities of other growth factor receptors, such as receptors for insulin, epidermal growth factor, platelet-derived growth factor, fibroblast growth factor and mast/stem cell growth factor (KIT). This agent shows potent activity in the suppression o f tumor cell proliferation and the induction of tumor cell apoptosis. IGF1R, a receptor tyrosine kinase overexpressed in a variety of human cancers, plays a critical role in the growth and survival of many types of cancer cells.
Picropodophyllotoxin is an organic heterotetracyclic compound that has a furonaphthodioxole skeleton bearing 3,4,5-trimethoxyphenyl and hydroxy substituents. It has a role as an antineoplastic agent, a tyrosine kinase inhibitor, an insulin-like growth factor receptor 1 antagonist and a plant metabolite. It is a lignan, a furonaphthodioxole and an organic heterotetracyclic compound.
Picropodophyllin has been investigated for the treatment of Non Small Cell Lung Cancer.
One of the largest challenges in pharmaceutical drug development is that drug compounds often are poorly soluble, or even insoluble, in aqeous media. Insufficient drug solubility means insufficient bioavailability, as well as poor plasma exposure of the drug when administered to humans and animals. Variability of plasma exposure in humans is yet a problem when developing drugs which are poorly soluble, or even insoluble, in aqeous media.
It is estimated that between 40% and 70 % of all new chemical entities identified in drug discovery programs, are insufficiently soluble in aqeous media (M. Lindenberg, S et al: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004). Scientists have investigated various ways of solving the problem with poor drug solubility in order to enhance bioavailability of poorly absorbed drugs, aiming at increasing their clinical efficacy when administered orally.
Technologies such as increase of the surface area and hence dissolution may sometimes solve solubility problems. Other techniques that may also solve bioavailability problems are addition of surfactants and polymers. However, each chemical compound has its own unique chemical and physical properties, and hence has its own unique challenges when being formulated into a pharmaceutical product that can exert its clinical efficacy.
Picropodophyllin is an insulin-like growth factor-1 receptor inhibitor fiGF-lR inhibitor) small-molecule compound belonging to the class of compounds denominated cyclolignans, having the chemical structure:
The patent applicant is presently entering clinical phase II development with its development compound picropodophyllin (AXL1717). However, picropodophyllin is poorly soluble in aqueous media. In a phase I clinical study performed by the applicant in 2012 (Ekman S et al; Acta Oncologica, 2016; 55: pp. 140-148), it was discovered that picropodophyllin, when administered as an oral suspension to lung cancer patients, resulted in unacceptable variability in drug exposure. A large variability in plasma exposure of the active drug picropodophyllin occurred not only within certain patients, but also between several patients.
Yet a problem with administering picropodophyllin as an aqeous solution, is that due to the poor solubility in aqueous media, it is difficult or even impossible to reach the required therapeutic doses.
The compound picropodophyllin is furthermore physically unstable, and transforms from amorphous picropodophyllin into crystalline picropodophyllin. Yet a stability problem with picropodophyllin is that it is chemically unstable in solution.

Product case, WO02102804
Patent
WO-2019130194
Novel amorphous forms of picropodophyllin , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating cancers, such as neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, and eye cancers. , claiming picropodophyllin derivatives as modulators of insulin-like growth factor-1 receptor (IGF-1), useful for treating cancers, assigned to Axelar AB ,
CLIP

CLIP
https://pubs.rsc.org/en/content/articlelanding/2004/cc/b312245j/unauth#!divAbstract
http://www.rsc.org/suppdata/cc/b3/b312245j/b312245j.pdf
dH(CDCl3; 300 MHz; Me4Si): 2.64-2.78 (1 H, m, 3-H), 3.23 (1 H, dd, J 4.4 and 8.2, 2-H), 3.81 (6 H, s, 2 x OMe), 3.85 (3 H, s, OMe), 4.09 (1 H, d, J 4.4, 1-H), 4.38–4.59 (3 H, m, 11-H2 and 4-H), 5.91 (1 H, d, J 1.5, OCH2O), 5.93 (1 H, d, J 1.5, OCH2O), 6.35 (1 H, s, 5-H/8-H), 6.46 (1 H, s, 2’-H and 6’-H) and 7.07 (1 H, s, 5-H/8-H).
CLIP
PAPER
Organic Letters (2018), 20(6), 1651-1654
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.8b00408

A nickel-catalyzed reductive cascade approach to the efficient construction of diastereodivergent cores embedded in podophyllum lignans is developed for the first time. Their gram-scale access paved the way for unified syntheses of naturally occurring podophyllotoxin and other members.
Synthesis of (−)-Podophyllotoxin (1)
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_001.pdf
The residue was purified by flash column chromatography (petroleum ether/EtOAc = 4 : 1 → petroleum ether/EtOAc = 2 : 1) on silica gel to afford 1 (8.6 mg, 87% yield) as a white solid; Rf = 0.23 (petroleum ether/EtOAc = 1 : 1); [α]20 D = –115.00 (c = 1.00, CHCl3) [ref.13: [α]20 D = –101.7 (c = 0.55, EtOH)]; Mp. 167–168 °C; 1H NMR (400 MHz, CDCl3): δ = 7.11 (s, 1H), 6.51 (s, 1H), 6.37 (s, 2H), 5.98 (s, 1H), 5.96 (s, 1H), 4.77 (t, J = 8.4 Hz, 1H), 4.60 (t, J = 8.0 Hz, 1H), 4.59 (d, J = 4.4 Hz, 1H), 4.08 (dd, J = 9.6, 8.8 Hz, 1H), 3.81 (s, 3H), 3.75 (s, 6H), 2.84 (dd, J = 14.0, 4.4 Hz, 1H), 2.83−2.74 (m, 1H), 2.13 (d, J = 8.0 Hz, 1H, −OH) ppm; 13C NMR (100 MHz, CDCl3): δ = 174.6, 152.5 (2C), 147.7, 147.6, 137.1, 135.5, 133.3, 131.0, 109.7, 108.4 (2C), 106.3, 101.4, 72.6, 71.4, 60.7, 56.2 (2C), 45.2, 44.1, 40.6 ppm.

https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_002.pdf


PAPER
Organic Letters (2017), 19(24), 6530-6533
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.7b03236

he first catalytic enantioselective total synthesis of (−)-podophyllotoxin is accomplished by a challenging organocatalytic cross-aldol Heck cyclization and distal stereocontrolled transfer hydrogenation in five steps from three aldehydes. Reversal of selectivity in hydrogenation led to the syntheses of other stereoisomers from the common precursor.
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.7b03236/suppl_file/ol7b03236_si_001.pdf
(-)-Picropodophyllin 4. The lactone 5 (0.2 g, 0.38 mmol) was taken in 1-pentanol (5 mL) in a double neck RB flask at rt. Water (0.14 mL, 7.6 mmol) was added to above mixture and it was then degassed with argon followed by addition of Pd/C (0.04 g, 20% by wt.) and HCO2Na (0.78g, 11.4 mmol). The reaction mixture was heated at 40 °C for 12 h. On completion, the reaction mixture was diluted with EtOAc (200 mL), filtered through a celite pad and solvent was removed under vacuum. This crude mixture was dissolved in THF (3.8 mL), TBAF (1.9 mL, 1.9 mmol, 1M in THF) was added and stirred for 6 h at 27 °C. On completion, EtOAc (250 mL) was added, washed with water (100 mL), brine and dried over Na2SO4. After removal of solvent, the crude product was purified by column chromatography (hexanes-EtOAc, 3:2) to get the title compound as a white solid (0.082 g, 52%): Rf 0.32 (hexanes/EtOAc, 1:1); [α]25 D = -10.6 (c = 0.4, CHCl3) [lit. -10 (c = 0.3, CHCl3), -11 (c = 0.41, CHCl3)]3a,b;
Mp 214-216 °C; 1H NMR (600 MHz, CDCl3) δ 7.05 (s, 1H), 6.47 (s, 2H), 6.41 (s, 1H), 5.95 (d, J = 14.1 Hz, 2H), 4.5 (m, 2H), 4.44 (t, J = 8.0 Hz, 1H), 4.15 (d, J = 4.1 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 6H), 3.24 (dd, J = 8.7, 5.0 Hz, 1H), 2.75 (m, 1H), 2.12 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 177.6, 153.7, 147.5, 147.1, 139.3, 137.4, 131.9, 130.6, 109.3, 105.9, 105.5, 101.2, 69.8, 69.6, 60.9, 56.3, 45.4, 44.1, 42.7; HRMS (ESI-TOF) m/z 437.1219 [(M+Na)+ ; calcd for C22H22O8Na+ : 437.1212].
PAPER
The Journal of organic chemistry (2000), 65(3), 847-60.
https://pubs.acs.org/doi/abs/10.1021/jo991582+

REF
Berichte der Deutschen Chemischen Gesellschaft [Abteilung] B: Abhandlungen (1932), 65B, 1846.
Justus Liebigs Annalen der Chemie (1932), 499, 59-76.
Justus Liebigs Annalen der Chemie (1932), 494, 126-42.
Journal of the American Chemical Society (1954), 76, 5890-1
Helvetica Chimica Acta (1954), 37, 190-202.
Journal of the American Chemical Society (1988), 110(23), 7854-8.
//////////////Picropodophyllin, AXL1717, NSC 36407, BRN 0099161, Picropodophyllotoxin, AXELAR, PHASE 1, CANCER, neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, eye cancers, NSCLC, malignant astrocytoma, SOLID TUMOUR
COC1=CC(=CC(=C1OC)OC)C2C3C(COC3=O)C(C4=CC5=C(C=C24)OCO5)O
Podofilox, Podophyllotoxin, Wartec, Condyline, Condylox
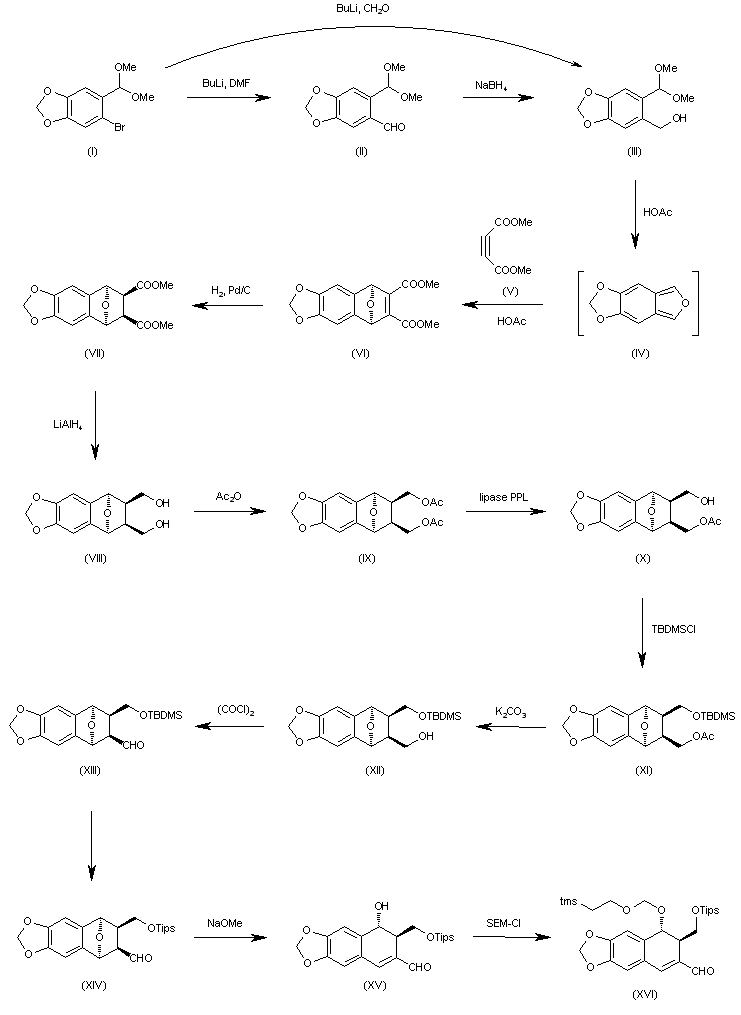
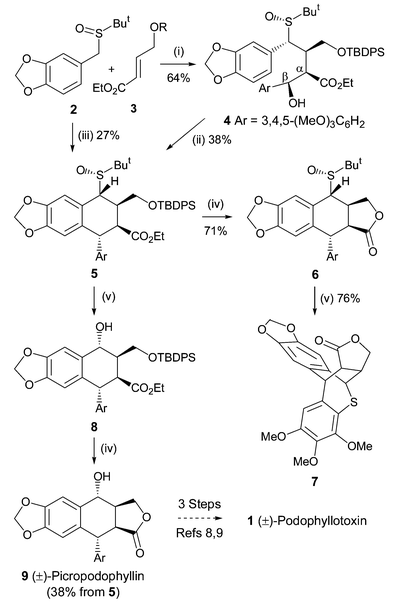
| J Org Chem 2000,65(3),847 |
The formylation of 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) with BuLi and DMF gives the 6-formyl derivative (II), which is reduced with NaBH4 in ethanol to yield the corresponding carbinol (III). The cyclization of (III) with dimethyl acetylenedicarboxylate (V) in hot acetic acid (through the nonisolated intermediate (IV)) affords dimethyl 1,4-epoxy-6,7-(methylenedioxy)naphthalene-2,3-dicarboxylate (VI), which is hydrogenated with H2 over Pd/C in ethyl acetate to give the (1R*,2S*,3R*,4S*)-tetrahydro derivative (VII). The reduction of (VII) with LiAlH4 in refluxing ethyl ether affords the corresponding bis carbinol (VIII), which is treated with acetic anhydride to afford the diacetate (IX). The enzymatic monodeacetylation of (VIII) with PPL enzyme in DMSO/buffer gives (1R,2R,3S,4S)-2-(acetoxymethyl)-1,4-epoxy-3-(hydroxymethyl)-6,7-(methylenedioxy)-1,2,3,4-tetrahydronaphthalene (X), which is silylated with TBDMS-Cl and imidazole in DMF yielding the silyl ether (XI). The hydrolysis of the acetoxy group of (XI) with K2CO3 in methanol affords the carbinol (XII), which is oxidized with oxalyl chloride in dichloromethane affording the carbaldehyde (XIII). The exchange of the silyl protecting group of (XIII) (for stability problems) provided the triisopropylsilyl ether (XIV), which is treated with sodium methoxide in methanol to open the epoxide ring yielding the hydroxy aldehyde (XV). The protection of the hydroxy group of (XV) with 2-(trimethylsilyl)ethoxymethyl chloride and DIEA in dichloromethane provides the corresponding ether (XVI). The carbinol (III) can also be obtained directly from 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) by reaction with formaldehyde and BuLi in THF.
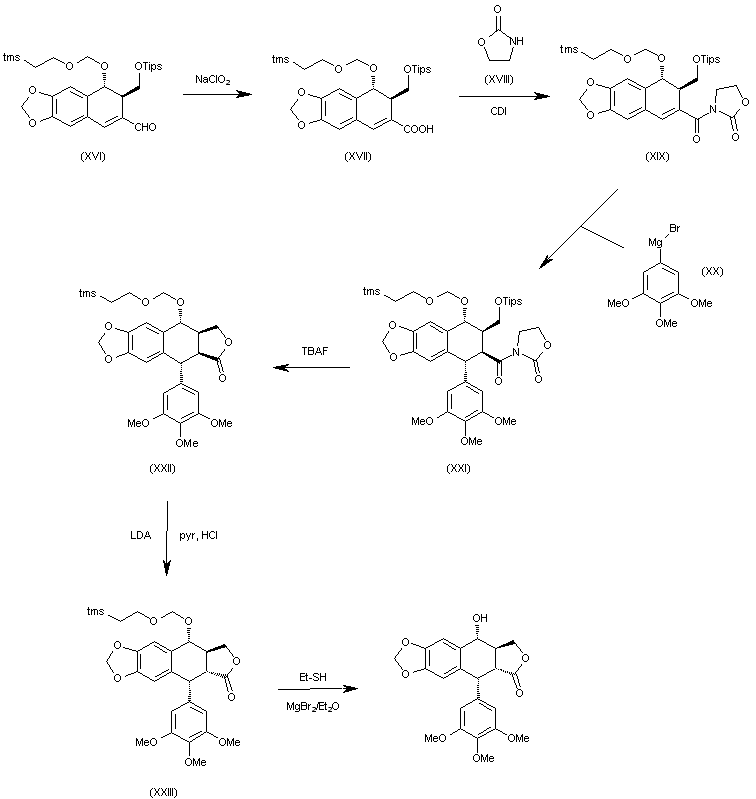
The oxidation of the aldehyde group of (XVI) with NaClO2 in tert-butanol affords the corresponding carboxylic acid (XVII), which is condensed with 2-oxazolidinone (XVIII) by means of carbonyldiimidazole (CDI) in THF to give the acyl imidazolide (XIX). The arylation of (XIX) with 3,4,5-trimethoxyphenylmagnesium bromide (XX) in THF yields the expected addition product (XXI), which is cyclized by means of TBAF in hot THF to afford the tetracyclic intermediate (XXII). Isomerization of the cis-lactone ring of (XXII) with LDA in THF affords intermediate (XXIII) with its lactone ring with the correct trans-conformation. Finally, this compound is deprotected with ethyl mercaptane and MgBr2 in ethyl ether to provide the target compound.
Synthesis 1992,719
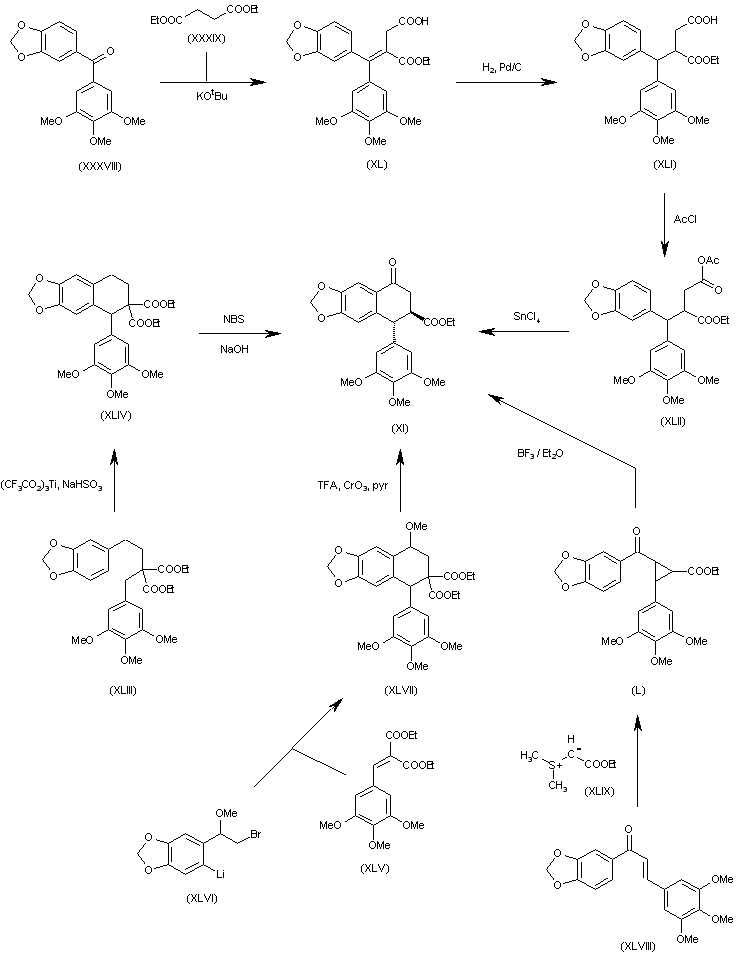
The intermediate trans-8-oxo-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetra-hydronaphtho[2,3-d][1,3]benzodioxole-6-carboxylic acid ethyl ester (XI) has been obtained by several different ways: (a) The condensation of benzophenone (XXXVIII) with diethyl malonate (XXXIX) by means of t-BuOK gives the alkylidenemalonate (XL), which is hydrogenated with H2 over Pd/C to the alkylmalonate hemiester (XLI). The reaction of (XLI) with acetyl chloride affords the mixed anhydride (XLII), which is finally cyclized to the target (XI) by means of SnCl4. (b) The cyclization of the malonic ester derivative (XLIII) by means of Ti(CF3–CO2)3 gives the 5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho [2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLIV), which is finally oxidized and decarboxylated with NBS and NaOH in methanol to afford the target intermediate (XI). (c) The cyclization of the benzylidenemalonate (XLV) with the aryllithium derivative (XLVI) gives the 8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLVII), which is demethylated with TFA and oxidized with CrO3 and pyridine to the target compound (XI). (d) The cyclopropanation of the chalcone (XLVIII) with (ethoxycarbonyl) (dimethylsulfonium)methylide (XLIX) gives the cyclopropanecarboxylate (L), which is finally rearranged with BF3/Et2O to the target intermediate (IX).
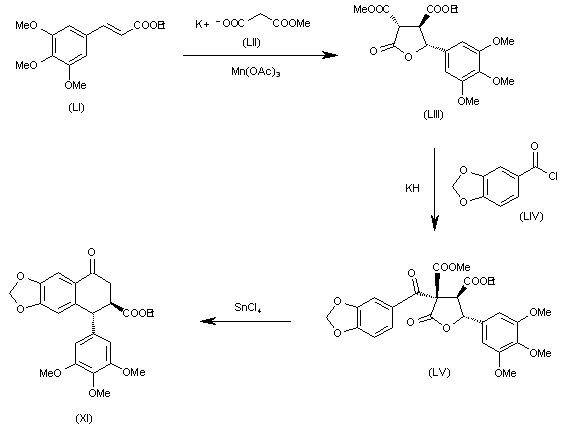
The cyclization of 3,4,5-trimethoxycinnamic acid ethyl ester (LI) with malonic acid ethyl ester potassium salt (LII) by means of Mn(OAc)3 gives the tetrahydrofuranone (LIII), which is acylated with 1,3-benzodioxol-5-ylcarbonyl chloride (LIV) yielding the tetrahydrofuranone (LV). Finally, this compound is rearranged and decarboxylated with SnCl4 to the target intermediate (XI).
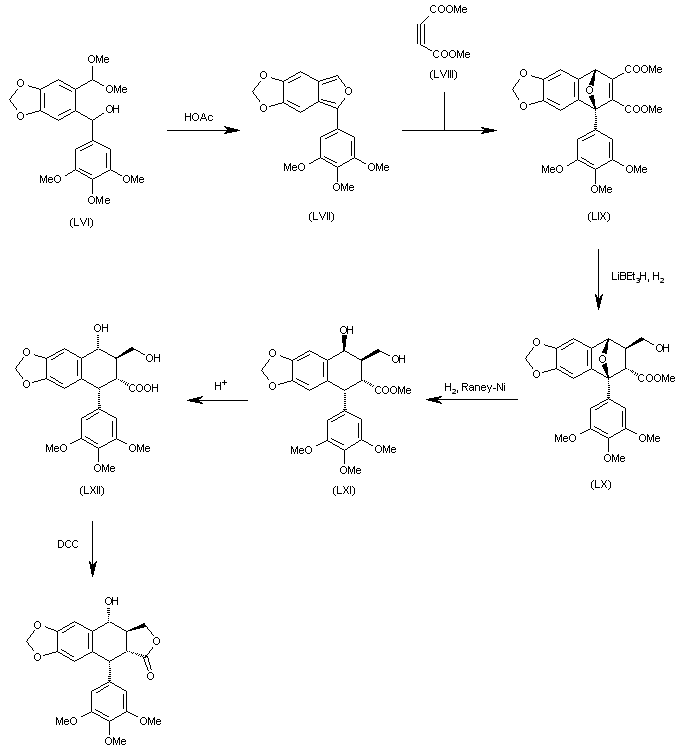
The cyclization of 6-[1-hydroxy-1-(3,4,5-trimethoxyphenyl)methyl]-1,3-benzodioxol-5-carbaldehyde dimethylacetal (LVI) by means of AcOH gives 5-(3,4,5-trimethoxyphenyl)-1,3-dioxolo[4,5-f]isobenzofuran (LVII), which is submitted to a Diels-Alder cyclization with acetylenedicarboxylic acid dimethyl ester (LVIII) yielding the epoxy derivative (LIX). The selective reduction of (LIX) with LiBEt3H and H2 affords the carbinol (LX), which is treated with H2 over RaNi in order to open the epoxide ring to give the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to (LXII) with the suitable configuration. Finally, this compound is cyclized with DCC to the desired target compound.
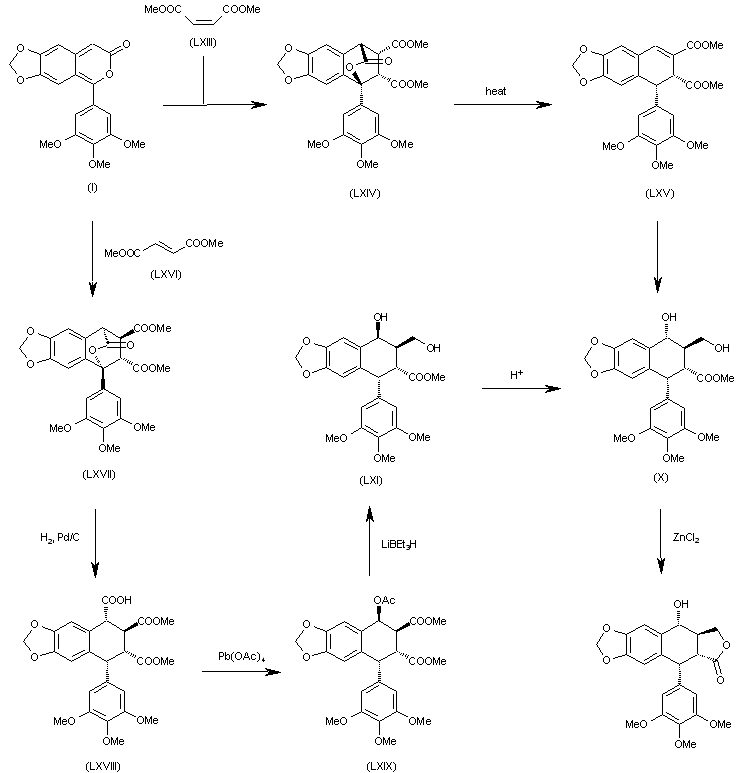
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl maleate (LXIII) gives the expected adduct (LXIV), which by thermal extrusion of CO2 yields the dihydronaphthodioxole (LXV). This compound is then converted to dihydroxycompound (X), which is finally cyclized by means of ZnCl2 to provide the target compound. The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl fumarate (LXVI) gives the expected adduct (LXVII), which by hydrogenation with H2 over Pd/C yields the tricarboxylic acid derivative (LXVIII). The reaction of (LXVIII) with Pb(OAc)4 affords the acetoxy derivative (LXIX), which is selectively reduced with LiBEt3H providing the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to give the previously described (X) with the suitable configuration.
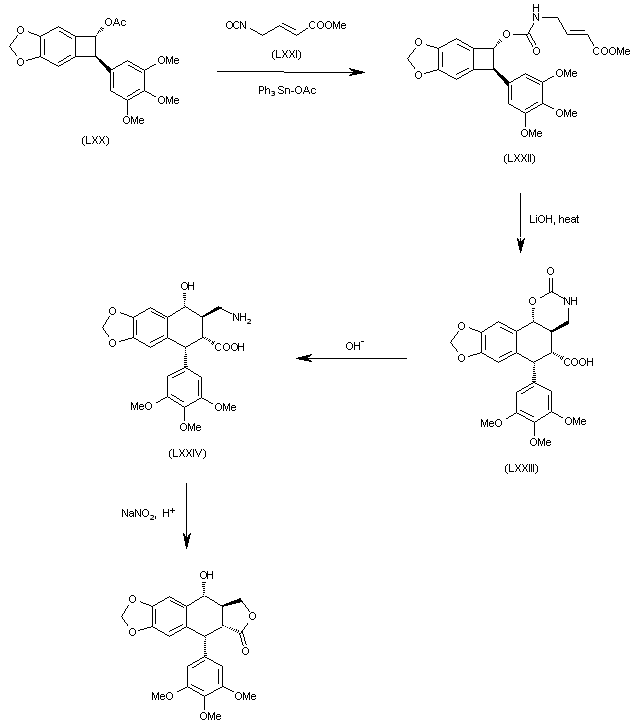
The reaction of benzocyclobutane derivative (LXX) with isocyanate (LXXI) by means of Ph3SnOAc gives the carbamate (LXXII), which is cyclized by a thermal treatment with LiOH yielding the tetracyclic carboxylic acid (LXXIII). The opening of the oxazinone ring of (LXXIII) in basic medium affords the tricyclic amino acid (LXXIV), which is finally cyclized to the target compound by reaction with sodium nitrite in acidic medium (pH = 4).
J Chem Soc Chem Commun 1993,1200
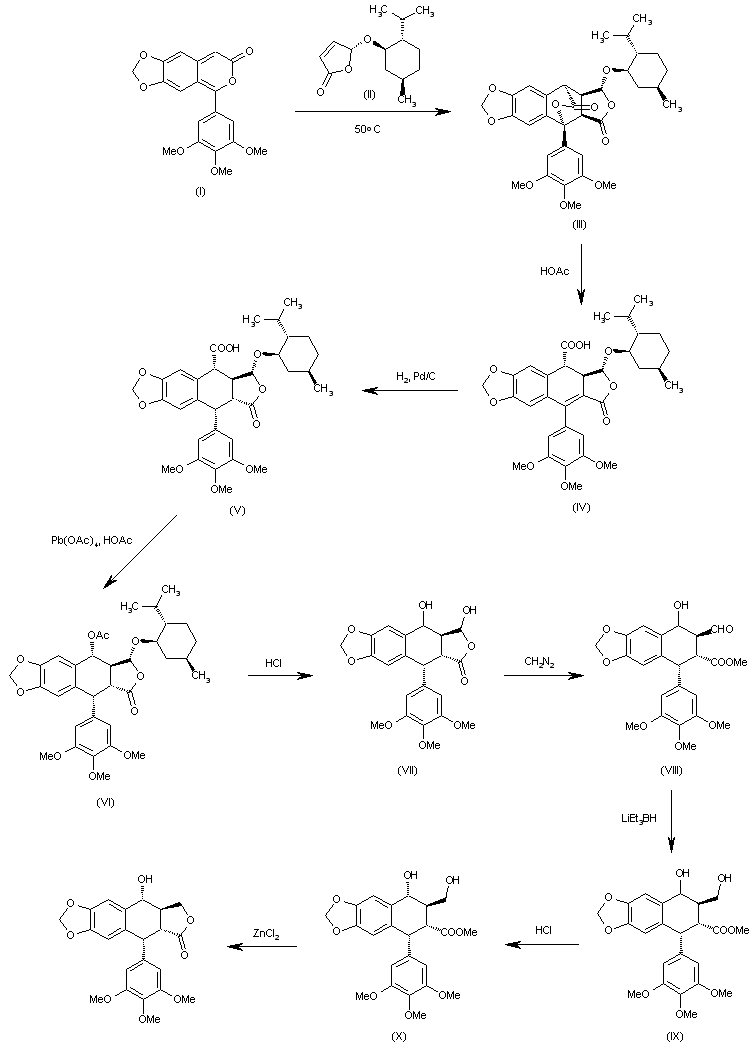
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with the chiral dihydrofuranone (II) in hot acetonitrile gives the pentacyclic anhydride (III), which is opened with warm acetic acid yielding the carboxylic acid (IV). Hydrogenation of the benzylic double bond of (IV) with H2 over Pd/C affords (V), which is treated with lead tetraacetate and acetic acid in THF to give the acetoxy compound (VI). The hydrolysis of the acetoxy group and the menthol hemiacetal group with HCl in hot dioxane yields the diol (VII), which is treated with diazomethane in ether/methanol affording the aldehyde (VIII). The reduction of the aldehyde group of (VIII) with LiEt3BH in THF gives the diol (IX) as a diastereomeric mixture, which is treated with HCl in THF to afford the diol (X) with the right conformation. Finally, this compound is lactonized to the target compound with ZnCl2 in THF.
//////////
SELPERCATINIB

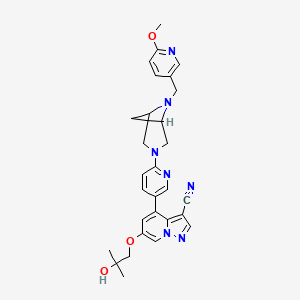
SELPERCATINIB
LOXO 292
CAS: 2152628-33-4
Chemical Formula: C29H31N7O3
Molecular Weight: 525.613
CEGM9YBNGD
UNII-CEGM9YBNGD
6-(2-hydroxy-2-methylpropoxy)-4-(6-{6-[(6-methoxypyridin- 3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl}pyridin-3- yl)pyrazolo[1,5-a]pyridine-3-carbonitrile
Selpercatinib is a tyrosine kinase inhibitor with antineoplastic properties.
A phase I/II trial is also under way in pediatric patients and young adults with activating RET alterations and advanced solid or primary CNS tumors.
Loxo Oncology (a wholly-owned subsidiary of Eli Lilly ), under license from Array , is developing selpercatinib, a lead from a program of RET kinase inhibitors, for treating cancer, including non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma and soft tissue sarcoma
In 2018, the compound was granted orphan drug designation in the U.S. for the treatment of pancreatic cancer and in the E.U. for the treatment of medullary thyroid carcinoma.

PATENT
WO2018071447
PATENT
US 20190106438
PATENT
WO 2019075108
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019075108&tab=PCTDESCRIPTION
Compounds of Formula I-IV, 4-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula I); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula II); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-(6-methoxynicotinoyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula III); and 6-(2-hydroxy-2-methylpropoxy)-4-(6-(4-hydroxy-4-(pyridin-2-ylmethyl)piperidin-1-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula IV) are inhibitors of RET kinase, and are useful for treating diseases such as proliferative diseases, including cancers.
[0007] Accordingly, provided herein is a compound of Formula I-IV:
and pharmaceutically acceptable salts, amorphous, and polymorph forms thereof.
PATENT
WO 2019075114
PATENT
WO-2019120194
Novel deuterated analogs of pyrazolo[1,5-a]pyrimidine compounds, particularly selpercatinib , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, inflammation, cancer and certain infectious diseases.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US10137124 | Substituted pyrazolo[1,5-a]pyridine compounds as RET kinase inhibitors | 2018-01-03 | |
| US10172851 | Substituted pyrazolo[1,5-A]pyridine compounds as RET kinase inhibitors | 2018-01-03 | |
| US10112942 | Substituted pyrazolo[1,5-A]pyridine compounds as RET kinase inhibitors | 2017-12-29 |
/////////////SELPERCATINIB, non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma, soft tissue sarcoma, LOXO, ELI LILY, ARRAY, LOXO 292, orphan drug designation
N#CC1=C2C(C3=CC=C(N4CC(C5)N(CC6=CC=C(OC)N=C6)C5C4)N=C3)=CC(OCC(C)(O)C)=CN2N=C1
Ceralasertib, AZD 6738
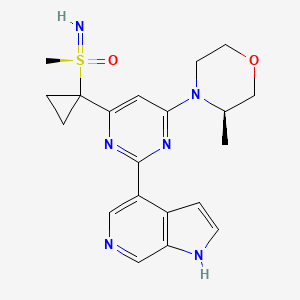
AZD-6738, Ceralasertib
- Molecular Formula C20H24N6O2S
- Average mass 412.509 Da

- 4-[4-[1-[[S(R)]-S-Methylsulfonimidoyl]cyclopropyl]-6-[(3R)-3-methyl-4-morpholinyl]-2-pyrimidinyl]-1H-pyrrolo[2,3-b]pyridine
- AZD 6738
- Ceralasertib
- Originator AstraZeneca; University of Pennsylvania
- Class Antineoplastics; Morpholines; Pyrimidines; Small molecules
- Mechanism of Action ATR protein inhibitors
- Phase II Breast cancer; Gastric cancer; Non-small cell lung cancer; Ovarian cancer
- Phase I/II Chronic lymphocytic leukaemia; Solid tumours
- Phase I Non-Hodgkin’s lymphoma
- Preclinical Diffuse large B cell lymphoma
- No development reported B-cell lymphoma; Lymphoid leukaemia
- 26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
- 18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
- 25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
- Kevin M. Foote


Patent
WO 2011154737
Example 1.01
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[((R)-S-methylsulfonimidoyl)methyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
(1 ml) to afford the title compound (34.6 mg, 49%); 1HNMR (400 MHz, CDCl3) 1.40 (3H, d), 3.17 (3H, s), 3.39 (1H, tt), 3.62 (1H, td), 3.77 (1H, dd), 3.85 (1H, d), 4.08 (1H, dd), 4.18 (1H, d), 4.37 – 4.48 (2H, q), 4.51 (1H, s), 6.59 (1H, s), 7.35 (1H, t), 7.46 (1H, d), 8.06 (1H, d), 8.42 (1H, d), 10.16 (1H, s); m/z: (ES+) MH+, 387.19.
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
3.29 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 3.99 (1H, dd), 4.02 (1H, s), 4.31 (1H, s), 6.41 (1H, s). Chiral HPLC: (HP1100 System 5, 20μm Chiralpak AD-H (250 mm × 4.6 mm) column eluting with Hexane/EtOH/TEA 50/50/0.1) Rf, 12.192 98.2%.
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
CDCI3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.23 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, d), 8.42 (1H, d); m/z: (ES+) MH+, 399.40.
Example 2.01 and example 2.02
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-blpyridine, and
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-blpyridine
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).


Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
(h) IBDA, trifluoroacetamide, MgO, DCM, Rh2(OAc)4 82%;
(i) 1,2-dibromoethane, sodium hydroxide, TOAB, 2-MeTHF, 47%;
(j) TsCl, tetrabutylammonium hydrogen sulfate, sodium hydroxide, DCM, 92%;
(k) bis(pinacolato)diboron, potassium acetate, 1,1′-bis(diphenylphosphino)ferrocene dichloro palladium(II), DMF, 62%;
(l) Pd(II)Cl2(PPh3)2, Na2CO3, DME, water, 80%;
(m) 2 N NaOH, DME, water, 92%.
Foote, K. M. N.; Johannes, W. M.; Turner, P.. Morpholino Pyrimidines and their use in therapy. WO 2011/154737 A1, 15 December 2011.
PAPER
Development and Scale-up of a Route to ATR Inhibitor AZD6738
- William R. F. Goundry et al




REFERENCES
1: Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN, Conrads TP, O’Connor MJ, Ferris RL, Tran PT, Delgoffe GM, Bakkenist CJ. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest. 2018 Jun 28. pii: 96519. doi: 10.1172/JCI96519. [Epub ahead of print] PubMed PMID: 29952768.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.
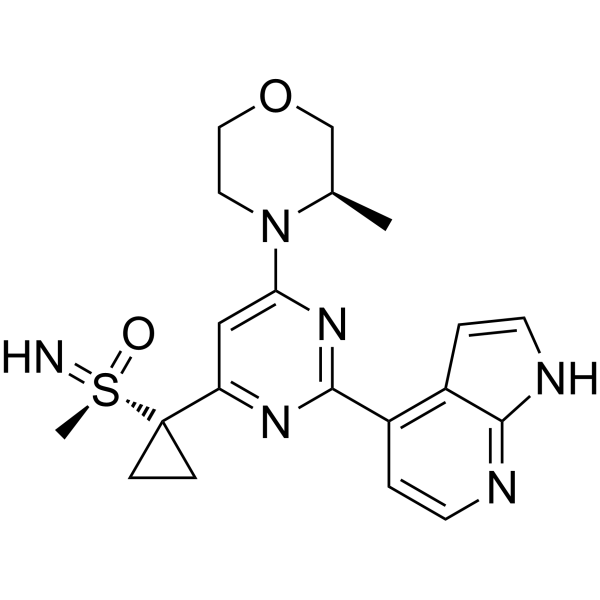



| Molecular Weight | 412.51 |
|---|---|
| Formula | C20H24N6O2S |
- Originator AstraZeneca; University of Pennsylvania
- Developer Acerta Pharma; AstraZeneca; Dana-Farber Cancer Institute; Gustave Roussy; National Cancer Institute (France); Samsung Medical Center; University of California at San Francisco; University of Pennsylvania
- Class Antineoplastics; Cyclopropanes; Imines; Ketones; Morpholines; Organic sulfur compounds; Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action ATR protein inhibitors
- Phase III Non-small cell lung cancer
- Phase IICholangiocarcinoma; Gynaecological cancer; Malignant melanoma; Osteosarcoma; Ovarian cancer; Pancreatic cancer; Prostate cancer; Small cell lung cancer; Solid tumours; Triple negative breast cancer
- Phase I/IIChronic lymphocytic leukaemia
- Phase IChronic myelomonocytic leukaemia; Myelodysplastic syndromes
- PreclinicalDiffuse large B cell lymphoma; Type 1 diabetes mellitus
- DiscontinuedHaematological malignancies; Non-Hodgkin’s lymphoma; Squamous cell cancer
- 22 Apr 2025AstraZeneca plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) (PO) in April 2025(NCT06929260)
- 23 Dec 2024AstraZeneca plans a phase I trial for Non-small Cell Lung Cancer, Ovarian Cancer, or Endometrial Cancer in United Kingdom(PO) In January 2025 (NCT06754761)
- 07 Dec 2024Updated efficacy and adverse event data from a phase I trial in Myelodysplastic syndrome presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)



Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry
- January 2022
- Organics 3(1):1-21

References
- ^ “Ceralasertib – AstraZeneca/University of Pennsylvania”. AdisInsight. Springer Nature Switzerland AG.
- ^ Mavroeidi D, Georganta A, Panagiotou E, Syrigos K, Souliotis VL (February 2024). “Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes”. International Journal of Molecular Sciences. 25 (5): 2767. doi:10.3390/ijms25052767. PMC 10932434. PMID 38474014.
- [1]. Vendetti FP, et al. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of CDDP to resolve ATM-deficient non-small cell lung cancer in vivo. [Content Brief][2]. Kim HJ, et al. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. [Content Brief]
| Clinical data | |
|---|---|
| Other names | AZD-6738 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1352226-88-0 |
| PubChem CID | 54761306 |
| IUPHAR/BPS | 9390 |
| DrugBank | DB14917 |
| ChemSpider | 58828171 |
| UNII | 85RE35306Z |
| KEGG | D11787 |
| ChEMBL | ChEMBL4285417 |
| PDB ligand | VJM (PDBe, RCSB PDB) |
| ECHA InfoCard | 100.232.607 |
| Chemical and physical data | |
| Formula | C20H24N6O2S |
| Molar mass | 412.51 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
TL 487
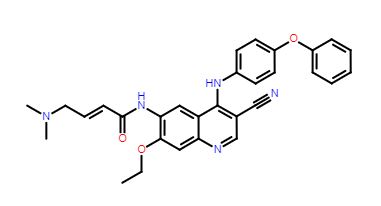
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
SEVITERONEL, севитеронел , سيفيتيرونيل , 赛维罗奈 ,
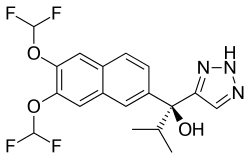
SEVITERONEL
CAS Registry Number 1610537-15-9
Molecular formulaC18 H17 F4 N3 O3, MW 399.34
1H-1,2,3-Triazole-5-methanol, α-[6,7-bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-, (αS)-
(αS)-α-[6,7-Bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-1H-1,2,3-triazole-5-methanol
8S5OIN36X4
- Mechanism of ActionAndrogen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- WHO ATC codeL01 (Antineoplastic Agents)L01X-X (Other antineoplastic agents)
- EPhMRA codeL1 (Antineoplastics)L1X9 (All other antineoplastics)
1H-1,2,3-Triazole-5-methanol, alpha-(6,7-bis(difluoromethoxy)-2-naphthalenyl)-alpha-(1-methylethyl)-, (alphaS)-
Seviteronel (developmental codes VT-464 and, formerly, INO-464) is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer.[1] It is a nonsteroidalCYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body.[1] As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer.[1] In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer.[1][2] In April 2017, seviteronel received fast-track designation for breast cancer as well.[1]
- Originator Viamet Pharmaceuticals
- Developer Innocrin Pharmaceuticals
- Clas sAntiandrogens; Antineoplastics; Fluorine compounds; Naphthalenes; Propanols; Small molecules; Triazoles
- Mechanism of Action Androgen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- Phase II Breast cancer; Prostate cancer; Solid tumours
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial in Prostate Cancer (Second-line therapy or greater, Hormone refractory) in the US (NCT02445976)
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial for Prostate Cancer (Hormone refractory) in the US, UK, Switzerland and Greece (NCT02012920)
- 31 Jan 2019 Innocrin Pharmaceuticals completes the phase I/II CLARITY-01 trial for Breast cancer (Late stage disease) in USA (NCT02580448)
- CYP-17 useful for treating fungal infections, prostate cancer, and polycystic ovary syndrome, assigned to Viamet Pharmaceuticals Inc , naming Hoekstra and Rafferty. Innocrin Pharmaceuticals , a spin-out of Viamet is developing oral seviteronel, the lead dual selective inhibitors of the 17,20-lyase activity of P450c17 (CYP17) and androgen receptor antagonist, which also includes VT-478 and VT-489, developed using the company’s Metallophile technology, for treating castration-resistant prostate cancer (CRPC) in men, breast cancer and androgen (AR) related cancers.
Pharmacology
Pharmacodynamics
Seviteronel is a nonsteroidal antiandrogen, acting specifically as an androgen synthesis inhibitor via inhibition of the enzyme CYP17A1, for the treatment of castration-resistant prostate cancer.[3][4][5][6][7][8] It has approximately 10-fold selectivity for the inhibition of 17,20-lyase (IC50 = 69 nM) over 17α-hydroxylase (IC50 = 670 nM), which results in less interference with corticosteroid production relative to the approved CYP17A1 inhibitor abiraterone acetate (which must be administered in combination with prednisone to avoid glucocorticoid deficiency and mineralocorticoid excess due to 17α-hydroxylase inhibition) and hence may be administerable without a concomitant exogenous glucocorticoid.[4][5][6][7][8] Seviteronel is 58-fold more selective for inhibition of 17,20-lyase than abiraterone (the active metabolite of abiraterone acetate), which has IC50 values for inhibition of 17,20-lyase and 17α-hydroxylase of 15 nM and 2.5 nM, respectively.[7] In addition, in in vitro models, seviteronel appears to possess greater efficacy as an antiandrogen relative to abiraterone.[6] Similarly to abiraterone acetate, seviteronel has also been found to act to some extent as an antagonist of the androgen receptor.[6]
Society and culture
Generic names
Seviteronel is the generic name of the drug and its INN.[9]
PATENT
WO2012064943
PATENT
WO-2019113312
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019113312&redirectedID=true
The present invention relates to a process for preparing compound 1 that is useful as an anticancer agent. In particular, the invention seeks to provide a new methodology for preparing compound 1 and substituted derivatives thereof.
Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-( 1,2, 4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes.
One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently-available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull. 1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable
Preparation of Compound 4:
de
Acetone (850 L), 2,3-dihydroxynaphthalene (85.00 kg, 530.7 moles), and potassium carbonate (219.3 kg, 1,586.7 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. Dimethyl sulfate (200.6 kg, 2131.09) was added to the stirred reaction at a rate that maintains the internal temperature of the exothermic reaction below 60 °C. This addition typically requires about 3 hours. At the end of the dimethyl sulfate addition, the reaction is continued to allow to stir while maintaining the internal temperature at 50 – 60 °C. After about 3 hours, the reaction was analyzed by HPLC. The reaction was concentrated by atmospheric pressure distillation of acetone. The distillation was continued until 340 – 425 L of distillate was collected. This represents 40 – 50 % of the initial charge of acetone. At the end of the distillation, the reaction mass is present as a thick suspension. While maintaining the internal temperature below 60 °C, the reactor contents were slowly diluted with water (850 L). When the addition is complete, the reaction was cooled to an internal temperature of 25 – 35 °C and stirring was continued for 1 – 2 hours after the designated internal temperature was reached. Compound 2 was isolated by filtration and the cake was washed with water (at least 3 X 85 L). Compound 2 was dried at 40 – 45 °C and full vacuum until the water content by Karl Fisher titration is found to be NMT 2.0 %. Typically, greater than 90 kg of dry product is obtained with an assay of >99.5% AUC by HPLC.
Dichloromethane (with a water content by Karl Fisher Titration of NMT 0.50%) (928 L) and 2,3-dimethoxynaphthalene (2, 116.00 kg, 616.3 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. The reactor contents were cooled to an internal temperature of -5 to 0 °C. Aluminum chloride (164.72 kg, 1235.3 moles, 2.00 molar equivalents) was carefully added in portions to the reaction, while maintaining the internal temperature at -5 to +5 °C. This addition typically requires 5 – 6 hours. At the end of the addition, the reactor contents were cooled to an internal temperature of -15 to -5 °C. Isobutyryl chloride (102.08 kg, 958.05 moles, 1.55 molar equivalents) was slowly added to the reaction while maintaining the internal temperature at -15 to -5 °C. The addition typically requires about 3 hours. At the end of the isobutyryl chloride addition, the reaction was warmed to an internal temperature of 20 – 35 °C. When the temperature was reached, these conditions were maintained for 2 – 3 hours until the IPC indicated a level of residual starting material of NMT 2.0 % AUC by HPLC. The reactor contents were then cooled to 0 – 5 °C. The reaction was quenched by adding the reaction to a precooled (0 – 5 °C) 3M aqueous solution of hydrochloric hcid (Water, 754 L: cone. HC1, 406 L). The mixture was vigorously stirred for 15 – 20 minutes then the layers were allowed to settle. The lower, dichloromethane, product-containing layer was washed sequentially with 10 % aqueous sodium bicarbonate (1044 L), water (1160 L), then 10 % aqueous sodium chloride (1044 L). The reaction was concentrated by distillation under full vacuum and at an internal temperature of NMT 40 °C. The reaction concentrate was cooled to 20 – 35 °C and diluted with hexanes (812 L). The resultant slurry was warmed to 45 – 50 °C and these conditions were maintained for 1 – 2 hours. The reactor contents were cooled to 20 – 35 °C for 1 – 2 hours. Compound 3 was isolated by filtration. The cake was washed with fresh hexanes (232 L) twice, the filter was cooled, and the cake was washed an additional two times with hexanes. Compound 3 was dried under full vacuum at a jacket temperature of 45 °C. Typically, about 95 kg of dry product was isolated with a product purity of >90% by HPLC.
Acetic acid (212.5 L L) and l-(6,7-dimethoxynaphthalene-2-yl)-2-methylpropane-l- one (42.5 kg, 164.5 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 25 – 45 °C. Concentrated hydrochloric acid (425.0 L) was added carefully to the stirring reactor contents while maintaining reactor contents at an internal temperature of 25 – 45 °C. When the addition was complete, the internal temperature of the reaction was raised to 100 – 105 °C. Note that the reaction is a heterogeneous mixture. The reaction was stirred under these conditions for 6 – 8 hours. The reaction was cooled to 85 – 90 °C to which was carefully added a fresh portion of hydrochloric acid (127.5 L). The reaction was warmed to 100 – 105 °C and stirred for another 6 – 8 hours. The reaction was cooled to 85 – 90 °C. The reaction was cooled further to 70 – 80 °C. Water (212.5 L) was added to the well stirred reaction and the reactor contents were cooled to an internal temperature of 35 – 45 °C and stirred for 3 – 4 hours. Compound 4 was collected by filtration. The wet cake was washed with water (212.5 L). The wet cake was added to a clean reactor with a 5% aqueous sodium bicarbonate solution and stirred at an internal temperature of 35 – 45 °C for 1 – 2 hours.
Compound 4 was collected by filtration and washed with water (212.5 L). Compound 4 was dried under full vacuum and a temperature of < 50 °C until the water content of the dried material was found to be NMT 5.0% by Karl Fisher Titration. The yield is typically >31 kg with a purity >99.5 %.
Preparation of Compound 5:
The following difluoromethylation conditions listed in Table 1 were investigated:
Preparation 1:
The reaction flask was dried under an argon flow at 120 °C. (lS,2R)-l-Phenyl-2-(l- pyrrolidinyl)propan-l-ol (ligand 45) (196.6 g, 0.96 mol, 2.2 eq.) was added into the flask and then toluene (195 mL) was added. The solution was cooled to <12 °C. A solution of diethyl zinc (716.4 g, 0.87 mol, 15 wt%, 2 eq.) in toluene was added through a septum over 30 min at 0-10 °C. Further, a solution of ((Trimethylsilyl)ethynyl)-magnesium bromide in THF (1.81 kg; 0.87 mol, 9.7 wt%, 2 eq.) was added over 30 min at 0-10 °C. Finally, trifluoroethanol (87.0 g; 0.87 mol; 2 eq.) was added over 10 min at 0-10 °C. The reaction solution was stirred at 10-12 °C for 3 h. Compound 5 (143.4 g; 0.434 mol; 1 eq.) was added (as a solid) at room
temperature. The reaction mixture was stirred at room temperature for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed with aqueous HC1 (3600 mL; 7.5 wt%) within 20 min. The temperature of the mixture was kept below 25 °C. Toluene (1250 mL) was added and the mixture was stirred at room temperature for 5 min. The aqueous phase was separated and stored for the recycling of ligand 45. The organic phases were washed with water (638 mL) and concentrated via distillation under reduced pressure (50 mbar). The residue (approx. 184 g) was treated with heptane (200 mL), which was removed
via distillation. The residue was dissolved in heptane (2050 mL) at 50 °C. The mixture was cooled to room temperature and subsequently to -8 °C within 2 hours. The obtained suspension was stirred at -8 °C for 1 h. Crystallized compound 5 (20.0 g; 14%) was isolated via filtration, washed twice with cold (0 °C) heptane (2×20 mL) and dried under vacuum at 50 °C for 12 hours. The combined heptane phases were concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (yield: 83.0%). The solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Preparation 2:
(7S,2R)-l-Phenyl-2-(l-pyrrolidinyl)propan-l-ol (ligand 45) (13.0 kg, 63.3 mol, 2.2 eq.) was charged into the reactor and toluene (60 L) was added. The solution was cooled to < 12 °C. A solution of diethyl zinc (35.6 kg, 57.3 mol, 20 wt%, 2 eq.) in toluene was added via mass flow controller at 8-16 °C. Further, a solution of ((trimethylsilyl)ethynyl)-magnesium bromide in THF (11.5 kg; 57.3 mol, 9.7 wt%, 2 eq.) was added at 8-16 °C. Finally, trifluoroethanol (5.7 kg; 57.3 mol; 2 eq.) was added over 10 min at 8-16 °C.The reaction solution was stirred at 22-25 °C for 3 h. A solution of compound 5 (9.5 kg; 28.7 mol; 1 eq.) in toluene (20 L) was added at room temperature. The reaction mixture was stirred at 25 °C for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed in aqueous HC1 (225L; 7.5 wt%) within 20 min. The temperature of the mixture should be kept below 25 °C. Toluene (80 L) was added and the mixture was stirred at room temperature for 5 min. The organic phases was washed with water (50 L) and concentrated via distillation under reduced pressure (50 mbar). The residue was treated with heptane (100 L), which was removed via distillation. The residue was dissolved in heptane (100 L) at 50°C, which was removed via distillation. The residue was dissolved in heptane (25 L). Heptane (110 L) was added, the mixture was cooled to room temperature and subsequently to 0-5 °C and seeded with compound 5 (0.15 kg). The obtained suspension was cooled to -8 °C within 1 h and stirred at this temperature for 2 h. Crystallized compound 5 was removed via filtration. The filtrate was concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (calculated 8.8 kg, 71.6%). This solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Recovery of the chiral ligand ( lS,2R)-l-Phenvl-2-
-l-ol from the
Preparation 1:
The above acidic aqueous phase was diluted with toluene (1000 mL) and the mixture was treated with sodium hydroxide (50 wt% solution) to adjust the pH to 12. The mixture was warmed to 50 °C and sodium chloride (100 g) was added. The aqueous phase was separated and washed with toluene (1000 mL). The combined organic phases were washed with water (200 mL). The combined toluene phases were treated with water (1000 mL) and the pH was adjusted to 2 by the addition of a cone. HC1 solution. The aqueous phase was separated and the mixture was treated with sodium hydroxide (50 wt% solution) at 5 °C to adjust the pH to 12. After seeding, the suspension was stirred at 5 °C for 30 min. The solids were isolated, washed with cold (0 °C) water (4×100 mL) and dried under vacuum at 30 °C for 24 hours. Ligand 45 (178.9g; 91%) was obtained as slightly yellow crystalline solid.
HPLC (purity): 99%.
Preparation 2:
The acidic aqueous phase containing ligand 45 (500 L) was diluted with toluene (125 L) and treated with“Kieselgur” (20 L). The mixture was treated with sodium hydroxide (40 L; 50 wt% solution) to adjust the pH to 12 whereas the temperature was kept <55 °C. The suspension was stirred for 15-20 min and filtered to remove all solids. Toluene (80 L) was added and the aqueous phase was separated. The organic phase was treated with water (150 mL) and the pH was adjusted to 1.5-2 by the addition of an aqueous HC1 solution (10 L; 32 wt%). The aqueous phase was separated, toluene (150 L) was added, and the mixture was treated with sodium hydroxide (5 L; 50 wt% solution) at 5 °C to adjust the pH to 12-12.5. The organic phase was separated, washed with water (30 L), and concentrated under reduced
pressure at 50 °C. Approx. 100L of distillate was removed. A sample of the solution of ligand 45 in toluene was analyzed:
The NMR results indicated a 21.6 wt% solution of ligand 45 in toluene which corresponds to a calculated amount of 118.4 kg (83.6%) of ligand 45.
Preparation of Compound 18a
Preparation 1:
A solution of tertiary alcohol 18b (320 g; 48 wt%; 0.36 mol; 1 eq.) in heptane was dissolved in methanol (800 mL). Potassium carbonate (219 g; 1.58 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at room temperature for 3 h. Water (1250 mL) was added and the mixture was treated with a cone. HC1 solution (approx. 130 mL) to adjust the pH to 7.8. The reaction mixture was extracted twice with methyl- /-butyl ether (MTBE; 2×465 mL). The combined MTBE phases were washed with water (155 mL). Water (190 mL) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (50 mbar). The obtained emulsion of compound 18a (yield: 99%) was directly used for the next step.
1H-NMR (600.6 MHz, CDC13) d: 0.87 (d, J = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / =
8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation 2:
The solution of tertiary alcohol 18b (48 wt%; 57.5 mol; 1 eq.) in heptane was dissolved in methanol (128 L). Potassium carbonate (35.0 kg; 253 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at 20-30 °C for 3 h. Water (200 L) was added and the mixture was treated with an aqueous HC1 solution (approx. 25 L; 32 wt%) to adjust the pH to 7.5 – 7.8. The reaction mixture was extracted twice with MTBE
(2×66.6 L). The combined MTBE phases were washed with water (25 L). Water (30 L) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (<80 mbar; 55°C). The residue was dissolved in tert-butanol (25 L). The resulting 18a was cooled to <30°C and used directly in the next step.
^-NMR (600.6 MHz, CDC13) d: 0.87 (d, / = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / = 8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation of Compound 31
Preparation 1:
Benzyl bromide (39.4 g; 0.23 mol; 1 eq.) was dissolved in water (177 mL) and t-BuOH (200 mL). Diisopropylethylamine (DIPEA; 59.4 g; 0.46 mol; 2 eq.) and sodium azide (15.0 g; 0.23 mol; 1 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of compound 18a (82 g; 0.23 mol; 1 eq.) in water (123 mL) was treated with t-BuOH (100 mL) and copper (I) iodide (8.8 g; 46 mmol; 0.2 eq.) was added and the temperature was kept below 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (5.0 g; 76 mmol) and ammonium chloride (7.4 g; 0.14 mol) were added and the reaction mixture was stirred at room temperature for 3 hours. The mixture was diluted with MTBE (800 mL), water (280 mL), and an aqueous ammonia solution (120 g; 25 wt%). Solids were removed by filtration and additional MTBE (200 mL) and brine (200 mL) were added. The aqueous phase was separated and extracted with MTBE (400 mL). The combined organic phases were treated with water (150 mL) and MTBE was distilled off under reduced pressure (100 mbar). The obtained suspension of compound 31 (113 g; 50 wt%) in water (approx. 113 mL) was directly used for the next step.
Ή-NMEI (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 2:
Benzyl bromide (11.0 kg g; 64.4 mol; 1,12 eq.) was dissolved in water (40 L) and t-BuOH (60 L). DIPEA (16.4 kg; 126.5 mol; 2,2 eq.) and sodium azide (4.12 kg; 63.3 mol; 1 eq.) were added. The suspension was stirred 5 min at room temperature. A mixture of compound 18a (20.5 kg; 57.5 mol; 1 eq.) in ieri-butanol (see previous step) was added together with water (5 L) and copper (I) iodide (2.2 kg; 11.5 mol; 0.2 eq.) at a temperature < 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (1.25 kg; 19 mol, 0.33 eq.) and an aqueous solution of ammonium chloride (2.14 kg; 20 wt%; 40 mol; 0.7 eq.) were added and the reaction mixture was stirred at 20-30 °C for 2 hours. The reaction mixture was concentrated under vacuum (<200 mbar, 55 °C). The residue was diluted with MTBE (200 L), water (30 L), and an aqueous ammonia solution (30 kg; 25 wt%). Solids were removed by filtration over a pad of“Kieselgur NF” (2 kg). Brine (50 L) was added for a better phase separation. The aqueous phase was separated and washed with MTBE (200 L). The combined organic phases were washed with an aqueous HC1 solution (1 N, 52 L) and water (50 L). MTBE was distilled off under reduced pressure (<400 mbar, 55°C; distillate min. 230L). The oily residue was dissolved in ethanol (150 L), which was distilled off under reduced pressure (<300 mbar; 55°C; distillate min. 150-155L) and the residue was dissolved in additional ethanol (60 L). To the resulting solution of compound 31 was added water (24 L) and the mixture was warmed to 50-55 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (2 x 12 L). The wet product was dissolved in ethanol (115L) at 60 °C and water (24 L) was added. The mixture was cooled to 40 °C and the crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for additional 2 hours. The solids were isolated and washed (without stirring) with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 was isolated as a white solid, which was used for the next step without drying. 14.0 kg of wet 31 were obtained with a 31 content of 81.6 wt%. Based on the determined content, the calculated amount of pure 31 was 11.4 kg with a yield of 41% over two steps (from 18b).
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 3: Synthesis of compound 31 directly from compound 18b
Benzyl bromide (1.64 g, 9.59 mmol, 1.12 eq) was dissolved in water (2.4 mL) and
MeOH (2.4 mL). K2CO3 (2.38 g, 17.2 mmol, 2.00 eq), sodium ascorbate (0.34 g, 1.72 mmol, 0.20 eq) and finally sodium azide (0.62 g, 9.40 mmol, 1.10 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of 18b (3.08 g; 8.64 mmol, 1.00 eq) in water (2.5 mL) and MeOH (2.5 mL) and the resulting mixture was stirred for 10 min.
CuS04 (0.21 g, 1.30 mmol, 0.15 eq) were added (slightly exothermic reaction). The reaction mixture was stirred for 19 h and the conversion was determined by HPLC (conv. 100%, purity of compound 31 by HPLC: 83 area%). To the yellow-green suspension was added zinc powder (0.24 g, 4.13 mmol, 0.43 eq) and ammonium chloride (0.34 g, 6.36 mmol, 0.74 eq) were added and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure (150 mbar, 50 °C). The mixture was diluted with MTBE (40 mL), water (15 mL), and an aqueous ammonia solution (6.5 mL). Solids were removed by filtration and brine (5.5 mL) was added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were treated with water (10 mL) and the pH was adjusted to a pH of 1 by addition of cone. HC1. After phase separation, the organic layer was washed with water (10 mL). MTBE was distilled off under reduced pressure (100 mbar, 50°C) to give the crude compound 31 as an oil. Water (2.5 mL) and EtOH (30 mL) were added and the mixture was warmed to 50 °C. After cooling to 30 °C, the mixture was seeded with compound 31 and compound 31 started to precipitate. The mixture was kept for 1 h at 30 °C, then cooled to 0 °C over 2 h and kept at 0 °C for 2 h. The resulting product, 31, was collected by filtration and the filter cake was washed with small portions of EtOH/water (1:1). After drying, the product (2.97 g) was obtained as a pale yellow, crystalline solid with an HPLC purity of 79 area% and a NMR content of ca. 70 wt%.
Recrystallization of
31
Preparation 1:
To a suspension of compound 31 (96 g; 0.196 mol; 50 wt%) in water (96 mL) was added ethanol (480 mL) and the mixture was warmed to 50 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 40 mL). The wet product was dissolved in ethanol (280 mL) at 60 °C and water (56 mL) was added. The mixture was cooled to 40 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 28 mL). Pure, wet compound 31 (46.8 g on dried basis; 49 % over 2 steps) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.5%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation 2:
14 kg of ethanol-wet 31 (content 81.6 wt%, calculated 11.4 kg, 23.7 mol) were suspended in ethanol (46 L) and the mixture was warmed to 50-55 °C, forming a homogenous solution at this temperature. Water (9 L) was added at 50-55 °C and the mixture was cooled to 40-45 °C. After the crystallization had started, the suspension was stirred at 40-45 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 (14.5 kg) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.8%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation of Azidomethyl Pivalate Protected Triazole (6) from Compound 18a
1
Azidomethyl pivalate (1.42 g, 9.00 mmol, 1.05 eq) was suspended in water (6.0 mL) and t-BuOH (7.2 mL) and the suspension was stirred for 5 min. Compound 18a (theor. 3.08 g, 8.64 mmol, 1.00 eq), sodium ascorbate (0.48 g, 2.4 mmol, 0.30 eq), and CuS04 (0.08 g, 0.40 mmol, 0.05 eq.) were added. The reaction mixture was stirred for 19 h and conversion was determined by HPLC (conv. 98%, purity of the product by HPLC: 81 area%). To the green suspension was added MTBE (20 mL), water (10 mL), and an aqueous ammonia solution (2 g). A biphasic turbid mixture was formed. To improve phase separation, additional MTBE (20 mL) and water (10 mL) were added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give the crude product as a brown oil that solidified upon standing. HPLC purity: ca. 65 area%; NMR content of ca. 73 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Azidomethyl Pivalate Protected Triazole (6) from 18b
In a reaction flask, sodium ascorbate (277 mg, 1.4 mmol, 1.20 eq) and CuS04 (37 mg, 0.23 mmol, 0.20 eq.) were suspended in MeOH (11 mL). Azidomethyl pivalate (183 mg, 1.16 mmol, 1.00 eq) and 18b (183 mg, 1.16 mmol, 1.00 eq) were added and the mixture was warmed to 60 °C. The reaction mixture was stirred for 19 h and worked up. To the green suspension was added an aq NH4Cl solution (2 mL) and zinc powder, and the mixture was stirred for 2 h. MTBE (2 mL) was added and the aqueous phase was separated and extracted with MTBE (2 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give 6 as a brown oil that solidified upon standing. HPLC purity: ca. 81 area%; NMR content of ca. 57 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Compound 1
Preparation 1:
Compound 31 (26 g; 53 mmol; 1 eq.) was dissolved in ethanol (260 mL) and Noblyst Pl 155 (2.2 g; 10 % Pd; 54 wt% water) was added. The autoclave was flushed with nitrogen and hydrogen (5 bar) was added. The reaction mixture was stirred at room temperature for 32 hours. The reaction mixture was treated with charcoal (2 g), stirred for 15 min, and the charcoal was filtered off. The filtrate was concentrated via distillation and the residue (approximately 42 g) was diluted with heptane (200 mL). The mixture was heated to reflux to
obtain a clear solution. The solution was cooled to room temperature within 1 h and the resulting suspension was cooled to 0 °C and stirred for 2 hours at 0 °C. The solids were isolated via filtration and washed with heptane/ethanol (10:1; v/v; 3×10 mL). Compound 1 (18.0 g; 85 %) was dried under vacuum at 60 °C for 24 hours and obtained as a white, crystalline solid.
1H-NMR (600 MHz) d: 0.80 (d, J = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 2:
Compound 31 (26.5 kg; 53.5 mol; 1 eq.) was dissolved in ethanol (265 L) and Pd/C (2.0 kg; 10 % Pd; 54 wt% water) was added. The reactor was flushed with nitrogen, and hydrogen (4.5 bar) was added. The reaction mixture was stirred at 28-32 °C until the reaction was complete. The reaction mixture was treated with charcoal (1.3 kg) at a temperature of <
33 °C, stirred for 10 min, and the charcoal was filtered off, and the filter was washed with ethanol (10 L).The filtrates from two reactions were combined and concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 480 L). The residue (approx. 50-60 L) was diluted with isopropylacetate (250 L). The mixture was again concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 240-245 L). The residue (approx. 60-70 L) was cooled to 35-40 °C and isopropylacetate (125 L) and heptane (540 L) were added. The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). Wet 1 was dried under vacuum at 60 °C and was obtained as a white, crystalline solid (35.4 kg, 81.9%).
1H-NMR (600 MHz) d: 0.80 (d, / = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 3: Preparation of Compound 1 from Compound 6
At room temperature, 6 (3.00 g, 5.84 mmol) was dissolved in MeOH (19.8 mL). NaOH (1.0 M, 19.8 mL) was added in one portion and the reaction mixture was stirred for 1 h at room temperature. The reaction progress was monitored by HPLC, which showed 98% conversion after 1 h. Aq. HC1 (19.8 mL) was added and the mixture was diluted with water (120 mL) and MTBE (60 mL), resulting in a clear biphasic solution. After phase separation, the organic phase was washed with aq NaHC03 (20 mL). The organic layer was concentrated under high vacuum (25 mbar, 45 °C) to yield 2.77 g of 1 as a greenish oil. The identity was confirmed by comparison of HPLC retention time with an authentic sample of 1 as well as by 1H NMR.
Recrystallization of Compound 1
Wet 1 (40 kg; isopropylacetate/heptane wet) was treated with isopropylacetate (110 L) and heptane (440 L). The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with
heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). A sample was taken for analysis
(criterion: a) purity; NLT 99.0 A% by HPLC; b) single impurities, NMT 0.15 A% by HPLC; c) enantiomer VT-463, NMT 1.0 A% by HPLC). Wet 1 was dried under vacuum at 60 °C for not less than 12 h. A sample was taken for analysis: criterion: a) LOD; NMT 0.5 wt% by gravimetry; b) residual toluene, NMT 890 ppm by HS-GC. 1 was obtained as a white, crystalline solid (28.5 kg, 66.7% from 31).
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(11), 2444-2447.
https://www.sciencedirect.com/science/article/pii/S0960894X14003606

PATENT
WO 2016040896
https://patents.google.com/patent/WO2016040896A1/en
References
- ^ Jump up to:a b c d e http://adisinsight.springer.com/drugs/800035241
- ^ http://www.pharmaceutical-technology.com/news/newsfda-grants-fast-track-status-innocrins-seviteronel-treat-metastatic-crpc-4770025
- ^ Yin L, Hu Q, Hartmann RW (2013). “Recent progress in pharmaceutical therapies for castration-resistant prostate cancer”. Int J Mol Sci. 14 (7): 13958–78. doi:10.3390/ijms140713958. PMC 3742227. PMID 23880851.
- ^ Jump up to:a b Stein MN, Patel N, Bershadskiy A, Sokoloff A, Singer EA (2014). “Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer”. Asian J. Androl. 16 (3): 387–400. doi:10.4103/1008-682X.129133. PMC 4023364. PMID 24759590.
- ^ Jump up to:a b Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ (2014). “Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors”. Bioorg. Med. Chem. Lett. 24 (11): 2444–7. doi:10.1016/j.bmcl.2014.04.024. PMID 24775307.
- ^ Jump up to:a b c d Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME (2015). “Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer”. Mol. Cancer Ther. 14 (1): 59–69. doi:10.1158/1535-7163.MCT-14-0521. PMID 25351916.
- ^ Jump up to:a b c Stephen Neidle (30 September 2013). Cancer Drug Design and Discovery. Academic Press. pp. 341–342. ISBN 978-0-12-397228-6.
- ^ Jump up to:a b Wm Kevin Kelly; Edouard J. Trabulsi, MD; Nicholas G. Zaorsky, MD (17 December 2014). Prostate Cancer: A Multidisciplinary Approach to Diagnosis and Management. Demos Medical Publishing. pp. 342–. ISBN 978-1-936287-59-8.
- ^ http://www.who.int/medicines/publications/druginformation/innlists/RL76.pdf
Further reading
- Gomez L, Kovac JR, Lamb DJ (2015). “CYP17A1 inhibitors in castration-resistant prostate cancer”. Steroids. 95: 80–7. doi:10.1016/j.steroids.2014.12.021. PMC 4323677. PMID 25560485.
- Bambury RM, Rathkopf DE (2015). “Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide”. Urol. Oncol. 34: 348–55. doi:10.1016/j.urolonc.2015.05.025. PMID 26162486.
External links[
 |
|
| Clinical data | |
|---|---|
| Synonyms | VT-464; INO-464 |
| Routes of administration |
By mouth |
| Drug class | Androgen biosynthesis inhibitor; Nonsteroidal antiandrogen |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C18H17F4N3O3 |
| Molar mass | 399.339 g/mol g·mol−1 |
| 3D model (JSmol) | |
References
-
Innocrin Pharmaceuticals Created as a Spin-out of the Prostate Cancer Program from Viamet Pharmaceuticals.
-
Viamet Pharmaceuticals and the Novartis Option Fund Enter Agreement for Development of Novel Metalloenzyme Inhibitors.
-
Innocrin Pharmaceuticals, Inc. Granted SME Status Designation by the European Medicines Agency.
-
A Single arm, open label, signal seeking, Phase II a trial of the activity of seviteronel in patients with androgen receptor (AR) positive solid tumours
-
Innocrin Pharmaceuticals and the Prostate Cancer Foundation (PCF) Join Forces for Innovative Phase 2 Clinical Study.
-
A Phase 2 Open-label Study to Evaluate the Efficacy and Safety of Seviteronel in Subjects With Castration-Resistant Prostate Cancer Progressing on Enzalutamide or Abiraterone
-
Innocrin Pharmaceuticals, Inc. Granted Fast Track Designation by FDA for VT-464 Treatment of Patients with Metastatic Castrate-resistant Prostate Cancer.
-
Innocrin Pharmaceuticals, Inc. Begins Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer and Expands Two Phase 2 Studies of Seviteronel in Men with Metastatic Castrate-resistant Prostate Cancer.
-
A Phase 2 Open-Label Study to Evaluate the Efficacy and Safety of VT-464 in Patients With Metastatic Castration Resistant Prostate Cancer Who Have Previously Been Treated With Enzalutamide, Androgen Receptor Positive Triple-Negative Breast Cancer Patients, and Men With ER Positive Breast Cancer
-
Innocrin Pharmaceuticals Inc. to Present Interim Results from Its Phase 1/2 Prostate Cancer Clinical Study and Preclinical Results That Demonstrate VT-464 Efficacy in a Clinically-Relevant Enzalutamide-Resistant Mouse Model.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of Seviteronel in Subjects With Castration-Resistant Prostate Cancer
-
A Phase 1/2 Open-Label, Multiple-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Once-Daily VT-464 in Patients With Castration-Resistant Prostate Cancer
-
Viamet Pharmaceuticals Appoints Former Novartis Executive Marc Rudoltz, M.D. as Chief Medical Officer.
-
VIAMET PHARMACEUTICALS AND THE NATIONAL INSTITUTES OF HEALTH TO JOINTLY DEVELOP NOVEL VIAMET COMPOUND.
-
Viamet Pharmaceuticals Initiates Phase 1/2 Clinical Trial of Novel Prostate Cancer Therapy, VT-464.
-
Viamet Pharmaceuticals to Present at the 32nd Annual J.P. Morgan Healthcare Conference.
-
VIAMET PHARMACEUTICALS TO PRESENT AT THE 31st Annual J.P. MORGAN HEALTHCARE CONFERENCE.
-
Innocrin Pharmaceuticals, Inc. Initiates Phase 2 Castration-Resistant Prostate Cancer (CRPC) Study in Men Who Have Failed Enzalutmaide or Abiraterone.
-
Innocrin Pharmaceuticals Appoints Fred Eshelman, PharmD as CEO and is Granted Fast Track Designation by FDA for Seviteronel Treatment of Women with Triple-negative Breast Cancer and Women or Men with Estrogen Receptor-positive Breast Cancer.
-
Gucalp A, Bardia A, Gabrail N, DaCosta N, Danso M, Elias AD, et al. Phase 1/2 study of oral seviteronel (VT-464), a dual CYP17-lyase inhibitor and androgen receptor (AR) antagonist, in patients with advanced AR positive triple negative (TNBC) or estrogen receptor (ER) positive breast cancer (BC). SABCS-2016 2016; abstr. P2-08-04.
Available from: URL:http://www.abstracts2view.com/sabcs/view.php?nu=SABCS16L_1479
-
Innocrin Pharmaceuticals Presents Data from the Ongoing Phase 2 Trial of Seviteronel in Estrogen Receptor-positive or Triple-negative Breast Cancer (CLARITY-01) at the San Antonio Breast Cancer Symposium.
-
Innocrin Pharmaceuticals, Inc. Appoints Edwina Baskin-Bey, MD as Chief Medical Officer and Expands the Ongoing Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer (TNBC).
-
Innocrin Pharmaceuticals, Inc. Raises $28 Million in Series D Financing.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Efficacy of Seviteronel in Subjects With Advanced Breast Cancer
-
Speers CW, Chandler B, Zhao S, Liu M, Wilder-Romans K, Olsen E, et al. Radiosensitization of androgen receptor (AR)-positive triple-negative breast cancer (TNBC) cells using seviteronel (SEVI), a selective CYP17 lyase and AR inhibitor. ASCO-2017 2017; abstr. e12102.
Available from: URL: http://abstracts.asco.org/199/AbstView_199_193240.html
-
Innocrin Pharmaceuticals, Inc. Appoints Charles F. Osborne Jr. as its Chief Financial Officer.
-
Viamet Pharmaceuticals Secures $18 Million Financing.
-
Viamet Pharmaceuticals Raises $4 Million Round of Financing.
///////////SEVITERONEL, VT-464, INO-464, VT 464, INO 464, Phase II, Breast cancer, Prostate cancer, Solid tumours, viamet, CANCER, севитеронел , سيفيتيرونيل , 赛维罗奈 ,
C1(=CN=NN1)C(C1=CC2=C(C=C1)C=C(C(=C2)OC(F)F)OC(F)F)(C(C)C)O
FDA approves first PI3K inhibitor Piqray (alpelisib) for breast cancer

FDA approves first PI3K inhibitor for breast cancer
syn https://newdrugapprovals.org/2018/06/25/alpelisib-byl-719/
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by
- May 24, 2019
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by the therascreen test using the liquid biopsy should undergo tumor biopsy for PIK3CA mutation testing.
“Piqray is the first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer. The ability to target treatment to a patient’s specific genetic mutation or biomarker is becoming increasingly common in cancer treatment, and companion diagnostic tests assist oncologists in selecting patients who may benefit from these targeted treatments,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For this approval, we employed some of our newer regulatory tools to streamline reviews without compromising the quality of our assessment. This drug is the first novel drug approved under the Real-Time Oncology Review pilot program. We also used the updated Assessment Aid, a multidisciplinary review template that helps focus our written review on critical thinking and consistency and reduces time spent on administrative tasks.”
Metastatic breast cancer is breast cancer that has spread beyond the breast to other organs in the body (most often the bones, lungs, liver or brain). When breast cancer is hormone-receptor positive, patients may be treated with anti-hormonal treatment (also called endocrine therapy), alone or in combination with other medicines, or chemotherapy.
The efficacy of Piqray was studied in the SOLAR-1 trial, a randomized trial of 572 postmenopausal women and men with HR-positive, HER2-negative, advanced or metastatic breast cancer whose cancer had progressed while on or after receiving an aromatase inhibitor. Results from the trial showed the addition of Piqray to fulvestrant significantly prolonged progression- free survival (median of 11 months vs. 5.7 months) in patients whose tumors had a PIK3CA mutation.
Common side effects of Piqray are high blood sugar levels, increase in creatinine, diarrhea, rash, decrease in lymphocyte count in the blood, elevated liver enzymes, nausea, fatigue, low red blood cell count, increase in lipase (enzymes released by the pancreas), decreased appetite, stomatitis, vomiting, weight loss, low calcium levels, aPTT prolonged (blood clotting taking longer to occur than it should), and hair loss.
Health care professionals are advised to monitor patients taking Piqray for severe hypersensitivity reactions (intolerance). Patients are warned of potentially severe skin reactions (rashes that may result in peeling and blistering of skin or mucous membranes like the lips and gums). Health care professionals are advised not to initiate treatment in patients with a history of severe skin reactions such as Stevens-Johnson Syndrome, erythema multiforme, or toxic epidermal necrolysis. Patients on Piqray have reported severe hyperglycemia (high blood sugar), and the safety of Piqray in patients with Type 1 or uncontrolled Type 2 diabetes has not been established. Before initiating treatment with Piqray, health care professionals are advised to check fasting glucose and HbA1c, and to optimize glycemic control. Patients should be monitored for pneumonitis/interstitial lung disease (inflammation of lung tissue) and diarrhea during treatment. Piqray must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Piqray is the first new drug application (NDA) for a new molecular entity approved under the Real-Time Oncology Review (RTOR) pilot program, which permits the FDA to begin analyzing key efficacy and safety datasets prior to the official submission of an application, allowing the review team to begin their review and communicate with the applicant earlier. Piqray also used the updated Assessment Aid (AAid), a multidisciplinary review template intended to focus the FDA’s written review on critical thinking and consistency and reduce time spent on administrative tasks. With these two pilot programs, today’s approval of Piqray comes approximately three months ahead of the Prescription Drug User Fee Act (PDUFA) VI deadline of August 18, 2019.
The FDA granted this application Priority Review designation. The FDA granted approval of Piqray to Novartis. The FDA granted approval of the therascreen PIK3CA RGQ PCR Kit to QIAGEN Manchester, Ltd.
//////////////FDA, PI3K inhibitor, breast cancer, fda 2019, Piqray, alpelisib, therascreen PIK3CA RGQ PCR Kit, QIAGEN Manchester, Priority Review, BYL719, BYL 719
Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

FDA approves Pfizer’s Ibrance (palbociclib) for postmenopausal women with advanced breast cancer
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
FDA approves Ibrance for postmenopausal women with advanced breast cancer
February 3, 2015
syn……….https://newdrugapprovals.org/2014/01/05/palbociclib/
The U.S. Food and Drug Administration today granted accelerated approval to Ibrance (palbociclib) to treat advanced (metastatic) breast cancer.
Breast cancer in women is the second most common type of cancer in the United States. It forms in the breast tissue and in advanced cases, spreads to surrounding normal tissue. The National Cancer Institute estimates that 232,670 American women were diagnosed with breast cancer and 40,000 died from the disease in 2014.
Ibrance works by inhibiting molecules, known as cyclin-dependent kinases (CDKs) 4 and 6, involved in promoting the growth of cancer cells. Ibrance is intended for postmenopausal women with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have not yet received an endocrine-based therapy. It is to be used in combination with letrozole, another FDA-approved product used to treat certain kinds of breast cancer in postmenopausal women.
“The addition of palbociclib to letrozole provides a novel treatment option to women diagnosed with metastatic breast cancer,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to expediting marketing approval of cancer drugs through our accelerated approval regulations.”
syn……….https://newdrugapprovals.org/2014/01/05/palbociclib/
The FDA granted Ibrance breakthrough therapy designation because the sponsor demonstrated through preliminary clinical evidence that the drug may offer a substantial improvement over available therapies. It also received a priority review, which provides for an expedited review of drugs intended to provide a significant improvement in safety or effectiveness in the treatment of a serious condition or meet an unmet medical need. Ibrance is being approved more than two months ahead of the prescription drug user fee goal date of April 13, 2015, the date when the agency was scheduled to complete its review of the application.
Ibrance is being approved under the FDA’s accelerated approval program, which allows approval of a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients. This program provides earlier patient access to promising new drugs while the company conducts confirmatory clinical trials.
The drug’s efficacy was demonstrated in 165 postmenopausal women with ER-positive, HER2-negative advanced breast cancer who had not received previous treatment for advanced disease. Clinical study participants were randomly assigned to receive Ibrance in combination with letrozole or letrozole alone. Participants treated with Ibrance plus letrozole lived about 20.2 months without their disease progressing (progression-free survival), compared to about 10.2 months seen in participants receiving only letrozole. Information on overall survival is not available at this time.
The most common side effects of the drug were a decrease in infection-fighting white blood cells called neutrophils (neutropenia), low levels of white blood cells (leukopenia), fatigue, low red blood cell counts (anemia), upper respiratory infection, nausea, inflammation of the lining of the mouth (stomatitis), hair loss (alopecia), diarrhea, low blood platelet counts (thrombocytopenia), decreased appetite, vomiting, lack of energy and strength (asthenia), damage to the peripheral nerves (peripheral neuropathy) and nosebleed (epistaxis). Healthcare professionals should inform patients of these risks.
It is recommended that treatment begin with a 125 milligram dose for 21 days, followed by seven days without treatment. Healthcare professionals are advised to monitor complete blood count prior to start of therapy and at the beginning of each cycle, as well as on Day 14 of the first two cycles, and as clinically indicated.
Ibrance is marketed by New York City-based Pfizer, Inc.
see synthesis……….https://newdrugapprovals.org/2014/01/05/palbociclib/
New York City-based Pfizer, Inc.


Pfizer World Headquarters building in New York City. Zoetis, based in Madison, N.J., traces its roots back to 1952 as a Pfizer unit and has made at least 10 …
 Pfizer’s NYC headquarters
Pfizer’s NYC headquarters

