
Home » Posts tagged 'breast cancer' (Page 2)
Tag Archives: breast cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PARP Inhibitor.. Veliparib (ABT-888) 维利帕尼

Veliparib
2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
CAS number: 912444-00-9 (Veliparib),
912445-05-7 (Veliparib dihydrochloride)
Mechanism of Action:poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) inhibitor
Indiction:cancer treatment
Development Status:Phase III
Drug Company: AbbVie
PARP Inhibitor Veliparib (ABT-888)
| Systematic (IUPAC) name | |
|---|---|
| 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide | |
| Clinical data | |
| Legal status | experimental |
| Identifiers | |
| ATC code | None |
| PubChem | CID 11960529 |
| DrugBank | DB07232 |
| ChemSpider | 10134775 |
| UNII | 01O4K0631N |
| ChEMBL | CHEMBL506871 |
| Chemical data | |
| Formula | C13H16N4O |
| Mol. mass | 244.29 g/mol |
|
2-10-2012
|
PARP1 TARGETED THERAPY
|
|
|
4-17-2009
|
2-{(R)-2-METHYLPYRROLIDIN-2-YL)-1H-BENZIMIDAZOLE-4-CARBOXAMIDE CRYSTALLINE FORM 1
|
Veliparib (ABT-888)[1] is a potential anti-cancer drug acting as a PARP inhibitor. It kills cancer cells by blocking a protein called PARP, thereby preventing the repair of DNA or genetic damage in cancer cells and possibly making them more susceptible to anticancer treatments. Veliparib may make whole brain radiation treatment work more effectively against brain metastases from NSCLC.
It inhibits both PARP1 and PARP2.[2][3]
AbbVie’s Veliparib (ABT-888,), an inhibitor of poly ADP-ribose polymerase 1 and 2 (PARP 1 and PARP 2), is being investigated in multiple tumor types, including 3 phase III studies, all initiated this year, in neoadjuvant treatment of triple-negative breast cancer (clinical trial number:NCT02032277), non-small cell lung cancer (NSCLC, clinical trial number:NCT02106546) and HER2-negative, BRCA1 and/or BRCA2-positive breast cancer (clinical trial number:NCT02163694).
AbbVie, which was spun off from Abbott Laboratories in early 2013, is currently looking to buy Irish drug maker Shire for $46 billion. The proposed deal follows Pfizer’s failed $120 billion attempt to buy AstraZeneca. Humira, AbbVie’s rheumatoid arthritis drug and the world’s top-selling drug last year, accounts for 60% of company revenue and is going off-patent in at the end of 2016. The threat of growing competition for Humira may be a major motivation for AbbVie.


Clinical trials
Numerous phase I clinical trials are in progress.[4]
A phase I/II clinical trial for use with/out doxorubicin (for Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma) started in 2008 and is due to complete in 2010.[5] Results (inc MTD) with topotecan.[6]
A phase II clinical trial for metastatic melanoma has started recruiting.[7] Due to end Dec 2011.
A phase II clinical trial for metastatic breast cancer has started recruiting.[8] Due to end Nov 2011.
A phase II clinical trial for add-on to Radiation Therapy for Patients with Brain Metastases from Non-Small Cell Lung Cancer.
It was included in the I-SPY2 breast cancer trial,[9] and there are encouraging data from that study [10]
A phase I clinical trial for prostate cancer in men who carry the BRCA mutation is underway and is now recruiting (as of May 2013).[11]


……………….
http://www.google.com/patents/US20060229289
EXAMPLE 1
2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide EXAMPLE 1A 1-benzyl 2-methyl 2-methylpyrrolidine-1,2-dicarboxylate
A solution of 1-benzyl 2-methyl pyrrolidine-1,2-dicarboxylate (15.0 g, 57 mmol) and iodomethane (7.11 ml, 114 mmol) in THF (100 mL) was treated with NaN(TMS)2 (1.0 M solution in THF, 114 mL, 114 mmol) at −75° C. under nitrogen. The temperature of the cooling bath was then slowly raised to −20° C. within 1 h and the mixture was stirred at the same temperature for another 3 h. After quenching with water, the mixture was acidified with 2 N HCl (˜100 mL) and was partitioned between water (400 mL) and EtOAc (400 mL). The organic phase was washed with brine and concentrated. The residue was purified by flash column chromatography (silica gel, EtOAc/hexane) to give Example 1A (15.15 g, Yield: 96%). MS (DCI/NH3) m/z 278 (M+H)+.
EXAMPLE 1B
1-[(benzyloxy)carbonyl]-2-methylpyrrolidine-2-carboxylic acid
A solution of Example 1A (15.15 g, 54.63 mmol) in a mixture of THF (100 mL) and water (50 mL) was treated with LiOH.H2O (4.58 g, 109.26 mmol) in water (50 mL). Methanol was added until a transparent solution formed (60 mL). This solution was heated at 60° C. for overnight and the organic solvents were removed under vacuum. The residual aqueous solution was acidified with 2 N HCl to pH 2 and was partitioned between ethyl acetate and water. The organic phase was washed with water, dried (MgSO4), filtered and concentrated to give Example 1B as a white solid (13.72 g, 95.4% yield). MS (DCI/NH3) m/z 264 (M+H)+.
EXAMPLE 1C
benzyl 2-({[2-amino-3-(aminocarbonyl)phenyl]amino}carbonyl)-2-methylpyrrolidine-1-carboxylate
A solution of Example 1B (13.7 g, 52 mmol) in a mixture of pyridine (60 mL) and DMF (60 mL) was treated with 1,1′-carbonyldiimidazole (9.27 g, 57.2 mmol) at 45° C. for 2 h. 2,3-Diamino-benzamide dihydrochloride (11.66 g, 52 mmol), which was synthesized as described in previous patent application WO0026192, was added and the mixture was stirred at rt overnight. After concentration under vacuum, the residue was partitioned between ethyl acetate and diluted sodium bicarbonate aqueous solution. The slightly yellow solid material was collected by filtration, washed with water and ethyl acetate, and dried to give Example 1C (16.26 g). Extraction of the aqueous phase with ethyl acetate followed by concentration, filtration and water-EtOAc wash, provided additional 1.03 g of Example 1C. Combined yield: 84%. MS (APCI) m/z 397 (M+H)+.
EXAMPLE 1D
benzyl 2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
A suspension of Example 1C (17.28 g, 43.6 mmol) in acetic acid (180 mL) was heated under reflux for 2 h. After cooling, the solution was concentrated and the residual oil was partitioned between ethyl acetate and sodium bicarbonate aqueous solution. The organic phase was washed with water and concentrated. The residue was purified by flash column chromatography (silica gel, 3-15% CH3OH in 2:1 EtOAc/hexane) to provide Example 1D (16.42 g, Yield: 99%).
MS (APCI) m/z 379 (M+H)+.
EXAMPLE 1E 2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
A solution of Example 1D (15.0 g, 40 mmol) in methanol (250 ml) was treated with 10% Pd/C (2.8 g) under 60 psi of hydrogen for overnight. Solid material was filtered off and the filtrate was concentrated. The residual solid was recrystallized in methanol to give 7.768 g of Example 1E as free base. The bis-HCl salt was prepared by dissolving the free base in warm methanol and treating with 2 equivalents of HCl in ether (10.09 g). MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 1.92 (s, 3 H), 2.00-2.09 (m, 1 H), 2.21-2.29 (m, 1 H), 2.35-2.41 (m, 1 H), 2.52-2.57 (m, 1 H), 3.54-3.65 (m, 2 H), 7.31 (t, J=7.93 Hz, 1 H), 7.68 (dd, J=8.24, 0.92 Hz, 1 H), 7.72 (dd, J=7.63, 0.92 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72N, 17.66. Found: C, 49.30; H, 5.60; N, 17.39.
EXAMPLE 3 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide EXAMPLE 3A benzyl(2R)-2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
Example 1D (1.05 g, 2.8 mmol) was resolved on chiral HPLC (Chiralcel OD, 80/10/10 hexane/EtOH/MeOH). The faster eluting peak was collected and concentrated to provide Example 3A (99.4% e.e., 500 mg). MS (APCI) m/z 379 (M+H)+.
EXAMPLE 3B 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
A solution of Example 3A (500 mg, 1.32 mmol) in methanol (10 ml) was treated with 10% Pd/C (150 mg) under hydrogen for overnight (balloon). Solid material was filtered off and the filtrate was concentrated. The residual solid was further purified by HPLC (Zorbax C-18, CH3CN/H2O/0.1%TFA) and was converted to bis-HCl salt to provide Example 4 as white solid (254 mg). Co-crystallization of the free base with 1 equivalent of L-tartaric acid in methanol gave a single crystal that was suitable for X-ray study. The X-ray structure with L-tartaric acid was assigned the R-configuration. MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 2.00 (s, 3 H), 2.10-2.19 (m, 1 H), 2.30-2.39 (m, 1 H), 2.45-2.51 (m, 1 H), 2.61-2.66 (m, 1 H), 3.64-3.73 (m, 2 H), 7.40 (t, J=7.95 Hz, 1 H), 7.77 (d, J=8.11 Hz, 1 H), 7.80 (d, J=7.49 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72; N, 17.66. Found: C, 49.10; H, 5.52; N, 17.61.
……………….
WO2009049111
http://www.google.com/patents/WO2009049111A1?cl=en
EXAMPLE 1 Preparation of ABT-888 Crystalline Form 1 A mixture of ABT-888 dihydrochloride (10 g) was stirred in saturated potassium bicarbonate (50 mL) and n-butanol (50 mL) until the ABT-888 dihydrochloride completely dissolved. The aqueous layer was extracted with a second portion of n-butanol then discarded. The extracts were combined, washed with 15% sodium chloride solution (50 mL) and concentrated. The concentrate was chase distilled three times with heptane (50 mL),dissolved in refluxing 2-propanol (45 mL) and filtered hot. The filtrate was cooled to ambient temperature with stirring over 18 hours, cooled to 0-50C, stirred for 1 hour, and filtered. The filtrant was washed with 2-propanol and dried in a vacuum oven at 45-500C with a slight nitrogen purge.
EXAMPLE 2
Preparation of ABT-888 Crystalline Form 2
A mixture of ABT-888 in methanol, in which the ABT-888 was completely dissolved, was concentrated at about 35 0C, and the concentrate was dried to a constant weight.
EXAMPLE 3 Preparation of ABT-888 Crystalline Form 1

15 16
Step 1 : 2-(2-methyl-2-pyrrolidino)-benzimidazole-4-carboxamide 2 HCl (15) is dissolved in water (3.5 kg / kg 15) at 20 + 5 0C. Dissolution of 15 in water results in a solution of pH 0 – 1.
Step 2: The reaction is run at 20 – 25 0C. One equivalent of sodium hydroxide is added, raising the pH to 2 – 3 with only a mild exotherm (100C observed with rapid addition of 1.0 equiv.). This generates a solution that remains clear for several days even when seeded with free base crystals. 3N NaOH (1.0 equiv., 1.25 kg / kg 15) is charged and the solution polish filtered into the crystallizer/ reactor.
Step 3: 5% Na2CO3 (1.5 equiv., 10.08 kg / kg 15) is then filtered into the crystallizer over 2 hours. Nucleation occurs after approximately l/6th of the Na2CO3 solution is added (-0.25 equiv.)
Step 4: The slurry is mixed for NLT 15 min before sampling (typically 1 to 4 hours (2.5 mg/mL product in the supernatant)). The slurry is filtered at 200C and washed with 6 portions of water (1.0 kg / kg 15 each). Each wash was applied to the top of the cake and then pressured through. No mixing of the wetcake was done.
Step 5 : The solids are then dried. Drying was performed at 500C keeping the Cogeim under vacuum while applying a slight nitrogen bleed. The agitator blade was left in the cake to improve heat transfer to the cake. It was rotated and lifted out of the cake once per hour of drying to speed the drying process while minimizing potential crystal attrition that occurs with continuous agitator use. In one embodiment of Step 1, the volume of water for dissolution of the Dihydrochloride (15) is about 1.3 g water/g 15. In another embodiment of Step 1,, the volume of water for dissolution is about 1.3 g to about 4 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 1.3 g to 3.5 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 3.5 g water/g 15.
……………………

(2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
excellent PARP enzyme potency as well as single-digit nanomolar cellular potency. These efforts led to the identification of 3a (2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT-888), currently in human phase I clinical trials. Compound 3a displayed excellent potency against both the PARP-1 and PARP-2 enzymes with a Ki of 5 nM and in a C41 whole cell assay with an EC50 of 2 nM. In addition, 3a is aqueous soluble, orally bioavailable across multiple species, and demonstrated good in vivo efficacy in a B16F10 subcutaneous murine melanoma model in combination with temozolomide (TMZ) and in an MX-1 breast cancer xenograft model in combination with either carboplatin or cyclophosphamide.
References
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models” May 2007
- http://www.cancer.gov/drugdictionary/?CdrID=496464
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models”, 2007
- http://clinicaltrialsfeeds.org/clinical-trials/results/term=Drug:+ABT-888
- “ABT-888 and Cyclophosphamide With Versus Without Doxorubicin in Treating Patients With Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma”
- Phase I Study of ABT-888, a PARP Inhibitor, in Combination with Topotecan Hydrochloride in Adults with Refractory Solid Tumors and Lymphomas.. July 2011. doi:10.1158/0008-5472.CAN-11-1227.
- “A Study Evaluating Efficacy of ABT-888 in Combination With Temozolomide in Metastatic Melanoma”
- “ABT-888 and Temozolomide for Metastatic Breast Cancer”
- “Breast cancer study aims to speed drugs, cooperation”, March 2010
- http://www.centerwatch.com/news-online/article/5737/new-presurgery-combination-therapy-for-triple-negative-breast-cancer
- “Veliparib in Treating Patients With Malignant Solid Tumors That Did Not Respond to Previous Therapy. Clinical Trial NCT00892736”
|
4-1-2013
|
Design, synthesis and biological evaluation of novel imidazo[4,5-c]pyridinecarboxamide derivatives as PARP-1 inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
8-15-2013
|
Discovery of novel benzo[b][1,4]oxazin-3(4H)-ones as poly(ADP-ribose)polymerase inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
|
8-1-2013
|
Identification of potent Yes1 kinase inhibitors using a library screening approach.
|
Bioorganic & medicinal chemistry letters
|
|
5-1-2010
|
A rapid and sensitive method for determination of veliparib (ABT-888), in human plasma, bone marrow cells and supernatant by using LC/MS/MS.
|
Journal of pharmaceutical and biomedical analysis
|
|
1-22-2009
|
Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer.
|
Journal of medicinal chemistry
|
External links
http://kdwn.com/2013/12/16/new-drug-study-method-show-breast-cancer-promise/
| US8013168 | Oct 10, 2008 | Sep 6, 2011 | Abbott Laboratories | Veliparib crystal structure; an anticancer PARP inhibitor |
| US8372987 | Oct 10, 2008 | Feb 12, 2013 | Abbvie Inc. | Title compound is Veliparib, a Poly(ADP-ribose) polymerase i.e. PARP inhibitor; anticancer agent |
| US20060229289 * | Apr 11, 2006 | Oct 12, 2006 | Gui-Dong Zhu | 2-(2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide, aka veliparib, for example; poly(ADP-ribose)polymerase inhibitors; antiinflammatory, antitumor agents; Parkinson’s disease |
Penning, Thomas D. et al. Discovery of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the Treatment of Cancer. Journal of Medicinal Chemistry, 52(2), 514-523; 2009
Zhu, Guidong. 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide crystalline form 2 compositions and preparation for treating cancer. PCT Int. Appl. (2009), WO2009049109 A1 20090416
Kolaczkowski, Lawrence . 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide (ABT-888) crystalline form I and its pharmaceutical composition for cancer treatment. PCT Int. Appl. (2009), WO2009049111 A1 20090416.
Zhu, Gui-Dong; Gong, Jianchun; Gandhi, Virajkumar B.; Penning, Thomas D.; Giranda, Vincent L. Preparation of 1H-benzimidazole-4-carboxamides as poly(ADP-ribose)polymerase (PARP) inhibitors. U.S. Pat. Appl. Publ. (2006), US20060229289 A1 20061012.

The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US
 GADOBUTROL
GADOBUTROL
gadolinium(III) 2,2′,2”-(10-((2R,3S)-1,3,4-trihydroxybutan-2-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate
Gadobutrol, SH-L-562, Gadovist,138071-82-6
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
The agency has approved the new indication for Gadavist injection for intravenous use with magnetic resonance imaging of the breast to assess the presence and extent of malignant breast disease.
Approval is based on priority review of two Phase III studies with identical design (GEMMA-1 and GEMMA-2).
Bayer HealthCare’s Gadavist (gadobutrol)
Bayer’s Gadavist injection cleared for breast cancer evaluation
UPDATE……. Gadoteridol 279.3 mg/ml for injection , CDSCO INDIA 29.07.2021
For intravenous use in magnetic
reasonance imaging (MRI) in adults and
pediatric patients over 2 years of age for
whole body MRI including the head, neck,
liver, breast, musculoskeletal system and
soft tissue pathologies
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
GADOBUTROL
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Licence data | US FDA:link |
| Pregnancy cat. | C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | IV |
| Identifiers | |
| CAS number | 138071-82-6 |
| ATC code | V08CA09 |
| PubChem | CID 72057 |
| DrugBank | DB06703 |
| UNII | 1BJ477IO2L |
| KEGG | D07420 |
| Chemical data | |
| Formula | C18H31GdN4O9 |
| Mol. mass | 604.710 g/mol |
………………………..
Gadobutrol (INN) (Gd-DO3A-butrol) is a gadolinium-based MRI contrast agent (GBCA).
It received marketing approval in Canada[1] and in the United States.[2][3][4]
As of 2007, it was the only GBCA approved at 1.0 molar concentrations.[5]
Gadobutrol is marketed by Bayer Schering Pharma as Gadovist, and by Bayer HealthCare Pharmaceuticals as Gadavist.[6]





 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL
……………………………………………….
http://www.google.com/patents/EP0988294B1?cl=en
-
This type of complexes with metal ions, in particular with paramagnetic metal ions; is used for the preparation of non-ionic contrast agents for the diagnostic technique known as magnetic resonance (MRI, Magnetic Resonance Imaging), among which are ProHance(R) (Gadoteridol, gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid), and Gadobutrol (gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid).

-
[0003]Two different synthetic approaches are described in literature for the preparation of this kind of complexes, said approaches differing in the strategy taken to discriminate one of the four nitrogen atoms: the first one (Dischino et al., Inorg. Chem., 1991, 30, 1265 or EP 448191, EP 292689, EP 255471) is based on the selective protection of one of the nitrogen atoms by formation of the compound of formula (III), 5H,9bH-2a,4a,7-tetraazacycloocta[cd]pentalene, and on the subsequent hydrolysis to compound of formula (IV), 1-formyl-1,4,7,10-tetraazacyclododecane, followed by the carboxymethylation of the still free nitrogen atoms and by the deprotection and alkylation of the fourth nitrogen atom, according to scheme 1.

-
[0004]The step from 1,4,7,10-tetraazacyclododecane disulfate (a commercially available product) to compound (III) is effected according to the conventional method disclosed in US 4,085,106, followed by formation of the compound of formula (IV) in water-alcohol medium.
-
[0005]This intermediate is subsequently tricarboxymethylated with tert-butyl bromoacetate (TBBA) in dimethylformamide at 2.5°C and then treated with a toluene-sodium hydroxide diphasic mixture to give the compound of formula (V), 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic, tris(1,1-dimethylethyl) ester, which is subsequently hydrolysed to compound of formula (II) in acidic solution.
-
[0006]In the process described in WO 93/24469 for the synthesis of Gadobutrol, at first one of the nitrogen atoms is alkylated in conditions such as to minimize the formation of polyalkylated derivatives, then the monoalkylderivative is purified and carboxymethylated, according to scheme 2.

-
[0007]The alkylation of 1,4,7,1,0-tetraazacyclododecane with the epoxide of formula (VI), 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]octane, is carried out in anhydrous n-BuOH under reflux and the reaction mixture is extracted with water, evaporated to dryness and the residue is subsequently diluted with water and extracted with methylene chloride.
-
[0008]The aqueous phase containing the mono-alkylated product (65% yield in Example 7 which reports the procedure for the preparation of 5 kg of Gadobutrol) is directly carboxymethylated at 70°C with chloroacetic acid, keeping pH 9.5 by addition of NaOH. The reaction mixture is adjusted to pH 1, concentrated to dryness and dissolved in methanol to remove the undissolved salts. The filtrate is then concentrated under vacuum, dissolved in water, and loaded onto a cation exchanger in the H+ form to fix the product. The subsequent elution with ammonia displaces the desired product, which is concentrated to small volume and subsequently complexed with gadolinium oxide according to conventional methods, and the resulting complex is purified by means of ion exchange resins. The overall yield is 42%.
-
[0009]Although the first of these two processes could theoretically provide a higher yield, in that all the single steps (protection, carboxymethylation and deprotection) are highly selective, the complexity of the operations required to remove salts and solvents and to purify the reaction intermediates makes such theoretical advantage ineffective: the overall yield is in fact, in the case of Gadoteridol, slightly higher than 37%.
-
[0010]The preparation of Gadobutrol according to the alternative process (WO 93/24469) provides a markedly better yield (72%) only on laboratory scale (example 2): example 7 (represented in the above Scheme 2) actually evidences that, when scaling-up, the yield of this process also remarkably decreases (42%).
-
[0011]In addition to the drawback of an about 40% yield, both processes of the prior art are characterized by troublesome operations, which often involve the handling of solids, the use of remarkable amounts of a number of different solvents, some of them having undesirable toxicological or anyway hazardous characteristics.
-
[0012]Moreover, the synthesis described by Dischino makes use of reagents which are extremely toxic, such as tert-butyl bromoacetate, or harmful and dangerous from the reactivity point of view, such as dimethylformamide dimethylacetal.
-
[0013]An alternative to the use of dimethyl formamide dimethylacetal is suggested by J. Am. Chem. Soc. 102(20), 6365-6369 (1980), which discloses the preparation of orthoamides by means of triethyl orthoformate.
-
[0014]EP 0596 586 discloses a process for the preparation of substituted tetraazacyclododecanes, among them compounds of formula (XII), comprising:
- formation of the tricyclo[5.5.1.0] ring;
- alkylation with an epoxide;
- hydrolysis of the 10-formyl substituent;
- reaction with an acetoxy derivative bearing a leaving group at the alpha-position.
-
[0015]Nevertheless, this method requires quite a laborious procedure in order to isolate the product of step b).
-
[0016]It is the object of the present invention a process for the preparation of the complexes of general formula (XII)

wherein
- R1 and R2
- are independently a hydrogen atom, a (C1-C20) alkyl containing 1 to 10 oxygen atoms, or a phenyl, phenyloxy group, which can be unsubstituted or substituted with a (C1-C5) alkyl or hydroxy, (C1-C5) alkoxy, carbamoyl or carboxylic groups,
- Me3+
- is the trivalent ion of a paramagnetic metal;
comprising the steps represented in the following Scheme 3:






-
The process of the present invention keeps the high selectivity typical of the protection/deprotection strategy described by Dischino in the above mentioned paper, while removing all its drawbacks, thus providing for the first time a reproducible industrial process for the preparation of the concerned compounds in high yields and without use of hazardous substances.
-
[0019]The preparation of the gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-tri-acetic) acid (Gadoteridol), according to scheme 4, is particularly preferred:

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) is propylene oxide.
-
[0020]The preparation of the gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic) acid (Gadobutrol), according to the scheme 5, is also preferred.

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) corresponds to the one of formula (VI), defined above.
-
[0021]On the other hand, step a) of the process of the present invention involves the use of triethyl orthoformate in the presence of an acid catalyst, instead of dialkylformamide-dialkylacetal.
-
[0022]Triethyl orthoformate can be added in amounts ranging from 105% to 200% on the stoichiometric value.
-
[0023]The reaction temperature can range from 110 to 150°C and the reaction time from 5 to 24 h.
-
[0024]The catalyst is a carboxylic acid having at least 3 carbon atoms, C3-C18, preferably selected from the group consisting of propionic, butyric and pivalic acids.
-
[0025]Triethyl orthoformate is a less toxic and less expensive product than N,N-dimethylformamide-dimethylacetal and does not involve the formation of harmful, not-condensable gaseous by-products. Moreover, triethyl orthoformate is less reactive than N,N-dimethylformamide-dimethylacetal, which makes it possible to carry out the loading procedures of the reactives as well as the reaction itself in utterly safe conditions even on a large scale, allows to better monitor the progress of the reaction on the basis of such operative parameters as time and temperature, without checking the progress by gas chromatography, and makes dosing the reactive less critical, in that it can be added from the very beginning without causing the formation of undesired by-products: all that rendering the process suitable for the production of compound (III) on the industrial scale in easily reproducible conditions.
-
[0026]The subsequent step b) involves the carboxymethylation of compound (III) in aqueous solution, using a haloacetic acid, to give compound (IX), i.e. the 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid salt with an alkali or alkaline-earth metal, the salts of compound (IX) with sodium, potassium or calcium being most preferred.




Example 2
-
[0065]

-
[0066]The procedure of Example 1 is followed until step C included, to obtain a solution of DO3A trisodium salt.
-
[0067]pH is adjusted to 12.3 with conc. HCl and 57.7 kg (0.4 kmol) of 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]-octane are added. After reaction for 4 h at 40°C and for 8 h at 80°C, the solution is cooled to 50°C, 120 kg of an aqueous solution containing 0.135 kmol of gadolinium trichloride are added. After 1 h the mixture is cooled at 17°C and acidified to pH 1.7 with conc. HCl, keeping this pH for 2 h. The solution is subsequently warmed to 50°C, pH is adjusted to 7 with sodium hydroxide, keeping these conditions for 1 h.
-
[0068]After that, the resulting crude Gadobutrol is purified repeating exactly the same process as in steps E and F of Example 1.

Recovery of the product (Gadobutrol)
-
[0069]The product-rich fraction is then thermally concentrated to a viscous residue and the residue is added with 350 kg of ethanol at 79°C.
-
[0070]The resulting suspension is refluxed for 1 h, then cooled, centrifuged and dried under reduced pressure to obtain 66.0 kg of Gadobutrol (0.109 kmol), HPLC assay 99.5% (A%).
Overall yield: 79.1% -
[0071]The IR and MS spectra are consistent with the indicated structure.

References
- Cheng, KT (2007). “Gadobutrol”. Molecular Imaging and Contrast Agent Database (MICAD) (Bethesda, MD: National Center for Biotechnology Information (NCBI)). PMID 20641787. NBK23589.
- http://bayerimaging.com/products/gadavist/index.php
- “FDA approves imaging agent for central nervous system scans” (Press release). U.S. Food and Drug Administration (FDA). March 15, 2011. Retrieved March 31, 2011.
- “U.S. FDA Approves Bayer’s Gadavist (Gadobutrol) Injection for MRI of the Central Nervous System” (Press release). Bayer HealthCare Pharmaceuticals. March 14, 2011. Retrieved March 31, 2011.
- “Gadobutrol 1.0-molar in Cardiac Magnetic Resonance Imaging (MRI) – Further Enhancing the Capabilities of Contrast-enhanced MRI in Ischaemic and Non-ischaemic Heart Disease?”
- “Gadavist full prescribing information”. Retrieved 2011-03-14.

Buparlisib in phase 3 for Breast tumor; Hematological neoplasm; Solid tumor

Buparlisib
5-[2,6-Di(4-morpholinyl)-4-pyrimidinyl]-4-(trifluoromethyl)-2-pyridinamine.
5-[2,6-Di(morpholin-4-yl)pyrimidin-4-yl]-4-(trifluoromethyl)pyridin-2-amine
5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine
944396-07-0
Chemical Formula: C18H21F3N6O2
Mass: 410.16781
NVP-BKM-120, BKM-120;
Novartis AG phase 3 for breast cancer
Phosphoinositide 3-kinase inhibitor
Buparlisib, also known as BKM120, is an orally bioavailable specific oral inhibitor of the pan-class I phosphatidylinositol 3-kinase (PI3K) family of lipid kinases with potential antineoplastic activity. PI3K inhibitor BKM120 specifically inhibits class I PIK3 in the PI3K/AKT kinase (or protein kinase B) signaling pathway in an ATP-competitive manner, thereby inhibiting the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate and activation of the PI3K signaling pathway. This may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis. Dysregulated PI3K signaling may contribute to tumor resistance to a variety of antineoplastic agents.
NVP-BKM-120 is an oral selective phosphatidylinositol 3-kinase (PI3K) inhibitor in phase III clinical development at Novartis for the treatment of breast cancer in combination with fulvestrant in postmenopausal women with hormone receptor-positive HER2-negative locally advanced or metastatic breast cancer which progressed on or after aromatase inhibitor treatment.
Early clinical development at Novartis Oncology, a division of Novartis, is also ongoing for the treatment of solid tumors, advanced endometrial carcinoma, non-small cell lung cancer (NSCLC), bladder cancer, gastrointestinal stromal cancer and for the treatment of metastatic castration-resistant prostate cancer.
Novartis is conducting phase II clinical trials for the treatment of follicular lymphoma, diffuse large B-cell lymphoma, mantle cell lymphoma and squamous cell carcinoma of head and neck.
The University of Kansas is evaluating the compound in phase I clinical trials for the treatment of advanced colorectal cancer in combination with irinotecan, while additional phase I trials are ongoing at the Dana-Farber Cancer Institute for the treatment of renal cell carcinoma. The Dana-Farber Cancer Institute is also conducting phase II clinical trials for the oral treatment of recurrent glioblastoma and preclinical studies for the treatment of ovarian cancer. Novartis is also conducting early clinical studies for the treatment of metastatic melanoma
pyrimidine derivative 5-(2,6-Di- 4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine (Compound A, see below), its hydrates, its salts and hydrates and solvates of its salts, to said specific solid forms thereof, to pharmaceutical compositions containing said solid forms, to processes for the preparation of pharmaceutical compositions containing said solid forms, to methods of using said solid forms and to pharmaceutical compositions for the therapeutic treatment of warm-blooded animals, especially humans. Background of the invention
WO 2007/084786 (priority date: January 20, 2006) describes certain pyrimidine derivatives having PI3 inhibiting properties, their use as pharmaceuticals and manufacturing processes thereof. One pyrimidine derivative disclosed in WO 2007/084786 is the selective
phosphatidylinositol 3-kinase inhibitor compound 5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine, hereinafter referred to as “Compound A” or “the compound of formula A”.

Compound A is described in WO 2007/084786 in free form and as the hydrochloric acid salt. The manufacturing process for preparing Compound A is described in Example 10 of this document. The manufacturing processes described therein are, although suitable, regarded as disadvantageous for commercial production.
Due to the high potency of pyrimidine derivatives, in particular PI3K inhibitors, there is a need for improved manufacturing methods of such compounds. In particular there is a need to provide processes that fulfill one or more of the following criteria: scalable, safer; simpler; higher yielding and more economical when compared to known.
…………………………………….
WO 2007084786
http://www.google.com/patents/WO2007084786A1?cl=en
Example 10
Preparation of 4-(“trifluoromethyπ-5-(2,6-dimorpholmoρyrirnidin-4-yπpyridin-2- amine
c

[0388] To a slurry of 2-moφholino-4,6-dichloropyrimidine (prepared as in
Method 22, 2.0 g, 8.54 mmol) in NMP (14 mL), triethylamine (1.43 mL, 10.25 mmol) was added. The heterogeneous mixture was stirred for 15 minutes, then treated with morpholine (0.75 mL, 8.54 mmol). Upon refluxing at 85 0C under argon for 2 hours, the solution was cooled, then added to EtOAc (160 mL). The organic solution was washed with 25 mL of NaHCO3(sat.) (2 x), water (2 x) and brine, dried over Na2SO4, filtered and concentrated. The crude material was dissolved in 200 mL EtOAc and filtered through a SiO2 pad, further eluting with EtOAc, yielding 2.2 g (93%) of 2,4-dimorpholino-6- chloropyrimidine as an off-white solid. LCMS (m/z): 285.0 (MH+), 1H NMR (CDCl3): δ 5.86 (s, IH), 3.71-3.76(m, 12H), 3.52-3.56(m, 4H).
[0389] 4-(trifluoromethyl)-5-(2,6-dimoφholmopyrimidin-4-yl)pyridin-2-amine 8

[0390] Argon gas was bubbled through a heterogeneous mixture of 2,4- dimoφholino-6-chloropyrimidine (4.1 g, 14.3 mmol) and 4-(trifluoromethyl)-5-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)pyridm-2-amine (16.5 g, 57.3 mmol) in 1,2- dimethoxyethane and 2M Na2Cθ3 (3:1) for 20 minutes. 1,1′-
Bis(diphenylphosphino)ferrocene palladium (IT) chloride (292 mg, 0.36 mmol) was added and the high pressure glass vessel containing the mixture was sealed. The reaction mixture was then heated at 900C for 15 hours, cooled and diluted with EtOAc (300 mL). The organic solution was washed with 300 mL of a mixture of water: Na2Cθ3(sat.):NH4θH(conc.) = 5:4:1, then NH4Cl(sat), and brine (2x), dried over Na2SO4, filtered and concentrated. The crude material was purified by SiO2 chromatography (50- 90% EtOAc/hexanes with 0.1% TEA) resulting in 5.62 g (95%) of 4-(trifluoromethyl)-5- (2,6-dimorpholinopyrimidin-4-yl)pyridin-2-amine as an off-white solid.
LCMS (m/z): 411.3 (MH+);
1H NMR (CDCl3): δ 8.27 (s, IH), 6.78 (s, IH), 5.97 (s, IH), 4.77 (bs, 2H), 3.59-3.80(m, 12H), 3.58-3.61(m, 4H).
…………….
WO2012044727 or equi as below
http://www.google.com/patents/EP2621908A2?cl=en
Example 1: 4,4′-(6-Chloropyrimidine-2,4-diyl)di[morpholine] (3) U 2011/053808
63
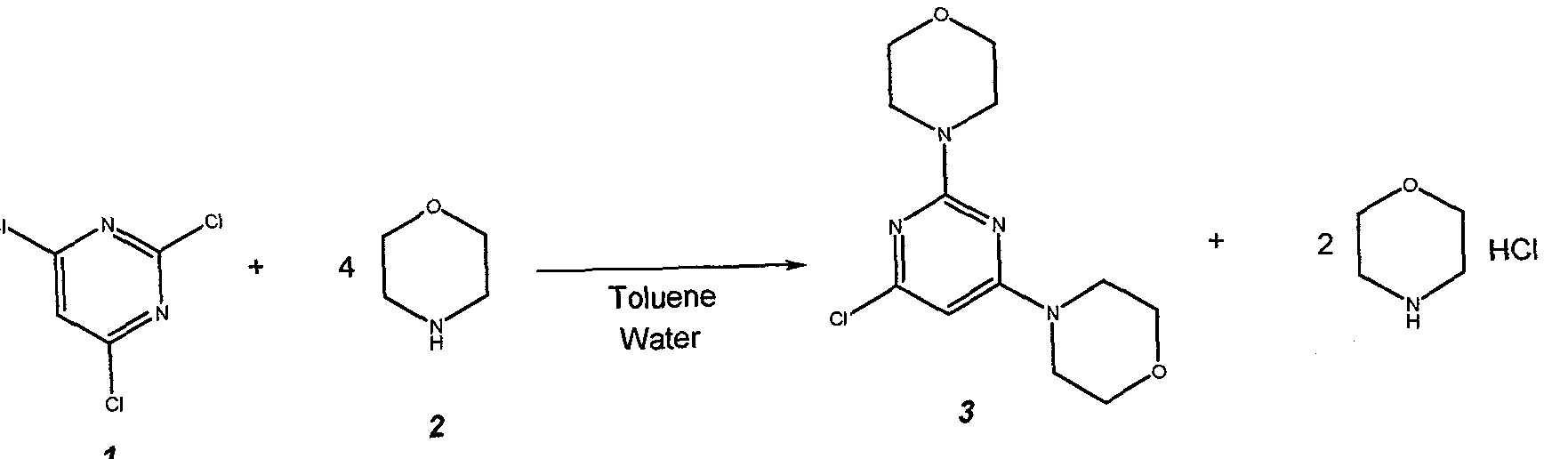
Prepare a solution of 22 g (0.12 mol) of 2,4,6-trichloropyrimidine 1 , in 95.2 g (110 mL) of toluene and charge it to the 25 mL addition funnel. Charge a nitrogen-flushed 500 mL round bottom 4- neck flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet with 62.7 g (63 mL, 0.72 mol) of morpholine 2, 95.2 g (110 mL) of toluene and 44 g (44 mL) of water. Add the toluene solution of 1 over 10 minutes. Heat the reaction mixture to 83 ± 3 °C. Stir at 83 ± 3 °C for 2 h. Check the progress of the reaction. Cool to 30 + 3 °C. Transfer the 2-phase mixture to a 1L separatory funnel.
Separate the phases. Wash the organic phase (top) twice with 200 mL (2 x 100 mL) of warm (30 °C) water. Separate the phases after each wash. Transfer the organic (top) phase back to the 500 mL reaction flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet. Stir and add 50.0 mL of 10.0 N aqueous hydrochloric acid solution. Heat the solution to 53 ± 3 °C and stir for 12 – 18 h. Check the progress of the reaction. Cool to 22 + 3 °C. Transfer the 2-phase mixture to a 1 L separatory funnel. Separate the phases. Transfer the aqueous (bottom) phase to a 500 mL round bottom 4-neck flask equipped with a cooling bath, thermocouple, addition funnel, pH probe, mechanical stirrer and nitrogen inlet / outlet. Stir and cool to 0 ± 3 °C. Add 85.0 g of 25% aqueous sodium hydroxide solution by drops over 30 minutes, maintaining a batch temperature of 10 ± 10 °C throughout the addition. Warm to 20 ± 3 °C and stir for 30 minutes. Isolate the solids by vacuum filtration. Wash the cake with 3 x 100 mL of water. Dry the solids (55°C, 30 mbar) for 24 hours to afford 30.9 g (91.9% yield) of 3 as a white crystalline solid.
Example 2:
4,4′-[6-(4>4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] (4)

Charge a nitrogen-flushed 2 L round bottom 4-neck flask that equipped with a condenser, heating mantle, thermocouple, rubber septum, mechanical stirrer and nitrogen inlet / outlet with 100.0 g (0.351 mol) of 4,4′-(6-chloropyrimidine -2,4-diyl)di[morpholine] 3 and 943 g (1200 mL) of acetonitrile. Stir and heat to 60 + 3 °C. Hold this solution at 60 + 3 °C for charge to batch. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 115.9 g (0.457 mol) of bis(pinacolato)- diboron, 51.7 g (0.527 mol) of potassium acetate, 12.9 g (0.014 mol) of tris(dibenzylideneacetone) – dipalladium(O), 7.9 g (0.029 mol) of tricyclohexylphosphine and 393 g (500 mL) of acetonitrile. Stir and heat the slurry to 84 ± 3 °C (reflux). Collect 00 mL of distillate. Transfer the warm 3 acetonitrile solution via peristaltic pump to the 3 L reactor containing the reaction mixture over 30 minutes and continue collecting distillate. Wash the 2 L flask and transfer lines with 79 g (100 mL) of acetonitrile and transfer the wash to the batch. Maintain distillation at 84 ± 3 °C and collect an additional 900 mL of distillate (batch volume ~ 1100 mL). Check the progress of the reaction 2 h from the start of the addition of 3. Cool the reaction mixture to 70 ± 3 °C and charge 693 g (800 mL) of toluene over 1-2 min. The batch will cool upon the addition of the toluene. Further cool the reaction mixture to 50 ± 3 °C. Charge to a clean 1 L flask, 347 g (400 mL) of toluene and warm it to 50 °C. This will be used as the cake wash. Filter the reaction mixture through a 15 g pad of Celite 545. Wash the filter cake with the warm (50 °C) toluene (400 mL) and collect this wash separately from the batch. This wash will be charged to the distillation residue later in the process. Transfer the filtrate back to the 3 L reactor. Concentrate the batch (25 °C to 40 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached.
Charge toluene cake wash held in reserve (~400 mL) and continue to concentrate the batch (37 °C to 43 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached. Check for complete removal of acetonitrile using the described Process Steering Control. Warm to 50 °C and stir for 15 min. Add 164 g (240 mL) of heptane over 30 minutes maintaining 50 °C throughout the addition. Stir the resulting suspension for 1 h. Cool the slurry to 23 ± 3 °C over 1 h and hold at this temperature for at least 1 h. Blanket the filtering funnel used for isolation of the product with nitrogen (to avoid moisture) and quickly filter the solids. Wash the filter cake twice with a mixture of 22 g (25 mL) of toluene and 51 g (75 mL) of heptane. Dry the solids at 50 °C, 35 mbar for 16 h to afford 4.4 g (72.7% corrected yield) of 4 as a sandy, beige solid. Example 3: 5-Bromo-4-(trifluoromethyl)pyridin-2-amine (4a)

4b 4a
Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 112.14 g (0.63 mol) of N-bromosuccinimide (NBS) and 645 g (725 mL) of tetrahydrofuran. Stir and cool the slurry to -5 ± 3 °C. Charge a nitrogen- flushed 1 L round bottom 4-neck flask that equipped with a thermocouple, mechanical stirrer and nitrogen inlet / outlet with 97.26 g (0.6 mol) of 2-amino-4-(trifluoromethyl)pyridine, 4b and 511 g (575 mL) of tetrahydrofuran. Stir to dissolve the 4b. Transfer the 4b solution to the addition funnel on the reactor and add the solution to the NBS slurry over 2 h maintaining an internal temperature of 0 ± 3 °C throughout the addition. Rinse the 1 L flask and addition funnel with 44 g (50 mL) of tetrahydrofuran and add the wash to the reaction mixture. Warm the solution to 20 + 3 °C over 30 minutes. Check for completeness of the reaction. Quench by charging a solution of 24.6 g of sodium thiosulfate pentahydrate dissolved in 475 mL of water over 10 minutes, maintaining a batch temperature of 20 ± 3 °C throughout the addition. Stir for 1 h after the quench. Concentrate (internal temp = 25 °C, 50 mbar) to remove tetrahydrofuran. Add 379 g (500 mL) of fert-butyl methyl ether. Stir and warm the resulting solution/suspension to 30 ± 3 °C and stir for 15 minutes. Separate the phases. Wash the extract four times with a solution of 32 g of sodium chloride dissolved in 768 g (768 mL) of water (4 x 200 mL per wash), separating the phases after each wash. Finally, wash the extract with 150 g (150 mL) of water. Separate the phases. Charge 152 g (200 mL) of terf-butyl methyl ether. Partially concentrate (57 ± 3 °C) to a volume of 350 mL. Cool to 50 °C and add 265 g (350 mL) of ferf-butyl methyl ether. Resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 265 g (350 mL) of fe/f-butyl methyl ether. Again, resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 103 g (150 mL) of terf-butyl methyl ether to raise the batch volume to 500 mL. Charge 1026 g (1500 mL) of heptane over 15 minutes maintaining 45 ± 3 °C throughout the addition. Slowly increase the vacuum and concentrate (internal temp = 40 °C to 50 °C) to a batch volume of 1000 mL. Release the vacuum and seed the batch. Resume the distillation, further increase the vacuum (slowly) and concentrate (internal temp = 25 °C to 40 °C) to a batch volume of 500 mL. Stir the resulting suspension at 0 °C for 30 min. Filter the solids. Wash the filter cake with 68 g (100 mL) of cold (0 °C) heptane (containing 30 ppm Octastat). Dry the solids (40 °C, 50 mbar) for 16 h to afford 109.8 g (78.0% yield) 4a as an orange solid.
Example 4: 5-(2,6-Di-4-morpholinyl^^yrimidinyl)-^trifluoromethylpyridin-2-ami^ (5)

Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 190.9 g (0.456 mol) of 4,4′-[6-(4,4,5,5- tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1,1′-bis(di-ferf-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of thf. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1 – 2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction. Cool to 22 ± 3 °C. Separate the phases. Partially concentrate the THF (25 °C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0. 25N aqueous N-acetyl-L- cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of 1 N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous HCI solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 ± 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
Alternative procedure:
Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 90.9 g (0.456 mol) of
4,4′[6(4,4,5,5tetramethyl1 ,3,2 dioxaborolan2yl)pyrimidine2,4diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1 ,1′-bis(di-fert-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of tetrahydrofuran. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1-2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction . Cool to 22 + 3 °C. Separate the phases. Partially concentrate the THF (25 C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0.125N aqueous N- acetyl-L-cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 + 3 °C C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 + 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N- acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous hydrochloric acid solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 + 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
…………..
Improved process for manufacturing 5-(2,6-di-4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine
Improved process for the preparation of buparlisib, an oral PI3K inhibitor Novartis is developing for the treatment of solid tumors, including breast cancer and hematological tumors. In January 2014, a phase III development was ongoing and Novartis expected to file for regulatory approval for breast cancer in 2015. Buparlisib was originally claimed in WO2007084786, protection for which expires in both the US and Europe in January 2027. Also see WO2012044727 for a more recent process case.
Burger, M.T.; Pecchi, S.; Wagman, A.; et al.
Discovery of BKM120, a pan class I PI3 kinase inhibitor in phase I/II clinical trials
240th ACS Natl Meet (August 22-26, Boston) 2010, Abst MEDI 489
Vu, A.T.; Morris, J.; Malhotra, S.V.
Efficient and improved synthesis of a PI3K inhibitor anticancer agent
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst ORGN 115
Capecitabine– treatment of metastatic breast and colorectal cancers.

Capecitabine, pentyl [1-(3,4-dihydroxy-5-methyltetrahydrofuran-2-yl)-5-fluoro-2-oxo-1H-pyrimidin-4-yl]carbamate
Capecitabine (Xeloda, Roche) is an orally-administered chemotherapeutic agent used in the treatment of metastatic breast and colorectal cancers. Capecitabine is a prodrug, that is enzymatically converted to 5-fluorouracil in the tumor, where it inhibits DNA synthesis and slows growth of tumor tissue.The activation of capecitabine follows a pathway with three enzymatic steps and two intermediary metabolites, 5′-deoxy-5-fluorocytidine (5′-DFCR) and 5′-deoxy-5-fluorouridine (5′-DFUR), to form 5-fluorouracil

Capecitabine is a chemotherapy drug that is administered as a treatment for a variety of cancer types, including bowel cancer, stomach cancer, breast cancer and oesophageal cancer. It acts as a prodrug, undergoing a three-step enzymic conversion into 5-fluorouracil in the tumour, where it inhibits DNA synthesis and thus slows growth of tumour tissue. Capecitabine can be synthesized from the readily available starting materials D-ribofuranose and cytosine [1].

When capecitabine is used for long-term treatment of recurrent cancers, one of the side-effects can be the onset of hand-foot syndrome, which eventually can lead to total eradication of the patients fingerprints [2]. This has recently proved rather inconvenient for one unsuspecting patient. A recent report [3] describes an instance where a patient on capecitabine for over three years went to the USA to visit relatives in December 2008. He was detained for 4 hours as his fingerprints could not be detected by immigration officials and was only allowed to enter after the officers were entirely satisfied that he posed no threat to security. As a result, all patients taking capecitabine long-term are being advised to travel with a letter from an oncologist stating condition and treatment being received to account for their lack of fingerprints.
As a final aside, recent reports suggest that the actual purpose of fingerprints is to enhance sensitivity rather than friction.
References
- Moon, B.S., Shim, A.Y., Lee, K.C., Lee, H.J., Lee, B.S., An, G.I., Yang, S. D., Chi, D.Y., Choi, C.W., Lim, S.M. and Chun, K.S. (2005) Synthesis of F-18 labeled capecitabine using [18F]F2 gas as a tumor imaging agent. Bull. Korean Chem. Soc. 26, 1865–1868.
- Chua, D., Wei, W.I., Sham, J.S.T. and Au, G.K.H. (2008) Capecitabine monotherapy for recurrent and metastatic nasopharyngeal cancer.Jpn. J. Clin. Oncol. 38, 244–249.
- Wong, M., Choo, S.-P. and Tan, E.-H. (2009) Travel warning with capecitabine. Annals of Oncology Advance Access May 26th 2009/doi:10.1093/annonc/mdp278.

http://promontory-science-education.webnode.com/animations/#.UbkbNtiNCS0
is link to below animation

Phase 3 , breast cancer, Ridaforolimus (MK-8669; AP23573; formerly Deforolimus) Merck, license,Ariad Pharmaceuticals

Ridaforolimus
572924-54-0
(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl dimethylphosphinate
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
42-(dimethylphosphinate) Rapamycin
- AP 23573
- AP23573
- Deforolimus
- MK 8669
- MK-8669
- MK8669
- Ridaforolimus
- Taltorvic
- UNII-48Z35KB15K
An mTOR inhibitor for the treatment of cancer.
Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME)
Ridaforolimus is an investigational small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN, known to be important to malignancy.
TARGET- mTOR
Ridaforolimus (also known as AP23573 and MK-8669; formerly known as Deforolimus[1]) is an investigational targeted and small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN known to be important to malignancy. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism, and angiogenesis.
It has had promising results in a clinical trial for advanced soft tissue and bone sarcoma.
RIDAFOROLIMUS
NMR….http://file.selleckchem.com/downloads/nmr/S102201-Deforolimus-HNMR-Selleck.pdf
HPLC . http://file.selleckchem.com/downloads/hplc/S102201-Deforolimus-HPLC-Selleck.pdf
MSDS..http://www.selleckchem.com/msds/Deforolimus-MSDS.html
| Commercial arrangements |
Ridaforolimus is being co-developed by Merck and ARIAD Pharmaceuticals. On May 5, 2010, Ariad Pharmaceuticals and Merck & Company announced a clinical development and marketing agreement. With this agreement, Ariad received $125 million in upfront payments from Merck and $53 million in milestone payments. Future payments are triggered upon acceptance of the NDA by the FDA with another payment when the drug receives marketing approval. There are similar milestones for acceptance and approval in both Europe and Japan. Other milestone payments are tied to revenue goals for the drug.[2] ARIAD has opted to co-promote ridaforolimus in the U.S. Merck plans to submit a New Drug Application (NDA) for ridaforolimus to the U.S. Food and Drug Administration (FDA) and a marketing application in the European Union in 2011.[3]
Clinical trials
Phase III SUCCEED
On June 6, 2011, Ariad and Merck announced detailed results from the largest randomized study ever in the soft tissue and bone sarcoma population, the Phase III SUCCEED clinical trial. SUCCEED evaluated oral ridaforolimus, in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. In this patient population, ridaforolimus improved progression-free survival (PFS) compared to placebo, the primary endpoint of the study. The complete study results were presented by Sant P. Chawla, M.D., director, Sarcoma Oncology Center, Santa Monica, CA, during the 2011 American Society of Clinical Oncology (ASCO) annual meeting.
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group. Furthermore, based on the full analysis of PFS determined by investigator assessment, there was a statistically significant (p<0.0001) 31 percent reduction by ridaforolimus in the risk of progression or death compared to placebo (hazard ratio=0.69). In the investigator assessment analysis, median PFS was 22.4 weeks for those treated with ridaforolimus compared to 14.7 weeks in the placebo group [4
EU WITHDRAWAL IN NOV 2012
Merck, known as MSD outside the U.S. and Canada, announced today that it has formally notified the European Medicines Agency (EMA) of Merck’s decision to withdraw the Marketing Authorisation Application (MAA) for ridaforolimus.
The application for Marketing Authorisation for ridaforolimus was accepted by the EMA in August 2011. At the time of the withdrawal it was under review by the Agency’s Committee for Medicinal Products for Human Use (CHMP). In its letter to the EMA, Merck said that the withdrawal of ridaforolimus was based on the provisional view of the CHMP that the data available to date and provided in the Marketing Authorisation Application were not sufficient to permit licensure of ridaforolimus in the European Union for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor.
Although the application for these uses was withdrawn, Merck is studying ridaforolimus in combination with other drugs in other tumor types. The withdrawal of the European application of ridaforolimus for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor does not change Merck’s commitment to the ongoing clinical trials with ridaforolimus.
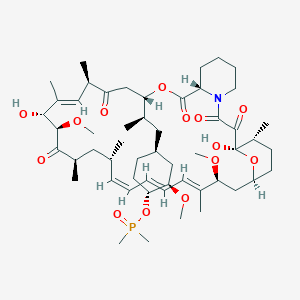
Description
42-(dimethylphosphinate) Rapamycin (Ridaforolimus) represented by the following formula I:

2. Description of RelatedArt
The mammalian target of Rapamycin (mTOR) is known as a mechanistic target of Rapamycin (H), which is found in the studies of Rapamycin. On the other hand, 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) is a derivative of Rapamycin (II), which is also a kind of mTOR inhibitor. Ridaforolimus (I) can inhibit cell division and possibly lead to tumor cell death. Hence, there are many studies related to solid tumor treatments and blood cancer treatments using Ridaforolimus (I). In addition, in 2011, Merck also applied a certification of this compound against soft tissue and bone cancer.
U.S. Pat. No. 7,091,213 discloses a process for preparing 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I), and the process thereof is shown in the following Scheme I.

In this process, a solution of Rapamycin (II) in dichloromethane (DCM) was respectively added with 2,6-di-tert-butyl-4-methylpyridine or 3,5-lutidine as a base, and followed by the addition of a solution of dimethylphosphinic chloride (DMP-Cl) to perform a phosphorylation reaction at 0° C., under a stream of N2(g). The crude product was purified by flash chromatography (eluted with MeOH/DCM/EtOAc/hexane=1:10:3:3) to provide 42-(dimethyl- phosphinate) Rapamycin (Ridaforolimus) (I), which is a phosphorylated compound at 42-hydroxyl position of Rapamycin (II). In addition, this patent also disclosed a side product of 31,42-bis(dimethyl phosphinate) Rapamycin (III), which is a phosphorylated compound at both 31- hydroxyl position and 42- hydroxyl position of Rapamycin (II).
…………………..
SYNTHESIS
Some additional transformations of potential interest to the practitioner are shown below, including the preparation of reagents for generating the described C-43 phosphorus-containing rapalogs:
Preparation of Diakyl/diaryl Chlorophoshates

Preparation of Alkyl Halide Phosphonates

Illustrative routes for using the foregoing sorts of reagents to prepare certain rapalogs of this invention are shown below.

The synthesis of compounds of this invention often involves preparation of an activated form of the desired moiety “J”, such as a phosphoryl chloride as shown above (e.g. (R)(RO)P—Cl or RR′P(═O)—Cl, etc), and reaction of that reagent with rapamycin (or the appropriate rapalog) under conditions yielding the desired product, which may then be recovered from residual reactants and any undesired side products. Protecting groups may be chosen, added and removed as appropriate using conventional methods and materials.
Purification of Compounds of the Invention
A variety of materials and methods for purifying rapamycin and various rapalogs have been reported in the scientific and patent literatures and may be adapted to purification of the rapalogs disclosed herein. Flash chromatography using a BIOTAGE prepacked cartridge system has been particularly effective. A typical protocol is disclosed in the Examples which follow.
Physicochemical Characterization of Compounds of the Invention
The identity, purity and chemical/physical properties of the rapalogs may be determined or confirmed using known methods and materials, including HPLC, mass spectral analysis, X ray crystallography and NMR spectroscopy. High resolution 1D 1H and 31P NMR spectra acquired using a typical relaxation delay of 3 seconds have proved useful, as has reverse phase HPLC analysis (analytical column, 3 micron particle size, 120 ansgstrom pore size, thermostatted to 50° C. with a mobile phase of 50% acetonitrile, 5% methanol and 45% water (all % s by volume), for example, in an isocratic elution system, with elution of product and impurity peaks followed by UV detection at 280 nanometers). Normal phase HPLC may also be used, especially to evaluate the level of residual rapamycin or rapalog by-products. The presence of residual solvent, heavy metals, moisture and bioburden may be assessed using conventional methods.
Example 9
Dimethyl-phosphinic Acid C-43 Rapamycin Ester

Dimethyl-phosphinic Acid C-43 Rapamycin Ester
To a cooled (0° C.) solution of rapamycin (0.1 g, 0.109 mmol) in 1.8 mL of dichloromethane was added 0.168 g (0.82 mmol) of 2,6-di-t-butyl-4-methyl pyridine, under a stream of N2, followed immediately by a solution of dimethylphosphinic chloride (0.062 g, 0.547 mmol) in 0.2 mL of dichloromethane. The slightly yellow reaction solution was stirred at 0° C., under an atmosphere of N2, for 3.5 h (reaction monitored by TLC). The cold (0° C.) reaction solution was diluted with ˜20 mL EtOAc then transferred to a separatory funnel containing EtOAc (150 mL) and saturated NaHCO3 (100 mL). Upon removing the aqueous layer, the organic layer was washed successively with ice cold 1N HCl (1×100 mL), saturated NaHCO3 (1×100 mL), and brine (1×100 mL), then dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (eluted with 1:10:3:3 MeOH/DCM/EtOAc/hexane) to provide 0.092 g of a white solid:
1H NMR (300 MHz, CDCl3) d 4.18 (m, 1H), 4.10 (m, 1H), 3.05 (m, 1H), 1.51 (m, 6H);
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
Example 9
Alternative Synthesis
Rapamycin and dichloromethane are charged into a nitrogen-purged reaction flask. The stirred solution is cooled to approximately 0° C. (an external temperature of −5±5° C. is maintained throughout the reaction). A solution of dimethylphosphinic chloride (2.0 molar equivalents) in dichloromethane is then added over a period of approximately 8–13 minutes.
This is followed immediately by the addition of a solution of 3,5-lutidine (2.2 molar equivalents) in dichloromethane over a period of approximately 15–20 minutes. Throughout both additions, the internal temperature of the reaction sssstays below 0° C. The cooled reaction solution is stirred for 1 hour and then transferred, while still cold, to an extractor containing saturated aqueous NaHCO3 and methyl-t-butyl ether (MTBE), ethyl acetate or diethyl ether. In-process samples are removed at 30 and 60 minute time points.
Samples are prepared in a similar fashion to that described for the reaction workup. Reaction progress is monitored by TLC (1:10:3:3 MeOH/DCM/EtOAc/hexanes) and reverse-phase HPLC analyses. The isolated organic layer is successively washed with ice cold 1N HCl, saturated aqueous NaHCO3 (2×), saturated aqueous NaCl, and dried over sodium sulfate. Upon filtration and solvent removal, the residue undergoes solvent exchange with acetone followed by concentration in vacuo to provide crude product, which may be analyzed for purity by normal- and reversed-phase HPLC.
…………………….
SYNTHESIS
The process of the present invention is shown in the following Scheme II.


- “ARIAD Reports First Quarter 2009 Development Progress and Financial Results- Ridaforolimus New USAN Name to Replace Deforolimus”. ARIAD Pharmaceuticals. 2009. Retrieved 2009-05-07.
- “ARIAD – News release”. Phx.corporate-ir.net. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-03-17. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-06-06. Retrieved 2012-10-07.
|
7-11-2012
|
FULLY HUMAN ANTI-VEGF ANTIBODIES AND METHODS OF USING
|
|
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
4-16-2004
|
Phosphorus-containing compounds and uses thereof
|

The European Medicines Agency (EMA) has approved Roche’s PERJETA (pertuzumab) for patients with previously untreated HER2-positive metastatic breast cancer (mBC)

The structure of HER2 and pertuzumab
march 4, 2013
Pertuzumab (also called 2C4, trade name Perjeta) is a monoclonal antibody. The first of its class in a line of agents called “HER dimerization inhibitors”. By binding to HER2, it inhibits the dimerization of HER2 with other HER receptors, which is hypothesized to result in slowed tumor growth.[1] Pertuzumab received US FDA approval for the treatment of HER2-positive metastatic breast cancer on June 8, 2012.[2] Pertuzumab was developed at Genentech and is now owned by Roche which acquired Genentech in 2009.
Clinical trials
Early clinical trials of pertuzumab in prostate, breast, and ovarian cancers have been met with limited success.[3]
The dosage of pertuzumab used in the pivotal phase III CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial was as follows: IV 840 mg loading dose followed by IV 420 mg every three weeks.[4]
The pharmacokinetics of intravenous pertuzumab appear to be unaffected by age and no drug-drug interaction has been reported with docetaxel. The pharmacokinetics and pharmacodynamics of pertuzumab were summarized in a Feb 2012 review by Gillian Keating.[4]
The combination of pertuzumab plus trastuzumab plus docetaxel, as compared with placebo plus trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged progression-free survival, with no increase in cardiac toxic effects in the randomized, double-blind, multinational, phase III CLEOPATRA trial.[5]
Intravenous pertuzumab is currently being evaluated in patients with breast cancer in the following trials: MARIANNE (advanced breast cancer), NEOSPHERE (early breast cancer), TRYPHAENA (HER2-positive stage II/III breast cancer) and APHINITY (HER2-positive nonmetastatic breast cancer).[4]
References
- de Bono, Johann S.; Bellmunt, J; Attard, G; Droz, JP; Miller, K; Flechon, A; Sternberg, C; Parker, C et al. (20 January 2007). “Open-Label Phase II Study Evaluating the Efficacy and Safety of Two Doses of Pertuzumab in Castrate Chemotherapy-Naive Patients With Hormone-Refractory Prostate Cancer”. Journal of Clinical Oncology 25 (3): 257–262. doi:10.1200/JCO.2006.07.0888. PMID 17235043.
- “FDA Approves Perjeta (Pertuzumab) for People With HER2-Positive Metastatic Breast Cancer” (Press release). Genentech. Retrieved 2012-06-09.
- Genentech press release – May 15, 2005
- Keating GM. Pertuzumab: in the first-line treatment of HER2-positive metastatic breast cancer. Drugs 2012 Feb 12; 72 (3): 353-60.Link text
- Baselga J, Cortés J, Kim SB, and the CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012 Jan 12; 366 (2): 109-19. Link text
