PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
To treat platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer after one to three prior systemic treatment regimens, at least one of which included bevacizumab
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Cushing syndrome
Phase IIICushing syndrome; Ovarian cancer; Pancreatic cancer
Phase IIFallopian tube cancer; Peritoneal cancer; Prostate cancer
Phase I/IISolid tumours
Phase IAdrenocortical carcinoma
Most Recent Events
09 Sep 2022Subgroup analysis efficacy data from a phase-II trial in Ovarian cancer presented at the 47th European Society for Medical Oncology Congress (ESMO-2022)
29 Jun 2022Phase-III clinical trials in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater) in USA (PO)
06 Jun 2022Corcept Therapeutics announces intentions to submit a NDA for Ovarian cancer
The drug was approved by the USFDA in 2026 for the treatment of platinum-resistant ovarian cancer.[3]
Relacorilant is an orally available antagonist of the glucocorticoid receptor (GR), with potential antineoplastic activity. Upon administration, relacorilant competitively binds to and blocks GRs. This inhibits the activity of GRs, and prevents both the translocation of the ligand-GR complexes to the nucleus and gene expression of GR-associated genes. This decreases the negative effects that result from excess levels of endogenous glucocorticoids, like those seen when tumors overproduce glucocorticoids. In addition, by binding to GRs and preventing their activity, inhibition with CORT125134 also inhibits the proliferation of GR-overexpressing cancer cells. GRs are overexpressed in certain tumor cell types and promote tumor cell proliferation.
OriginatorCorcept Therapeutics
DeveloperCorcept Therapeutics; University of Chicago
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Ovarian cancer; Cushing syndrome
RegisteredFallopian tube cancer; Ovarian cancer; Peritoneal cancer
PreregistrationCushing syndrome
Phase IIIAdenocarcinoma
Phase IIProstate cancer
DiscontinuedAdrenocortical carcinoma
27 Mar 2026Discontinued – Phase-I for Adrenocortical carcinoma (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Combination therapy) in USA (PO), before March 2026 (Corcept Therapeutics pipeline, March 2026)
27 Mar 2026Corcept Therapeutics plans the phase II STELLA trial for Cervical cancer (Combination therapy, Second-line therapy or greater) in first quarter of 2026
25 Mar 2026Registered for Fallopian tube cancer (Combination therapy, Second-line therapy or greater) in USA (PO) – First global approval
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)
118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017). [Abstract] [Google Scholar]
120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
The nonselective glucocorticoid receptor (GR) antagonist mifepristone has been approved in the U.S. for the treatment of selected patients with Cushing’s syndrome. While this drug is highly effective, lack of selectivity for GR leads to unwanted side effects in some patients. Optimization of the previously described fused azadecalin series of selective GR antagonists led to the identification of CORT125134, which is currently being evaluated in a phase 2 clinical study in patients with Cushing’s syndrome.
Cushing’s syndrome (CS) is a metabolic disorder caused by chronic hypercortisolism. CS is associated with cardiovascular, metabolic, skeletal and psychological dysfunctions and can be fatal if left untreated. The first-line treatment for all forms of CS is a surgery. However, medical therapy has to be chosen if surgical resection is not an option or is deemed ineffective. Currently available therapeutics are either not selective and have side effects or are only available as an injection (pasireotide).
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonateOTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
An orally available inhibitor of poly(ADP-ribose) polymerase 1 and 2 (PARP-1/2) for treatment of solid tumors (Jiangsu Hengrui Medicine Co. Ltd., Lianyungang, China)
Fluazolepali, developed by Hengrui and Howson, is intended for the treatment of recurrent ovarian cancer, triple-negative breast cancer, advanced gastric cancer and other advanced solid tumors. Currently, the drug has been introduced into China for recurrent ovarian cancer. Clinical stage.
In February 2019, a randomized, double-blind, controlled, multicenter, phase III clinical study (CTR20190294) of flazopril capsule versus placebo for maintenance of recurrent ovarian cancer was initiated in China and was sponsored by Hengrui Medicine.
Jiangsu Hansoh Pharmaceutical , in collaboration with Jiangsu Hengrui Medicine , is developing an oral capsule formulation of fluazolepali (fluzoparib; SHR-3162), a small molecule inhibitor to PARP-1 and PARP-2, for the treatment of solid tumors including epithelial ovarian, fallopian tube or primary peritoneal, breast and gastric cancer.
Phase I Breast cancer; Fallopian tube cancer; Gastric cancer; Peritoneal cancer; Solid tumours
09 Jul 2019 Jiangsu HengRui Medicine initiates a phase I trial in Solid tumors in China (NCT04013048) [14C]-Fluzoparib
01 Jul 2019 Jiangsu HengRui Medicine plans a phase I drug-drug interaction trial (In volunteers) in China (PO) (NCT04011124)
12 Jun 2019 Jiangsu HengRui Medicine completes a phase I trial in Gastric cancer (Combination therapy, Recurrent, Metastatic disease, Second-line therapy or greater, Late-stage disease) in China (PO) (NCT03026881)
Fluzoparib (SHR 3162) is a selective poly [ADP-ribose] polymerase 1 (PARP1) and poly [ADP-ribose] polymerase 2 inhibitor (PARP2), being developed by Jiangsu HengRui Medicine, for the treatment of cancer. PARP enzymes play a vital role in repair of DNA damage and maintaining genomic stability. Fluzoparib inhibits PARP enzymes and induces DNA-double strands breaks, G2/M arrest and apoptosis in homologous recombination repair (HR)-deficient cells. Clinical development for ovarian cancer, breast cancer, fallopian tube cancer, peritoneal cancer, gastric cancer and solid tumours is underway in China and Australia.
An orally available inhibitor of poly (ADP-ribose) polymerase (PARP) types 1 and 2, with potential antineoplastic activity. Upon oral administration, fluzoparib inhibits PARP 1 and 2 activity, which inhibits PARP-mediated repair of damaged DNA via the base excision repair (BER) pathway, enhances the accumulation of DNA strand breaks, promotes genomic instability, and leads to an induction of apoptosis. The PARP family of proteins catalyze post-translational ADP-ribosylation of nuclear proteins, which then transduce signals to recruit other proteins to repair damaged DNA. PARP inhibition may enhance the cytotoxicity of DNA-damaging agents and may reverse tumor cell chemoresistance and radioresistance. Check for active clinical trials using this agent. (NCI Thesaurus)
Process for preparing heterocyclic compounds (presumed to be fluazolepali ) and its intermediates as PARP inhibitors useful for treating cancer.
Example 1
The compound and 5.0kg of 10% palladium on carbon 250g, 80L of methanol was added to the kettle at 0.4MPa, 24h 25 ℃ hydrogenation reaction. The palladium carbon was removed by filtration, the filter cake was washed with methanol, and the filtrate was collected, evaporated to dryness under reduced pressure, and ethyl acetate (20 L) was added to the concentrate, and the mixture was stirred and evaporated, and then cooled to 0° C. ~3, stirring, filtration, filter cake and then adding 20 L of ethyl acetate, pulping at room temperature for 3 to 4 h, filtration, vacuum drying at 45 ° C for 6-8 h to obtain 5.5 kg of compound 3 solid, yield 91.7%, HPLC purity 99.69%.
Example 2
According to the method of Example 19 of CN102686591A, 2 g of the compound 3 and 2.79 g of the compound 4 were charged to obtain 3.6 g of the compound of the formula I in a yield of 87.8%.
Example 3
At room temperature, 2.0 g of compound 2 (prepared according to the method disclosed in WO2009025784) was dissolved in 30 mL of isopropanol, and concentrated sulfuric acid was added dropwise with stirring to adjust the pH to 3, and stirred at room temperature without solid precipitation; the reaction solution was poured into 150 mL of n-hexane. After stirring at room temperature, no solid precipitated, and the sulfate solid of Compound 2 could not be obtained.
Example 4
1. At room temperature, 1.11 g of compound 2 was dissolved in 10 mL of isopropanol, and 15% phosphoric acid/isopropanol solution was added dropwise with stirring to adjust the pH to 3, stirred at room temperature, filtered, and the filter cake was washed with isopropyl alcohol and dried under vacuum. Compound 2 phosphate solid 1.46 g, yield 87.1%, HPLC purity 99.72%.
Example 5
At room temperature, 1.28 g of compound 2 was dissolved in 10 mL of isopropanol, and 20% acetic acid/isopropanol solution was added dropwise with stirring to adjust the pH to 3, and stirred at room temperature without solid precipitation; the reaction solution was poured into 100 mL of n-hexane, and continued. After stirring at room temperature, no solid precipitated, and the acetate solid of Compound 2 could not be obtained.
Example 6
1.05g of compound 2 was dissolved in 10mL of isopropanol at room temperature, and the pH was adjusted to 3 by adding 15% citric acid/isopropanol solution while stirring. At room temperature, no solid precipitated; the reaction solution was poured into 100 mL of n-hexane. After stirring at room temperature, no solid precipitated, and the citrate solid of Compound 2 could not be obtained.
Example 7
1.12 g of compound 2 was dissolved in 10 mL of isopropanol at room temperature, and 0.74 g of maleic acid was added thereto with stirring. The mixture was stirred at room temperature, filtered, and the filter cake was washed with isopropyl alcohol and dried in vacuo to obtain the maleate salt of compound 2. 1.51 g, yield 84.6%.
No development reported B-cell lymphoma; Lymphoid leukaemia
26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
The kinase ataxia telangiectasia mutated and rad3 related (ATR) is a key regulator of the DNA-damage response and the apical kinase which orchestrates the cellular processes that repair stalled replication forks (replication stress) and associated DNA double-strand breaks. Inhibition of repair pathways mediated by ATR in a context where alternative pathways are less active is expected to aid clinical response by increasing replication stress. Here we describe the development of the clinical candidate 2(AZD6738), a potent and selective sulfoximine morpholinopyrimidine ATR inhibitor with excellent preclinical physicochemical and pharmacokinetic (PK) characteristics. Compound 2 was developed improving aqueous solubility and eliminating CYP3A4 time-dependent inhibition starting from the earlier described inhibitor 1 (AZ20). The clinical candidate 2 has favorable human PK suitable for once or twice daily dosing and achieves biologically effective exposure at moderate doses. Compound 2 is currently being tested in multiple phase I/II trials as an anticancer agent.
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).
Scheme 1. Medicinal Chemistry Route to AZD6738
Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
AZD6738 is currently being tested in multiple phase I/II trials for the treatment of cancer. Its structure, comprising a pyrimidine core decorated with a chiral morpholine, a cyclopropyl sulfoximine, and an azaindole, make it a challenging molecule to synthesize on a large scale. We describe the evolution of the chemical processes, following the manufacture of AZD6738 from the initial scale-up through to multikilos on plant scale. During this evolution, we developed a biocatalytic process to install the sulfoxide with high enantioselectivity, followed by introduction of the cyclopropyl group first in batch, then in a continuous flow plate reactor, and finally through a series of continuous stirred tank reactors. The final plant scale process to form AZD6738 was operated on 46 kg scale with an overall yield of 18%. We discuss the impurities formed throughout the process and highlight the limitations of this route for further scale-up.
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) (30.0 g) were added at 75 °C, and the reaction mixture was held for 2 h. The mixture was cooled to 20 °C, and n-heptane (141.9 kg) was added at the rate of 40 kg/h. The solid was collected by filtration, washed with a mixture of 1-butanol and n-heptane (9.3 and 22.4 kg respectively), and then given a further wash with n-heptane (32.2 kg). The solid was dried at 40 °C to give imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) as a whit solid (41.4 kg, 92% yield): Assay (HPLC) 99.9%; Assay (NMR) 99% wt/wt.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.
//////AZD6738, AZD-6738, AZD 6738, AstraZeneca, University of Pennsylvania, Phase II, Breast cancer, Gastric cancer, Non-small cell lung cancer, Ovarian cancer, Ceralasertib
Ceralasertib
CAS 1352226-88-0
(R)-imino(methyl)(1-{6-[(3R)-3-methylmorpholin-4-yl]-2-{1H-pyrrolo[2,3-c]pyridin-4-yl}pyrimidin-4-yl}cyclopropyl)-lambda6-sulfanone
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane
UNII 85RE35306Z
Molecular Weight
412.51
Formula
C20H24N6O2S
AZD 6738, ATR KINASE INHIBITOR AZD6738, AZD-6738, AZD6738, WHO 10842
Ceralasertib (AZD6738) is an orally active and bioavailable inhibitor of ATR kinase with an IC50 of 1 nM.
Ceralasertib is an investigational new drug that is being evaluated for the treatment of cancer.[1] It is an ATR kinase inhibitor.[2]
Originator AstraZeneca; University of Pennsylvania
Developer Acerta Pharma; AstraZeneca; Dana-Farber Cancer Institute; Gustave Roussy; National Cancer Institute (France); Samsung Medical Center; University of California at San Francisco; University of Pennsylvania
Class Antineoplastics; Cyclopropanes; Imines; Ketones; Morpholines; Organic sulfur compounds; Pyridines; Pyrimidines; Pyrroles; Small molecules
Mechanism of Action ATR protein inhibitors
Phase III Non-small cell lung cancer
Phase IICholangiocarcinoma; Gynaecological cancer; Malignant melanoma; Osteosarcoma; Ovarian cancer; Pancreatic cancer; Prostate cancer; Small cell lung cancer; Solid tumours; Triple negative breast cancer
PreclinicalDiffuse large B cell lymphoma; Type 1 diabetes mellitus
DiscontinuedHaematological malignancies; Non-Hodgkin’s lymphoma; Squamous cell cancer
22 Apr 2025AstraZeneca plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) (PO) in April 2025(NCT06929260)
23 Dec 2024AstraZeneca plans a phase I trial for Non-small Cell Lung Cancer, Ovarian Cancer, or Endometrial Cancer in United Kingdom(PO) In January 2025 (NCT06754761)
07 Dec 2024Updated efficacy and adverse event data from a phase I trial in Myelodysplastic syndrome presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
SCHEME
PATENT
WO2011154737
CN112939966
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00482
4-{4-[(3R)-3-methyl-4-morpholinyl]-6-[1-(S-methylsulfonimidoyl)cyclopropyl]-2-
pyrimidinyl}-1H-pyrrolo[2,3-b]pyridine (1).
Recent Applications of Pd-Catalyzed Suzuki–Miyaura and Buchwald–Hartwig Couplings in Pharmaceutical Process Chemistry
AG014699, the phosphate salt of AG14447, which has improved aqueous solubility, has been selected for clinical trial.AG014699 is a tricyclic indole poly(ADP-Ribose) polymerase (PARP) inhibitor with potential antineoplastic activity.
M.Wt: 421.3593
Formula: C19H21FN3O5P
CAS No: 459868-92-9
Rucaparib, PF-01367338283173-50-2 cas 6H-Pyrrolo[4,3,2-ef][2]benzazepin-6-one, 8-fluoro-1,3,4,5-tetrahydro-2-[4-[(methylamino)methyl]phenyl]-6H- Azepino[5,4,3-cd]indol-6-one, 8-fluoro-1,3,4,5-tetrahydro-2-[4-[(methylamino)methyl]phenyl] -8-Fluoro-2-[4-[(methylamino)methyl]phenyl]-1,3,4,5- tetrahydro-6H-azepino[5,4,3-cd]indol-6-one;8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one8-Fluoro-2-(4-methylaminomethyl-phenyI)-l,3,4,5-tetrahydro-azepino[5,4,3- cd]indol-6-one
WO 2014052550, WO 2014037313, WO 2000042040WO 2004087713WO 2005012305
Rucaparib (AG 014699) is a PARP inhibitor being investigated as a potential anti-cancer agent.
Rucaparib inhibits “the contraction of isolated vascular smooth muscle, including that from the tumours of cancer patients. It also reduces the migration of some cancer and normal cells in culture.”[1]
It is thought that 20% of women with ovarian cancer who are not BRCA positive might also benefit from PARP inhibitors. Clinical trials are beginning (as of April, 2014)
As of November 2012 four clinical trials of rucaparib were recruiting patients.[5]
Inhibition of poly(ADP ribose) polymerase, or PARP, is an exciting new mechanism for the treatment of cancer.(1) The PARP enzyme is responsible for repair of damaged DNA in both normal and tumor cells, and inhibition of this repair mechanism is expected to make the cell more likely to undergo apoptosis. Preclinical work has shown that PARP inhibitors coadministered with a standard chemotherapuetic agent are more effective than the standard treatment aloneRucaparib is a NAD+ ADP-ribosyltransferase inhibitor in phase II clinical development at Cancer Research UK for the treatment of patients with advanced ovarian cancer and in patients with locally advanced or metastatic breast cancer. Clovis Oncology is conducting early clinical evaluation of rucaparib for the treatment of triple negative breast cancer or ER/PR +, HER2 negative with known BRCA1/2 mutations p2 and for the treatment of gBRCA mutation breast cancer.. Pfizer discontinued development of rucaparibin 2011.In 2011, the compound was licensed to Clovis Oncology by Pfizer for the treatment of cancer. In 2012, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of ovarian cancer.
The compound 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3- cd]indol-6-one represented by formula
is a small molecule inhibitor of poly(ADP-ribose) polymerase (PARP). 8-Fluoro-2-{4- [(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one and salts thereof, is disclosed in U.S. Patent No. 6,495,541 and PCT Application No. PCT/IB2004/000915, International Publication No. WO 2004/087713, the disclosures of which are incorporated herein by reference in their entireties. U.S. Provisional Patent Applications No. 60/612,459 and 60/679,296, entitled “Polymorphic Forms of the Phosphate Salt of 8-Fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H- azepino[5,4,3-cd]indol-6-one,” the disclosures of which are incorporated herein by reference in their entireties, describe novel polymorphic forms of the phosphate salt of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one, and methods for their preparation. U.S. Provisional Patent Applications No. 60/612,458; and 60/683,006, entitled “Therapeutic Combinations Comprising Poly(ADP-Ribose) Polymerases Inhibitor,” the disclosures of which are incorporated herein by reference in its entirety, describe pharmaceutical combinations of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one.
Example IIII:8-Fluoro-2-(4-methylaminomethyl-phenyI)-l,3,4,5-tetrahydro-azepino[5,4,3- cd]indol-6-one
4-(8-fluoro-6-oxo-3,4,5,6-tetrahydro-lH-azepino[5,4,3-cd]indol-2-yl)- benzaldehyde (100 mg, 0.32 mmol; prepared in a manner similar to that described for compound 12 for 2-bromo-8-fluoro-l,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one and 4-formylphenylboronic acid) was reacted with methylamine (1.62 mmol) as described for Compound PPP to yield 8-fluoro-2-(4-methylaminomethyl-phenyl)- l,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one, 32 mg (31%) as a yellow solid: m.p. 1543-155 °C; Η NMR (300 MHz, d6-DMSO) 2.28 (s, 3H), 3.04 (m, 2H), 3.40 (m, 2H), 3.69 (s, 2H), 7.32 (dd, 7= 9.0, 2.4 Hz, IH), 7.44 (m, 3H), 7.57 (d, 7= 8.1 Hz, 2H), 8.25 (br t, IH), 11.67 (br s, IH). HRMS (MALDI MH+) Calcd for C19H18N3OF: 324,1512. Found: 325.1524. Anal. (C19H18N3OF03 H2O) C, H, N.
PAPER
Org. Process Res. Dev., 2012, 16 (12), pp 1897–1904
DOI: 10.1021/op200238p
http://pubs.acs.org/doi/full/10.1021/op200238pNovel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
To a solution of aqueous sodium hydroxide (40% w/w, 3.6 kg, 2.0 equiv) in water (88 L, 14 L/kg) and methanol (35 L, 5.5 L/kg) was added 12 ……………………………………………………deleted……………………..and dried at 45 °C under vacuum to give 1 as a 1:1 THF solvate (5.57 kg, 14.08 mol, 84% yield);
8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one (S)-camphorsulfonate Salt (21)
To a slurry of 1 (5.32 kg, 13.48 mol) in isopropanol (30 L, 5.5 L/kg) and water (39 L, 7.3 L/kg) was added a solution of (S)-camphorsulfonic acid (3.75 kg, 16.18 mol, 1.2 equiv) in water (10.6 L, 2 L/kg). The resultant slurry was then heated to 70 °C and held for 1 h to ensure dissolution. …………………………..deleted…………………..C to give 21 as a white crystalline solid (7.09 kg, 12.76 mol, 95% yield); mp (IPA/water) 303 °C;
The compound 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3- cd]indol-6-one represented by formula
is a small molecule inhibitor of poly(ADP-ribose) polymerase (PARP). 8-Fluoro-2-{4- [(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one and salts thereof, is disclosed in U.S. Patent No. 6,495,541 and PCT Application No. PCT/IB2004/000915, International Publication No. WO 2004/087713, the disclosures of which are incorporated herein by reference in their entireties.
U.S. Provisional Patent Applications No. 60/612,459 and 60/679,296, entitled “Polymorphic Forms of the Phosphate Salt of 8-Fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H- azepino[5,4,3-cd]indol-6-one,” the disclosures of which are incorporated herein by reference in their entireties, describe novel polymorphic forms of the phosphate salt of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one, and methods for their preparation. U.S. Provisional Patent Applications No. 60/612,458; and 60/683,006, entitled “Therapeutic Combinations Comprising Poly(ADP-Ribose) Polymerases Inhibitor,” the disclosures of which are incorporated herein by reference in its entirety, describe pharmaceutical combinations of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one.
Example 13. Synthesis of 8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3.4.5-tetrahvdro-azepinor5.4.3- ccflindol-6-one (15) i
Lactam 14 (14.42 g, 0.038 mol) was dissolved in hydrobromic acid in acetic acid (30%-32%, 140 ml). The reaction solution was stirred for 46 hours at room temperature in a 500ml flask that was connected to an ethanolamine scrubber system. HPLC analysis indicated the completion of the reaction. Ice (30 g) was added to the reaction solution followed by addition of aqueous NaOH (327 ml, 10 M, 3.27 mol) while the temperature was maintained between 25 0C and 35 0C. When addition of NaOH was complete, the pH was 10. The resulting solid was collected by filtration, washed with water (2 x 50 ml). The filter cake was then suspended in water (125 ml) and stirred for 2 hours. The solid was collected by filtration, washed with water (2 x 25 ml) and dried to afford 10.76 g of product (88% yield). 1H NMR (300 MHz, DMSO-d6) δ 2.577(s, 3H), 3.053(m, 2H), 3.406(m, 2H), 4.159(s, 2H), 7.36(dd, 1 H, J= 2.4 Hz and J= 9.3 Hz), 7.44(dd, 1 H, J= 2.4 Hz and J= 11.1 Hz), 7.63(d, 2H, J=8.1 Hz), 7.70(d, 2H, J= 8.1 Hz), 8.265(t, 1H, J= 5.7 Hz), 11.77(s, 1 H). Exact mass calculated for C19H19FN3O: 324.1512. Found: 324.1497.
UPDATES
OriginatorClovis Oncology; Foundation Medicine
ClassDiagnostic agents
Highest Development Phases
RegisteredOvarian cancer
Phase IIIFallopian tube cancer; Peritoneal cancer
Clinical Phase UnknownCancer
Most Recent Events
19 Dec 2016Registered for Ovarian cancer (Diagnosis) in USA
23 Aug 2016Preregistration for Ovarian cancer (Diagnosis) in USA (unspecified route)
05 May 2016Clovis Oncology announces intention to submit PMA application to US FDA
Rucaparib phosphateis in phase Ⅲ clinical trials for the treatment of patients with advanced ovarian cancer, fallopian tube cancer and ovarian cancer. It was granted breakthrough therapy designation by FDA for the treatment of ovarian cancer in 2015.
The compound was originally developed by Pfizer, then licensed to Clovis Oncology by Pfizer in 2011 for the treatment of cancer.
Clovis Oncology receives Breakthrough Therapy designation for rucaparib for treatment of advanced ovarian cancer in patients with BRCA-mutated tumours
7 April 2015 • Author: Victoria White
Clovis Oncology has announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation for the Company’s investigational agent rucaparib as monotherapy treatment of advanced ovarian cancer in patients who have received at least two lines of prior platinum-containing therapy, with BRCA-mutated tumours, inclusive of both germline BRCA (gBRCA) and somatic BRCA (sBRCA) mutations.
2525 28th Street
Suite 100
Boulder, CO 80301 Tel: 303.625.5000
Fax: 303.245.0360
are a biopharmaceutical company focused on acquiring, developing and commercializing cancer treatments in the United States, Europe and other international markets. Our development programs are targeted at specific subsets of cancer, combining personalized medicine with companion diagnostics to direct therapeutics to those patients most likely to benefit from them.
We have three product candidates in clinical development: rociletinib (CO-1686), which is in Phase II development for the treatment of non-small cell lung cancer; rucaparib, which is in Phase II and Phase III clinical trials for the treatment of ovarian cancer; and lucitanib, which is in Phase II clinical trials for the treatment of breast and lung cancers. We have received Breakthrough Therapy designation from the FDA for rociletinib and rucaparib. We maintain global rights for rociletinib and rucaparib, and U.S. and Japanese rights to lucitanib.
Women suffering from advanced relapsed BRCA-mutated ovarian cancer could gain access to a new treatment option after European regulators waved through AstraZeneca’s Lynparza (olaparib).
The European Commission has approved the first-in-class PARP inhibitor for the maintenance treatment of adults with platinum-sensitive relapsed BRCA-mutated high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete response or partial response to platinum-based chemotherapy.
Olaparib is an oral poly-ADP-ribose polymerase (PARP) enzyme inhibitor developed by AstraZeneca. The product is awaiting registration in the E.U. and US as a maintenance treatment of patients with BRCA mutated platinum-sensitive relapsed serous ovarian cancer. In 2014, positive opinion was received in the E.U. recommending Lynparza approval for the maintanance treatment of BRCA mutated platinum-sensitive relapsed serous ovarian cancer.
An oral poly (ADP ribose) polymerase (PARP) inhibitor being investigated by British drug company AstraZeneca, is seeking approval from the U.S. Food and Drug Administration (FDA) for the treatment of BRCA mutated platinum-sensitive relapsed ovarian cancer. AstraZeneca filed the US regulatory submission for olaparib in February 2014. Olaparib, one of several cancer drugs AstraZeneca flagged as having strong potential in its defense of a $118 billion take-over bid by Pfizer,was accepted for priority review on April 30, 2014 by the U.S. Food and Drug Administration (FDA). The NDA filing was based on Phase II study 19 data, a randomized, double-blind, placebo-controlled, Phase II study.
On June 25, 2014, FDA Oncologic Drugs Advisory Committee (ODAC), an advisory panel to the U.S. Food and Drug Administration (FDA), voted 11 to two against the accelerated approval of the PARP inhibitor olaparib as a maintenance therapy for women with platinum-sensitive relapsed ovarian cancer who have the germline BRCA (gBRCA) mutation, and who are in complete or partial response to platinum-based chemotherapy. By voting no, the committee recommended waiting for results from the larger confirmatory phase III SOLO-2 trial, which began enrolling in September 2013. According to clincialtrials.gov, the SOLO-2 study (NCT01874353) is slated to wrap in July 2015.
In terms of clinical development, phase III trials are ongoing at AstraZeneca for the treatment of gastric cancer and metastatic breast cancer. Olaparib is also in phase II clinical studies for several indications, including breast cancer, pancreatic cancer and castration-resistant prostate cancer. In March 2014, a phase II was also initiated in GB for the treatment of patients with stage IIIB or stage IV NSCLC that is not amenable to curative therapy. A phase I clinical trial for the treatment of melanoma has been completed. Phase II clinical trials are ongoing at General Hospital Corp. for the treatment of sarcoma. The drug had been in phase II clinical trials for the treatment of colorectal cancer; however no recent developments have been reported.
Discovered by KuDOS Pharmaceuticals, has experienced several twists and turns during its clinical development. Promising results for the drug were reported at the 2011 ASCO Annual Meeting, based on impressive early phase II results, only to have clinical development discontinued later that year after disappointing phase II trial results in a more generalized group of ovarian cancer patients. However, a re-analysis of the data in BRCA-positive patients – coupled with a reformulation of the drug – convinced the British drugmaker to think again and keep it going. AstraZeneca initiates Phase III clinical studies (SOLO 1 and SOLO 2) for olaparib in the U.S. in September 2013. AstraZeneca has filed Marketing Authorisation Application (MAA) for olaparib in EU in September 2013 based on Phase II study 19 data. The U.S. Food and Drug Administration has already granted olaparib orphan drug status for ovarian cancer and will hold an advisory panel hearing on the company’s application on June 25, 2014.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of ovarian cancer. The compound was originally developed by Kudos Pharmaceuticals, which was acquired by AstraZeneca in 2006.
Early Phase I trials were promising, and olaparib underwent Phase II trials. However, in December 2011, AstraZeneca announced following interim analysis of a phase-II study which indicated that the previously reported progression free survival benefit was unlikely to translate into an overall survival benefit, that it would not progress into Phase III development for the maintenance treatment of serous ovarian cancer,[2] and took a charge of $285 million. The decision to discontinue development of the drug was reversed in 2013,[3] with AstraZeneca posting a new Phase III trial of Olaparib for patients with BRCA mutated ovarian cancer in April 2013.[4]
Mechanism of action
Olaparib acts as an inhibitor of the enzymePoly ADP ribose polymerase (PARP) and is one of the first PARP inhibitors. Patients with BRCA1/2 mutations may be genetically predisposed to developing some forms of cancer, and are often resistant to other forms of cancer treatment, but this also sometimes gives their cancers a unique vulnerability, as the cancer cells have increased reliance on PARP to repair their DNA and enable them to continue dividing. This means that drugs which selectively inhibit PARP may be of significant benefit in patients whose cancers are susceptible to this treatment.[5][6][7][8][9][10]
Trial results
Phase I clinical trials, in patients with BRCA-mutated tumors including ovarian cancer, were encouraging.[11] In one of these studies, it was given to 19 patients with inherited forms of advanced breast, ovarian and prostate cancers caused by mutations of the BRCA1 and BRCA2 genes. In 12 of the patients, none of whom had responded to other therapies, tumours shrank or stabilised.[12] One of the first patients to be given the treatment (who had castration-resistant prostate cancer) was as of July 2009 still in remission after two years.
In 2009 Phase II clinical trials examining the efficacy of Olaparib in treating breast, ovarian and colorectal cancer were initiated.[13][14] A phase II trial that included 63 cases of ovarian cancer concluded that olaparib is promising for women with ovarian cancer. [7 responses in 17 patients with BRCA1 or BRCA2 mutations and 11 responses in the 46 who did not have these mutations.][15]
4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1
J Med Chem 2008, 51(20): 6581
3- (4-0x0-3 , 4-dihydrophthalazin-l -ylmethyl) benzoic a cid (A)
A mixture of 27% sodium methoxide solution in methanol (400 g, 2 mol) and methanol (150 ml) was added dropwise between ambient temperature and 30°C over 15 minutes to a stirred mixture of phthalide (67 g, 0.5 mol), 3-formylbenzonitrile (65.5 g, 0.5 mol) and ethyl propionate (250 ml) , the mixture was stirred at ambient temperature for 40 minutes and at reflux temperature for 1 hour, then it was allowed to cool to ambient temperature. The resulting red solid was collected by filtration, washed with ethyl acetate (2 x 50 ml) and dissolved in water (1800 ml) . The solution was acidified by the addition of acetic acid (60 ml) and the resulting red solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give 3- (1,3- dioxoindan-2-yl) benzonitrile (83.2 g) as a dark red solid, m.pt. 179- 182°C, m/z (M+H)+‘ 248, which was used without further purification.
3- (1, 3-Dioxoindan-2-yl) benzonitrile (74.18 g, 0.3 mol) was added in portions to a solution of sodium hydroxide (36 g, 0.9 mol) in water (580 ml), the resulting dark red suspension was stirred at reflux temperature for 5 hours, then it was cooled to ambient temperature and washed with ethyl acetate (3 x 300 ml) . The aqueous solution was acidified by the dropwise addition of concentrated hydrochloric acid (110 ml), the mixture was stirred at ambient temperature for 1 hour, then the resulting solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give a 1:1 mixture of 3- (1,3- dioxoindan-2-yl)benzoic acid, (M+H)+” 267, and 2- [2- (3- carboxyphenyl) acetyl] benzoic acid, (M+H)+‘ 285, (69.32 g) , which was used without further purification.
The mixture obtained in the previous step (52.8 g) was added to a solution of triethylamine (37.55 g, 0.372 mol) in industrial methylated spirit (500 ml) and the resulting cloudy solution was filtered through a pad of filter-aid to give a clear solution. Hydrazine monohydrate (9.3 g, 0.186 mol) was added in one portion at ambient temperature, the stirred mixture was heated under reflux for 1 hour, then it was concentrated in vacuo to approximately 250 ml and added to a solution of sodium acetate (41 g, 0.5 mol) in water (500 ml) . The mixture was brought to pH 7 by the dropwise addition of concentrated hydrochloric acid, then it was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a white solid (15.62 g) . The combined filtrate and washings were acidified to pH 6 by the addition of hydrochloric acid, then the mixture was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a second crop of off-white solid (17.57 g) . The combined filtrate and washings from the second crop were readjusted to pH 6 and treated as before to give a third crop of pale orange solid (6.66 g) . The three crops were combined to give essentially pure 3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A), (M+H)+‘ 281, δH 4.4 (2H, s), 7.2-7.4 (IH, m) , 7.5-7.6 (IH, ) , 7.7-8.0 (5H, m) , 8.1- 8.2 (IH, m) , 12.6 (IH, s)
b . 2-Fluoro-5- (4-oxo-3 , 4-dihydro-phthalazin -l -ylmethyl) benzoi c a cid (B)
Dimethyl phosphite (22.0 g, 0.2 mol) was added drop-wise to a solution of sodium methoxide (43.0 g) in methanol (100 ml) at 0°C. 2- Carboxybenzaldehyde (21.0 g, 0.1 mol) was then added portion-wise to the reaction mixture as a slurry in methanol (40 ml), with the temperature kept below 5°C. The resulting pale yellow solution was warmed to 20°C over 1 hour. Methanesulphonic acid (21.2 g, 0.22 mol) was added to the reaction drop-wise and the resulting white suspension was evaporated in va cuo . The white residue was quenched with water and extracted into chloroform (3 x 100 ml) . The combined organic extracts were washed with water (2 x 100 ml) , dried over MgS04, and evaporated in va cuo to yield (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester as a white solid (32.0 g, 95 %, 95 % purity) . This was then used without further purification in the next stage.
To a mixture of (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester (35.0 g, 0.14 mol) in tetrahydrofuran (200 ml) and 2- fluoro-5-formylbenzonitrile (20.9 g, 0.14 mol) in tetrahydrofuran (130 ml) was added triethylamine (14 ml, 0.14 mol) drop-wise over 25 min, with the temperature kept below 15°C. The reaction mixture was warmed slowly to 20°C over 1 hour and concentrated in vacuo . The white residue was slurried in water (250 ml) for 30 minutes, filtered, washed with water, hexane and ether, and dried to yield 2-fluoro-5- (3- oxo-3H-isobenzofuran-l-ylidenemethyl) benzonitrile as a 50:50 mixture of E and Z isomers (37.2 g, 96 %); m/z [M+l]+ 266 (98 % purity) To a suspension of 2-fluoro-5- (3-oxo-3H-isobenzofuran-l- ylidenemethyl) benzonitrile in water (200 ml) was added aqueous sodium hydroxide (26.1 g in 50 ml water) solution and the reaction mixture was heated under nitrogen to 90 °C for 30 minutes. The reaction mixture was partially cooled to 70°C, and hydrazine hydrate (100 ml) was added and stirred for 18 hours at 70°C. The reaction was cooled to room temperature and acidified with 2M HC1 to pH 4. The mixture was stirred for 10 min and filtered. The resulting solid was washed with water, hexane, ether, ethyl acetate and dried to yield 2-fluoro-5- (4-oxo-3, 4- dihydrophthalazin-l-ylmethyl)benzoic acid as a pale pink powder (30.0 g, 77 %) . m/z [M+l]+ 299 (96 % purity), δH 4.4 (2H, s) , 7.2-7.3 (IH, m) , 7.5-7.6 (IH, m) , 7.8-8.0 (4H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s).
c . 1 – [3- (4-Oxo-S , 4-dihydrophthalazin-l -ylmethyl) benzoyl]piperidine-4- carboxylic a cid (C)
undesried????????
(A) (C)
3- (4-Oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoic acid (A) (7.0 g, 0.25 mol), ethyl isonipecotate (5 ml, 0.32 mol), 2- (lH-benzotriazol-1-yl) – 1, 1, 3, 3-tetramethyluronium hexafluorophosphate (HBTU) (12.3 g, 0.32 mol) and N, N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (40 ml) and stirred for 18 h. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were washed with water (3 x 100 ml), dried over MgS0, filtered and evaporated in va cuo to yield an oil. To a solution of the oil in tetrahydrofuran (100 ml) was added 10 % aqueous sodium hydroxide solution (20 ml) and the reaction was stirred for 18 hours. The reaction was concentrated, washed with ethyl acetate (2 x 30 ml) and acidified with 2M HCl to pH 2. The aqueous layer was extracted with dichloromethane (2 x 100 ml), then the extracts were dried over MgS04, filtered and evaporated to yield 1- [3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoyl]piperidine- 4-carboxylic acid (C) as a yellow solid (7.0 g, 65 %), m/z [M+l]+ 392
(96 % purity), δH 1.3-1.8 (5H, m) , 2.8-3.1 (4H, m) , .4 (2H, s), 7.2- 7.3 (IH, m) , 7.3-7.4 (IH, ) , 7.7-8.0 (5H, m) , 8.2-E 3 (IH, m) , 12.6 (IH, s) .
d . 1 – [2-Fluoro-5- (4 -oxo-3 , 4-dihydrophthala zin-l – ylmethyl) benzoyl]piperidine-4~carboxylic a cid (D)
(B) (D)
2-Fluoro-5- ( -oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (B) (3.1 g, 0.14 mol), ethyl isonipecotate (1.7 ml, 0.11 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (5.1 g, 0.13 mol) and N,N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (15 ml) and stirred for 18 hours. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were, filtered, washed with water (3 x 100 ml), dried over MgS04, filtered and evaporated in vacuo to yield an orange oil. The oil was purified by flash chromatography (ethyl acetate) to yield l-[2- fluoro-5- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoyl] piperidine-4- carboxylic acid as the methyl ester (1.5 g, 33 %, 96 % purity) . To a solution of the methyl ester in tetrahydrofuran: water (2:1, 40 ml) was added sodium hydroxide (0.3 g, 0.075 mol) and the reaction was stirred for 18 h. The reaction was concentrated, washed with ethyl acetate (2 x 20 ml) and acidified with 2M HC1 to pH 2. The aqueous layer was extracted with dichloromethane (2 x 20 ml) , and the combined extracts were dried over MgS04 and evaporated to yield 1- [3- ( 4-oxo-3, 4- dihydrophthalazin-1-ylmethyl) benzoyl] piperidine- -carboxylic acid (D) as a yellow solid (0.6 g, 65 %), m/z [M+l]+ 392 (96 % purity) Example 1 – Synthesis of Key Compounds
a. Synthesis of 4- [3- (piperazine-1-carfoonyl)benzyl] -2H-phthalasin-l- one (1)
undesired????????
(A) (1)
3- (4-0xo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A) (5.0g, 0.17mol), tert-butyl 1-piperazinecarboxylate (3.9 g, 0.21 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (8.6 g, 0.22 mol) and N, , -diisopropylethylamine (6.7 ml, 0.38 mol) were added to dimethylacetamide (40 ml) and stirred for 18 hours. Water (100 ml) was added and the reaction mixture was heated to 100°C for 1 hour. The suspension was cooled to room temperature, filtered and dried to yield a white solid. The solid was dissolved in a solution of 6M HC1 and ethanol (2:1, 50 ml) and stirred for 1 hour. The reaction was concentrated, basified with ammonia to pH 9, and the product was extracted into dichloromethane (2 x 50 ml). The combined organic layers were washed with water (2 x 50 ml), dried over MgS04, and evaporated in va cuo to yield 4- [3- (piperazine-1-carbonyl) benzyl] – 2H-phthalazin-l-one (1) as a yellow crystalline solid (4.0 g, 77 %); m/z [M+l]+ 349 (97 % purity), δH 2.6-3.8 (8H, ) , 4.4 (2H, s), 7.2-7.5 (4H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s)
b . Synthesis of 4 – [4-Fluoro-3- (piperazine-1 -carbonyl) benzyl ] -2H- phthala zin ~l -one (2)
desired……
(β) (2)
The synthesis was carried out according to the method described in (a) above using 2-fluoro-5- (4-oxo-3, -dihydrophthalazin-l-ylmethyl) benzoic acid (B) to yield 4- [4-fluoro-3- (piperazine-1-carbonyl) benzyl] -2H- phthalazin-1-one (2) as a white crystalline solid (4.8 g, 76 %); m/z [M+l]+ 367 (97 % purity), δH 2.6-3.8 (8H, m) , 4.4 (2H, s), 7.2-7.5 (3H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s) .
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:
is of particular interest.
A crystalline form of compound A (Form A) is disclosed in co-pending applications, which claim priority from U.S. 60/829,694, filed 17 Oct. 2006, entitled “Phthalazinone Derivative”, including U.S. Ser. No. 11/873,671 and WO 2008/047082.
Form A
References(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (I)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. 0-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:
is of particular interest.
In WO 2004/080976, compound A was synthesised as one of a number of library compounds from 4-[4-fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (compound B):
by the addition of cyclopropanecarbonyl chloride:
to a solution of (B) in dichloromethane, followed by Hünig’s base (N,N-diisopropylethyl amine). This reaction is carried out with stirring at room temperature for 16 hours, and the resulting compound being purified by preparative HPLC.
The piperazine derivative (B) was prepared by deprotecting 4-[2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (compound C):
by the use of 6M HCl and ethanol for 1 hour, followed by basification with ammonia to pH 9, and extraction into dichloromethane.
The Boc-protected piperazine derivative (C) was prepared from 2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoic acid (compound D):
by the addition of piperazine-1-carboxylic acid tert-butyl ester:
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N,-diisopropylethylamine in dimethylacetamide, followed by stirring for 18 hours.
In WO 2004/080976, the following route to compound D is disclosed:
The method of synthesising compound D may further comprise the step of:
from compound E by reaction with hydrazine hydrate; and
(d) synthesising compound D from compound ED by reaction with sodium hydroxide.
Step (c) may be achieved by using between 1.1 and 1.3 equivalents of hydrazine hydrate in tetrahydrofuran followed by neutralisation of the excess hydrazine hydrate using acetic acid.
A sixth aspect of the present invention provides the compound ED:
and its use in the synthesis of compound D.
EXAMPLES
Example 1Synthesis of Compound A
Starting material (D) was synthesised by the method disclosed in WO 2004/080976
Methods
Preparative HPLC
Samples were purified with a Waters mass-directed purification system utilising a Waters 600 LC pump, Waters Xterra C18 column (5 μm 19 mm×50 mm) and Micromass ZQ mass spectrometer, operating in positive ion electrospray ionisation mode. Mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) were used in a gradient; 5% B to 100% over 7 min, held for 3 min, at a flow rate of 20 ml/min.
Analytical HPLC-MS
Analytical HPLC was carried out with a Spectra System P4000 pump and Jones Genesis C18 column (4 μm, 50 mm×4.6 mm). Mobile phases A (0.1% formic acid in water) and B (acetonitrile) were used in a gradient of 5% B for 1 min rising to 98% B after 5 min, held for 3 min at a flow rate of 2 ml/min. Detection was by a TSP UV 6000LP detector at 254 nm UV and range 210-600 nm PDA. The Mass spectrometer was a Finnigan LCQ operating in positive ion electrospray mode.
To a stirred solution of the starting material D (850 g) in dimethylacetamide (DMA) (3561 ml) at room temperature under nitrogen was added HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (1402 g) in one portion. Hünig’s base (iPr2NEt, 1096 ml) was then added with the temperature kept between 15 to 25° C. followed by a solution of 1-Boc-piperazine (637 g) in DMA (1428 ml) with the temperature kept between 15 to 25° C.
The solution was stirred at room temperature for 2 hours and sampled for completion (HPLC). Upon completion the solution was added to vigorously stirred water (17085 ml) with the temperature kept between 15 to 25° C. and the solid filtered off, washing with water (2×7131 ml), hexane (2×7131 ml) and methyl tert-butyl ether (MTBE) (2×3561 ml). The solid was then dried overnight and then sampled for water content and chemical purity.
This reaction was then repeated, see table:
Purity
Water Content
Batch
Yield (g)
(HPLC Area %)
(K.F.)
Corrected yield
1
1571.3
86.80
24.3
1032.5 g (78%)
2
2781.6
85.00
40.3
1411.5 g (106%)
a. Greater than 100% yield attributed to non-representative sampling
To a stirred solution of industrial methylated spirits (IMS) (2200 ml) and concentrated HCl (4400 ml) was added compound C (2780.2 g) in portions at room temperature under nitrogen, the foaming was controlled by the addition rate. The solution was then stirred at 15 to 25° C. for 30 minutes and sampled for completion (HPLC).
Upon completion the solution was evaporated to remove any IMS and the aqueous extracted with CH2Cl2 (2×3500 ml) before the pH was adjusted to >8 using concentrated ammonia. The resultant slurry was then diluted with water (10000 ml) and extracted with CH2Cl2 (4×3500 ml), washed with water (2×2000 ml), dried over MgSO4 (250 g) and evaporated. The crude product was then slurried in CH2Cl2 (3500 ml) and added to MTBE (5000 ml). The resultant suspension was filtered and dried at 50° C. overnight yielding 611.0 g (58.5% yield) of material with a purity of 94.12%
To a stirred suspension of compound B (1290 g) in CH2Cl2 (15480 ml) under nitrogen was added a pre-mixed solution of triethylamine (470 ml) and cyclopropane carbonyl chloride (306 ml) in CH2Cl2 (1290 ml) dropwise with the temperature kept below 20° C. The solution was then stirred at 10-15° C. for 15 minutes and sampled for completion. The reaction mixture was found to contain only 1.18% of starting material B and so the reaction was deemed complete and the batch was then worked-up.
The reaction mixture was washed with water (7595 ml), 5% citric acid solution (7595 ml), 5% sodium carbonate solution (7595 ml) and water (7595 ml). The organic layer was then dried over magnesium sulfate (500 g).
The CH2Cl2 containing product layer was then isolated, filtered through Celite and charged to a 251 vessel. CH2Cl2 (8445 ml) was then distilled out at atmospheric pressure and ethanol (10000 ml) added. Distillation was then continued with every 4000 ml of distillate that was removed being replaced with ethanol (4000 ml) until the head temperature reached 73.7° C. The reaction volume was then reduced (to 7730 ml) by which time the head temperature had reached 78.9° C. and the solution was allowed to cool to 8° C. overnight. The solid was then filtered off, washed with ethanol (1290 ml) and dried at 70° C. overnight. Yield=1377.3 g (90%). HPLC purity (99.34% [area %]). Contained 4.93% ethanol and 0.45% CH2Cl2 by GC.
(d) Water Treatment of Compound A
A suspension of compound A (1377.0 g), as produced by the method of Example 1, in water (13770 ml) was heated to reflux for 4 hours, cooled to room temperature and filtered. The solid was washed with water (2754 ml) and dried at 70° C. overnight. Yield=1274.8 g (92.6%). HPLC purity (99.49% [area %]). Contained 0.01% ethanol and 0.01% CH2Cl2 by GC.
1H NMR spectrum of compound A (DMSO-d6) following the water treatment is shown in FIG. 1.
The powder XRD pattern of Compound A following the water treatment is shown in FIG. 2, which shows the compound is as Form A.
Example 2
Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine
Methods (also for Examples 3 & 4)
NMR
1H NMR spectra were recorded using Bruker DPX 400 spectrometer at 400 MHz. Chemical shifts were reported in parts per million (ppm) on the δ scale relative to tetramethylsilane internal standard. Unless stated otherwise all samples were dissolved in DMSO-d6.
Mass Spectra
Mass spectra were recorded on an Agilent XCT ion trap mass spectrometer using tandem mass spectrometry (MS/MS) for structural confirmation. The instrument was operated in a positive ion elctrospray mode.
(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (1)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. O-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Example 3Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine HCl salt
(a) 1-(Cyclopropylcarbonyl)piperazine HCl salt (I′)
Acetic acid (700 ml) was treated with piperazine (50.00 g, 0.581 mol) portionwise over 15 minutes with stirring under nitrogen The reaction mixture was warmed to 40° C. and maintained at this temperature until a complete solution was obtained. Cyclopropanecarbonyl chloride 59.2 ml, 0.638 mol) was added over 15 minutes. The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered and the filtrate distilled under reduced pressure until ˜430 ml of distillates had been collected. Toluene (550 ml) was charged to the reaction mixture and reduced pressure distillation continued until a further 400 ml of distillates were collected. A further charge of toluene (550 ml) was added and reduced pressure distillation continued until 350 ml of distillates were collected. The resulting slurry was diluted with toluene (200 ml) and stirred overnight. Further toluene (500 ml) was added in order to mobilise the slurry. The slurry was filtered, washed with toluene (100 ml) and dried in vacuo at 40° C. to give the title compound as an off white solid (86.78 g).
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(0.95 g, 3.19 mmol) was suspended with stirring under nitrogen in acetonitrile (4 ml). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.45 g, 3.83 mmol) was added followed by 1-cyclopropylcarbonylpiperazine HCl salt (I′)(0.73 g, 3.83 mmol). Diisopropylethylamine (1.39 ml, 7.98 mmol) was added over 3 minutes and the reaction mixture was stirred for overnight at room temperature. The reaction mixture was cooled to 5° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (2 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (0.93 g).
Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, Douglas-Jones A, Smith GC, Martin NM, O’Connor M, Clarke AR (May 2009). “Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin”. Cancer Research69 (9): 3850–5. doi:10.1158/0008-5472.CAN-08-2388. PMID19383921.
http://clinicaltrials.gov/ct2/show/NCT00912743 Efficacy and Safety of Olaparib in Pretreated Patients With Measurable Colorectal Cancer, Stratified by Microsatellite Instability (MSI) Status
Olaparib would act against BRCA1 or BRCA2 mutations. AZD2281 is not sensitive to tankyrase-1 (IC50 >1 μM). Olaparib could ablate the PARP-1 activity at concentrations of 30-100 nM in SW620 cells. Olaparib is hypersensitive to BRCA1-deficient cell lines (MDA-MB-463 and HCC1937), compared with BRCA1- and BRCA2-proficient cell lines (Hs578T, MDA-MB-231, and T47D). [1] Olaparib is strongly sensitive to KB2P cells due to suppression of base excision repair by PARP inhibition, which may result in the conversion of single-strand breaks to double-strand breaks during DNA replication, thus activating BRCA2-dependent recombination pathways. [2]
In Vivo
Combining with temozolomide, Olaparib (10 mg/kg, p.o.) significantly suppresses tumor growth in SW620 xenografts. [1] Olaparib shows great response to Brca1-/-;p53-/- mammary tumors (50 mg/kg i.p. per day), while no responses to HR-deficient Ecad-/-;p53-/- mammary tumors. Olaparib even does not show dose-limiting toxicity in tumor-bearing mice. [3] Olaparib has been used to treat with BRCA mutated tumors, such as ovarian, breast and prostate cancers. Moreover, Olaparib shows selectively inhibition to ATM (Ataxia Telangiectasia Mutated)-deficient tumor cells, which indicates to be a potential agent for treating ATM mutant lymphoid tumors. [4]
Clinical Trials
Combining with cediranib, Olaparib is currently in Phase I/II study for treatment of recurrent papillary-serous ovarian, fallopian tube or peritoneal cancer or treatment of recurrent triple-negative breast cancer.
To columns 1 through 10, 1 μL of Olaparib (in DMSO) is added, and 1 μL DMSO only is added to the positive (POS) and negative (NEG) control wells (columns 11 and 12, respectively) of a pretreated FlashPlate. PARP-1 is diluted 1:40 in buffer (buffer B: 10% glycerol (v/v), 25 mM HEPES, 12.5 mM MgCl2,50 mM KCl, 1 mM DTT, 0.01% NP-40 (v/v), pH 7.6) and 40 μL added to all 96 wells (final PARP-1 concentration in the assay is ~1 ng/μL). The plate is sealed and shaken at RT for 15 min. Following this, 10 μL of positive reaction mix (0.2 ng/μL of double-stranded oligonucleotide [M3/M4] DNA per well, 5 μM of NAD+ final assay concentration, and 0.075 μCi 3H-NAD+ per well) is added to the appropriate wells (columns 1-11). The negative reaction mix, lacking the DNA oligonucleotide, is added to column 12 (with the mean negative control value used as the background). The plate is resealed and shaken for a further 60 min at RT to allow the reaction to continue. Then, 50 μL of ice-cold acetic acid (30%) is added to each well to stop the reaction, and the plate is sealed and shaken for a further 60 min at RT. Tritiated signal bound to the FlashPlate is then determined in counts per minute (CPM) using the TopCount plate reader.
In vitro isolated enzyme assay
PARP-2 activity inhibition uses a variation of the PARP-1 assay in which PARP-2 protein (recombinant) is bound down by a PARP-2 specific antibody in a 96-well white-walled plate. PARP-2 activity is measured following 3H-NAD+ DNA additions. After washing, scintillant is added to measure 3H-incorporated ribosylations. For tankyrase-1, a α-Screen assay is developed in which HIS-tagged recombinant TANK-1 protein is incubated with biotinylated NAD+in a 384-well ProxiPlate assay. Alpha beads are added to bind the HIS and biotin tags to create proximity signal, whereas the inhibition of TANK-1 activity is directly proportional to the loss of this signal.
Breast cancer cell lines including SW620 colon, A2780 ovarian, HCC1937, Hs578T, MDA-MB-231, MDA-MB-436, and T47D
Concentrations
1-300 nM
Incubation Time
7-14 days
Method
The cytotoxicity of Olaparib is measured by clonogenic assay. Olaparib is dissolved in DMSO and diluted by culture media before use. The cells are seeded in six well plates and left to attach overnight. Then Olaparib is added at various concentrations and the cells are incubated for 7-14 days. After that the surviving colonies are counted for calculating the IC50.
Janssen seeks FDA approval for Yondelis drug to treat advanced STS
Janssen Research & Development is seeking approval from US Food and Drug Administration (FDA) for its Yondelis (trabectedin) to treat patients with advanced soft tissue sarcoma (STS).
Trabectedin, also referred as ET-743 during its development, is a marine derived antitumoral agent discovered in the Carribean tunicate _Ecteinascidia turbinata_ and now produced synthetically. Trabectedin has a unique mechanism of action. It binds to the minor groove of DNA interfering with cell division and genetic transcription processes and DNA repair machinery.It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have granted orphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Trabectedin (also known as ecteinascidin 743 or ET-743) is an anti-tumor drug. It is sold by Zeltia and Johnson and Johnson under the brand name Yondelis. It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have granted orphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Discovery and development
The ecteinascidins (herein abbreviated ETs) are exceedingly potent antitumor agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example U.S. Pat. No. 5,089,273, which describes novel compounds of matter extracted from the tropical marine invertebrate Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumor agents in mammals. U.S. Pat. No. 5,478,932 describes other novel ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo antitumor activity against P388 lymphoma, B16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- I human lung and MX- 1 human mammary carcinoma xenografts.
One of the ETs, ecteinascidin 743 (ET-743), is a tetrahydroisoquinoline alkaloid with considerable in vitro and in vivo antitumor activity in murine and human tumors, and potent antineoplastic activity against a variety of human tumor xenografts grown in athymic mice, including melanoma, ovarian and breast carcinoma.
ET-743 is a natural compound with the following structure:
ET-743 is also known with the generic name trabectedin and the trademark Yondelis®, and it is currently approved in Europe for the treatment of soft tissue sarcoma. The clinical development of trabectedin continues in phase 11/ III clinical trials in breast, ovarian and prostate cancer. A clinical development program of ET-743 in cancer patients was started with phase I studies investigating 1- hour, 3-hour, 24-hour, and 72-hour intravenous infusion schedules and a 1 hour daily x 5 (dx5) schedule. Promising responses were observed in patients with sarcoma, breast and ovarian carcinoma.
Therefore this new drug is currently under intense investigation in several phase 11/ III clinical trials in cancer patients with a variety of neoplastic diseases. Further information regarding the dosage, schedules, and administration of ET-743 for the treatment of cancer in the human body, either given alone or in combination is provided in WO 00/69441 , WO 02/36135, WO 03/39571 , WO 2004/ 105761 , WO 2005/039584, WO 2005/049031 , WO 2005/049030, WO 2005/049029, WO 2006/046080, WO 2006/005602, and PCT/US07/98727, which are incorporated by reference herein in their entirety.
A review of ET-743, its chemistry, mechanism of action and preclinical and clinical development can be found in Kesteren, Ch.
Van et al., Anti-Cancer Drugs, 2003, 14 (7), 487-502: “ET-743 (trabectedin, ET-743): the development of an anticancer agent of marine origin”, and references therein.
During the past 30 years medical oncologists have focused to optimise the outcome of cancer patients and it is just now that the new technologies available are allowing to investigate polymorphisms, gene expression levels and gene mutations aimed to predict the impact of a given therapy in different groups of cancer patients to tailor chemotherapy. Representative examples include the relationship between the Thymidylate Synthase (TS) mRNA expression and the response and the survival with antifolates, beta tubulin III mRNA levels and response to tubulin interacting agents, PTEN gene methylation and resistance to CPT- I l and, STAT3 over expression and resistance to Epidermal Growth Factor (EGF) interacting agents.
A molecular observation of potential clinical impact relates to the paradoxical relation between the efficiency of the Nucleotide Excision Repair (NER) pathway and the cytotoxicity of ET-743. In fact, tumour cells that are efficient in this DNA repair pathway appear to be more sensitive to ET-743. This evidence is in contrast with the pattern noted with platin based therapeutic regimens which are highly dependent on the lack of activity of this repair pathway (ie. an increase in ERCCl expression has been associated to clinical resistance to platinum-based anti-cancer therapy).
There are evidences on the key role of NER pathways on the cytotoxicity of ET-743 in cell lines. ET-743 binds to G residues in the minor groove of DNA forming adducts that distort the DNA helix structure and they are recognised by NER mechanisms (Pourquier, P. et al., 2001 , Proceedings of the American Association for Cancer Research Annual Meeting, Vol. 42, pp. 556. 92nd Annual Meeting of the American Association for Cancer Research. New Orleans, LA, USA. March 24-28, 2001. ISSN: 0197-016X). Takebayasi et al. (Nature Medicine, 2001 , 7(8), 961-966) have proposed that the presence of these DNA adducts in transcribed genes, blocks the Transcription Coupled NER (TC-NER) system by stalling the cleavage intermediates and producing lethal Single Strand Breaks (SSBs). It is known from Grazziotin et al (Proc.Natl.Acad.Sic.USA, 104: 13062- 13067) that the DNA adducts formed by exposure to ET-743 are transformed into double strand DNA breaks.
The fact that NER mediates ET-743 ‘s cytotoxicity has also been found in the yeast Saccharomyces cerevisae by Grazziotin et al. (Biochemical Pharmacology, 2005, 70, 59-69) and in the yeast Schizosaccharomyces pombe by Herrero et al. (Cancer Res. 2006, 66(16), 8155-8162).
In addition, Bueren et al. (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have been shown that ET-743 induces double-strand breaks in the DNA in early S phase that are detected and repaired by the Homologous Recombination Repair (HRR) pathway. In addition, Erba et al (Eur. J. Cancer, 2001 , 37(1), 97- 105) and Bueren et al (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have shown that inactivation/ mutations of genes related to the Double Strand Break detection such as DNA-PK, ATM and ATR and of genes related to Homologous Recombination Repair pathway, such as Fanconi Anemia genes, BRCAl , BRCA2 and RAD51 make cells more sensitive to trabectedin. Such unique finding is the opposite to the pattern with conventional DNA interacting agents, like in the case of microtubule poisons such as taxanes and vinorelbine.
Finally, pharmacogenomic studies prior have demonstrated that increased expression of the NER genes ERCCl and XPD in the tumor tissue does not impact the outcome of patients treated with
ET-743. However, the low expression of BRCAl in the tumor tissue is correlated with a better outcome in cancer patents treated with
ET-743. Further information can be found in WO 2006/005602, which is incorporated by reference herein in its entirety.
Three rare, autosomal recessive inherited human disorders are associated with impaired NER activity: xeroderma pigmentosum (XP), Cockayne Syndrome (CS), and trichothiodystrophy (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245- 274). XP patients exhibit extreme sensitivity to sunlight, resulting in a high incidence of skin cancers (Kraemer et al. Arch. Dermatol. 123, 241-250, and Arch. Dermatol. 130, 1018- 1021). About 20% of XP patients also develop neurologic abnormalities in addition to their skin problems. These clinical findings are associated with cellular defects, including hypersensitivity to killing and mutagenic effects of UV, and inability of XP cells to repair UV-induced DNA damage (van Steeg et al. MoI. Med. Today, 1999, 5, 86-94).
Seven different NER genes, which correct seven distinct genetic XP complementation groups (XPA-XPG), have been identified (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245-274). The human gene responsible for XP group G was identified as ERCC5 (Mudgett et al. Genomics, 1990, 8, 623-633; O’Donovan et al. Nature, 1993, 363, 185- 188; and Nouspikel et al. Hum. MoI. Genet. 1994, 3, 963-967). The XPG gene codes for a structure-specific endonuclease that cleaves damaged DNA ~5 nt 3′ to the site of the lesion and is also required non-enzymatically for subsequent 5’ incision by the XPF/ ERCCl heterodimer during the NER process (Aboussekhra et al. Cell, 1995, 80, 859-868; Mu et al. J. Biol. Chem. 1996, 271 , 8285-8294; and Wakasugi et al. J. Biol. Chem. 1997, 272, 16030- 16034). There is also evidence suggesting that XPG is also involved in transcription-coupled repair of oxidative DNA lesions (Le Page et al. Cell, 101 , 159- 171).
Takebayashi et al. (Cancer Lett., 2001 , 174: 1 15- 125) have observed an increase in heterozygosity loss and microsatellite instability in a substantial percentage of samples of ovarian, lung and colon carcinoma. Le Moirvan et al, (Int.J. Cancer, 2006,1 19: 1732- 1735) have described the presence of polymorphisms in the XPG gene in sarcoma patients. It is also known from Takebayashi et al. (Proceedings of the American Association forCancer Research Annual Meeting, March, 2001 , Vol. 42, pp. 813.92nd Annual Meeting of the American Association for Cancer
Research. New Orleans, LA, USA. March 24-28, 2001) that cells deficient in the NER system are resistant to treatment with ET-743 (Zewail-Foote, M. et al., 2001 , Chemistry and Biology, 8: 1033- 1049 and Damia, G. et al., 2001 , Symposium AACR NCI EORTC) and that the antiproliferative effects of ET-743 require a functional XPG gene.
Since cancer is a leading cause of death in animals and humans, several efforts have been and are still being undertaken in order to obtain an antitumor therapy active and safe to be administered to patients suffering from a cancer. Accordingly, there is a need for providing additional antitumor therapies that are useful in the treatment of cancer.
Trabectedin is a tetrahydroisoquinoline, a novel marine-derived antitumor agent isolated from the colonial tunicate Ecteinascidia turbinate. The drug binds to the minor groove of the DNA, bending the DNA towards the major groove, blocking the activation of genes in a unique way via several pathways, including selective inhibition of the expression of key genes (including oncogenes) involved in cell growth and drug resistance, inhibition of genetic repair pathways and inhibition of cell cycle progression leading to p53-independent programmed cell death.
In July 2003, the European Committee of Proprietary Medicinal Products (CPMP) recommended against granting marketing authorization to trabectedin for soft tissue sarcoma. PharmaMar appealed the decision in September 2003. Later that year, the CPMP rejected the company’s appeal. In 2006, the company filed another regulatory application for this indication and, finally, in 2007, a positive opinion was received in the E.U. for the treatment of metastatic soft tissue sarcoma. First commercialization of the product in the E.U. took place in October 2007 in the U.K. and Germany.
The compound is also available in several other countries. In 2008, the compound was filed for approval in the U.S. and the E.U. for the treatment of relapsed advanced ovarian cancer in combination with liposomal doxorubicin, and in 2009 approval was received in both countries. Trabectedin is available in several European countries, including the U.K. and Germany. Also in 2009 the drug candidate was approved in Philippines for the ovarian cancer indication.
The compound had been in phase II development by Johnson & Johnson for the treatment of prostate cancer; however, no recent development has been reported for this research. PharmaMar is evaluating the compound in phase II trials for the treatment of breast cancer. Additional early clinical trials are ongoing at the National Cancer Institute (NCI) to evaluate trabectedin for potential use in the treatment of advanced, persistent or recurrent uterine leiomyosarcomas and solid tumors.
In 2011, a regulatory application that had been filed in the U.S. seeking approval for the treatment of relapsed advanced disease in combination with liposomal doxorubicin was withdrawn by the company based on the FDA’s recommendation that an additional phase III study be conducted to obtain approval. In 2014, Janssen Research & Development, LLC submitted an NDA for trabectedin to the FDA for the treatment of patients with advanced soft tissue sarcoma (STS), including liposarcoma and leiomyosarcoma subtypes, who have received prior chemotherapy including an anthracycline.
Trabectedin was developed by PharmaMar, a subsidiary of Zeltia. The drug was being codeveloped and comarketed in partnership with Ortho Biotech, a subsidiary of Johnson & Johnson pursuant to an agreement signed in 2001. However, in 2008 the license agreement between the two companies was terminated.
The compound was granted orphan drug designation for the treatment of soft tissue sarcoma and for the treatment of ovarian cancer by the FDA and the EMEA. In 2011, orphan drug designation was granted in Japan for the treatment of malignant soft tissue tumor accompanied with chromosomal translocation. In 2009, the product was licensed to Taiho by PharmaMar in Japan for the treatment of cancer.
During the 1950s and 1960s, the National Cancer Institute carried out a wide ranging program of screening plant and marine organism material. As part of that program extract from the sea squirtEcteinascidia turbinata was found to have anticancer activity in 1969.[1] Separation and characterisation of the active molecules had to wait many years for the development of sufficiently sensitive techniques, and the structure of one of them, Ecteinascidin 743, was determined by KL Rinehart at the University of Illinois in 1984.[2] Rinehart had collected his sea squirts by scuba diving in the reefs of the West Indies.[3]
Recently, the biosynthetic pathway responsible for producing the drug, has been determined to come from Candidatus Endoecteinascidia frumentensis, a microbial symbiont of the tunicate.[4] The Spanish company PharmaMar licensed the compound from the University of Illinois before 1994 and attempted to farm the sea squirt with limited success.[3]
Yields from the sea squirt are extremely low – it takes 1 tonne of animals to isolate 1 gram of trabectedin – and about 5 grams were believed to be needed for a clinical trial[5] so Rinehart asked the Harvard chemist E. J. Corey to search for a synthetic method of preparation. His group developed such a method and published it in 1996.[6] This was later followed by a simpler and more tractable method which was patented by Harvard and subsequently licensed to PharmaMar.[3] The current supply is based on a semisynthetic process developed by PharmaMar starting from Safracin B, an antibiotic obtained by fermentation of the bacterium Pseudomonas fluorescens.[7] PharmaMar have entered into an agreement with Johnson and Johnson to market the compound outside Europe.
Trabectedin was first dosed in humans in 1996.In 2007, the EMEA gave authorisation for the marketing of trabectedin, under the trade name Yondelis, for the treatment of patients with advanced soft tissue sarcoma, after failure of anthracyclines and ifosfamide, or who are unsuited to receive these agents. The agency’s evaluating committee, the CHMP observed that trabectedin had not been evaluated in an adequately designed and analyzed randomized trial against current best care, and that the clinical efficacy data was mainly based on patients with liposarcoma and leiomyosarcoma. However the pivotal study did show a significant difference between two different trabectedin treatment regimens, and due to the rarity of the disease the CHMP considered that marketing authorisation could be granted under exceptional circumstances.[8] As part of the approval PharmaMar agreed to conduct a further trial to identify whether any specific chromosomal translocations could be used to predict responsiveness to trabectedin.[9] Trabectedin is also approved in South Korea[10] and Russia.
In 2008 the submission was announced of a registration dossier to the European Medicines Agency (EMEA) and the FDA for Yondelis when administered in combination with pegylated liposomal doxorubicin (Doxil, Caelyx) for the treatment of women with relapsed ovarian cancer. In 2011, Johnson&Johnson voluntarily withdrew the submission in the United States following a request by the FDA for an additional Phase III study to be done in support of the submission.[11]
Trabectedin is also in phase II trials for prostate, breast and paediatric cancers.[12]
Structure
Trabectedin is composed of 3 tetrahydroisoquinolinemoieties, 8 rings including one 10-membered heteocyclic ring containing a cysteine residue, and 7 chiral centers.
Biosynthesis
The biosynthesis of Trabectedin in Candidatus Endoecteinascidia frumentensis starts with a fatty acid loading onto the acyl-ligase domain of the EtuA3 module. A cysteine and glycine are then loaded as canonical NRPS amino acids. A tyrosine residue is modified by the enzymes EtuH, EtuM1, and EtuM2 to add a hydroxyl at the meta position of the phenol, and adding two methyl groups at the para-hydroxyl and the meta carbon position. This modified tyrosine reacts with the original substrate via a Pictet-Spangler reaction, where the amine group is converted to an imine by deprotonation, then attacks the free aldehyde to form a carbocation that is quenched by electrons from the methyl-phenol ring. This is done in the EtuA2 T-domain. This reaction is done a second time to yeid a dimer of modified tyrosine residues that have been further cyclized via Pictet-spangler reaction, yielding a bicyclic ring moiety. The EtuO and EtuF3 enzymes continue to post-translationally modify the molecule, adding several functional groups and making a sulfide bridge between the original cysteine residue and the beta-carbon of the first tyrosine to form ET-583, ET-597, ET-596, and ET-594 which have been previously isolated.[4] A third o-methylated tyrosine is added and cyclized via Pictet-Spangler to yield the final product.[4]
Proposed biosynthetic scheme for the biosynthesis of Trabecteden (ET-743)
The previously reported synthesis of 139221 (scheme 13922101a) has been investigated in order to find a more efficient, reproducible and economical route to work in the mutikilogram scale. Herein it is reported a new process which is simpler and proceeds with an overall yield of 54% (the original process, 35%). The condensation of intermediate aminolactone (I) (scheme 13922101a, intermediate (VII)) with acid (XLII) (the acid derived from scheme 13922101a, intermediate ester (IX)) by means of 2-chloro-1,3-dimethylimidazolidinium hexafluorophosphate (CIP), and 1-hydroxy-7-azabenzotriazole (HOAt) in THF/dichloromethane gives the coupling product (XLIII), which is allylated with allyl bromide (XLIV) and Cs2CO3 in DMF yielding the allyl ether (XLV). The reduction of the lactone group of (XLV) with LiAlH2(OEt)2 in ethyl ether affords the lactol (XLVI), which is desilylated with KF in methanol to provide the phenolic compound (XLVII). The opening of the lactol ring of (XLVII) with simultaneous cyclization by means of Tf-OH in water/trifluoroethanol gives the hexacyclic intermediate (XLVIII), which is finally reductocondensed with KCN by means of LiAlH2(OEt)2 in THF to furnish the previously reported pentacyclic intermediate (XI) (scheme 13922101a, intermediate (XI)).
……………………………………………
Reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino-protected compound (II), which is treated with methoxymethyl bromide (MOM-Br), DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF to provide compound (V). Reaction of (V) with allyl bromide (VI) and Cs2CO3 in DMF gives the allyl ether (VII), which first is treated with TFA, phenyl isothiocyanate and HCl to yield the primary amine (VIII) and then protected at the free NH2 group with Troc-Cl and pyridine, to afford the amino protected compound (IX).Org Lett 2000,2(16),2545
……………………………….
Reaction of (IX) with MOM-Br and DIEA as before affords the ether (X), which is treated with Zn/HOAc in order to regenerate the primary amino group giving (XI). The reaction of (XI) with NaNO2 and HOAc eliminates the NH2 group, affording the primary alcohol (XII), which is esterified with the protected (S)-cysteine (XIII) by means of EDC and DMAP in dichloromethane furnishing the cysteine ester (XIV). Reaction of (XIV) with Bu3SnH and PdCl2(PPh3)2, followed by oxidation with (PhSeO)2O in dichloromethane gives the hydroxyketone (XV), which is cyclized with Tf2O and Ac2O yielding the heptacyclic compound (XVI). Elimination of the MOM protecting group with TMSCl and NaI in CH3CN/CH2Cl2 affords the phenolic compound (XVII).
…………………….
Intermediate (XVII) by a treatment with Zn and HOAc eliminates the Troc protecting group, giving the primary amine (XVIII). This compound by treatment with 4-formyl-1-methylpyridinium iodide (NMPC), DBU and oxalic acid in order to convert the nitrile group into an alcohol, provides compund (XIX), which is finally cyclized with 2-(3-hydroxy-4-methoxyphenyl)ethylamine (XX) by means of SiO2 / EtOH, followed treatment with and AgNO3 in acetonitrile/water.
……………………….
The reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino protected compound (II), which is treated with Mom-Br, DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF providing the methylenedioxy compound (V). The reaction of (V) with acetyl chloride and pyridine in dichloromethane gives the acetate (VI), which is treated with TFA, phenyl isothiocyanate and HCl yielding the primary amine (VII). Finally, this compound is treated with phthalic anhydride (VIII) and CDI in dichloromethane to afford the target phthalimide (phthalascidin Pt-650)
Ecteinascidins are a group of marine alkaloid having antineoplasticity which is isolated from the extracted products from the marine tunicate habitat of the Caribbean sea by a very small amount. Arming the ecteinascidins, Et 743 has a very strong antineoplastic activity, studies to put it into practical use as a carcinostatic agent are limited, and the phase II clinical tests are now being carried out in ten countries in Europe and America. It is known that Et 743 has an effect of depressing the proliferation of cancer cells by 10 to 100 times more potent than (IC50=0.1-1 nM) Toxol, Camptotesin, Adriamycin or Mitomycin which are currently used carcinostatic agents.
From the background mentioned above, various studies for synthesis were carried out; however, the complete synthesis was only reported by Prof. E. J. Corey of Harvard University in the U.S.A. (J. Am. Chem. Soc. 1996, 118, 9202-9203, reference document A).
In the process of the total synthesis disclosed in Document A (refer to page 9202), the main feature of the process is that Et 743 is synthesized from the analogous compound to the compound represented by general formula 1 of the present invention via intermediates 4 and 8. That is, according to said process, the C4 site of ring B (regarding the location of rings, and the sites of atoms comprising the 6 membered ring, refer to general formula 1), which composes a 6 membered ring, is formed from the intermediate 4 at the first step. Since the atom C4 composing the ring B of the 6-membered ring H, which lacks reactivity, is bonded, it becomes necessary to perform an oxidation reaction at the C4 site on the B ring. This oxidation reaction is not effective and is carried out under harsh conditions; therefore production on an industrial scale is difficult, and also the yield is not good. Further, since the atom N12 site of the synthesized intermediate is substituted by an alkyl group which lacks reactivity, in this case substituted by a methyl group, it is not suited to the synthesis of various compounds. Although total synthesis was reported, the supplying source of Et 743 still depends on the natural sample whose supply is very scarce. Therefore, the establishment of the method for a large scale production of Et 743 is desired and requires accomplishing an effective synthesizing process.
Since ET 743 is known as a medicine having high antineoplasticity, and phthalascidin induced from the intermediate product at the synthesis of Et 743 displays the same activity to ET 743, the establishment of an effective and mild method for synthesis of ET 743 and analogous compounds thereof is strongly desired.
Therefore, the subject of the present invention is to accomplish the effective method for total synthesis of Et 743, and further, to provide not only Et 743 but also analogous compounds.
To dissolve the subject, the present invention uses retrosynthetic analysis for easy synthesis. It will be possible to form a B ring by a ring forming reaction at the ortho position of phenol, which binds an A ring to inner molecular aldehyde in a compound generated by the 4-8 reaction. Further, the present invention contemplates that the generated compound by the 4-8 reaction can be synthesized based on the polycondensation reaction of general formula 4, and general formula 5 via a compound of general formula 3. Then the total synthesis of Et 743, which is the aimed compound, can be accomplished by way of the compounds represented by general formulae 5, 4, 3, 2 and 1 and the specific structure of general formulae 1 and 2. This synthetic route provides for the analogous compounds of Et 743.
Mechanism of action
The biological mechanism of action is believed to involve the production of superoxide near the DNA strand, resulting in DNA backbone cleavage and cell apoptosis. The actual mechanism is not yet known, but is believed to proceed from reduction of molecular oxygen into superoxide via an unusual auto-redox reaction on a hydroxyquinone moiety of the compound following. There is also some speculation the compound becomes ‘activated’ into its reactive oxazolidine form.
Schematic of the unique and complex mode of action of trabectedin. The antitumor effects of trabectedin are due to multiple mechanisms involving DNA binding in the minor groove, interactions with DNA repair mechanisms, modulation of transcription regulation, and induction of microenvironment changes.
References
Lichter et al. Worthen LW, ed. “Food-drugs from the sea. Proc: Aug 20–23, 1972.” 173. Marine Tech Soc. pp. 117–127.
E. J. Corey, David Y. Gin, and Robert S. Kania (1996). “Enantioselective Total Synthesis of Ecteinascidin 743”. J. Am. Chem. Soc.118 (38): 9202–9203. doi:10.1021/ja962480t.
C. Cuevas et al. (2000). “Synthesis of ecteinascidin ET-743 and phthalascidin PT-650 from cyanosafracin”. B. Org. Lett. 2: 2545–2548.
Tohma, “Synthesis of Optically Active alpha-Arylglycines: Stereoselective Mannich-Type Reaction with a New Chiral Template“, Synlett 2001, No. 7, 1179-1181.Hamprecht, D.W.; Berge, J.M.; Copley, R.C.B.; Eggleston, D.S.; Houge-Frydrych, C.S.V.; Jarvest, R.L.; Mensah, L.M.; O’Hanlon, P.J.; Pope, A.J.; Rittenhouse, S.
Derivatives of the natural product SB-219383 and synthetic analogues: Potent inhibitors of bacterial tyrosyl tRNA synthetase
16th Int Symp Med Chem (September 18-22, Bologna) 2000, Abst PA-155Cuevas, C.; Perez, M.; Martin, M.J.; et al.
Synthesis of ecteinascidin ET-743 and phathalascidin Pt-650 from cyanosafracin B
Org Lett 2000, 2(16): 2545
Patent
Submitted
Granted
Assay for identifying biological targets of polynucleotide-binding compounds [US2008096201]
2008-04-24
Compounds of the saframycin-ecteinascidin series, uses, and synthesis thereof [US6936714]
2004-07-01
2005-08-30
Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7807833]
2009-08-06
2010-10-05
Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7820838]
2009-02-05
2010-10-26
Assay for identifying biological targets of polynucleotide-binding compounds [US7183054]
Array Starts First Phase 3 Trial, Shifts to Late-Stage Development Xconomy Array Biopharma said Tuesday it has received a $5 million milestone payment from Novartis for beginning Phase 3 testing of a drug it hopes can treat ovarian cancer. The milestone is just part of a larger transition for the Boulder, CO-based biopharmaceutical company.
A novel ovarian cancer treatment made from tumour cells has cured a woman in the US with an advanced form of the disease, scientists at the Perelman School of Medicine at the University of Pennsylvania have announced.
During a preliminary trial of the two-step immunotherapy, the patient achieved complete remission, while seven other women had no measurable disease at the end of the study.
The therapy includes a personalised immune cell vaccination made from the patients’ live tumour cells and adoptive T-cell therapy.
Both treatments are given in conjunction with Avastin (bevacizumab), a drug developed by Roche that controls the blood vessel growth that feeds tumours.
“This is the first time such a combination immunotherapy approach has been used for patients with ovarian cancer, and we believe the results are leading us toward a completely new way to treat this disease.”
The second step in the study involved the isolation of immune cells, known as dendritic cells, from the patients’ blood through a process called apheresis, similar to the process used for blood donation.
Announcing its findings at the American Association of Cancer Research (AACR) Annual Meeting in Washington DC on Saturday, the research team reported that in the study of 31 patients, vaccination therapy alone showed a 61% clinical benefit, and the combination of both therapies benefited around 75% of participants.
Ovarian cancer is the fifth leading cause of cancer-related deaths among women in the US, taking the lives of 14,000 people each year.
Lead author of the study Lana Kandalaft said; “Given these grim outcomes, there is definitely a vast unmet need for the development of novel, alternate therapies.”
“This is the first time such a combination immunotherapy approach has been used for patients with ovarian cancer, and we believe the results are leading us toward a completely new way to treat this disease.”
The vaccine trial is still open to accrual to test new combinatorial strategies.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











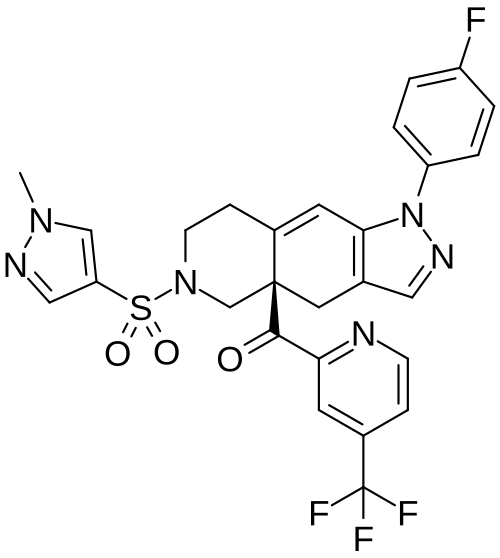


















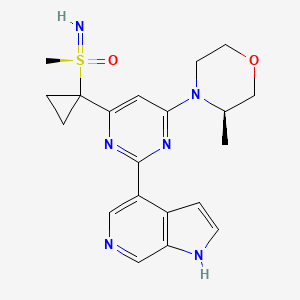















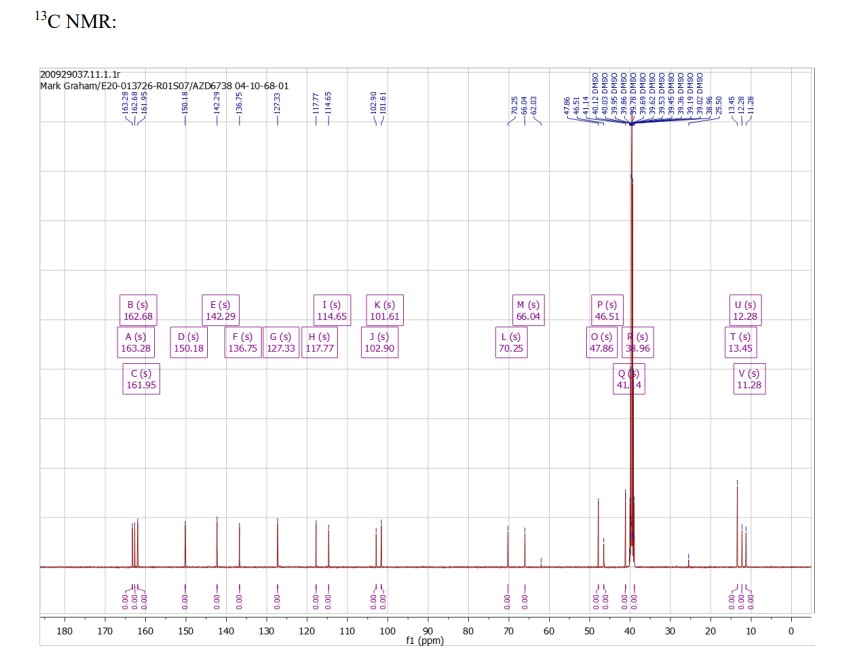





 Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.



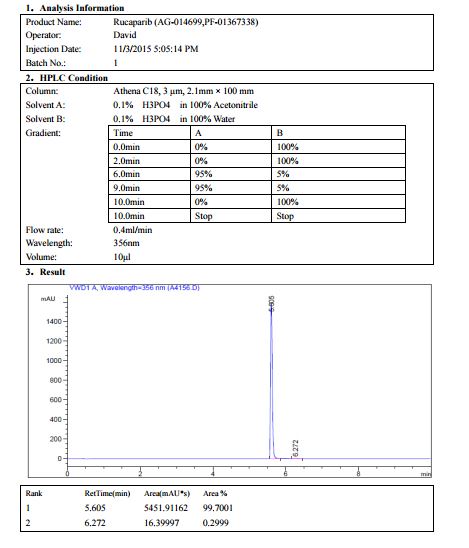
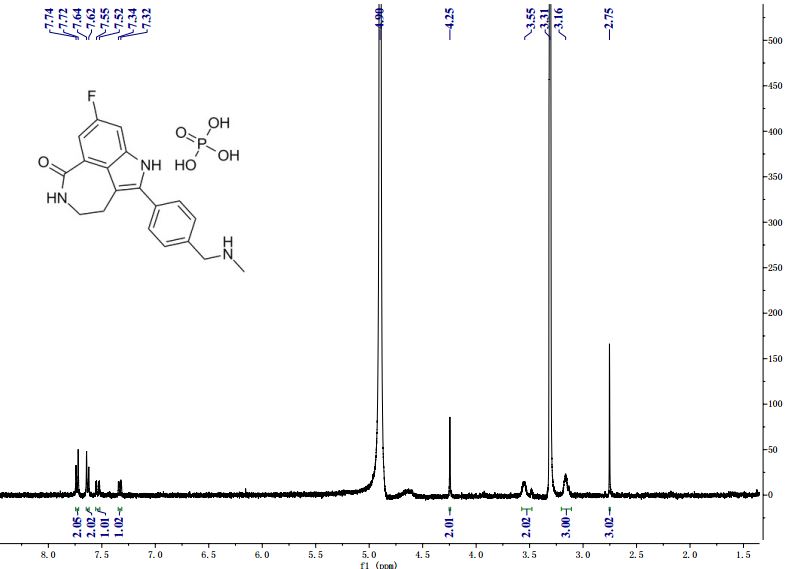


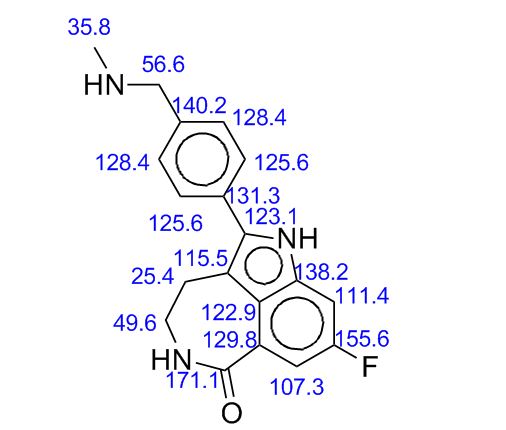
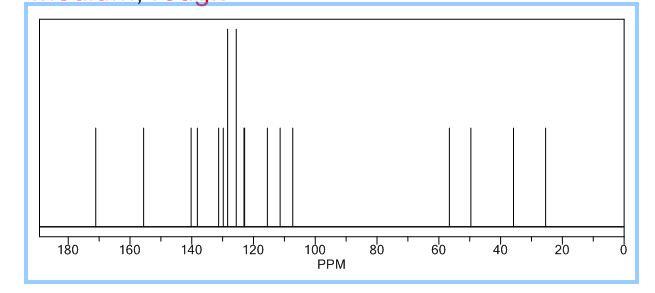





































































{kind=link}
{kind=link}
{kind=link}