
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
VADADUSTAT
| AKB-6548, PG-1016548 |
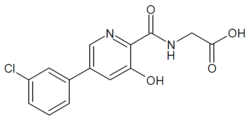
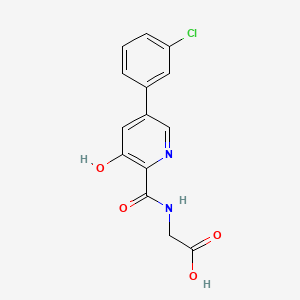
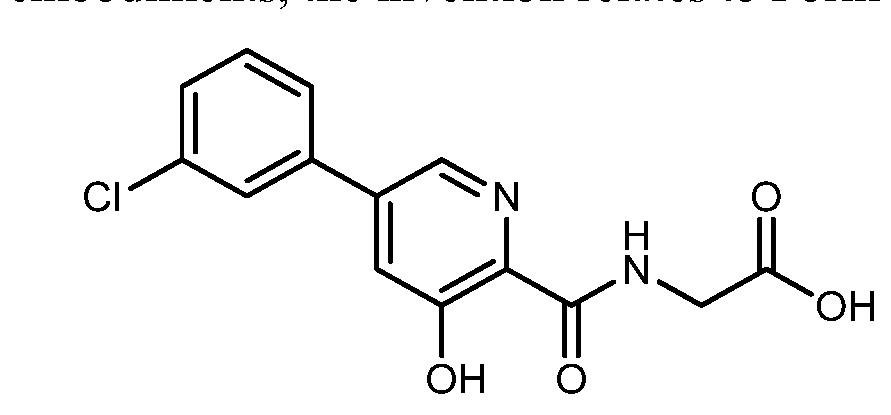
[5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxamido]acetic acid
N-[[5-(3-Chlorophenyl)-3-hydroxy-2-pyridinyl]carbonyl]glycine
MF C14H11ClN2O4 , 306.0407
| Inventors | Richard Kawamoto |
| Original Assignee | The Procter & Gamble Company |
for Treatment of Anemia associated with Chronic Kidney Disease (CKD)
USFDA APPROVED 3/27/2024 Vafseo, To treat anemia due to chronic kidney disease
Treatment of anemia due to chronic kidney disease
Akebia Therapeutics, under license from Procter & Gamble Pharmaceuticals, and licensees Mitsubishi Tanabe Pharma and Otsuka,
![]()
- Originator Procter & Gamble
- Developer Akebia Therapeutics
- Class Antianaemics; Chlorophenols; Pyridines; Small molecules
- Mechanism of Action Hypoxia-inducible factor-proline dioxygenase inhibitors
- Phase III Anaemia
- 01 Aug 2016 Akebia Therapeutics initiates the phase III INNO2VATE trial for Anaemia in USA (NCT02865850)
- 23 May 2016 Interim drug interactions and adverse events data from a phase I trial (In volunteers) Chronic kidney disease released by Akebia
- 05 May 2016 Akebia completes a clinical trial (ethnobridging study) in Healthy volunteers
Vadadustat (also known as AKB-6548) in anemia secondary to chronic kidney disease (CKD)
We are developing our lead product candidate, vadadustat, to be the potential best-in-class hypoxia inducible factor–prolyl hydroxylase inhibitor for the treatment of anemia secondary to CKD.
PATENT
CN 105837502
https://patents.google.com/patent/CN105837502A/sv
HIF inhibitor Vadadustat (Code AKB-6548) The chemical name N- [5- (3- chlorophenyl) -3-hydroxypyridine-2-carbonyl] glycine,
Vadadustat is a treatment for anemia associated with chronic kidney disease oral HIF inhibitor, is an American biopharmaceutical company Akebia Therapeutics invention in the research of new drugs, has completed Phase II pivotal clinical trial treatment studies, successfully met the researchers set given the level of hemoglobin in vivo target and good security, a significant effect, and phase III clinical trials.
U.S. Patent Publication US20120309977 synthetic route for preparing a Vadadustat: A 3-chlorophenyl boronic acid and 3,5_-dichloro-2-cyanopyridine as starting materials, by-catalyzed coupling methoxy substituted, cyano hydrolysis and condensation and ester hydrolysis reaction Vadadustat, process route is as follows:

Since the entire synthetic route 12 steps long, complicated operation, high cost.U.S. Patent No. 1 2 ^ ¥ disclosed 20070299086 & (^ (Scheme 3 1118 seven seven to 3,5-dichloro-2-cyanopyridine starting material, first-dichloro substituted with benzyloxy, then cyano hydrolysis, condensation, hydrogenation and deprotection trifluorosulfonyl, to give N- [5- trifluoromethanesulfonyloxy-3-hydroxypyridine-2-carbonyl) glycine methyl ester, 3-chlorophenyl and then boronic acid catalyzed coupling reactions, the final ester hydrolysis reaction Vadadustat, process route is as follows:

The synthesis steps long, intermediate products and final products contain more impurities and byproducts, thus purified requires the use of large amounts of solvents, complicated operation, low yield, and because the hydrogenation reaction is a security risk on the production, not conducive to the promotion of industrial production, it is necessary to explore a short process, simple operation, low cost synthetic method whereby industrial production Vadadus tat fit.

Example 1
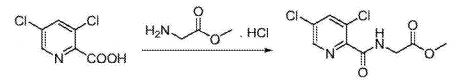
A) Preparation of N- (3,5_-dichloro-2-carbonyl) glycine methyl ester:
3,5-dichloro-2-pyridinecarboxylic acid (19.2g, 0.10mol) and N, N’_ carbonyldiimidazole (24.3g, 0.15mol) was dissolved in N, N- dimethylformamide (100 mL ), was added glycine methyl ester hydrochloride (15.18,0.12111〇1), 11 was added dropwise diisopropylethylamine (51.7g, 0.40mol), the reaction mixture was stirred 35 ° C for 8 hours, TLC determined the completion of reaction gussets The reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was adjusted to neutral by adding ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give N- (3,5- dichloro-pyridin-2 – carbonyl) glycine methyl ester, an off-white solid (21.6g), a yield of 82.0%, this reaction step is as follows:
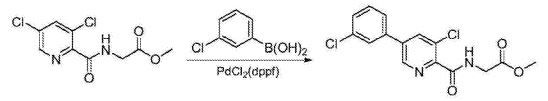
1234567 B) Preparation of N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester: 2
1 (3,5-dichloro-2-carbonyl) glycine methyl ester (20 (^, 〇1 76111111), 3-chlorophenyl boronic acid (13.18, 3 83.7mmol), [l, l’- bis (diphenylphosphino) ferrocene] dichloropalladium (2.8g, 3.8mmol), potassium carbonate (14.2g, 4 0. lmo 1) and N, N- dimethylformamide (75mL) was added The reaction flask, the reaction mixture was heated to 60 ° C for 20 hours the reaction was stirred for 5:00, point TLC plates to determine completion of the reaction, the reaction solution was cooled to room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, sulfuric acid 6 magnesium dried and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane was recrystallized to give N- [5- (3- chlorophenyl) -3-7-chloro-2-carbonyl] glycine methyl ester, white solid (19.7g), yield 76.4%, this reaction step is as follows:
C) Preparation of N_ [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine:
N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester (19 (^, 56111 111〇1) and sodium methoxide (7.6g, 0.14mol) was dissolved in methanol (150 mL), the reaction mixture was heated to 65 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution was cooled to room temperature, water (300mL) was stirred for 3h, cooled to 0 ° C, stirred for 2h, precipitated solid was filtered, the filter cake was dried to give N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine, off-white solid (17.4 g of), a yield of 96.5%, of the reaction steps are as follows:
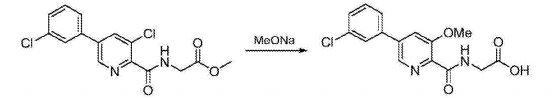
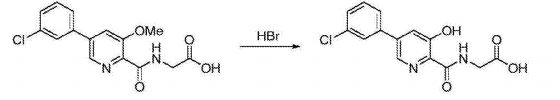
D) Preparation Vadadustat:
N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine (16.68,51.7111111〇1) and 48% hydrobromic acid solution (52mL, 0.46mol) added to the reaction bottle, the reaction mixture was heated to 100 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution cooled square ~ 5 ° C, was slowly added 50% sodium hydroxide solution was adjusted to pH 2 at 0 -5 ° C under crystallization 3h, the filter cake washed with ethyl acetate and n-hexane mixed solvent of recrystallization, in finished Vadadustat, off-white solid (15.6g), a yield of 98.0%, this reaction step is as follow
PATENT
WO-2016153996
Lanthier et al. (U.S. Patent Application 2012/0309977) described a procedure for synthesizing a compound of Formula (II) starting from 3-chloroboronic acid and 3,5-dichloropicolinonitrile, as shown in the scheme below:
Scheme 1

Scheme 2

PATENT
WO 2015073779
FORM A, B C REPORTED
https://www.google.com/patents/WO2015073779A1?cl=en
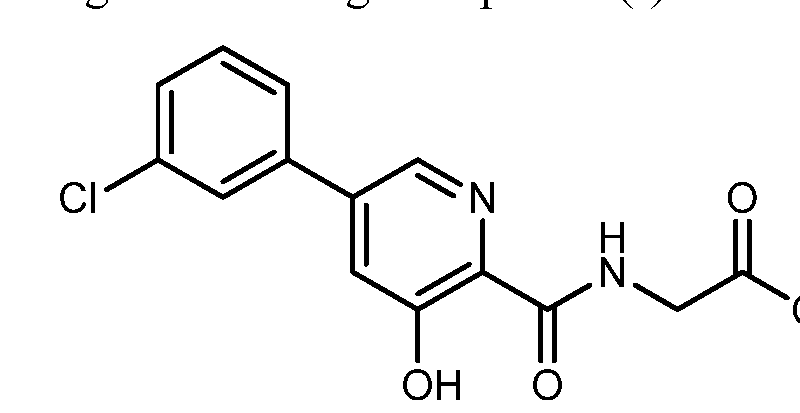
Form A of Compound (I):
(I),
which has an X-ray powder diffraction pattern as shown in FIG. 1. In certain embodiments, Form A of Compound (I) has an X-ray powder diffraction pattern comprising one, two, three, four, or five peaks at approximately 18.1 , 20.3, 22.9, 24.0, and 26.3 °2Θ; and wherein the crystalline Compound (I) is substantially free of any other crystalline form of Compound (I).
Compound (I) as prepared according to e.g., U.S. 7,811,595 and/or U.S. Patent Application No. 13/488,554 and then subjecting the resulting Compound (I)
(I),
to a procedure comprising
a) preparing a solution of Compound (I) in 2-methyltetrahydrofuran;
b) adding n-heptane;
c) heating the suspension {e.g., to about 40-50 °C);
d) cooling the suspension {e.g., to about 0-10 °C); and
c) isolating the crystals.
SYNTHESIS
US 2015361043

Synthesis of vadadustat and its intermediates is described. The process involves Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile with (3-chlorophenyl)boronic acid, selective chloride displacement, simultaneous hydrolysis of nitrile and methyl ether, activation with CDI, condensation with methyl glycinate hydrochloride and finally ester hydrolysis. The process is simple and provides high product yield with high quality. Vadadustat is expected to be useful for the treatment of renal failure anemia (1). Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile (I) with (3-chlorophenyl)boronic acid (II) in the presence of PdCl2(dppf) and K2CO3 in DMF yields 3-chloro-5-(3-chlorophenyl)pyridine-2-carbonitrile (III), which upon selective chloride displacement with NaOMe in refluxing MeOH affords methyl ether (IV). Hydrolysis of nitrile and methyl ether in intermediate (IV) with HBr or HCl at 100 °C furnishes 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (V). After activation of carboxylic acid (V) with CDI or pivaloyl chloride and DIEA in DMSO, condensation with methyl glycinate hydrochloride (VI) in the presence of DIEA provides vadadustat methyl ester (VII). Finally, hydrolysis of ester (VII) with NaOH in H2O/THF produces the target vadadustat (1).
PATENT
-
FIG. 1 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitors.FIG. 2 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor ester prodrugs.FIG. 3 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor amide prodrugs.




Example 1 describes a non-limiting example of the disclosed process for the preparation of a prolyl hydroxylase ester pro-drug


EXAMPLE 1Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): To a 100 mL round bottom flask adapted for magnetic stirring and equipped with a nitrogen inlet was charged (3-chlorophenyl)boronic acid (5 g, 32 mmol), 3,5-dichloro-2-cyanopyridine (5.8 g, 34 mmol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (0.1 g, 0.13 mmol), dimethylformamide (50 mL) and water (5 mL). The reaction solution was agitated and heated to 45° C. and held at that temperature for 18 hours after which the reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to room temperature and the contents partitioned between ethyl acetate (250 mL) and saturated aqueous NaCl (100 mL). The organic phase was isolated and washed a second time with saturated aqueous NaCl (100 mL). The organic phase was dried for 4 hours over MgSO4, the MgSO4 removed by filtration and the solvent removed under reduced pressure. The residue that remained was then slurried in methanol (50 mL) at room temperature for 20 hours. The resulting solid was collected by filtration and washed with cold methanol (50 mL) then hexanes (60 mL) and dried to afford 5.8 g (73% yield) of an admixture containing a 96:4 ratio of the desired regioisomer. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H)
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): To a 500 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (10 g, 40 mmol), sodium methoxide (13.8 mL, 60 mmol) and methanol (200 mL). With stirring, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to room temperature and combined with water (500 mL). A solid began to form. The mixture was cooled to 0° C. to 5° C. and stirred for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 9.4 g (96% yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (1 g, 4 mmol) and a 48% aqueous solution of HBr (10 mL). While being stirred, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction contents was then cooled to 0° C. to 5° C. with stirring and the pH was adjusted to approximately 2 by the slow addition of 50% aqueous NaOH. Stirring was then continued at 0° C. to 5° C. for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 1.03 g (quantitative yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a nitrogen inlet tube was charged 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (1 gm, 4 mmol), N,N′-carbonyldiimidazole (CDI) (0.97 g, 6 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at 45° C. for about 1 hour then cooled to room temperature. Glycine methyl ester hydrochloride (1.15 g, 12 mmol) is added followed by the dropwise addition of diisopropylethylamine (3.2 mL, 19 mmol). The mixture was then stirred for 2.5 hours at room temperature after which water (70 mL) was added. The contents of the reaction flask was cooled to 0° C. to 5° C. and 1N HCl was added until the solution pH is approximately 2. The solution was extracted with dichloromethane (100 mL) and the organic layer was dried over MgSO4 for 16 hours. Silica gel (3 g) is added and the solution slurried for 2 hours after which the solids are removed by filtration. The filtrate is concentrated to dryness under reduced pressure and the resulting residue was slurried in methanol (10 mL) for two hours. The resulting solid was collected by filtration and washed with cold methanol (20 mL) then hexane and the resulting cake is dried to afford 0.85 g of the desired product as an off-white solid. The filtrate was treated to afford 0.026 g of the desired product as a second crop. The combined crops afford 0.88 g (68% yield) of the desired product. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use
EXAMPLE 2Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): A 20 L reactor equipped with a mechanical stirrer, dip tube, thermometer and nitrogen inlet was charged with (3-chlorophenyl)boronic acid (550 g, 3.52 mol), 3,5-dichloro-2-cyanopyridine (639 g, 3.69 mol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (11.5 g, 140 mmol), and dimethylformamide (3894 g, 4.125 L). The reaction solution was agitated and purged with nitrogen through the dip-tube for 30 minutes. Degassed water (413 g) was then charged to the reaction mixture while maintaining a temperature of less than 50° C. 25 hours. The reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to 5° C. and charged with heptane (940 g, 1.375 L) and agitated for 30 minutes. Water (5.5 L) was charged and the mixture was further agitated for 1 hour as the temperature was allowed to rise to 15° C. The solid product was isolated by filtration and washed with water (5.5 L) followed by heptane (18881 g, 2750 ML). The resulting cake was air dried under vacuum for 18 hours and then triturated with a mixture of 2-propanol (6908 g, 8800 mL0 and heptane (1 g, 2200 mL0 at 50° C. for 4 hours, cooled to ambient temperature and then agitated at ambient temperature for 1 hour. The product was then isolated by filtration and washed with cold 2-propanol (3450 g, 4395 mL) followed by heptane (3010 g, 4400 mL). The resulting solid was dried under high vacuum at 40° C. for 64 hours to afford 565.9 g (65% yield) of the desired product as a beige solid. Purity by HPLC was 98.3. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H).
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): A 20 L reactor equipped with a mechanical stirred, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (558 g, 2.24 mol) and sodium methoxide (25% solution in methanol, 726.0 g, 3.36 mol). With agitation, the reaction solution was heated to reflux for 24 hours, resulting in a beige-colored suspension. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to 5° C. and then charged with water (5580 mL). The resulting slurry was agitated for 3 hours at 5° C. The solid product was isolated by filtration and washed with water (5580 mL) until the filtrate had a pH of 7. The filter cake was air dried under vacuum for 16 hours. The filter cake was then charged back to the reactor and triturated in MeOH (2210 g, 2794 mL) for 1 hour at ambient temperature. The solid was collected by filtration and washed with MeOH (882 g, 1116 mL, 5° C.) followed by heptane (205 mL, 300 mL), and dried under high vacuum at 45° C. for 72 hours to afford 448 g (82% yield) of the desired product as an off-white solid. Purity by HPLC was 97.9%. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer, nitrogen inlet and 25% aqueous NaOH trap was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (440.6 g, 1.8 mol) and 37% aqueous solution of HCl (5302 g). While being agitated, the reaction solution was heated to 102° C. for 24 hours. Additional 37% aqueous HCl (2653 g) was added followed by agitation for 18 hours at 104° C. The reaction contents was then cooled to 5° C., charged with water (4410 g) and then agitated at 0° C. for 16 hours. The resulting precipitated product was isolated by filtration and washed with water until the filtrate had a pH of 6 (about 8,000 L of water). The filter cake was pulled dry under reduced pressure for 2 hours. The cake was then transferred back into the reactor and triturated in THF (1958 g, 2201 mL) at ambient temperature for 2 hours. The solid product was then isolated by filtration and washed with THF (778 g, 875 mL) and dried under reduced pressure at 5° C. for 48 hours to afford 385 g (89% yield) of the desired product as an off-white solid. HPLC purity was 96.2%. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (380 g, 1.52 mol) and diisopropylethylamine (DIPEA) (295 g, 2.28 mol). With agitation, the solution was cooled to 3° C. and charged with trimethylacetyl chloride (275.7 g, 2.29 mol) while maintaining a temperature of less than 11° C., The mixture was then agitated at ambient temperature for 2 hours. The mixture was then cooled to 10° C. and charged with a slurry of glycine methyl ester HCl (573.3 g, 4. 57 mol) and THF (1689 g, 1900 mL), then charged with DIPEA (590.2 g, 4.57 mol) and agitated at ambient temperature for 16 hours. The mixture was then charged with EtOH (1500 g, 1900 mL) and concentrated under reduced pressure to a reaction volume of about 5.8 L. The EtOH addition and concentration was repeated twice more. Water (3800 g) was then added and the mixture was agitated for 16 hours at ambient temperature. The resulting solid product was isolated by filtration and washed with a mixture of EtOH (300 g, 380 mL) and water (380 g), followed by water (3800 g), dried under reduced pressure for 18 hours at 50° C. to afforded 443 g (91% yield) of the desired product as an off-white solid. Purity by HPLC was 98.9%. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
Scheme II herein below outlines and Example 2 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase inhibitor from an ester prodrug.
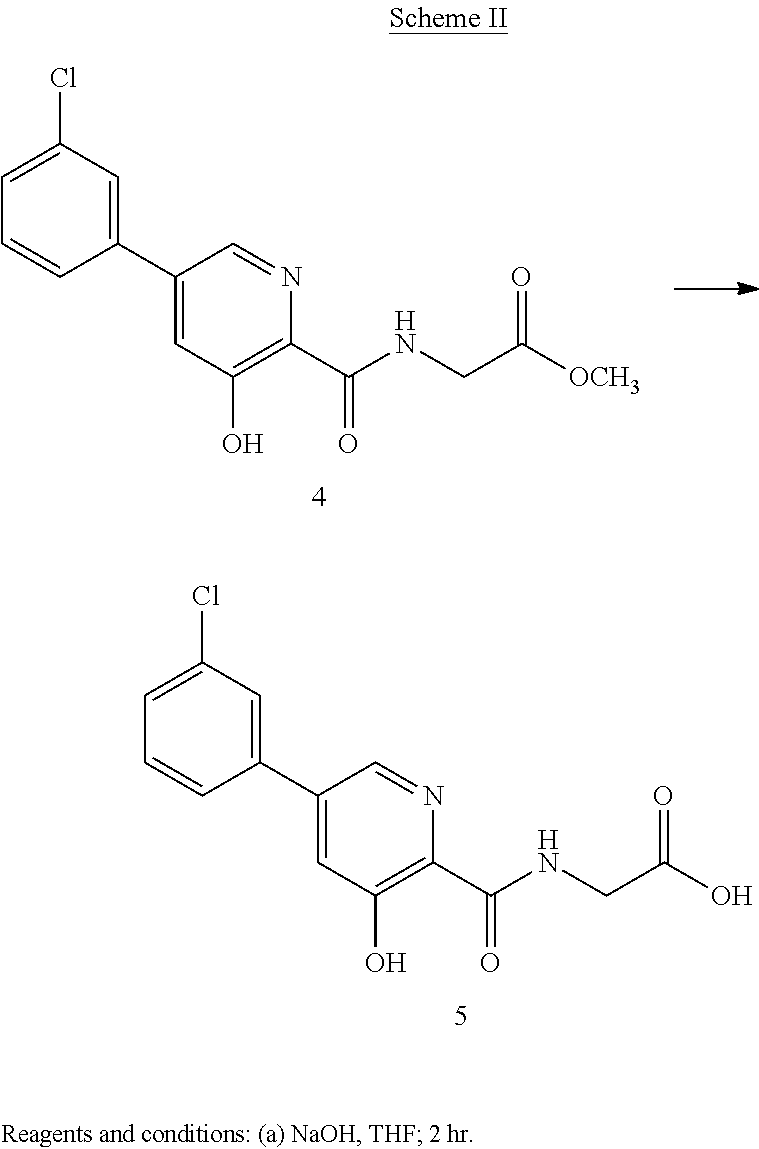
EXAMPLE 3{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3 -chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 50 mL flask is charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (0.45 g, 1.4 mmol), tetrahydrofuran (4.5 mL) and 1 M NaOH (4.5 mL, 4.5 mmol). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was adjusted to pH 1 with concentrated HCl and the solution was heated at 35° C. under vacuum until all of the tetrahydrofuran had been removed. A slurry forms as the solution is concentrated. With efficient stirring the pH is adjusted to ˜2 with the slow addition of 1 M NaOH. The solid which forms was collected by filtration, washed with water, followed by hexane, then dried under vacuum to afford 0.38 g (88% yield) of the desired product as a white solid. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use.
EXAMPLE 4{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (440 g, 1.42 mol), tetrahydrofuran (3912 g, 4400 mL) and 1 M NaOH (4400 mL). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was acidified to a pH of 2 with slow addition of 2M HCl (2359 g). The resulting mixture was concentrated under reduced pressure to a volume of about 7.5 L. Ware (2210 g) was added and the solution cooled to ambient temperature and agitated for 18 hours. The solid product was isolated by filtration and washed with water (6 L). the crude product was transferred back into the reactor and triturated with 2215 g o deionized water at 70° C. for 16 hours. The mixture was cooled to ambient temperature, The solid product was isolated by filtration and washed with water (500 mL) and dried under reduced pressure at 70° C. for 20 hours to afford 368 g (87% yield) of the desired product as an off-white solid. Purity by HPLC was 99.3%. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
Scheme III herein below outlines and Example 3 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase amide prodrug.
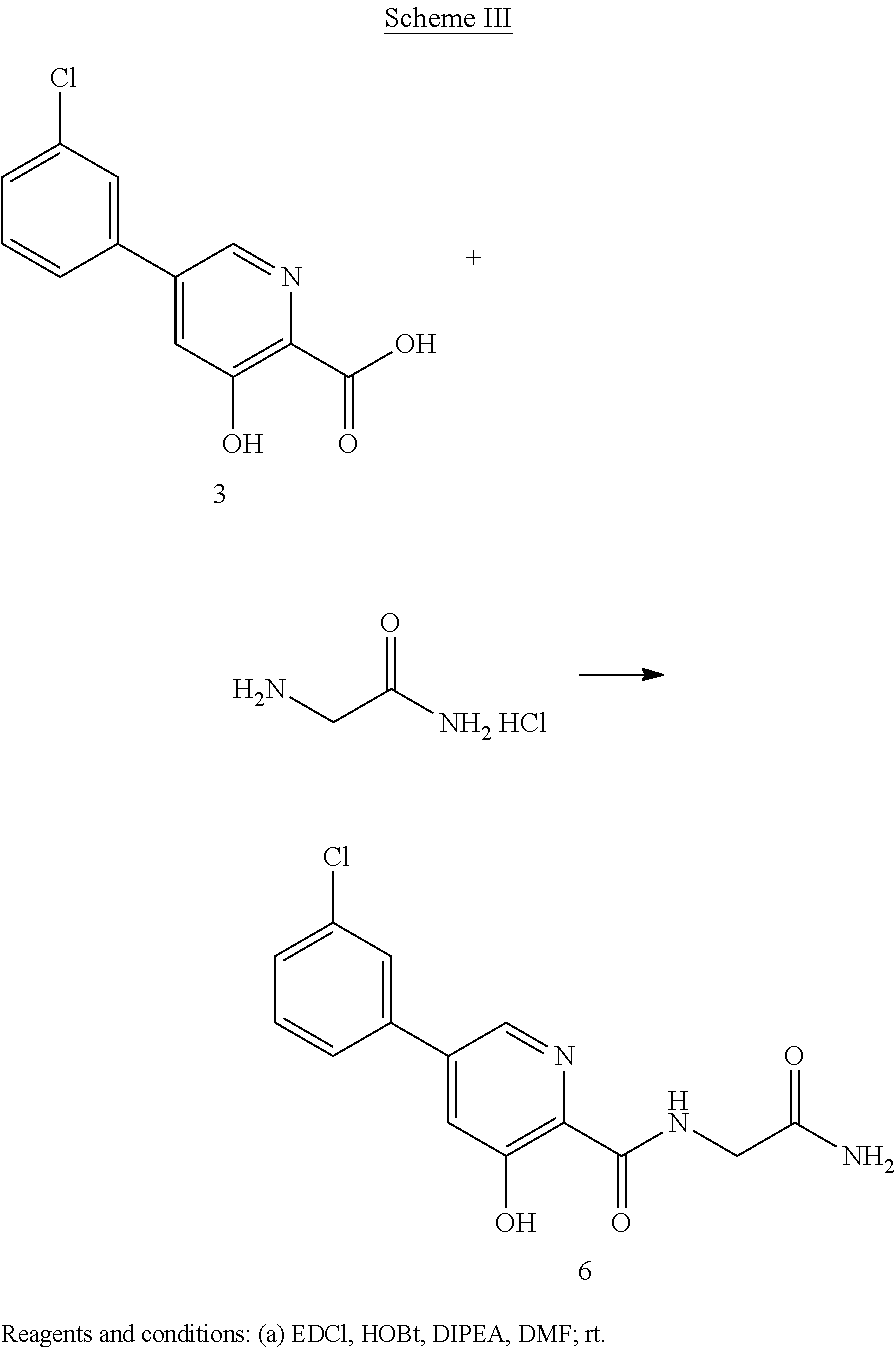
EXAMPLE 55-(3-Chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide
Preparation of 5-(3-chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide (6): To a solution of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (749 mg, 3 mmol) in DMF (20 mL) at room temperature under N2 is added 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) (0.925 g, 5.97 mmol) and 1-hydroxybenzo-triazole (HOBt) (0.806 g, 5.97 mmol). The resulting solution is stirred for 15 minutes then 2-aminoacetamide hydrochloride (0.66 g, 5.97 mmol) and diisopropylethylamine (1.56 ml, 8.96 mmol) are added. The reaction is monitored by TLC and when the reaction is complete the reaction mixture is concentrated under reduced pressure and H2O added. The product can be isolated by normal work-up: The following data have been reported for compound (6). 1H NMR (250 MHz, DMSO-d6) δ ppm 12.46 (1H, s), 9.17 (1H, t, J=5.9 Hz), 8.55 (1H, d, J=2.0 Hz), 7.93 (1H, d, J=0.9 Hz), 7.75-7.84 (2H, m), 7.49-7.60 (3H, m), 7.18 (1H, s), 3.91 (2H, d, J=5.9 Hz). HPLC-MS: m/z 306 [M+H]+.
Scheme IV herein below depicts a non-limiting example the hydrolysis of an amide pro-drug to a prolyl hydroxylase inhibitor after removal of a R10 protecting group

PATENT
US 20070299086
https://www.google.com/patents/US20070299086
REF
Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other
small-molecule erythropoiesis-stimulating agents in current and preventive doping
analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012
Feb 24. Review. PubMed PMID: 22362605.
Abstracts, posters, and presentations
2016 ERA-EDTA: Poster
A Drug-Drug Interaction Study to Evaluate the Effect of Vadadustat on the Pharmacokinetics of Celecoxib—a CYP2C9 Substrate—in Healthy Volunteers
Dose Exposure Relationship of Vadadustat is Independent of the Level of Renal Function
Variability in Hemoglobin Levels in Hemodialysis Patients in the Current Era
2014 ERA-EDTA: Oral presentation
Controlled Hemoglobin Response in a Double-Blind, Placebo-Controlled Trial of AKB-6548 in Subjects with Chronic Kidney Disease
2012 ASN: Oral presentation
AKB-6548, A New Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor, Increases Hemoglobin in Chronic Kidney Disease Patients Without Increasing Basal Erythropoietin Levels
2011 ASN: Oral presentation
AKB-6548, A Novel Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Reduces Hepcidin and Ferritin while It Increases Reticulocyte Production and Total Iron Binding Capacity In Healthy Adults


| WO2013013609A1 * | Jul 23, 2012 | Jan 31, 2013 | Zhejiang Beta Pharma Incorporation | Polymorphic forms of compounds as prolyl hydroxylase inhibitor, and uses thereof |
| US20070299086 * | Jun 26, 2007 | Dec 27, 2007 | The Procter & Gamble Company | Prolyl hydroxylase inhibitors and methods of use |
| US20100331303 * | Aug 20, 2010 | Dec 30, 2010 | Richard Masaru Kawamoto | Prolyl hydroxylase inhibitors and methods of use |
| US20130203816 * | Nov 20, 2012 | Aug 8, 2013 | Akebia Therapeutics Inc. | Prolyl hydroxylase inhibitors and methods of use |
| WO2016118858A1 * | Jan 22, 2016 | Jul 28, 2016 | Akebia Therapeutics, Inc. | Solid forms of 2-(5-(3-fluorophenyl)-3-hydroxypicolinamido)acetic acid, compositions, and uses thereof |
clip
![]()
Akebia Reaches Agreement with FDA and EMA on Vadadustat Global Phase 3 Program
Plans to Initiate Phase 3 PRO2TECT™ Clinical Program by Year-End
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Akebia Therapeutics, Inc. (NASDAQ: AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced the successful completion of the End-of-Phase 2 Meeting process with the United States Food and Drug Administration (FDA) and the Scientific Advice Process with the European Medicines Agency (EMA) for its lead product, vadadustat (formerly AKB-6548), for patients with anemia related to non-dialysis dependent chronic kidney disease (NDD-CKD). The company has reached agreement with both the FDA and EMA regarding key elements of the Phase 3 program, known as the PRO2TECT™ program, and expects to launch the program later this year.
The PRO2TECT™ program includes two separate studies and will collectively enroll approximately 3,100 NDD-CKD patients across 500 sites globally. The correction study will address anemia patients not currently being treated with recombinant erythropoiesis stimulating agents (rESAs). The conversion study includes patients currently receiving rESA who will be converted to either vadadustat or the active control with the goal of maintaining their baseline hemoglobin levels. Both studies will include a 1:1 randomization and an open label, active-control, non-inferiority design. Primary endpoints include an efficacy assessment of the hemoglobin response and an assessment of cardiovascular safety measured by major adverse cardiovascular events.
“Akebia’s Phase 3 program is designed to provide the medical community and regulators with a clear understanding of vadadustat’s potential benefit and safety advantages over rESAs, the current standard of care worldwide and, with a positive outcome, to establish vadadustat as the best-in-class treatment option for patients with renal anemia,” stated John P. Butler, President and Chief Executive Officer of Akebia. “We are pleased that the regulators are in agreement regarding the importance of an active-control trial as this design is the most clinically relevant and commercially valuable, and will allow us the quickest path to full enrollment. We are now moving rapidly to launch these studies and advance our goal of bringing forward new treatment options for patients suffering from renal anemia.”
“This Phase 3 program builds on the positive data from our Phase 2 program in NDD-CKD patients which demonstrated that once-daily vadadustat can control and maintain hemoglobin levels in a clinically relevant range while minimizing fluctuations in hemoglobin levels that are associated with increased cardiovascular safety risks,” stated Brad Maroni, M.D., Chief Medical Officer at Akebia. “These two Phase 3 event-driven studies are designed to establish the safety and efficacy of vadadustat in the setting of contemporary clinical practice patterns, and support regulatory approvals globally.”
In addition, Akebia discussed with the FDA and EMA a parallel Phase 3 program, known as the INNO2VATE™ program, for vadadustat in patients with anemia related to chronic kidney disease who are undergoing dialysis (DD-CKD). Akebia expects to formalize its Phase 3 program in DD-CKD patients after presenting the results from its recently completed Phase 2 study to both regulatory agencies.
About Vadadustat (Formerly AKB-6548)
Vadadustat is an oral therapy currently in development for the treatment of anemia related to chronic kidney disease (CKD). Vadadustat is designed to stabilize HIF, a transcription factor that regulates the expression of genes involved with red blood cell (RBC) production in response to changes in oxygen levels, by inhibiting the hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzyme. Vadadustat exploits the same mechanism of action used by the body to naturally adapt to lower oxygen availability associated with a moderate increase in altitude. At higher altitudes, the body responds to lower oxygen availability with increased production of HIF, which coordinates the interdependent processes of iron mobilization and erythropoietin (EPO) production to increase RBC production and, ultimately, improve oxygen delivery.
As a HIF stabilizer with best-in-class potential, vadadustat raises hemoglobin levels predictably and sustainably, with a dosing regimen that allows for a gradual and controlled titration. Vadadustat has been shown to improve iron mobilization, potentially eliminating the need for intravenous iron administration and reducing the overall need for iron supplementation.
About Anemia Related to CKD
Approximately 30 million people in the United States have CKD, with an estimated 1.8 million of these patients suffering from anemia. Anemia results from the body’s inability to coordinate RBC production in response to lower oxygen levels due to the progressive loss of kidney function, which occurs in patients with CKD. Left untreated, anemia significantly accelerates patients’ overall deterioration of health with increased morbidity and mortality. Renal anemia is currently treated with injectable rESAs, which are associated with inconsistent hemoglobin responses and well-documented safety risks.
About Akebia Therapeutics
Akebia Therapeutics, Inc. is a biopharmaceutical company headquartered in Cambridge, Massachusetts, focused on delivering innovative therapies to patients with kidney disease through HIF biology. The company has completed Phase 2 development of its lead product candidate, vadadustat, an oral therapy for the treatment of anemia related to CKD in both non-dialysis and dialysis patients.
clip
Akebia Announces Positive Top-Line Results from its Phase 2 Study of Vadadustat in Dialysis Patients with Anemia Related to Chronic Kidney Disease
-Treatment with Vadadustat Successfully Maintained Mean Hemoglobin Levels Following Conversion from rESA Therapy-
-Vadadustat Demonstrated a Favorable Safety Profile with Once Daily and Three Times per Week Dosing-
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Akebia Therapeutics, Inc. (NASDAQ:AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced positive top-line results from its Phase 2 study of vadadustat (formerly AKB-6548) in dialysis patients with anemia related to chronic kidney disease (CKD). The study achieved its primary objective, indicating that vadadustat maintained stable hemoglobin (HGB) levels throughout the 16-week treatment period following conversion from recombinant erythropoiesis-stimulating agent (rESA) therapy. Vadadustat demonstrated a favorable safety profile with no drug-related serious adverse events and no deaths. The results highlight the potential of vadadustat, dosed either once daily or three times per week, to safely and predictably manage and sustain HGB levels in CKD patients undergoing dialysis.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy”
The open-label, multi-center, 94 patient study was designed to evaluate the ability of vadadustat to maintain hemoglobin levels in patients undergoing hemodialysis who were previously being treated with rESAs. Patients were assigned to one of three dose cohorts: once daily vadadustat at a starting dose of 300mg, once daily vadadustat at a starting dose of 450mg, or vadadustat three times per week in conjunction with the patient’s hemodialysis schedule at a starting dose of 450mg. The study achieved its primary endpoints of maintaining stable hemoglobin levels over 16 weeks of treatment in all three cohorts of patients converting from rESAs to vadadustat.
| Mean Hemoglobin Levels (g/dL)* | Baseline | Week 7/8 | Week 15/16 | |||||||||||||
| 300mg Daily Dose | 10.4 | 10.4 | 10.3 | |||||||||||||
| 450mg Daily Dose | 10.6 | 10.3 | 10.5 | |||||||||||||
| 450mg Three Times per Week Dose | 10.5 | 10.2 | 10.4 | |||||||||||||
|
* Modified intent-to-treat (MITT) population, n=94 |
||||||||||||||||
Vadadustat was well tolerated among patients in all three dose cohorts. Treatment-emergent adverse events (TEAEs) with vadadustat were balanced across the cohorts. Serious adverse events (SAEs) were reported in 13 subjects (13.8%), well within the expected range for this patient population. There were no drug-related SAEs and no deaths reported in the study.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy,” said Brad Maroni, M.D., Chief Medical Officer at Akebia. “We are impressed with the consistency in hemoglobin levels across the duration of the study, which highlights the ability of vadadustat to control and maintain hemoglobin levels in this patient population. Furthermore, the results indicate that daily and three times per week dosing regimens are both viable options for patients on dialysis.”
John P. Butler, President and Chief Executive Officer of Akebia, stated, “These results further confirm vadadustat as a potential best-in-class anemia treatment for CKD patients, and reinforce our confidence in this product candidate as we advance toward our Phase 3 program. Adding these results to the 12 other clinical studies we have completed, we are confident in the potential for vadadustat to treat anemia in a broad array of patients with CKD. We are pleased to have successfully completed this stage of our drug development and look forward to initiating Phase 3 studies.”
Complete efficacy and safety data from this Phase 2 study will be presented at an upcoming medical meeting.
About the Phase 2 Study Design of Vadadustat in Dialysis Patients with Anemia Related to CKD
The Phase 2 multi-center, open-label study evaluated 94 patients over 16 weeks of treatment, at 20 dialysis centers in the United States, including an assessment of HGB response to the starting dose of vadadustat during the first 8 weeks, followed by an assessment of HGB response to algorithm-guided dose adjustments of vadadustat during the subsequent 8 weeks of treatment. The study enrolled three cohorts, each consisting of approximately 30 CKD patients with anemia undergoing dialysis who were switched from injectable rESA therapy to vadadustat. Patients in the first two cohorts received once daily doses of vadadustat, while patients in the third cohort received vadadustat three times per week in conjunction with their hemodialysis schedule.
References
- Jump up^ Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016 Nov;90(5):1115-1122. . doi:10.1016/j.kint.2016.07.019. PMID 27650732.Missing or empty
|title=(help) - Jump up^ Gupta N, Wish JB. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am J Kidney Dis. 2017 Jun;69(6):815-826. . doi:10.1053/j.ajkd.2016.12.011. PMID 28242135. Missing or empty
|title=(help) - Jump up^ Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 2017;45(5):380-388. . doi:10.1159/000464476. PMID 28343225. Missing or empty
|title=(help)
 |
|
| Clinical data | |
|---|---|
| Synonyms | AKB-6548, PG-1016548 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C14H11ClN2O4 |
| Molar mass | 306.701 g/mol |
| 3D model (JSmol) | |
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Vadadustat (Vafseo). Vadadustat (28) is a hypoxia inducible factor prolyl hydroxylase (HIF-PH) inhibitor
developed by Akebia Therapeutics. It was approved by the European Commission in April 2023, and recently also by the USFDA, for the treatment of symptomatic anemia associated with chronic kidney disease in adults receiving chronic maintenance dialysis. Vadadustat acts by inhibiting HIFPH, 214
which results in increases of endogenous erythropoietin production, red blood cell synthesis, and iron mobilization. 215 While a number of syntheses of vadadustat (28) have been published in previous patents 216−228 and a journal article, 229 Akebia Therapeutics has published two patents regarding the
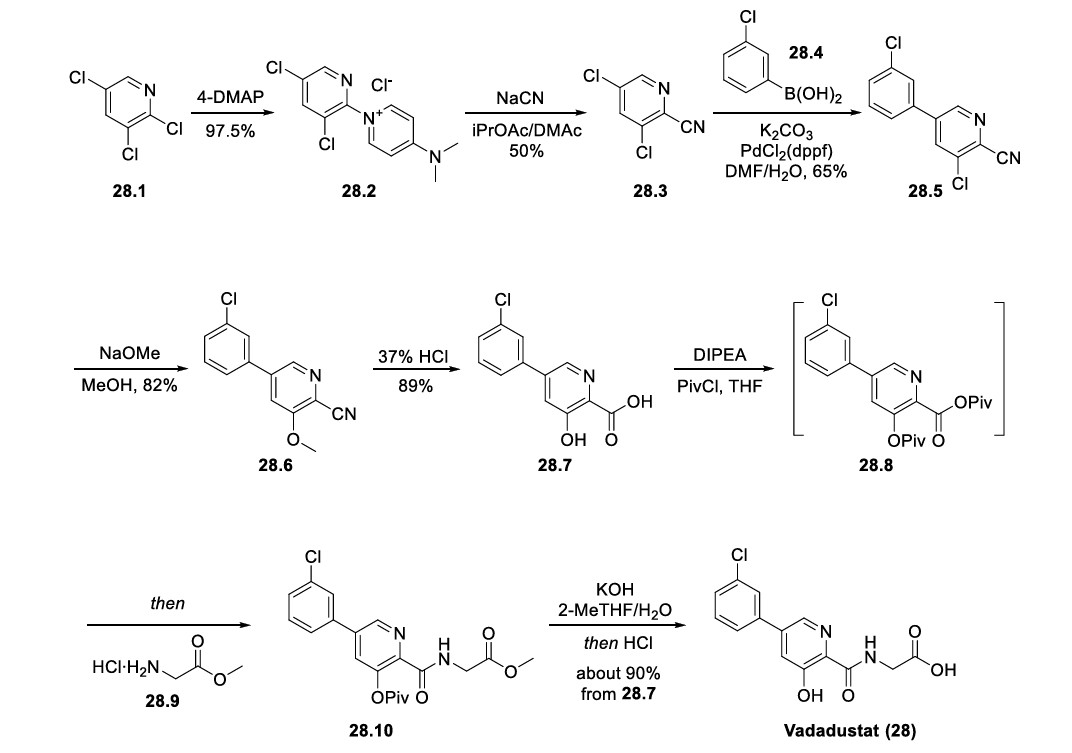
large-scale preparation of vadadustat (Scheme 52). 218,226 The key intermediate nitrile 28.3 could be accessed in two steps: the neat SNAr reaction between commercially available 2,3,5trichloropyridine (28.1) and 4-DMAP to generate pyridinium salt 28.2, followed by a second SNAr reaction of 28.2 with
NaCN. The Suzuki coupling between 28.3 and 3-chlorophenyl boronic acid (28.4) gave the biaryl 28.5, and the subsequent SNAr reaction of 28.5 with NaOMe replaced the 3-chloro
substitution on the pyridine ring with a methoxy group, generating intermediate 28.6. Global acidic hydrolysis of both methyl ether and nitrile group in 28.6 gave the 3 hydroxypicolinic acid 28.7. Treatment of 28.7 with DIPEA and excess pivaloyl chloride (PivCl) resulted in the formation of mixed anhydride 28.8 with concomitant acylation of the 3 hydroxy group. Without isolation of 28.8, glycine methyl ester
hydrochloride (28.9) was then charged with additional DIPEA to generate the corresponding amide 28.10. The residual amount (∼0.5%) of 28.7 in 28.10 was hard to remove, but this impurity could be effectively rejected with an extra amount of DIPEA during workup and solvent switch. Finally, the Opivaloyl group and methyl ester were both removed via basic hydrolysis, giving vadadustat (28) in about 90% yield from 28.7.
REF
(215) Pergola, P. E.; Spinowitz, B. S.; Hartman, C. S.; Maroni, B. J.; Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease.
Kidney Int. 2016, 90, 1115−1122.
(216) Lanthier, C. M.; Gorin, B.; Oudenes, J.; Dixon, C. E.; Lu, A. Q.; Copp, J. D.; Janusz, J. M. Preparation of [(3-hydroxypyridine-2carbonyl)amino]alkanoic acids, esters and amides as prolyl hydroxylase
inhibitors. US 20120309977, 2012.
(217) Li, X.; Chen, J. Process for the preparation of vadadustat. CN105837502, 2016.
(218) Gorin, B. I.; Lanthier, C. M.; Luong, A. B. C.; Copp, J. D.; Gonzalez, J. Process for preparing 2-[[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino]acetic acid. WO 2019217550, 2019.
(219) Kou, J.; Li, Y.; Xiao, Q.; Lin, B.; Sun, J.; Wang, Z.; Luo, Z.;Huang, F. Preparation method of vadadustat. CN 110903238, 2020.
(220) Machida, K.; Yasukouchi, H.; Nishiyama, A. Method for producing vadadustat intermediate. WO 2020217733, 2020.
(221) Xiao, Q.; Lin, B.; Kou, J.; Sun, J.; Qiu, X.; Wang, Z.; Luo, Z.;Huang, F. Preparation of vadadustat intermediate. CN 111848505,2020
(222) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Li, Y.; Sun, J.; Jin, L.; Luo,
Z.; Huang, F. Preparation of vadadustat and intermediate thereof. CN
111205222, 2020.
(223) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Luo, Z.; Huang, F.; Li, Y.
Preparation of vadadustat and intermediate thereof. CN 111423367,
2020.
(224) Xiao, Q.; Qiu, X.; Lin, B.; Kou, J.; Li, Y.; Sun, J.; Wang, Z.; Luo,
Z.; Huang, F. Preparation of vadadustat. CN 111320577, 2020.
(225) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Qiu, X.; Cai, X.; Li, Y.; Luo,
Z.; Huang, F. Method for preparing vadadustat and intermediate
thereof. WO 2021179540, 2021.
(226) Jurkauskas, V.; Jung, Y. C.; Kwon, T.; Kannan, A.; Gondi, V. B.
Manufacturing process for 3,5-dichloropicolinonitrile for synthesis of
vadadustat. WO 2022006427, 2022.
(227) Chen, Z.; Zheng, Y.; Zhang, L.; Yu, C.; Liu, L.; He, B.
Preparation of a pyridine compound used for the preparation of
vadadustat. CN 117843565, 2024.
(228) Patel, K. R.; Thakrar, V. H.; Mehta, T. B.; Wagh, A. G.; Patel, J.
A.; Patil, R. R.; Solanki, Y. U.; Ladumor, C. B. A process for the
preparation of Vadadustat or salts thereof. WO 2024079708, 2024.
(229) Lin, B. Y.; Kou, J. P.; Wu, S. M.; Cai, X. R.; Xiao, Q. B.; Li, Y. L.;
Hu, J.; Li, J. B.; Wang, Z. Q. Development of a robust and scalable
process for the large-scale preparation of Vadadustat. Org. Process Res.
Dev. 2021, 25, 960−968.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
///////////VADADUSTAT, PHASE 3, AKB-6548, PG-1016548, B-506, AKB 6548, Akebia Therapeutics, Procter & Gamble Pharmaceuticals, Mitsubishi Tanabe Pharma, Otsuka, вададустат , فادادوستات , 伐达度司他 , PG1016548, UNII:I60W9520VV, MT-6548 , MT 6548 , APPROVALS 2024, FDA 2024
c1cc(cc(c1)Cl)c2cc(c(nc2)C(=O)NCC(=O)O)O

