PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
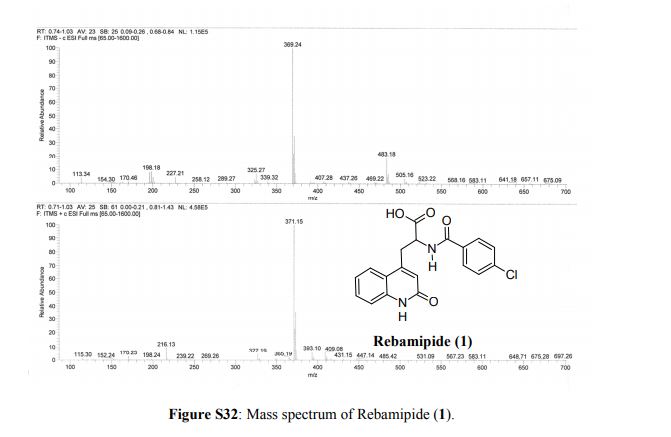
obtain the white powder from dimethylformamide-water with its hemihydrate m.p. being 288-290°C (decomposition).
(-)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20-116.7 ° (C = 1.0, dimethylformamide).
(+)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20 + 116.9 ° (C = 1.0, dimethylformamide).
Rebamipide is a quinolone derivative that was launched in 1990 by Otsuka in Japan for the oral treatment of Helicobacter pylori-induced gastric inflammation after eradication therapy and peptic ulcer
Title: Rebamipide
CAS Registry Number: 90098-04-7
CAS Name: a-[(4-Chlorobenzoyl)amino]-1,2-dihydro-2-oxo-4-quinolinepropanoic acid
Percent Composition: C 61.55%, H 4.08%, Cl 9.56%, N 7.56%, O 17.26%
Literature References: Gastric cytoprotectant. Prepn: M. Uchida et al.,DE3324034; eidem,US4578381; (1984, 1986 both to Otsuka). Synthesis and pharmacology: M. Uchida et al.,Chem. Pharm. Bull.33, 3775 (1985); of enantiomers: eidem,ibid.35, 853 (1987). Antiulcer activity in rats: K. Yamasaki et al.,Eur. J. Pharmacol.142, 23 (1987); K. Yamasaki et al.,Jpn. J. Pharmacol.49,441 (1989). HPLC determn in plasma and urine: Y. Shioya, T. Shimizu, J. Chromatogr.434, 283 (1988).
Properties: White powder from DMF-water, mp 288-290° (dec) as hemihydrate.
Melting point: mp 288-290° (dec) as hemihydrate
Derivative Type: (-)-Form
Properties: Colorless needles from DMF, mp 305-306° (dec). [a]D20 -116.7° (c = 1.0 in DMF).
Melting point: mp 305-306° (dec)
Optical Rotation: [a]D20 -116.7° (c = 1.0 in DMF)
Derivative Type: (+)-Form
Properties: Colorless needles from DMF, mp 305-306° (dec). [a]D20 +116.9° (c = 1.0 in DMF).
Rebamipide has been investigated for the treatment of Stomach Ulcer, Keratoconjunctivitis Sicca, and Gastric Adenoma and Early Gastric Cancer.
Rebamipide is a quinolinone derivative that stimulates endogenous PGE2 generation in gastric mucosa, enhancing gastric mucosal defense in a COX-2-dependent manner.
Rebamipide has been shown to inhibit the production of reactive oxygen species and to decrease cytokine release induced by H. pylori infection.
A daily oral dose of 100 mg/kg was found to be protective against the development of pyloric channel ulcers in Mongolian gerbils infected with H. pylori.
In addition to the stomach, rebamipide can also enhance secretion of mucin covering the conjunctiva and cornea, which is important for tear film adhesion.
Rebamipide, a gastroprotective drug, was developed in Japan and was proven to be superior to cetraxate, the former most prescribed drug of the same category, in 1989 in the treatment for gastric ulcers. The initially discovered basic mechanisms of action of rebamipide included its action as a prostaglandin inducer and oxygen free-radical scavenger. In the last 5 years, several basic and clinical studies have been performed for functional dyspepsia, chronic gastritis, NSAID-induced gastrointestinal injuries, gastric ulcer following eradication therapy for Helicobacter pylori, gastric ulcer after endoscopic surgery and ulcerative colitis. In addition, several molecules have been identified as therapeutic targets of rebamipide to explain its pleiotropic pharmacological actions.
Rebamipide, an amino acid derivative of 2-(1H)-quinolinone, is used for mucosal protection, healing of gastroduodenal ulcers, and treatment of gastritis. It works by enhancing mucosal defense, scavenging free radicals, and temporarily activating genes encoding cyclooxygenase-2.
Rebamipide is used in a number of Asian countries including Japan (marketed as Mucosta), South Korea, China[1] and India (where it is marketed under the trade name Rebagen). It is also approved in Russia under the brand name Rebagit.[2] It is not approved by the Food and Drug Administration for use in the United States.
Studies have shown that rebamipide can fight the damaging effects of NSAIDs on the GIT mucosa, and more recently, the small intestine.[citation needed] It has also been studied for the treatment of Behçet’s disease.[3] It was shown to successfully treat pouchitis in a single-N study after first-line therapies for the condition were unsuccessful.[4] Some studies have shown effectiveness in presbyacusis(age-related hearing loss).[citation needed]
It has also been shown to alleviate signs and symptoms of dry eyes in a randomised controlled trial although this is not yet widely available clinically.[5]
SYN
Rebamipide (CAS NO.: 111911-87-6), with its systematic name of 4-Quinolinepropanic acid, alpha-((4-chlorobenzoyl)amino)-1,2-dihydro-2-oxo-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:
4-(Bromomethyl)quinolin-2(1H)-one (I) could react with hot phosphorus oxychloride to produce a mixture of 4-(bromomethyl)-2-chloroquinoline (II) and 2-chloro-4-(chloromethyl)quinoline (III), and then the mixture without separation is ondensed with 2(S)-isopropyl-3,6-dimethoxy-2,5-dihydropyrazine (IVs) in the presence of butyllithium in hexane, affording (-)-2-chloro-4-[6(S)-isopropyl-2,5-dimethoxy-3,6-dihydropyrazin-3(R)-yl methyl]quinoline (Vr). The hydrolysis of (Vr) with HCl produces 3-(2-chloroquinolin-4-yl)-(R)-alanine methyl ester (VIr), which is treated with HCl and propylene oxide to afford 3-(2-oxo-2,3-dihydroquinolin-4-yl)-(R)-alanine (VIIr). At last, this compound is acylated with 4-chlorobenzoyl chloride (VIII) by means of K2CO3in acetone, affording (R)-OPC-12759.
Figure 2 The synthetic route of Rebamipide.
DE 3324034; US 4578381 ABOVE
The condensation of 4-(bromomethyl)quinolin-2(1H)-one (I) with diethyl acetamidomalonate (II) by means of sodium ethoxide in refluxing ethanol gives ethyl 2-acetamido-2-(ethoxycarbonyl)-3-(2-oxo-1,2-dihydroquinolin-4yl)propionate (III), which is submitted to a decarboxylative hydrolysis with refluxing 20% HCl yielding 3-(2-oxo-1,2-dihydroquinolin-4yl)alanine (IV). Finaily this compound is acylated with 4-chlorobenzoyl chloride by means of K2CO3 in acetone water.
SYN
Chem Pharm Bull 1991,39(11),2906 ABOVE
The synthesis of (R)- and (S)-isomers of OPC-12759 has been described: These optical isomers can be obtained in three different ways: 1) The reaction of 4-(bromomethyl)quinolin-2(1H)-one (I) with hot phosphorus oxychloride gives a mixture of 4-(bromomethyl)-2-chloroquinoline (II) and 2-chloro-4-(chloromethyl)quinoline (III), which, without separation, is condensed with 2(S)-isopropyl-3,6-dimethoxy-2,5-dihydropyrazine (IVs) by means of butyllithium in hexane, yielding (-)-2-chloro-4-[6(S)-isopropyl-2,5-dimethoxy-3,6-dihydropyrazin-3(R)-yl methyl]quinoline (Vr). The hydrolysis of (Vr) with HCl affords 3-(2-chloroquinolin-4-yl)-(R)-alanine methyl ester (VIr), which is treated with HCl and propylene oxide to give 3-(2-oxo-2,3-dihydroquinolin-4-yl)-(R)-alanine (VIIr). Finally, this compound is acylated with 4-chlorobenzoyl chloride (VIII) by means of K2CO3 in acetone, affording (R)-OPC-12759.
SYN
3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
Rebamipide
Synonyms:Proamipide
ATC:A02BX
Use:ulcer therapeutic
Chemical name:α-[(4-chlorobenzoyl)amino]-1,2-dihydro-2-oxo-4-quinolinepropanoic acid
Magic Bullet! Rebamipide, a Superior Anti-ulcer and Ophthalmic Drug and Its Large-Scale Synthesis in a Single Organic Solvent via Process Intensification Using Krapcho Decarboxylation
Chemical Research Division, API R&D Centre, Micro Labs Ltd., Plot No.43-45, KIADB Industrial Area, fourth phase, Bommasandra-Jigani Link Road, Bommasandra, Bangalore 560 105, Karnataka, India
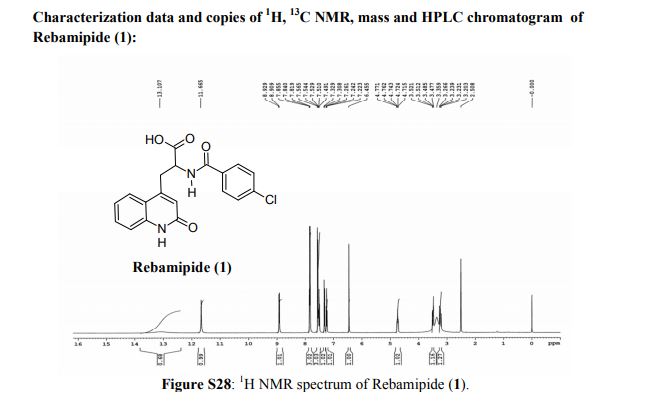
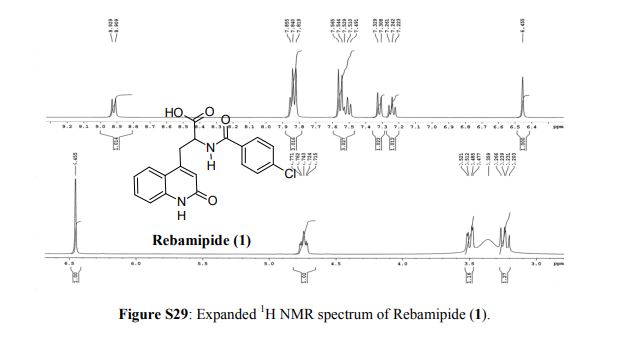
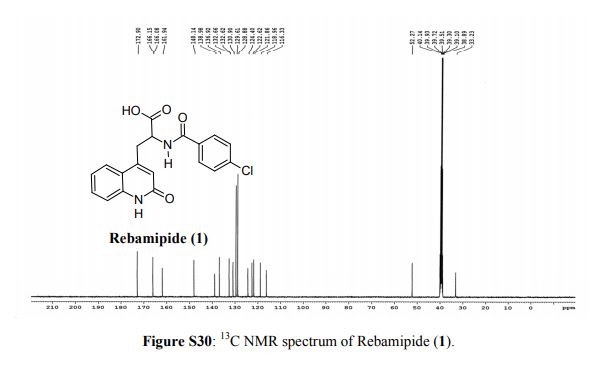
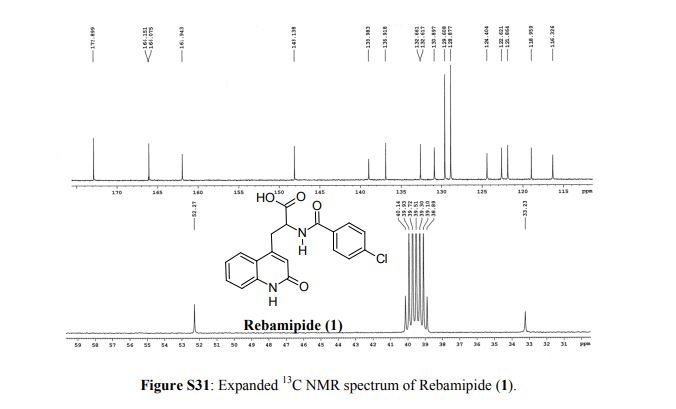
Rebamipide (1) is a superior drug compared to existing drugs for use in healing of peptic ulcers, gastrointestinal bleeding, and dyspepsia. It is also useful as an ophthalmic drug for the treatment of dry eye syndrome. Process intensification for its synthesis was achieved by (i) averting uncontrollable frothing using Krapcho decarboxylation instead of conventional acid hydrolysis, where uncontrollable frothing became chaotic, (ii) minimizing organic waste generation by using a single organic solvent, and (iii) avoiding anti-foaming agents (n-octanol, acetophenone) and acetic acid. With these trifling modifications, the overall yield of active pharmaceutical ingredient (API) was ≥83% with excellent purity (≥99.89%), and the process meets the metrics of “green” chemistry with an E-factor = 11.5. The developed hassle-free commercial process is viable for multi-kilogram synthesis of Rebamipide (1) as the key step, Krapcho decarboxylation is safe to run at 130–140 °C in DMSO, and it was proved to be effective by differential scanning calorimetry thermal screening studies. The characterization data of intermediates, process-related impurities, and API are reported. The carryover and process-related impurities were controlled efficiently. The present work can enhance the scope and worldwide adoptability of Rebamipide (1), which is currently limited to Asian countries.
Arakawa T, Watanabe T, Fukuda T, Yamasaki K, Kobayashi K (1995). “Rebamipide, novel prostaglandin-inducer accelerates healing and reduces relapse of acetic acid-induced rat gastric ulcer. Comparison with cimetidine”. Dig Dis Sci. 40 (11): 2469–72. doi:10.1007/BF02063257. PMID7587834.
Arakawa T, Kobayashi K, Yoshikawa T, Tarnawski A (1998). “Rebamipide: overview of its mechanisms of action and efficacy in mucosal protection and ulcer healing”. Dig Dis Sci. 43 (9 Suppl): 5S–13S. PMID9753220.
Tarnawski AS, Chai J, Pai R, Chiou SK (2004). “Rebamipide activates genes encoding angiogenic growth factors and Cox2 and stimulates angiogenesis: a key to its ulcer healing action?”. Dig Dis Sci. 49 (2): 202–9. doi:10.1023/B:DDAS.0000017439.60943.5c. PMID15104358.
Takumida M, Anniko M (2009). “Radical scavengers for elderly patients with age-related hearing loss”. Acta Otolaryngol. 129 (1): 36–44. doi:10.1080/00016480802008215. PMID18607930.
Jump up^Matsuda T, Ohno S, Hirohata S, Miyanaga Y, Ujihara H, Inaba G, Nakamura S, Tanaka S, Kogure M, Mizushima Y (2003). “Efficacy of rebamipide as adjunctive therapy in the treatment of recurrent oral aphthous ulcers in patients with Behcet’s disease: a randomised, double-blind, placebo-controlled study”. Drugs R D. 4 (1): 19–28. doi:10.2165/00126839-200304010-00002. PMID12568631.
Jump up^Kinoshita, S.; K. Oshiden; S. Awamura; H. Suzuki; N. Nakamichi (2013). “A randomized, multicenter phase 3 study comparing 2% rebamipide (OPC-12759) with 0.1% sodium hyaluronate in the treatment of dry eye”. Ophthalmology. 120 (6): 1158–65. doi:10.1016/j.ophtha.2012.12.022. PMID23490326.
Vadadustat, also known as AKB-6548 and PG-1016548, is a potent Hypoxia-inducible factor-proline dioxygenase inhibitor. AKB-6548 works by inhibiting hypoxia inducible factor-prolyl hydroxylase (HIP-PH), leading to stabilization and increased levels of HIFα. In turn HIFα improves production of hemoglobin and red blood cells (RBCs), while maintaining normal levels of erythropoietin (EPO) in patients. We believe this differentiated mechanism of action has the potential to be safer than that of injectable recombinant erythropoietin stimulating agents (rESAs), avoiding supra-physiological levels of EPO and saturation of EPO receptors for prolonged periods of time.
Akebia Therapeutics, under license from Procter & Gamble, and sublicensee Mitsubishi Tanabe Pharma are developing vadadustat, an orally active small-molecule hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitor that stabilize HIF2-α, as a once-daily formulation, for treating anemia. Also the company is investigating AKB-6899, an oral HIF-PH inhibitor, for treating cancer and ocular diseases. In March 2016, the IND application was opened. Aerpio Therapeutics, a spinoff of Akebia, is investigating AKB-4924, a HIF2-α stabilizer, which inhibits HIF prolyl hydroxylase-2, for treating inflammatory bowel disease and wound healing
Hypoxia-inducible factor (HIF) is a transcription factor that is a key regulator of responses to hypoxia. In response to hypoxic conditions, i.e., reduced oxygen levels in the cellular environment, HIF upregulates transcription of several target genes, including those encoding erythropoietin. HIF is a heteroduplex comprising an alpha and beta subunit. While the beta subunit is normally present in excess and is not dependent on oxygen tension, the HIF-alpha subunit is only detectable in cells under hypoxic conditions. In this regard, the accumulation of HIF-alpha is regulated primarily by hydroxylation at two proline residues by a family of prolyl hydroxylases known as HIF prolyl hydroxylases, wherein hydroxylation of one or both of the proline residues leads to the rapid degradation of HIF-alpha. Accordingly, inhibition of HIF prolyl hydroxylase results in stabilization and accumulation of HIF-alpha {i.e., the degradation of HIF-alpha is reduced), thereby leading to an increase in the amount of HIF-alpha available for formation of the HIF heterodimer and upregulation of target genes, such as the Erythropoietin gene. Conversely, activation of HIF prolyl hydroxylase results in destabilization of HIF-alpha {i.e., the degradation of HIF-alpha is increased), thereby leading to a decrease in the amount of HIF-alpha available for formation of the HIF heterodimer and downregulation of target genes, such as VEGF.
The family of hypoxia inducible factors includes HIF- 1 -alpha, HIF-2-alpha, and HIF-3 -alpha.
A new class of prolyl hydroxylase inhibitors and their use to treat or prevent diseases ameliorated by modulation of hypoxia-inducible factor (HIF) prolyl hydroxylase are described in U.S. Patent No. 7,811,595, which is incorporated herein by reference in its entirety. The synthesis of such prolyl hydroxylase inhibitors is described in U.S. Patent Publication No.2012/0309977, which is incorporated herein by reference in its entirety. Such compounds inhibit HIF prolyl hydroxylase, thereby stabilizing HIF-alpha. As a consequence of stabilizing HIF-alpha, endogenous erythropoietin (EPO) production is increased. As with all drugs, proper doses and dosing regimens for treating patients having diseases such as anemia are essential for achieving a desired or optimal therapeutic effect without adverse effects or unwanted side-effects. Indeed, many active compounds fail in clinical trials because an effective and safe dosing regimen cannot be found.
Vadadustat (also known as AKB-6548) in anemia secondary to chronic kidney disease (CKD)
We are developing our lead product candidate, vadadustat, to be the potential best-in-class hypoxia inducible factor–prolyl hydroxylase inhibitor for the treatment of anemia secondary to CKD.
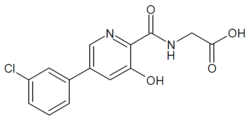
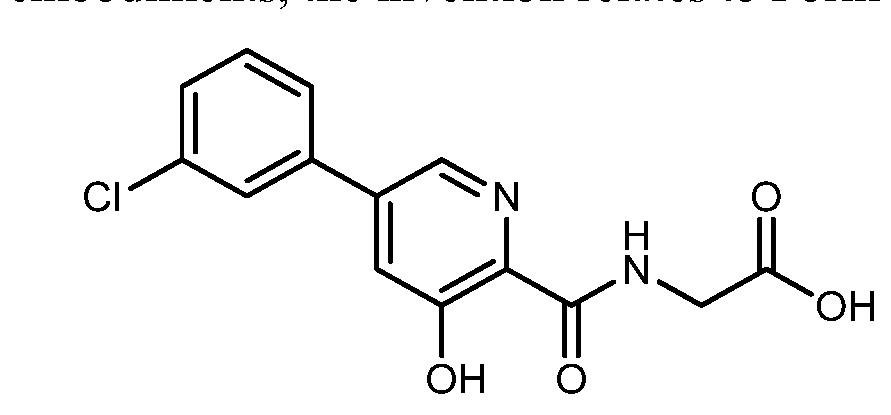
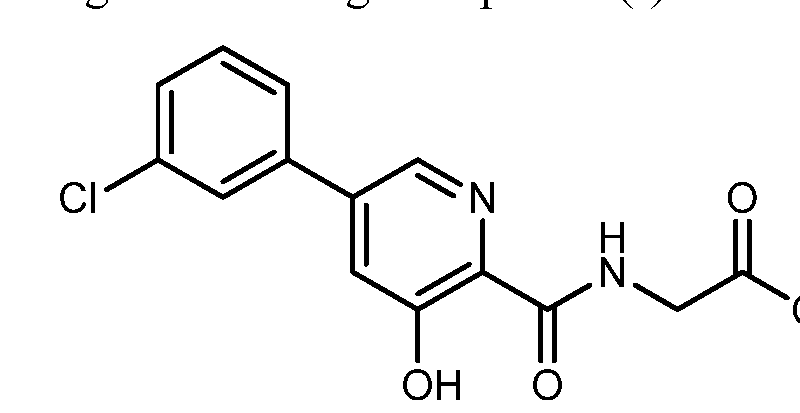
HIF inhibitor Vadadustat (Code AKB-6548) The chemical name N- [5- (3- chlorophenyl) -3-hydroxypyridine-2-carbonyl] glycine,
Vadadustat is a treatment for anemia associated with chronic kidney disease oral HIF inhibitor, is an American biopharmaceutical company Akebia Therapeutics invention in the research of new drugs, has completed Phase II pivotal clinical trial treatment studies, successfully met the researchers set given the level of hemoglobin in vivo target and good security, a significant effect, and phase III clinical trials.
U.S. Patent Publication US20120309977 synthetic route for preparing a Vadadustat: A 3-chlorophenyl boronic acid and 3,5_-dichloro-2-cyanopyridine as starting materials, by-catalyzed coupling methoxy substituted, cyano hydrolysis and condensation and ester hydrolysis reaction Vadadustat, process route is as follows:
Since the entire synthetic route 12 steps long, complicated operation, high cost.U.S. Patent No. 1 2 ^ ¥ disclosed 20070299086 & (^ (Scheme 3 1118 seven seven to 3,5-dichloro-2-cyanopyridine starting material, first-dichloro substituted with benzyloxy, then cyano hydrolysis, condensation, hydrogenation and deprotection trifluorosulfonyl, to give N- [5- trifluoromethanesulfonyloxy-3-hydroxypyridine-2-carbonyl) glycine methyl ester, 3-chlorophenyl and then boronic acid catalyzed coupling reactions, the final ester hydrolysis reaction Vadadustat, process route is as follows:
The synthesis steps long, intermediate products and final products contain more impurities and byproducts, thus purified requires the use of large amounts of solvents, complicated operation, low yield, and because the hydrogenation reaction is a security risk on the production, not conducive to the promotion of industrial production, it is necessary to explore a short process, simple operation, low cost synthetic method whereby industrial production Vadadus tat fit.
Example 1
A) Preparation of N- (3,5_-dichloro-2-carbonyl) glycine methyl ester:
3,5-dichloro-2-pyridinecarboxylic acid (19.2g, 0.10mol) and N, N’_ carbonyldiimidazole (24.3g, 0.15mol) was dissolved in N, N- dimethylformamide (100 mL ), was added glycine methyl ester hydrochloride (15.18,0.12111〇1), 11 was added dropwise diisopropylethylamine (51.7g, 0.40mol), the reaction mixture was stirred 35 ° C for 8 hours, TLC determined the completion of reaction gussets The reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was adjusted to neutral by adding ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give N- (3,5- dichloro-pyridin-2 – carbonyl) glycine methyl ester, an off-white solid (21.6g), a yield of 82.0%, this reaction step is as follows:
1234567 B) Preparation of N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester: 2
1 (3,5-dichloro-2-carbonyl) glycine methyl ester (20 (^, 〇1 76111111), 3-chlorophenyl boronic acid (13.18, 3 83.7mmol), [l, l’- bis (diphenylphosphino) ferrocene] dichloropalladium (2.8g, 3.8mmol), potassium carbonate (14.2g, 4 0. lmo 1) and N, N- dimethylformamide (75mL) was added The reaction flask, the reaction mixture was heated to 60 ° C for 20 hours the reaction was stirred for 5:00, point TLC plates to determine completion of the reaction, the reaction solution was cooled to room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, sulfuric acid 6 magnesium dried and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane was recrystallized to give N- [5- (3- chlorophenyl) -3-7-chloro-2-carbonyl] glycine methyl ester, white solid (19.7g), yield 76.4%, this reaction step is as follows:
C) Preparation of N_ [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine:
N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester (19 (^, 56111 111〇1) and sodium methoxide (7.6g, 0.14mol) was dissolved in methanol (150 mL), the reaction mixture was heated to 65 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution was cooled to room temperature, water (300mL) was stirred for 3h, cooled to 0 ° C, stirred for 2h, precipitated solid was filtered, the filter cake was dried to give N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine, off-white solid (17.4 g of), a yield of 96.5%, of the reaction steps are as follows:
D) Preparation Vadadustat:
N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine (16.68,51.7111111〇1) and 48% hydrobromic acid solution (52mL, 0.46mol) added to the reaction bottle, the reaction mixture was heated to 100 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution cooled square ~ 5 ° C, was slowly added 50% sodium hydroxide solution was adjusted to pH 2 at 0 -5 ° C under crystallization 3h, the filter cake washed with ethyl acetate and n-hexane mixed solvent of recrystallization, in finished Vadadustat, off-white solid (15.6g), a yield of 98.0%, this reaction step is as follow
Lanthier et al. (U.S. Patent Application 2012/0309977) described a procedure for synthesizing a compound of Formula (II) starting from 3-chloroboronic acid and 3,5-dichloropicolinonitrile, as shown in the scheme below:
which has an X-ray powder diffraction pattern as shown in FIG. 1. In certain embodiments, Form A of Compound (I) has an X-ray powder diffraction pattern comprising one, two, three, four, or five peaks at approximately 18.1 , 20.3, 22.9, 24.0, and 26.3 °2Θ; and wherein the crystalline Compound (I) is substantially free of any other crystalline form of Compound (I).
Compound (I) as prepared according to e.g., U.S. 7,811,595 and/or U.S. Patent Application No. 13/488,554 and then subjecting the resulting Compound (I)
(I),
to a procedure comprising
a) preparing a solution of Compound (I) in 2-methyltetrahydrofuran;
b) adding n-heptane;
c) heating the suspension {e.g., to about 40-50 °C);
d) cooling the suspension {e.g., to about 0-10 °C); and
c) isolating the crystals.
SYNTHESIS
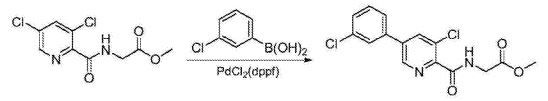
US 2015361043
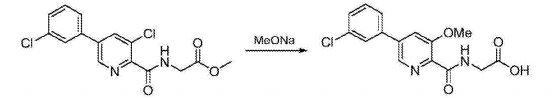
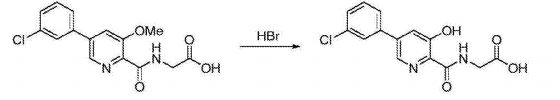
Synthesis of vadadustat and its intermediates is described. The process involves Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile with (3-chlorophenyl)boronic acid, selective chloride displacement, simultaneous hydrolysis of nitrile and methyl ether, activation with CDI, condensation with methyl glycinate hydrochloride and finally ester hydrolysis. The process is simple and provides high product yield with high quality. Vadadustat is expected to be useful for the treatment of renal failure anemia (1). Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile (I) with (3-chlorophenyl)boronic acid (II) in the presence of PdCl2(dppf) and K2CO3 in DMF yields 3-chloro-5-(3-chlorophenyl)pyridine-2-carbonitrile (III), which upon selective chloride displacement with NaOMe in refluxing MeOH affords methyl ether (IV). Hydrolysis of nitrile and methyl ether in intermediate (IV) with HBr or HCl at 100 °C furnishes 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (V). After activation of carboxylic acid (V) with CDI or pivaloyl chloride and DIEA in DMSO, condensation with methyl glycinate hydrochloride (VI) in the presence of DIEA provides vadadustat methyl ester (VII). Finally, hydrolysis of ester (VII) with NaOH in H2O/THF produces the target vadadustat (1).
FIG. 1 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitors.
FIG. 2 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor ester prodrugs.
FIG. 3 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor amide prodrugs.
Example 1 describes a non-limiting example of the disclosed process for the preparation of a prolyl hydroxylase ester pro-drug
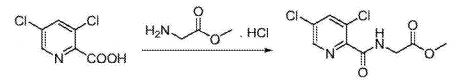
EXAMPLE 1Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): To a 100 mL round bottom flask adapted for magnetic stirring and equipped with a nitrogen inlet was charged (3-chlorophenyl)boronic acid (5 g, 32 mmol), 3,5-dichloro-2-cyanopyridine (5.8 g, 34 mmol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (0.1 g, 0.13 mmol), dimethylformamide (50 mL) and water (5 mL). The reaction solution was agitated and heated to 45° C. and held at that temperature for 18 hours after which the reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to room temperature and the contents partitioned between ethyl acetate (250 mL) and saturated aqueous NaCl (100 mL). The organic phase was isolated and washed a second time with saturated aqueous NaCl (100 mL). The organic phase was dried for 4 hours over MgSO4, the MgSO4 removed by filtration and the solvent removed under reduced pressure. The residue that remained was then slurried in methanol (50 mL) at room temperature for 20 hours. The resulting solid was collected by filtration and washed with cold methanol (50 mL) then hexanes (60 mL) and dried to afford 5.8 g (73% yield) of an admixture containing a 96:4 ratio of the desired regioisomer. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H)
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): To a 500 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (10 g, 40 mmol), sodium methoxide (13.8 mL, 60 mmol) and methanol (200 mL). With stirring, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to room temperature and combined with water (500 mL). A solid began to form. The mixture was cooled to 0° C. to 5° C. and stirred for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 9.4 g (96% yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (1 g, 4 mmol) and a 48% aqueous solution of HBr (10 mL). While being stirred, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction contents was then cooled to 0° C. to 5° C. with stirring and the pH was adjusted to approximately 2 by the slow addition of 50% aqueous NaOH. Stirring was then continued at 0° C. to 5° C. for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 1.03 g (quantitative yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a nitrogen inlet tube was charged 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (1 gm, 4 mmol), N,N′-carbonyldiimidazole (CDI) (0.97 g, 6 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at 45° C. for about 1 hour then cooled to room temperature. Glycine methyl ester hydrochloride (1.15 g, 12 mmol) is added followed by the dropwise addition of diisopropylethylamine (3.2 mL, 19 mmol). The mixture was then stirred for 2.5 hours at room temperature after which water (70 mL) was added. The contents of the reaction flask was cooled to 0° C. to 5° C. and 1N HCl was added until the solution pH is approximately 2. The solution was extracted with dichloromethane (100 mL) and the organic layer was dried over MgSO4 for 16 hours. Silica gel (3 g) is added and the solution slurried for 2 hours after which the solids are removed by filtration. The filtrate is concentrated to dryness under reduced pressure and the resulting residue was slurried in methanol (10 mL) for two hours. The resulting solid was collected by filtration and washed with cold methanol (20 mL) then hexane and the resulting cake is dried to afford 0.85 g of the desired product as an off-white solid. The filtrate was treated to afford 0.026 g of the desired product as a second crop. The combined crops afford 0.88 g (68% yield) of the desired product. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use
EXAMPLE 2Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): A 20 L reactor equipped with a mechanical stirrer, dip tube, thermometer and nitrogen inlet was charged with (3-chlorophenyl)boronic acid (550 g, 3.52 mol), 3,5-dichloro-2-cyanopyridine (639 g, 3.69 mol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (11.5 g, 140 mmol), and dimethylformamide (3894 g, 4.125 L). The reaction solution was agitated and purged with nitrogen through the dip-tube for 30 minutes. Degassed water (413 g) was then charged to the reaction mixture while maintaining a temperature of less than 50° C. 25 hours. The reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to 5° C. and charged with heptane (940 g, 1.375 L) and agitated for 30 minutes. Water (5.5 L) was charged and the mixture was further agitated for 1 hour as the temperature was allowed to rise to 15° C. The solid product was isolated by filtration and washed with water (5.5 L) followed by heptane (18881 g, 2750 ML). The resulting cake was air dried under vacuum for 18 hours and then triturated with a mixture of 2-propanol (6908 g, 8800 mL0 and heptane (1 g, 2200 mL0 at 50° C. for 4 hours, cooled to ambient temperature and then agitated at ambient temperature for 1 hour. The product was then isolated by filtration and washed with cold 2-propanol (3450 g, 4395 mL) followed by heptane (3010 g, 4400 mL). The resulting solid was dried under high vacuum at 40° C. for 64 hours to afford 565.9 g (65% yield) of the desired product as a beige solid. Purity by HPLC was 98.3. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H).
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): A 20 L reactor equipped with a mechanical stirred, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (558 g, 2.24 mol) and sodium methoxide (25% solution in methanol, 726.0 g, 3.36 mol). With agitation, the reaction solution was heated to reflux for 24 hours, resulting in a beige-colored suspension. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to 5° C. and then charged with water (5580 mL). The resulting slurry was agitated for 3 hours at 5° C. The solid product was isolated by filtration and washed with water (5580 mL) until the filtrate had a pH of 7. The filter cake was air dried under vacuum for 16 hours. The filter cake was then charged back to the reactor and triturated in MeOH (2210 g, 2794 mL) for 1 hour at ambient temperature. The solid was collected by filtration and washed with MeOH (882 g, 1116 mL, 5° C.) followed by heptane (205 mL, 300 mL), and dried under high vacuum at 45° C. for 72 hours to afford 448 g (82% yield) of the desired product as an off-white solid. Purity by HPLC was 97.9%. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer, nitrogen inlet and 25% aqueous NaOH trap was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (440.6 g, 1.8 mol) and 37% aqueous solution of HCl (5302 g). While being agitated, the reaction solution was heated to 102° C. for 24 hours. Additional 37% aqueous HCl (2653 g) was added followed by agitation for 18 hours at 104° C. The reaction contents was then cooled to 5° C., charged with water (4410 g) and then agitated at 0° C. for 16 hours. The resulting precipitated product was isolated by filtration and washed with water until the filtrate had a pH of 6 (about 8,000 L of water). The filter cake was pulled dry under reduced pressure for 2 hours. The cake was then transferred back into the reactor and triturated in THF (1958 g, 2201 mL) at ambient temperature for 2 hours. The solid product was then isolated by filtration and washed with THF (778 g, 875 mL) and dried under reduced pressure at 5° C. for 48 hours to afford 385 g (89% yield) of the desired product as an off-white solid. HPLC purity was 96.2%. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (380 g, 1.52 mol) and diisopropylethylamine (DIPEA) (295 g, 2.28 mol). With agitation, the solution was cooled to 3° C. and charged with trimethylacetyl chloride (275.7 g, 2.29 mol) while maintaining a temperature of less than 11° C., The mixture was then agitated at ambient temperature for 2 hours. The mixture was then cooled to 10° C. and charged with a slurry of glycine methyl ester HCl (573.3 g, 4. 57 mol) and THF (1689 g, 1900 mL), then charged with DIPEA (590.2 g, 4.57 mol) and agitated at ambient temperature for 16 hours. The mixture was then charged with EtOH (1500 g, 1900 mL) and concentrated under reduced pressure to a reaction volume of about 5.8 L. The EtOH addition and concentration was repeated twice more. Water (3800 g) was then added and the mixture was agitated for 16 hours at ambient temperature. The resulting solid product was isolated by filtration and washed with a mixture of EtOH (300 g, 380 mL) and water (380 g), followed by water (3800 g), dried under reduced pressure for 18 hours at 50° C. to afforded 443 g (91% yield) of the desired product as an off-white solid. Purity by HPLC was 98.9%. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
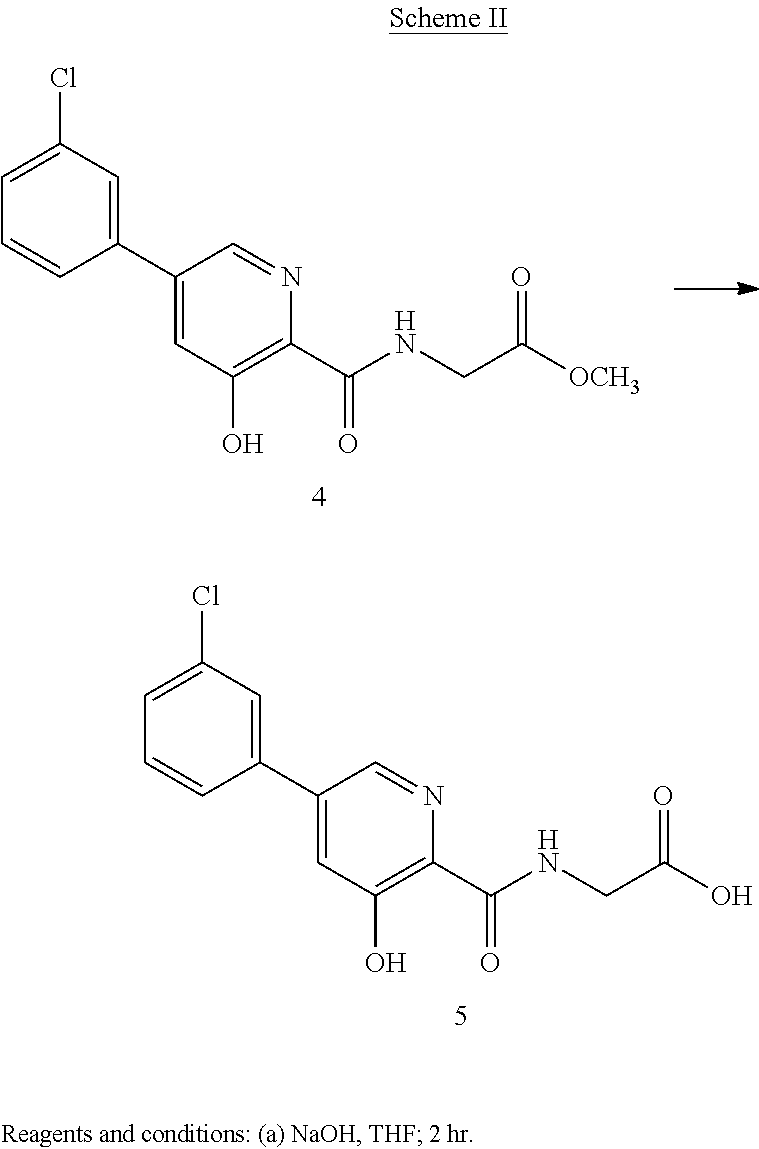
Scheme II herein below outlines and Example 2 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase inhibitor from an ester prodrug.
EXAMPLE 3{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3 -chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 50 mL flask is charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (0.45 g, 1.4 mmol), tetrahydrofuran (4.5 mL) and 1 M NaOH (4.5 mL, 4.5 mmol). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was adjusted to pH 1 with concentrated HCl and the solution was heated at 35° C. under vacuum until all of the tetrahydrofuran had been removed. A slurry forms as the solution is concentrated. With efficient stirring the pH is adjusted to ˜2 with the slow addition of 1 M NaOH. The solid which forms was collected by filtration, washed with water, followed by hexane, then dried under vacuum to afford 0.38 g (88% yield) of the desired product as a white solid. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use.
EXAMPLE 4{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (440 g, 1.42 mol), tetrahydrofuran (3912 g, 4400 mL) and 1 M NaOH (4400 mL). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was acidified to a pH of 2 with slow addition of 2M HCl (2359 g). The resulting mixture was concentrated under reduced pressure to a volume of about 7.5 L. Ware (2210 g) was added and the solution cooled to ambient temperature and agitated for 18 hours. The solid product was isolated by filtration and washed with water (6 L). the crude product was transferred back into the reactor and triturated with 2215 g o deionized water at 70° C. for 16 hours. The mixture was cooled to ambient temperature, The solid product was isolated by filtration and washed with water (500 mL) and dried under reduced pressure at 70° C. for 20 hours to afford 368 g (87% yield) of the desired product as an off-white solid. Purity by HPLC was 99.3%. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
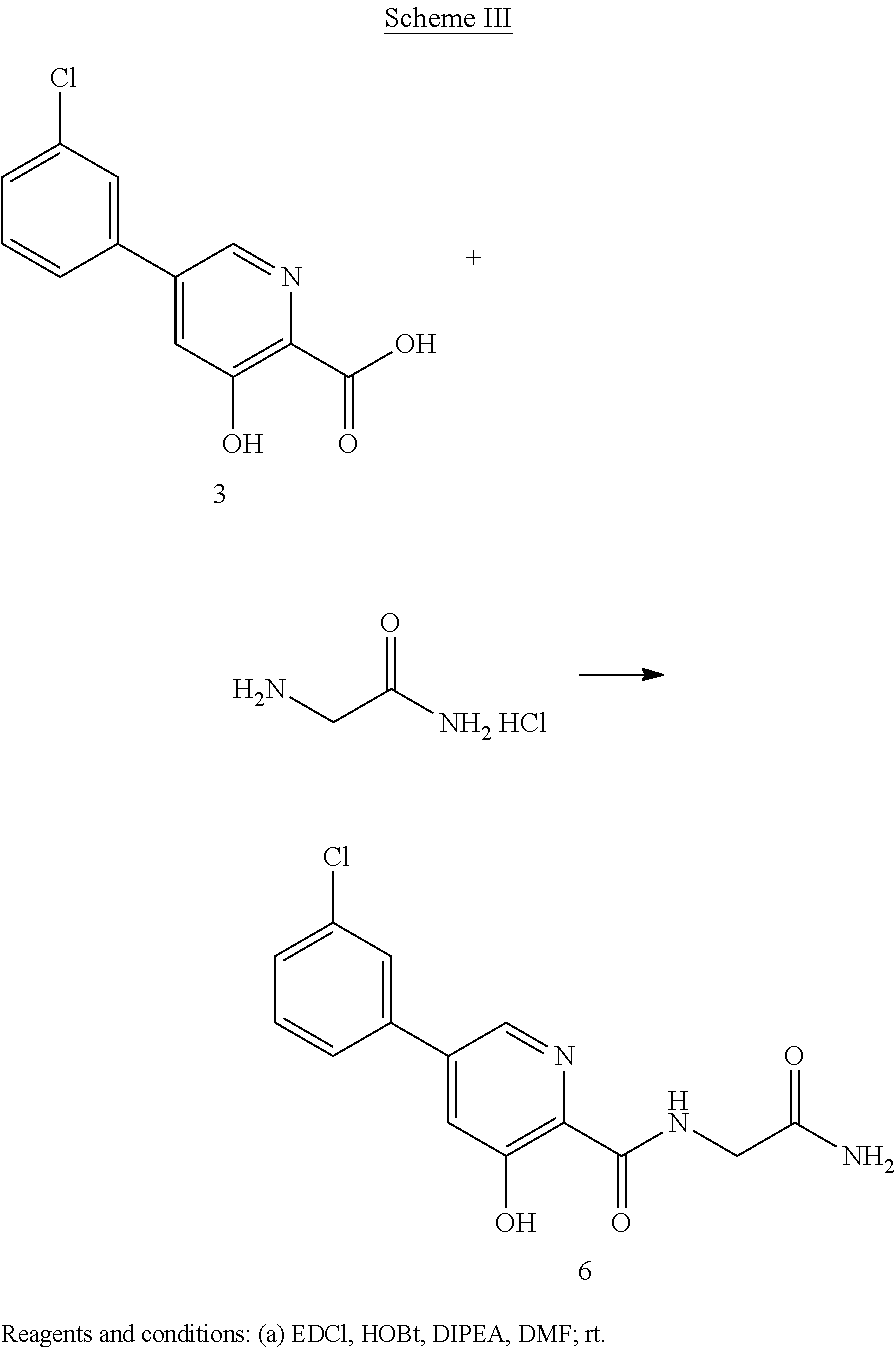
Scheme III herein below outlines and Example 3 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase amide prodrug.
EXAMPLE 55-(3-Chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide
Preparation of 5-(3-chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide (6): To a solution of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (749 mg, 3 mmol) in DMF (20 mL) at room temperature under N2 is added 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) (0.925 g, 5.97 mmol) and 1-hydroxybenzo-triazole (HOBt) (0.806 g, 5.97 mmol). The resulting solution is stirred for 15 minutes then 2-aminoacetamide hydrochloride (0.66 g, 5.97 mmol) and diisopropylethylamine (1.56 ml, 8.96 mmol) are added. The reaction is monitored by TLC and when the reaction is complete the reaction mixture is concentrated under reduced pressure and H2O added. The product can be isolated by normal work-up: The following data have been reported for compound (6). 1H NMR (250 MHz, DMSO-d6) δ ppm 12.46 (1H, s), 9.17 (1H, t, J=5.9 Hz), 8.55 (1H, d, J=2.0 Hz), 7.93 (1H, d, J=0.9 Hz), 7.75-7.84 (2H, m), 7.49-7.60 (3H, m), 7.18 (1H, s), 3.91 (2H, d, J=5.9 Hz). HPLC-MS: m/z 306 [M+H]+.
Scheme IV herein below depicts a non-limiting example the hydrolysis of an amide pro-drug to a prolyl hydroxylase inhibitor after removal of a R10 protecting group
Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
Solid forms of 2-(5-(3-fluorophenyl)-3-hydroxypicolinamido)acetic acid, compositions, and uses thereof
clip
Oct 6, 2015
Akebia Reaches Agreement with FDA and EMA on Vadadustat Global Phase 3 Program
Plans to Initiate Phase 3 PRO2TECT™ Clinical Program by Year-End
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Akebia Therapeutics, Inc. (NASDAQ: AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced the successful completion of the End-of-Phase 2 Meeting process with the United States Food and Drug Administration (FDA) and the Scientific Advice Process with the European Medicines Agency (EMA) for its lead product, vadadustat (formerly AKB-6548), for patients with anemia related to non-dialysis dependent chronic kidney disease (NDD-CKD). The company has reached agreement with both the FDA and EMA regarding key elements of the Phase 3 program, known as the PRO2TECT™ program, and expects to launch the program later this year.
The PRO2TECT™program includes two separate studies and will collectively enroll approximately 3,100 NDD-CKD patients across 500 sites globally. The correction study will address anemia patients not currently being treated with recombinant erythropoiesis stimulating agents (rESAs). The conversion study includes patients currently receiving rESA who will be converted to either vadadustat or the active control with the goal of maintaining their baseline hemoglobin levels. Both studies will include a 1:1 randomization and an open label, active-control, non-inferiority design. Primary endpoints include an efficacy assessment of the hemoglobin response and an assessment of cardiovascular safety measured by major adverse cardiovascular events.
“Akebia’s Phase 3 program is designed to provide the medical community and regulators with a clear understanding of vadadustat’s potential benefit and safety advantages over rESAs, the current standard of care worldwide and, with a positive outcome, to establish vadadustat as the best-in-class treatment option for patients with renal anemia,” stated John P. Butler, President and Chief Executive Officer of Akebia. “We are pleased that the regulators are in agreement regarding the importance of an active-control trial as this design is the most clinically relevant and commercially valuable, and will allow us the quickest path to full enrollment. We are now moving rapidly to launch these studies and advance our goal of bringing forward new treatment options for patients suffering from renal anemia.”
“This Phase 3 program builds on the positive data from our Phase 2 program in NDD-CKD patients which demonstrated that once-daily vadadustat can control and maintain hemoglobin levels in a clinically relevant range while minimizing fluctuations in hemoglobin levels that are associated with increased cardiovascular safety risks,” stated Brad Maroni, M.D., Chief Medical Officer at Akebia. “These two Phase 3 event-driven studies are designed to establish the safety and efficacy of vadadustat in the setting of contemporary clinical practice patterns, and support regulatory approvals globally.”
In addition, Akebia discussed with the FDA and EMA a parallel Phase 3 program, known as the INNO2VATE™ program, for vadadustat in patients with anemia related to chronic kidney disease who are undergoing dialysis (DD-CKD). Akebia expects to formalize its Phase 3 program in DD-CKD patients after presenting the results from its recently completed Phase 2 study to both regulatory agencies.
About Vadadustat (Formerly AKB-6548)
Vadadustat is an oral therapy currently in development for the treatment of anemia related to chronic kidney disease (CKD). Vadadustat is designed to stabilize HIF, a transcription factor that regulates the expression of genes involved with red blood cell (RBC) production in response to changes in oxygen levels, by inhibiting the hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzyme. Vadadustat exploits the same mechanism of action used by the body to naturally adapt to lower oxygen availability associated with a moderate increase in altitude. At higher altitudes, the body responds to lower oxygen availability with increased production of HIF, which coordinates the interdependent processes of iron mobilization and erythropoietin (EPO) production to increase RBC production and, ultimately, improve oxygen delivery.
As a HIF stabilizer with best-in-class potential, vadadustat raises hemoglobin levels predictably and sustainably, with a dosing regimen that allows for a gradual and controlled titration. Vadadustat has been shown to improve iron mobilization, potentially eliminating the need for intravenous iron administration and reducing the overall need for iron supplementation.
About Anemia Related to CKD
Approximately 30 million people in the United States have CKD, with an estimated 1.8 million of these patients suffering from anemia. Anemia results from the body’s inability to coordinate RBC production in response to lower oxygen levels due to the progressive loss of kidney function, which occurs in patients with CKD. Left untreated, anemia significantly accelerates patients’ overall deterioration of health with increased morbidity and mortality. Renal anemia is currently treated with injectable rESAs, which are associated with inconsistent hemoglobin responses and well-documented safety risks.
About Akebia Therapeutics
Akebia Therapeutics, Inc. is a biopharmaceutical company headquartered in Cambridge, Massachusetts, focused on delivering innovative therapies to patients with kidney disease through HIF biology. The company has completed Phase 2 development of its lead product candidate, vadadustat, an oral therapy for the treatment of anemia related to CKD in both non-dialysis and dialysis patients.
clip
Akebia Announces Positive Top-Line Results from its Phase 2 Study of Vadadustat in Dialysis Patients with Anemia Related to Chronic Kidney Disease
-Treatment with Vadadustat Successfully Maintained Mean Hemoglobin Levels Following Conversion from rESA Therapy-
-Vadadustat Demonstrated a Favorable Safety Profile with Once Daily and Three Times per Week Dosing-
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Akebia Therapeutics, Inc. (NASDAQ:AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced positive top-line results from its Phase 2 study of vadadustat (formerly AKB-6548) in dialysis patients with anemia related to chronic kidney disease (CKD). The study achieved its primary objective, indicating that vadadustat maintained stable hemoglobin (HGB) levels throughout the 16-week treatment period following conversion from recombinant erythropoiesis-stimulating agent (rESA) therapy. Vadadustat demonstrated a favorable safety profile with no drug-related serious adverse events and no deaths. The results highlight the potential of vadadustat, dosed either once daily or three times per week, to safely and predictably manage and sustain HGB levels in CKD patients undergoing dialysis.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy”
The open-label, multi-center, 94 patient study was designed to evaluate the ability of vadadustat to maintain hemoglobin levels in patients undergoing hemodialysis who were previously being treated with rESAs. Patients were assigned to one of three dose cohorts: once daily vadadustat at a starting dose of 300mg, once daily vadadustat at a starting dose of 450mg, or vadadustat three times per week in conjunction with the patient’s hemodialysis schedule at a starting dose of 450mg. The study achieved its primary endpoints of maintaining stable hemoglobin levels over 16 weeks of treatment in all three cohorts of patients converting from rESAs to vadadustat.
Vadadustat was well tolerated among patients in all three dose cohorts. Treatment-emergent adverse events (TEAEs) with vadadustat were balanced across the cohorts. Serious adverse events (SAEs) were reported in 13 subjects (13.8%), well within the expected range for this patient population. There were no drug-related SAEs and no deaths reported in the study.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy,” said Brad Maroni, M.D., Chief Medical Officer at Akebia. “We are impressed with the consistency in hemoglobin levels across the duration of the study, which highlights the ability of vadadustat to control and maintain hemoglobin levels in this patient population. Furthermore, the results indicate that daily and three times per week dosing regimens are both viable options for patients on dialysis.”
John P. Butler, President and Chief Executive Officer of Akebia, stated, “These results further confirm vadadustat as a potential best-in-class anemia treatment for CKD patients, and reinforce our confidence in this product candidate as we advance toward our Phase 3 program. Adding these results to the 12 other clinical studies we have completed, we are confident in the potential for vadadustat to treat anemia in a broad array of patients with CKD. We are pleased to have successfully completed this stage of our drug development and look forward to initiating Phase 3 studies.”
Complete efficacy and safety data from this Phase 2 study will be presented at an upcoming medical meeting.
About the Phase 2 Study Design of Vadadustat in Dialysis Patients with Anemia Related to CKD
The Phase 2 multi-center, open-label study evaluated 94 patients over 16 weeks of treatment, at 20 dialysis centers in the United States, including an assessment of HGB response to the starting dose of vadadustat during the first 8 weeks, followed by an assessment of HGB response to algorithm-guided dose adjustments of vadadustat during the subsequent 8 weeks of treatment. The study enrolled three cohorts, each consisting of approximately 30 CKD patients with anemia undergoing dialysis who were switched from injectable rESA therapy to vadadustat. Patients in the first two cohorts received once daily doses of vadadustat, while patients in the third cohort received vadadustat three times per week in conjunction with their hemodialysis schedule.
Jump up^Gupta N, Wish JB. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am J Kidney Dis. 2017 Jun;69(6):815-826. . doi:10.1053/j.ajkd.2016.12.011. PMID28242135. Missing or empty |title= (help)
Jump up^Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 2017;45(5):380-388. . doi:10.1159/000464476. PMID28343225. Missing or empty |title= (help)
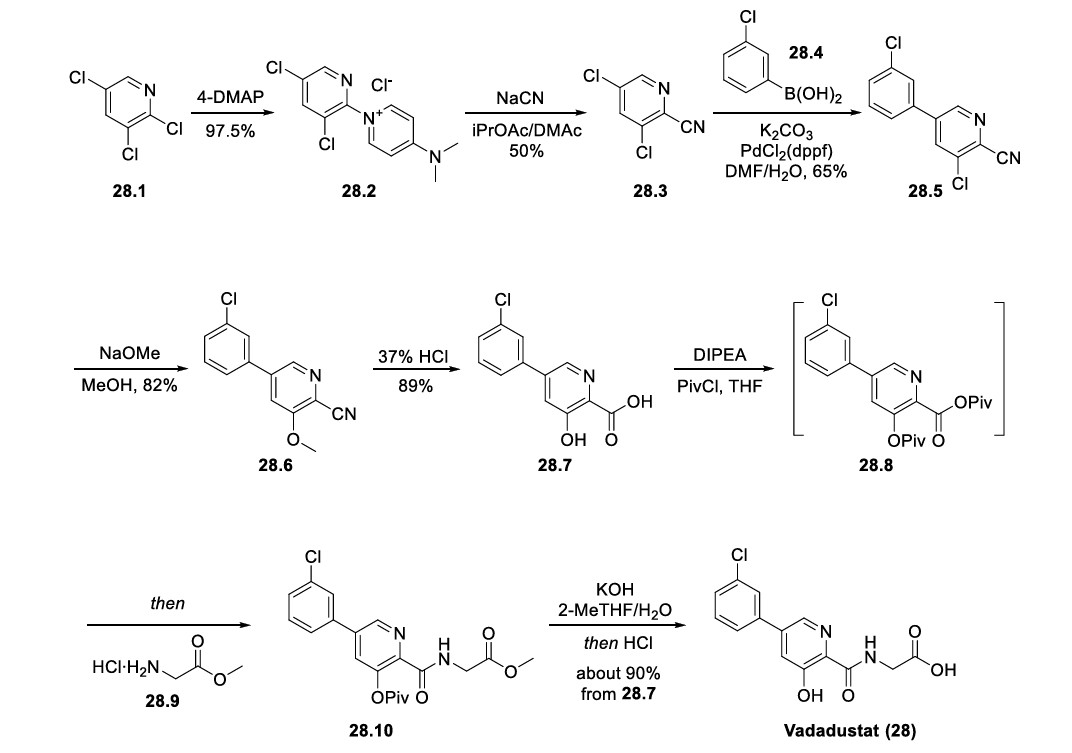
Vadadustat (Vafseo). Vadadustat (28) is a hypoxia inducible factor prolyl hydroxylase (HIF-PH) inhibitor developed by Akebia Therapeutics. It was approved by the European Commission in April 2023, and recently also by the USFDA, for the treatment of symptomatic anemia associated with chronic kidney disease in adults receiving chronic maintenance dialysis. Vadadustat acts by inhibiting HIFPH, 214 which results in increases of endogenous erythropoietin production, red blood cell synthesis, and iron mobilization. 215 While a number of syntheses of vadadustat (28) have been published in previous patents 216−228 and a journal article, 229 Akebia Therapeutics has published two patents regarding the large-scale preparation of vadadustat (Scheme 52). 218,226 The key intermediate nitrile 28.3 could be accessed in two steps: the neat SNAr reaction between commercially available 2,3,5trichloropyridine (28.1) and 4-DMAP to generate pyridinium salt 28.2, followed by a second SNAr reaction of 28.2 with NaCN. The Suzuki coupling between 28.3 and 3-chlorophenyl boronic acid (28.4) gave the biaryl 28.5, and the subsequent SNAr reaction of 28.5 with NaOMe replaced the 3-chloro substitution on the pyridine ring with a methoxy group, generating intermediate 28.6. Global acidic hydrolysis of both methyl ether and nitrile group in 28.6 gave the 3 hydroxypicolinic acid 28.7. Treatment of 28.7 with DIPEA and excess pivaloyl chloride (PivCl) resulted in the formation of mixed anhydride 28.8 with concomitant acylation of the 3 hydroxy group. Without isolation of 28.8, glycine methyl ester hydrochloride (28.9) was then charged with additional DIPEA to generate the corresponding amide 28.10. The residual amount (∼0.5%) of 28.7 in 28.10 was hard to remove, but this impurity could be effectively rejected with an extra amount of DIPEA during workup and solvent switch. Finally, the Opivaloyl group and methyl ester were both removed via basic hydrolysis, giving vadadustat (28) in about 90% yield from 28.7.
REF
(215) Pergola, P. E.; Spinowitz, B. S.; Hartman, C. S.; Maroni, B. J.; Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115−1122. (216) Lanthier, C. M.; Gorin, B.; Oudenes, J.; Dixon, C. E.; Lu, A. Q.; Copp, J. D.; Janusz, J. M. Preparation of [(3-hydroxypyridine-2carbonyl)amino]alkanoic acids, esters and amides as prolyl hydroxylase inhibitors. US 20120309977, 2012. (217) Li, X.; Chen, J. Process for the preparation of vadadustat. CN105837502, 2016. (218) Gorin, B. I.; Lanthier, C. M.; Luong, A. B. C.; Copp, J. D.; Gonzalez, J. Process for preparing 2-[[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino]acetic acid. WO 2019217550, 2019. (219) Kou, J.; Li, Y.; Xiao, Q.; Lin, B.; Sun, J.; Wang, Z.; Luo, Z.;Huang, F. Preparation method of vadadustat. CN 110903238, 2020. (220) Machida, K.; Yasukouchi, H.; Nishiyama, A. Method for producing vadadustat intermediate. WO 2020217733, 2020. (221) Xiao, Q.; Lin, B.; Kou, J.; Sun, J.; Qiu, X.; Wang, Z.; Luo, Z.;Huang, F. Preparation of vadadustat intermediate. CN 111848505,2020
(222) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Li, Y.; Sun, J.; Jin, L.; Luo, Z.; Huang, F. Preparation of vadadustat and intermediate thereof. CN 111205222, 2020. (223) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Luo, Z.; Huang, F.; Li, Y. Preparation of vadadustat and intermediate thereof. CN 111423367, 2020. (224) Xiao, Q.; Qiu, X.; Lin, B.; Kou, J.; Li, Y.; Sun, J.; Wang, Z.; Luo, Z.; Huang, F. Preparation of vadadustat. CN 111320577, 2020. (225) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Qiu, X.; Cai, X.; Li, Y.; Luo, Z.; Huang, F. Method for preparing vadadustat and intermediate thereof. WO 2021179540, 2021. (226) Jurkauskas, V.; Jung, Y. C.; Kwon, T.; Kannan, A.; Gondi, V. B. Manufacturing process for 3,5-dichloropicolinonitrile for synthesis of vadadustat. WO 2022006427, 2022. (227) Chen, Z.; Zheng, Y.; Zhang, L.; Yu, C.; Liu, L.; He, B. Preparation of a pyridine compound used for the preparation of vadadustat. CN 117843565, 2024. (228) Patel, K. R.; Thakrar, V. H.; Mehta, T. B.; Wagh, A. G.; Patel, J. A.; Patil, R. R.; Solanki, Y. U.; Ladumor, C. B. A process for the preparation of Vadadustat or salts thereof. WO 2024079708, 2024. (229) Lin, B. Y.; Kou, J. P.; Wu, S. M.; Cai, X. R.; Xiao, Q. B.; Li, Y. L.; Hu, J.; Li, J. B.; Wang, Z. Q. Development of a robust and scalable process for the large-scale preparation of Vadadustat. Org. Process Res. Dev. 2021, 25, 960−968.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
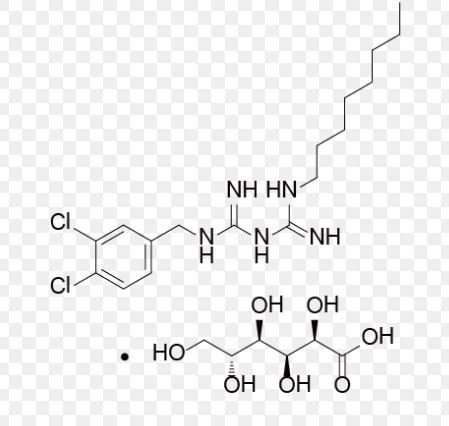
Olanexidine gluconate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jul 03, 2015. It was developed and marketed as Olanedine® by Otsuka in Japan.
Olanexidine gluconate is an antiseptic/disinfectant compound with potent bactericidal activity against Gram-negative and Gram-positive bacteria, for use in preparing patients for surgery and preventing of postoperative bacterial infections.
Olanedine® is available as topical solution (1.5%), containing 3 g/200 mL, 0.15 g/10 mL and 0.375 g/25 mL, and the recommendation is applying appropriate amount of the drug.
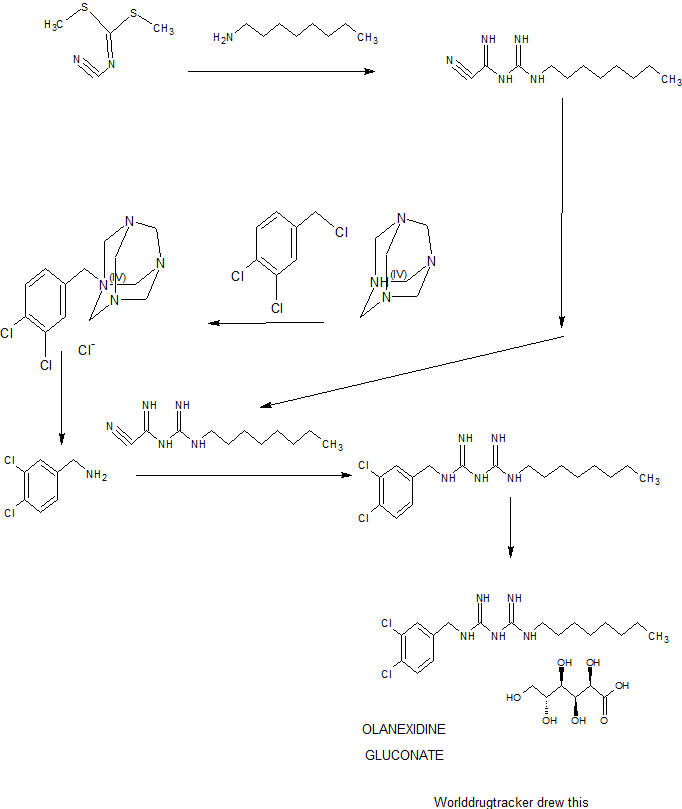
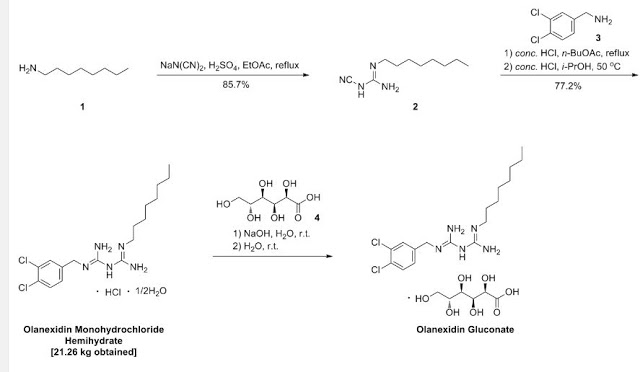
A 7.00-kg quantity of Compound (4) (54.16 mol) was dissolved in 105 liters of ethyl acetate, and the resulting mixture was cooled to 5°C or below. A 2.66-kg quantity of concentrated sulfuric acid (27.12 mol) was added thereto dropwise at a temperature of 4O0C or below while stirring. To the thus- obtained suspension of 1/2 sulfate of Compound (4) was added 5.06 kg of sodium dicyanamide (56.83 mol), and the resulting suspension was heated under reflux for 7 hours. The reaction solution was cooled to 400C or below, and 70 liters of water was added thereto. Subsequently, the resulting solution was heated to 80 to 900C (internal temperature) to distill the ethyl acetate off. The remaining liquid was cooled to 400C or below, and 70 liters of toluene was then added thereto, followed by the extraction of 1-cyano — 3-n-octyl guanidine at about 500C. The extracted toluene layer was washed with 35 liters of water at about 500C and cooled to 100C or below, followed by stirring for about 30 minutes. The resulting precipitated crystals were separated and washed with 7 liters of toluene. The resulting crystals were dried at 400C for 7.5 hours, yielding l-cyano-3-n- octylguanidine. 2007/067107
-16-
Yield: 9.11 kg (The yield was 85.7% based on the Compound(4).) White crystals having a melting point of 69 to 740C (no clear melting point was observed)
Reference Example 2: Acidolysis of 1- (3,4-dichlorobenzyl) -5- octylbiguanide dihydrochloride
A 1-g quantity of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide dihydrochloride was dissolved in 15 ml of 10% ethanol, followed by refluxing for 5 hours. HPLC analysis was conducted under the conditions described below.
The yield of 1-[N- (3,4-dichlorobenzyl) carbamoyl-3- octyl]guanidine (holding time: 9.84 minutes) was 0.91%, and the yield of 1- (N-octyl-carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine
(holding time: 10.54 minutes) was 0.22%.
HPLC analysis conditions:
Column: YMC AM302 4.6 mm I. D. x 150 mm
Eluate: MeCN/0.05 M aqueous solution of sodium 1- octanesulfonate/acetic acid = 700/300/1
Detector: UV 254 nm
The physical property values of the resulting 1-[N- (3,4- dichlorobenzyl) carbamoyl-3-octyl] guanidine were as follows: NMR (DMSO-de) δ: 0.86 (3H, t, J = 6.0 Hz), 1.07-1.35 (1OH, m) , 1.35-1.49 (2H, m) , 2.95-3.15 (2H, m) , 4.12 (2H, d, J = 6.3 Hz), 6.78-7.40 (4H, m) , 7.23 (IH, dd, J = 2.1 Hz, J = 8.4 Hz), 7.46 (IH, d, J = 2.1 Hz), 7.54 (IH, d, J = 8.4 Hz)
The physical property values of the resulting 1- (N-octyl- carbamoyl) -3- (3, 4-dichlorobenzyl) guanidine were as follows: NMR (DMSO-d6) δ: 0.85 (3H, t, J = 6.6 Hz), 1.02-1.40 (12H, m) , 2.89-2.95 (2H, m) , 4.33 (2H, bs) , 5.76-7.00 (4H, m) , 7.28 (IH, dd, J = 2.1 Hz, J = 8.1 Hz), 7.52 (IH, d, J = 2.1 Hz), 7.58 (IH, d, J = 8.1 Hz)
Example 1: 1- (3, 4-dichlorobenzyl) -5-octylbiguanide monohydrochloride 1/2 hydrate
A 9.82-g quantity of Compound (2) (0.05 mol) and 10.63 g of 3, 4-dichlorobenzylamine (0.05 mol) were added to 49 ml of butyl acetate, followed by refluxing for 6 hours. The reaction solution was concentrated under reduced pressure, and a mixture of 12 ml of water and 47 ml of isopropyl alcohol was added and dissolved into the remainder. To the thus-obtained solution was added, dropwise, 10.13 g of concentrated hydrochloric acid. The resulting mixture was stirred at 28 to 300C for 30 minutes, and the precipitated crystals were then filtered out. The thus- obtained crystals were washed with a small amount of isopropyl alcohol, yielding 23.42 g of (non-dried) 1- (3, 4-dichlorobenzyl) – 5-octylbiguanide dihydrochloride. The resulting crystals were suspended in 167 ml of water without drying, the suspension was then stirred at 25 to 27°C for 2 hours, followed by separation of the crystals by filtration. The thus-obtained crystals were washed with a small amount of water and dried at 400C for 20 hours, yielding 17.05 g of 1- (3, 4-dichlorobenzyl) -5-octyl biguanide monohydrochloride 1/2 hydrate having a purity of 99.9% at a yield of 81.6%.
Example 2 : 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride
A 100-g quantity of Compound (4) (0.774 mol) was dissolved in 1 liter of n-butyl acetate, and 37.6 g of concentrated sulfuric acid (0.383 mol) was added thereto while stirring. To the thus-obtained suspension of 1/2 sulfate of Compound (4) was added 68.9 g of sodium dicyanamide (0.774 mol), 7107
-18- and the resulting suspension was heated under reflux for 3 hours. The reaction solution was cooled to about 200C, and the organic layer thereof was sequentially washed with about 500 ml each of (i) 5% hydrochloric acid, (ii) 5% aqueous caustic soda solution, (iii) 5% aqueous sodium bicarbonate solution, and (iv) water.
To the thus-obtained n-butyl acetate solution of Compound (2) were added 118.5 g of Compound (3) (0.673 mol) and then 58.4 ml of concentrated hydrochloric acid while stirring. The reaction solution was heated, and about 800 ml of n-butyl acetate was distilled off under atmospheric pressure (ordinary pressure) , followed by heating the reaction solution under reflux for 3.5 hours . Subsequently, the reaction solution was cooled to about 400C, and 900 ml of isopropanol, 100 ml of water, and 134 ml of concentrated hydrochloric acid were added thereto. The mixture was stirred at 60 to 70°C for 1 hour and cooled to 100C or below and the precipitated crystals were then separated. The resulting crystals were washed with 200 ml of isopropanol and dried at 6O0C, yielding 1- (3, 4-dichlorobenzyl) -5-octylbiguanide dihydrochloride. Yield: 243.8 g (The yield was 81.3% based on the Compound (3).) Melting point: 228.90C IR(KBr) spectrum: 2920, 1682, 1634, 1337, 1035, 820, and 640 cm“1
Olanexidine is a compound with high bactericidal activity having the chemical name 1-(3,4-dichlorobenzyl)-5-octylbiguanide. Research has been carried out into bactericides containing, olanexidine hydrochloride as an active ingredient (see Japanese Patent No. 2662343, etc.).
Olanexidine has very poor solubility in water, and hitherto known salts of olanexidine are also poorly soluble in water. For example, the solubility at 0° C. of olanexidine hydrochloride in water has been measured to be less than 0.05% (W/V), and the solubility of free olanexidine is a further order of magnitude less than this. Consequently, sufficient bactericidal activity cannot be expected of an aqueous solution merely having olanexidine dissolved therein, and moreover, depending on the conditions the olanexidine may precipitate out.
In the case of making an aqueous preparation of olanexidine in particular, to make the concentration of the olanexidine sufficient for exhibiting effective bactericidal activity, and to reduce the possibility of the olanexidine precipitating out, it has thus been considered necessary to use a dissolution aid such as a surfactant.
EXAMPLE 1 Preparation of an Aqueous Solution Aqueous Solution 1
20.9 g (50 mmol) of olanexidine hydrochloride hemihydrate was added to 250 mL of a 1 N aqueous sodium hydroxide solution, and the suspension was stirred for 1.5 hours at room temperature (25° C.). The solid was filtered off, and washed with water. The solid obtained was further suspended in 250 mL of purified water, the suspension was stirred for 5 minutes at room temperature, and the solid was filtered off, and washed with water. This operation was carried out once more to remove sodium chloride formed. The solid obtained (free olanexidine) was put into purified water in which 8.9 g (50 mmol) of gluconolactone had been dissolved, and the mixture was stirred at room temperature until the solid dissolved, and then purified water was further added to give a total volume of 300 mL. The concentration of olanexidine in the aqueous solution obtained was measured by using high performance liquid chromatography to be 6% in terms of free olanexidine.
This aqueous solution was still transparent and colorless even after being left for several months at room temperature.
ZSTK474 is a cell permeable and reversible P13K inhibitor with an IC₅₀ at 6nm. It was identified as part of a screening library, selected for its ability to block tumor cell growth. ZSTK474 has shown strong antitumor activities against human cancer xenographs when administered orally to mice without a significant toxic effect.
Phosphatidylinositol 3-kinase (PI3K) has been implicated in a variety of diseases including cancer. A number of PI3K inhibitors have recently been developed for use in cancer therapy. ZSTK474 is a highly promising antitumor agent targeting PI3K. We previously reported that ZSTK474 showed potent inhibition against four class I PI3K isoforms but not against 140 protein kinases.
However, whether ZSTK474 inhibits DNA-dependent protein kinase (DNA-PK), which is structurally similar to PI3K, remains unknown. To investigate the inhibition of DNA-PK, we developed a new DNA-PK assay method using Kinase-Glo. The inhibition activity of ZSTK474 against DNA-PK was determined, and shown to be far weaker compared with that observed against PI3K. The inhibition selectivity of ZSTK474 for PI3K over DNA-PK was significantly higher than other PI3K inhibitors, namely NVP-BEZ235, PI-103 and LY294002.
Kawashima, S.; Matsuno, T.; Yaguchi, S.; Sasahara, H.; Watanabe, T.Preparation of Heterocyclic Compounds as Antitumor Agents. PCT Int. Appl. WO 02088112, 2002;
Chem. Abstr.2002, 137, 370113.
………………………………….
2-(difluoromethyl)-1H-benzimidazole
A mixture of o-phenylenediamine (5.41 g, 50 mmol) and difluoroacetic acid (9.6 g, 100 mmol) in 4 M HCl (20 mL) was heated under reflux for 1 h and diluted with hot water (50 mL). The solution was treated with charcoal and filtered through Celite before being neutralized with aqueous NH3. The resulting white precipitate was collected, washed with water, and dried to give 2-(difluoromethyl)-1H-benzimidazole (6.07 g, 72% yield): mp 156–158 °C; 1H NMR (DMSO-d6) δ 13.28 (br, 1H), 7.76–7.68 (m, 1H), 7.61–7.54 (m, 1H), 7.36–7.26 (m, 2H), 7.26 (t,JHF= 53.3 Hz, 1H).
Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J.One-pot synthesis of 2-trifluoromethyl and 2-difluoromethyl substituted benzo-1,3-diazolesTetrahedron Lett.2007, 48, 3251–3254
TRIAZINE, PYRIMIDINE AND PYRIDINE ANALOGS AND THEIR USE AS THERAPEUTIC AGENTS AND DIAGNOSTIC PROBES [US2011275762]2011-11-10
Patent
Submitted
Granted
Heterocyclic compound and antitumor agent containing the same as active ingredient [US7071189]
2004-06-17
2006-07-04
Treatment of prostate cancer, melanoma or hepatic cancer [US2007244110]
2007-10-18
Heterocyclic compound and antitumor agent containing the same as effective ingredient [US7307077]
2006-11-02
2007-12-11
IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US7750001]
2008-05-15
2010-07-06
PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLES AND THEIR USE IN CANCER THERAPY [US2011009405]
2011-01-13
SUBSTITUTED PYRIMIDINES AND TRIAZINES AND THEIR USE IN CANCER THERAPY [US2011053907]
2011-03-03
IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US2010267700]
2010-10-21
AMORPHOUS BODY COMPOSED OF HETEROCYCLIC COMPOUND, SOLID DISPERSION AND PHARMACEUTICAL PREPARATION EACH COMPRISING THE SAME, AND PROCESS FOR PRODUCTION OF THE SAME [US8227463]
2010-09-30
2012-07-24
PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY [US2010226881]
2010-09-09
PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLE SULFONAMIDES AND THEIR USE IN CANCER THERAPY [US2010249099]
2010-09-30
…………..
Zenyaku Kogyo
Sector: Health Care
Industry: Biotech & Pharma
Sub-Industry: Specialty Pharma
Zenyaku Kogyo Co. Ltd. produces pharmaceuticals. The Company manufactures and sells over-the-counter drugs, health foods, and prescription medicines, as well as skin care products.
Tolvaptan was discovered by Otsuka in Japan, and its primary results from a global clinical trial involving 1,400 ADPKD patients from 15 countries, which demonstrated a statistically significant reduction in the rate of total kidney volume, were published in New England Journal of Medicine in 2012. It is also currently under a fast track review in the US, following our announcement of FDA accepting to review the application in April 2013.
ADPKD is a hereditary and often physically and mentally burdensome disease characterized by the development of multiple cysts in the kidneys. ADPKD is often associated with pain, hypertension, decreased kidney function and ultimately, kidney failure that may result in hemodialysis or kidney transplantation.
There are estimated to be approximately 31,000 ADPKD patients in Japan, and the diagnosed prevalence is estimated to be between 1:1000 and 1:4000 globally.
(Tokyo, Japan, May 30, 2013) – Otsuka Pharmaceutical Co., Ltd. Today announced it filed an application with the Pharmaceutical and Medical Devices Agency in Japan (PMDA) to market its novel compound tolvaptan for the treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD). Phase III clinical trial results that form the basis of the regulatory filing were published in the New England Journal of Medicine in November 2012. The MHLW has designated tolvaptan as an Orphan Drug.http://www.otsuka.co.jp/en/release/2013/0603_02.html
Tolvaptan is also in fast-track clinical trials[2] for polycystic kidney disease. In a 2004 trial, tolvaptan, when administered with traditional diuretics, was noted to increase excretion of excess fluids and improve blood sodium levels in patients with heart failure without producing side effects such as hypotension (low blood pressure) or hypokalemia(decreased blood levels of potassium) and without having an adverse effect on kidney function.[3] In a recently published trial (TEMPO 3:4 ClinicalTrials.gov number, NCT00428948) the study met its primary and secondary end points. Tolvaptan, when given at an average dose of 95 mg per day over a 3-year period, slowed the usual increase in kidney volume by 50% compared to placebo (2.80% per year versus 5.51% per year, respectively, p<0.001) and reduced the decline in kidney function when compared with that of placebo-treated patients by approximately 30% (reciprocal serum creatinine, -2.61 versus -3.81 (mg/mL)-1 per year, p <0.001)[4]
Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther10 (3): 165–71. doi:10.1177/107424840501000304. PMID16211205.
Gheorghiade M, Gattis W, O’Connor C, et al. (2004). “Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial”. JAMA291 (16): 1963–71. doi:10.1001/jama.291.16.1963. PMID15113814.
Gheorghiade M, Niazi I, Ouyang J et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04.PMID12742979.
Otsuka Pharmaceutical Co., Ltd. announced today that the U.S. Food and Drug Administration (FDA) has accepted for priority review the company’s new drug application (NDA) for the potential use of tolvaptan for the treatment of autosomal dominant polycystic kidney disease (ADPKD). Phase III clinical trial results………..read more at financial post
Tolvaptan (INN), also known as OPC-41061, is a selective, competitive vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca and in India is manufactured & sold by MSN laboratories Ltd. under the trade name Tolvat & Tolsama.
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
Combination of
Trifluridine
cytotoxin
Tipiracil
thymidine phosphorylase inhibitor
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
February 28, 2013,
Taiho Pharmaceutical Co., Ltd. has submitted an application to the Japanese Ministry of Health, Labour and Welfare for approval of the manufacture and marketing of the novel oral nucleoside anti-tumour agent TAS-102 (combination of trifluorothymidine [FTD] and tipiracil hydrochloride [TPI]). Taiho is seeking approval of TAS-102 for the indication of unresectable, advanced, recurrent colorectal cancer.
The application for approval is based on the results of a phase II clinical trial (Study 10040030) conducted at 20 facilities throughout Japan. It was a randomized, double-blind comparative study of TAS-102 and a placebo involving 172 patients with unresectable, advanced, recurrent colorectal cancer that was refractory to the standard chemotherapy of at least two or more regimens containing fluoropyrimidine, irinotecan, and oxaliplatin.
The results indicated that the group administered TAS-102 had improved overall survival rates (median overall survival: 9.0 months vs. 6.6 months) and a significantly reduced risk of mortality (HR: 0.56, p=0.0011). The most frequently reported adverse drug reaction with a CTCAE grade of 3 or higher was neutropenia. Grade 3 or higher diarrhea, fatigue, nausea, and other adverse reactions were no more than 10 per cent.
Taiho Pharmaceutical is currently proceeding with a global phase III clinical trial of TAS-102 in a similar colorectal cancer population (RECOURSE) with the ultimate goal of global registration and commercialization of the agent.
Taiho Pharmaceutical believes that TAS-102 will make a significant contribution to cancer patients and will continue its development efforts to broaden its use.
TAS-102 is an anti-tumour agent composed of a combination of trifluorothymidine (FTD), a nucleoside that incorporates into DNA and inhibits a variety of genetic functions required for the proliferation of cancer cells, and tipiracil hydrochloride (TPI), an inhibitor of thymidine phosphorylase (which degrades FTD) that maintains an effective blood concentration of FTD. TAS-102 is administered twice daily to achieve a total daily dose of 70mg/m2 for five days followed by two days of rest and then repeated a second time. This is followed by a 14-day rest period to make a 28-day schedule for one course.
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
Taiho Pharmaceutical, a subsidiary of Otsuka Holdings Co., Ltd., is an R&D-driven specialty pharma focusing on the three fields of oncology, allergies and immunology, and urology.
Otsuka Pharmaceutical Co., Ltd. (Otsuka) and H. Lundbeck A/S (Lundbeck) announced the U.S. Food and Drug Administration (FDA) has approved ABILIFY MAINTENA™ (aripiprazole) for extended- release injectable suspension, an intramuscular (IM) depot formulation indicated for the treatment of schizophrenia.
ABILIFY MAINTENA is the first dopamine D2 partial agonist approved as a once- monthly injection. It contributes a new treatment option to address the ongoing need for relapse prevention in patients with schizophrenia – a chronic, debilitating disease.
Efficacy was demonstrated in a 52-week, placebo-controlled, double-blind, randomized-withdrawal, Phase 3 maintenance trial of ABILIFY MAINTENA in patients with schizophrenia. The time to relapse was the primary endpoint. In the trial, ABILIFY MAINTENA>1 In a key secondary endpoint, the percentage of subjects experiencing relapse (i.e., meeting clinical trial criteria for exacerbation of psychotic symptoms/relapse) was also significantly lower with ABILIFY MAINTENA compared to placebo at the end of the study (10% vs. 40%, respectively; p<0.0001). Additional support for efficacy was derived from oral aripiprazole trials.
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. ABILIFY MAINTENA is not approved for the treatment of patients with dementia-related psychosis. ABILIFY MAINTENA is contraindicated in patients with a known hypersensitivity reaction to aripiprazole. Reactions have ranged from pruritus/urticaria to anaphylaxis (see Important Safety Information below).
ABILIFY MAINTENA will be the first commercialized product from the long-term global alliance between Otsuka and Lundbeck to develop CNS medicines worldwide. The companies expect the product will start becoming available in the U.S. on March 18.
Aripiprazolebrand names: Abilify, Aripiprex) is a partial dopamine agonist of the second generation class of atypical antipsychoticswith additional antidepressant properties that is used in the treatment of schizophrenia,bipolar disorder, and clinical depression. It was approved by the U.S. Food and Drug Administration (FDA) for schizophrenia on November 15, 2002 and the European Medicines Agency on 4 June 2004; for acute manic and mixed episodes associated with bipolar disorder on October 1, 2004; as an adjunct for major depressive disorder on November 20, 2007; and to treat irritability in children with autism on 20 November 2009.[1][2] Aripiprazole was developed by Otsuka in Japan, and in the United States,Otsuka America markets it jointly with Bristol-Myers Squibb.
Aripiprazole (OPC-14597, OPC-31, BMS-337039) cas no 129722-12-9
Aripiprazole is a psychotropic drug that is available as ABILIFY® (aripiprazole) Tablets, ABILIFY DISCMELT® (aripiprazole) Orally Disintegrating Tablets, ABILIFY® (aripiprazole) Oral Solution, and ABILIFY® (aripiprazole) Injection, a solution for intramuscular injection.
Aripiprazole is 7-[4-[4-(2,3-dichlorophenyl)-1- piperazinyl]butoxy]-3,4-dihydrocarbostyril. The empirical formula is C23H27Cl2N3O2 and its molecular weight is 448.38.
ABILIFY Tablets are available in 2 mg, 5 mg, 10 mg, 15 mg, 20 mg, and 30 mg strengths. Inactive ingredients include cornstarch, hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. Colorants include ferric oxide (yellow or red) and FD&C Blue No. 2 Aluminum Lake.
ABILIFY DISCMELT Orally Disintegrating Tablets are available in 10 mg and 15 mg strengths. Inactive ingredients include acesulfame potassium, aspartame, calcium silicate, croscarmellose sodium, crospovidone, crème de vanilla (natural and artificial flavors), magnesium stearate, microcrystalline cellulose, silicon dioxide, tartaric acid, and xylitol. Colorants include ferric oxide (yellow or red) and FD&C Blue No. 2 Aluminum Lake.
ABILIFY Oral Solution is a clear, colorless to light yellow solution available in a concentration of 1 mg/mL. The inactive ingredients for this solution include disodium edetate, fructose, glycerin, dl-lactic acid, methylparaben, propylene glycol, propylparaben, sodium hydroxide, sucrose, and purified water. The oral solution is flavored with natural orange cream and other natural flavors.
ABILIFY Injection is available in single-dose vials as a ready-to-use, 9.75 mg/1.3 Ml (7.5 mg/mL) clear, colorless, sterile, aqueous solution for intramuscular use only. Inactive ingredients for this solution include 150 mg/mL of sulfobutylether β-cyclodextrin (SBECD), tartaric acid, sodium hydroxide, and water for injection.
Wednesday, February 6, 2013
Otsuka Pharmaceutical Co. Ltd. announced today that the European Medicines Agency (EMA) has approved a label extension for aripiprazole for the treatment up to 12 weeks of moderate to severe manic episodes in Bipolar I Disorder in adolescents aged 13 and older.
Aripiprazole was studied in a 30-week placebo controlled trial involving 296 children and adolescents, who met DSM-IV criteria for Bipolar I Disorder with manic or mixed episodes with or without psychotic features and had a Y-MRS score ≥ 20 at baseline. Aripiprazole was superior to placebo in change from baseline at week 4 and at week 12 on the Y-MRS total score.
The recommended dose for aripiprazole in this indication is 10mg/day administered on a once-a-day schedule without regard to meals. Treatment should be initiated at 2mg (using aripiprazole oral solution 1mg/ml) for 2 days, titrated to 5mg for 2 additional days to reach the recommended daily dose of 10 mg.
The treatment duration should be the minimum necessary for symptom control and must not exceed 12 weeks. The frequency and type of undesirable effects in adolescents with Bipolar I Disorder were similar to those in adults except for somnolence, extrapyramidal disorder, akathisia, and fatigue, abdominal pain upper, heart rate increased, weight increased, increased appetite, muscle twitching, and dyskinesia. Younger patients are at increased risk of experiencing adverse events associated with aripiprazole. Therefore, aripiprazole is not recommended for use in patients below 13 years of age
Aripiprazole can be synthesized beginning with a dichloroaniline and bis(2-chloroethyl)amineU.S. Patent 5,006,528
Aripiprazole, 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro carbostyril or 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro-2 (1H)-quinolinone, is an atypical antipsychotic agent useful for the treatment of schizophrenia (U.S. Pat. No. 4,74,416 and U.S. Pat. No. 5,006,528). Schizophrenia is a common type of psychosis characterized by delusions, hallucinations and extensive withdrawal from others. Onset of schizophrenia typically occurs between the age of 16 and 25 and affects 1 in 100 individuals worldwide. It is more prevalent, than Alzheimer’s disease, multiple sclerosis, insulin-dependent diabetes and muscular dystrophy. Early diagnosis and treatment can lead to significantly improved recovery and outcome. Moreover, early therapeutic intervention can avert costly hospitalization.
Aripiprazole (Aripiprazole) is an atypical antipsychotic, on 15 November 2002 by the U.S. FDA clearance to market, its efficacy is through the dopamine D2 receptor and serotonin 5HT1A receptor partial agonist activity and serotonin 5HT2A receptor antagonism activity mediated common. With its unique mechanism of action and safety assessment, aripiprazole known as third-generation antipsychotic drugs.
[0003] Aripiprazole is a quinolinone derivative, developed by the Japanese company Otsuka Pharmaceutical, the chemical name
Is: 7 – {4 – [4 – (2,3 – dichlorophenyl)-1_ piperazinyl] butoxy} -3,4 – dihydro-quinolone, the following structural formula:
[0004]
[0005] For the preparation of aripiprazole, Japanese OtsukaPharmaceutical’s patent EP 0367141A2, and related patents US4234585, CN89108934 preparation methods described in 5. In addition, the patent CN1450056A, CN1562973A, CN1784385A, CN1680328A, CN1576273A, etc. describe some of these five Preparation
Method is very similar way. These preparation methods are direct or indirect use of 7 – hydroxy -3,4 – dihydro – quinolin-2 – one (HCS) that the key to higher prices of raw materials, and some methods involve harsh reaction conditions, poor selectivity, low yield, but also increases the cost of industrial production of the product.
[0006] Chinese patent CN1304373C preparation method is not described in the 7 – hydroxy-3 ,4 _ dihydro-2_ (1H) – quinoline
Quinolone intermediates for their preparation of the core reaction is as follows:
[0007]
[0008] This reaction is Friedel-Crafts alkylation reaction, there is a harsh reaction conditions, the yield is low, the reaction selectivity is poor, the shortcomings of high emissions, is not conducive to industrial mass production. SUMMARY OF THE INVENTION
[0009] In order to solve the above problems, the present invention provides a simple, high selectivity, high yield, low cost, environmentally friendly, easy to prepare industrialization aripiprazole and intermediates thereof.
[0010] The technical solution of the present invention, the present invention provides in one aspect a process for preparingaripiprazole novel intermediates.
[0011] The present invention, on the other hand provides a method for the preparation of intermediates.
[0012] The present invention provides the use of the other intermediates for preparing aripiprazole two new preparation methods.
[0013] Specifically, the present invention relates to novel intermediates, compounds of formula ⑴:
[0014]
[0015] wherein, R is selected from methyl, ethyl, propyl, isopropyl, butyl, t-butyl, benzyl and other common alkyl groups in any one, and preferably is ethyl.
[0016] Compound of formula ⑴: 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate, is the following prepared by the procedure:
[0017] Step one, the acylation reaction: with 4 – methyl – 3 – nitro-phenol (VIII) and acetic anhydride as the raw material, DMAP as catalyst, to give 4 – methyl – 3 – nitrophenyl acetate ( VII).
[0018] wherein 4 – methyl – 3 – nitro-phenol (VIII), acetic anhydride, DMAP molar ratio is preferably 1: 1.0 to 1.4: 0.05, at room temperature, the reaction time is preferably 0.5 to 3 hours.
[0019] Step two, the bromination reaction: The resulting product, 4 to Step one – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide and benzoyl peroxide as a raw material , carbon tetrachloride solvent reflux, to give 4 – bromomethyl-3 – nitrophenyl acetate (VI).
[0020] wherein 4 – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide, benzoyl peroxide molar ratio is preferably 1: 1 to 1.2: 0.05, reaction time is preferably 4-18 hours.
[0021] Step three, instead of the reaction: in an appropriate solvent, adding an alkaline agent and diethyl malonate was stirred in an ice bath, was added dropwise step two the resulting product, 4 – bromomethyl-3 – nitrophenyl yl acetate (VI) solution after completion of the addition reaction of 1 to 3 hours to obtain a brown liquid product, 2 – (4_ acetoxy-2 – nitrobenzyl) malonate (V).
[0022], wherein the alkali agent is a common organic or inorganic base selected from sodium methoxide, sodium ethoxide, sodium hydride, sodium tert-butoxide or potassium tert-butoxide, preferably sodium tert-butoxide; the solvent is selected from tetrahydrofuran, methanol, ethanol, butanol, tert-butanol, toluene or N, N-dimethylformamide; 4 – bromomethyl-3 – nitrophenyl acetate (VI), alkaline agent and lipid diethyl molar ratio is preferably 1: 1.0 to 1.8: 1.0 to 1.4.
[0023] Step 4 Hydrolysis decarboxylation: the product obtained in Step Three 2 – (4_ acetoxy-2 – nitro-benzyl)-malonic acid diethyl ester (V) was added concentrated hydrochloric acid and a suitable solvent, heating and stirring reflux, to give a yellow solid product 3 – (4_ hydroxy-2 – nitrophenyl) propionic acid (IV).
[0024] wherein the solvent is selected from water, methanol, ethanol or acetic acid, water soluble solvent, was heated with stirring under reflux time is preferably 3 to 18 hours. [0025] Step five, the esterification reaction: the product obtained in step 4, 3 – (4 – hydroxy-2 – nitrophenyl) propionic acid (IV) was added to an appropriate solvent, the mixture was stirred in an ice bath, was added dropwise thionyl sulfone, after completion of the addition reaction of 1 to 3 hours, to give a pale brown liquid product 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III).
[0026] wherein the solvent is selected from anhydrous methanol, ethanol, propanol, isopropanol, butanol, t-butanol, benzyl alcohol, alcohol and other common solvents.
[0027] Step VI substitution reaction: 1,4 – dibromobutane was added to an appropriate solvent and an alkaline reagent, heated to 50 ~ 100 ° C, the product obtained was added dropwise Step Five 3 – (4_ hydroxy – nitrophenyl) propionate (III) solution, after the addition was complete the reaction was kept 2 to 4 hours to obtain a brown liquid product 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II).
[0028] wherein the solvent is selected from methanol, 95% ethanol, ethanol, acetonitrile and N, N-dimethylformamide, and the like; said alkaline agent is a common organic or inorganic weak base, such as triethylamine, pyridine, potassium carbonate, sodium carbonate, etc..
[0029] Step 7 condensation reaction: the product obtained in Step Six 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II) adding a suitable solvent, (2,3 – dichlorophenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, to obtain
[0030] Among them, 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate (I), (2, 3 – dichloro-phenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, the four molar ratio is preferably 1: 0.9 to 1.0: 2.0 to 2.2: 0.05 to 0.5. The solvent is selected from methanol, ethanol and N, N-dimethylformamide, acetonitrile and the like. Step six of the alkaline reagent and alkaline reagent used in the same, said catalyst is a common low-iodine salts, such as sodium iodide, potassium iodide.
[0031] The present invention provides two other hand, the use of a compound of formula ⑴ preparing aripiprazole new method.
[0032] Method one: ⑴ intermediate compound of formula in an appropriate solvent in the acid or salt or a base in the presence of a reducing agent under the action of restoring ring closure reaction to obtain aripiprazole.
[0033] Method one reductive cyclization of the reducing agent used is iron, zinc, sodium sulfide, stannous chloride, and preferably iron; reaction solvent is selected from water, methanol, ethanol, ethyl acetate or in one or more of the mixed solvent; said acid is a common organic or inorganic acid, preferably acetic acid or hydrochloric acid; said salt is a common inorganic or organic salts selected from chloride, ferrous chloride, , ammonium sulfate, calcium chloride, zinc chloride, sodium chloride, sodium bromide or sodium acetate and the like; common said base is an inorganic base selected from sodium hydroxide, potassium hydroxide or sodium bicarbonate; the reduction ring-closing reaction temperature range of 30 ~ 140 ° C, preferably about 80 ° C; reaction time ranges from about 0.5 to 8 hours, preferably 2 hours.
[0034] Method two: ⑴ intermediate compound of formula in an appropriate solvent in the first catalyst, the reduction reaction, and then carried out in a suitable solvent can be prepared by cyclization of aripiprazole.
[0035] The reduction reaction of the second approach, the reducing agent is hydrogen or a carboxylic acid; the catalyst is selected from molybdenum, molybdenum dioxide or Raney nickel, preferably Raney nickel; the solvent is selected from methanol, ethanol, ethyl acetate or acetic acid, preferably ethanol; said ring-closing reaction of the solvent is selected from N, N-dimethylformamide, trichlorobenzene or xylene; reaction temperature range of 50 ~ 180 ° C, preferably about 70 ~ 150 ° C; reaction time the range of about 1 to 8 hours.
[0036] In summary, the present invention is described for preparing aripiprazole method in 4– methyl – 3 – nitro-phenol (VIII) as a starting material, by acetylation protected hydroxy, radical instead of 4 – bromomethyl-3 – nitrophenyl acetate (VI), the diethyl malonate and a nucleophilic substitution reaction to obtain 2 – (4_ acetoxy-2 – nitrobenzyl ) malonic acid diethyl ester (V), which is decarboxylated by hydrolysis, esterification, to give 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III), the reaction product with dibromobutane an ether compounds, and with (2,3 – dichlorophenyl)-piperazine hydrochloride 1_ condensation, to give 3 – (4 – (4 – (4 – (2,3 – dichlorophenyl) piperazine -1 – yl) butoxy) -2 – nitrophenyl) propionate (I), and then by reductive cyclization step, or first reduced and then ring-closing reaction of aripiprazole. The synthetic route of the present invention is as follows: [0037]
According to Example 1 of Japanese Unexamined Patent Publication No. 191256/1990, anhydrous aripiprazole crystals are manufactured for example by reacting 7-(4-bromobutoxy)-3,4-dihydrocarbostyril with 1-(2,3-dichlorophenylpiperadine and recrystallizing the resulting raw anhydrousaripiprazole with ethanol. Also, according to the Proceedings of the 4th Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996), anhydrousaripiprazole crystals are manufactured by heating aripiprazole hydrate at 80° C. However, the anhydrous aripiprazole crystals obtained by the aforementioned methods have the disadvantage of being significantly hygroscopic.
The hygroscopicity of these crystals makes them difficult to handle since costly and burdensome measures must be taken in order ensure they are not exposed to moisture during process and formulation. Exposed to moisture, the anhydrous form can take on water and convert to a hydrous form. This presents several disadvantages. First, the hydrous forms of aripiprazole have the disadvantage of being less bioavailable and less dissoluble than the anhydrous forms ofaripiprazole. Second, the variation in the amount of hydrous versus anhydrousaripiprazole drug substance from batch to batch could fail to meet specifications set by drug regulatory agencies. Third, the milling may cause the drug substance, Conventional Anhydrous Aripiprazole, to adhere so manufacturing equipment which may further result in processing delay, increased operator involvement, increased cost, increased maintenance, and lower production yield. Fourth, in addition to problems caused by introduction of moisture during the processing of these hygroscopic crystals, the potential for absorbance of moisture during storage and handling would adversely affect the dissolubility of aripiprazole drug substance. Thus shelf-life of the product could be significantly decreased and/or packaging costs could be significantly increased. It would be highly desirable to discover a form of aripiprazole that possessed low hygroscopicity thereby facilitating pharmaceutical processing and formulation operations required for producing dosage units of an aripiprazole medicinal product having improved shelf-life, suitable dissolubility and suitable bioavailability.
Also, Proceedings of the 4 the Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996) state that, anhydrous aripiprazole crystals exist as type-I crystals and type-II crystals; the type-I crystals of anhydrous aripiprazolecan be prepared by recrystallizing from an ethanol solution of aripiprazole, or by heating aripiprazole hydrate at 80° C.; and the type-II crystals of anhydrousaripiprazole can be prepared by heating the type-I crystals of anhydrousaripiprazole at 130 to 140° C. for 15 hours.
By the aforementioned methods, anhydrous aripiprazole type-II crystals having high purity can not be easily prepared in an industrial scale with good repeatability.
Chemical Synthesis of Aripiprazole (active ingredient for Abilify)
Experimental Procedures for the preparation of Aripiprazole (Abilify, aripiprazole)
US 5,006,528 discloses process for the preparation of Aripiprazole in two steps The first step comprises synthesis of 7 -. (4-bromobutoxy) -3,4-dihydrocarbostyril (7-BBQ) by alkylating the hydroxy group of 7-hydroxy-3, 4 -dihydrocarbostyril (7-HQ) with 1 ,4-dibromobutane using potassium carbonate in water at reflux temperature for 3 hours to obtain 7-BBQ in 68% yield The resulting 7-BBQ is further reacted with 1 -. (2,3 – dichlorophenyl)-piperazine to obtain Aripiprazole.
7-Hydroxy-3 ,4-dihydro-2 (1H)-quinolinone (aka 7-Hydroxy-3 ,4-dihydrocarbostyril, 60gm) and potassium carbonate (76.3 gm) were taken in acetonitrile (1200ml) at room temperature. To this tetra butyl ammonium iodide (13.7 gm) and 1 ,4-dibromobutane (238.5gm) were added and heated at 40 – 45 ° C for 24 hours Reaction mass was cooled upto room temperature and was filtered off The resulting filtrate was distilled off.. under vacuum. The resultant mass was cooled to 25-30 ° C and cyclohexane (300 ml) was added under stirring. The resulting solid was filtered off and was dried. The resulting solid was taken in water and was stirred for few minutes. The . solid was filtered and dried under vacuum at 55-60 ° C for 20 hours to obtain title compound mp 110.5-111 ° C; 1H NMR (DMSO-d6) ä 1.81 (2H, m,-CH2-), 1.95 (2H , m,-CH2-), 2.41 (2H, t, J) 7 Hz,-CH2CO-), 2.78 (2H, t, J) 7 Hz,-CH2-C-CO-), 3.60 (2H, t, J) 6 Hz,-CH2Br), 3.93 (2H, t, J) 6 Hz, O-CH2-), 6.43 (1H, d, J) 2.5 Hz), 6.49 (1H, dd, J) 2.5, 8 Hz ), 7.04 (1H, d, J) 8 Hz), 9.98 (1H, s, NHCO). Anal. (C13H16NO2Br) C, H, N.
7 – (4-Bromobutoxy)-l ,2,3,4-tetrahydroquinolin-2-one (50 gm) was taken in acetonitrile (500 ml) at 25-30 ° C. To this potassium carbonate (67.2 gm) and l – (2,3 – dichlorophenyl). piperazine hydrochloride (44.9gm) were added under stirring The reaction mixture was refluxed at 80-85 ° C for 8 hours The reaction mass was cooled to room temperature, filtered and the resulting solid was washed. with acetonitrile. To the resulting solid, water was added and was stirred. The solid was filtered off, washed with water and dried under vacuum at 75-80 ° C for 15 hrs. The resulting crude aripiprazole was crystallized from isopropyl alcohol and water to . obtain title compound Yield: 75-80%; Dimer Impurity: <0.1% 1H NMR:. DMSO-d6 d 9.96 [1H, s, NH]; 7.29 [2H, m, Ar]; 7.13 [1H, q, Ar ]; 7.04 [1H, d, Ar]; 6.49 [1H, dd, Ar]; 6.45 [1H, d, Ar]; 3.92 [2H, t,-CH2-O-]; 2.97 [4H, bb, 2 ( -CH2-)]; 2.78 [2H, t,-CH2-N2-)]; 2.39 [4H, m, 2 (-CH2-)]; 1.73 [2H, m, – CH2-]; 1.58 [2H, m .,-CH2-] IR: cm-1 3193; 2939; 2804; 1680; 1627; 1579; 1520; 1449; 1375; 1270; 1245; 1192; 1169; 1045; 965; 649; 869; 780; 712; 588 .
Preparation of aripiprazole anhydrous Type I using isopropyl alcohol and water
Crude aripiprazole (30 g) was taken in isopropyl alcohol (600 ml) and was heated to 80-85 ° C. Water (90 ml) was added at the same temperature. Activated carbon was added and the mixture was stirred for 30 minutes at the same temperature. The resulting hot solution was filtered and the bed was washed with hot isopropyl alcohol. The resulting filtrate was cooled to 25-30 ° C for 4 hours. The resulting solid was filtered, washed with isopropyl alcohol and dried under suction for 1 hour. The resulting wet solid was dried in preheated oven maintained at 100-105 ° C for 6 hours to obtain title compound.
Yield: 87-89% HPLC Purity: 99.89
Anhydrous crystal D: Below detectable limit (BDL) at limit of detection 1%.
Hydrate A: Below detectable limit (BDL) at limit of detection 1%.
Particle Size Distribution: d 10 = 15.83 m, d 50 = 60.12 m, d 90 = 144.99 m
Preparation of aripiprazole anhydrous Type I using ethanol and water
Crude aripiprazole (15 g) was taken in ethanol (300 ml) and water (45 ml) and was heated to 80-85 ° C for 1-2 hours. The resulting mixture was cooled to 25-30 ° C within 4 hours and . stirred for 3 hours The resulting solid was filtered and dried under suction for 1 hour The resulting wet solid was dried in preheated oven maintained at 100-105 ° C for 3 hours to obtain title compound Yield:.. 90% HPLC Purity: 99.9 %
Anhydrous crystal D: Below detectable limit (BDL) at limit of detection 1%.
Hydrate A: Below detectable limit (BDL) at limit of detection 1%.
Particle Size Distribution: d 10 = 22.01 m, d 50 = 105.10 m, d 90 = 232.97 m
For the Process of references Aripiprazole (Abilify, Japanese: Oh, Bldg re phi, Ann reピplastic AKZO have suitable; Chinese: Ann-law who, aripiprazole)
Yasuo Oshiro, Seiji Sato, Nobuyuki Kurahashi, Tatsuyoshi Tanaka, Tetsuro Kikuchi, Katsura Tottori, Yasufumi Uwahodo, and Takao Nishi; Novel Antipsychotic Agents with Dopamine autoreceptor Agonist Properties: Synthesis and Pharmacology of 7 – [4 – (4-Phenyl-1- piperazinyl) butoxy] – 3,4-dihydro-2 (1H)-quinolinone Derivatives ; J. Med Chem. 1998, 41, 658-667.
BANDO, Takuji, YANO, Katsuhiko, FUKANA, Makoto, AOKI, Satoshi; Method for producing fine particles of aripiprazole anhydride crystals b; OTSUKA PHARMACEUTICAL CO, LTD, WO 2013002420 A1..
Yuanqiu Hui, Chen Hongwen, Qian Wen, firewood rain column, Xu Dan, Yang Zhimin, Tian Zhoushan; method for preparing high purity of aripiprazole; NJCTT Pharmaceutical Co., Ltd.; application number: 201210292382.0; Publication Number: CN102863377A; Publication date: 2013.01.09 After (The invention relates to the field of medicine and chemical industry, in particular to a method for preparing high purity of aripiprazole would join aripiprazole A solvent is heated, filtered, and the filtrate was added to a solvent B, low temperature mixing, filtration, the filter cake is suspended in water, adjusted to alkaline pH of the aqueous solution, filtration, high temperature vacuum dried to obtain a high-purity refined product Aripiprazole This method is simple, high purity, suitable for the industrial the large-scale application)
ZHENG Siji, LIU Xiaoyi, FU Linyong, TAN Bo, ZHOU Min:.. ARIPIPRAZOLE MEDICAMENT FORMULATION AND PREPARATION METHOD THEREFOR / FORMULATION DE MÉDICAMENT ARIPIPRAZOLE ET SON PROCÉDÉ DE PRÉPARATION / a aripiprazole pharmaceutical formulation and preparation method SHANGHAI ZHONGXI. PHARMACEUTICAL January 2013: WO 2013/000391
Zheng Si Ji, Liu Xiaoyi, Fulin Yong, Tan Bo, Zhou Min: A aripiprazole pharmaceutical formulation and preparation method; Shanghai Pharmaceutical Co., Ltd. and Western; Publication date: 2013.01.02: Application Number: CN 201210235157.3; Publication Number: CN102846543A (the invention provides a method for preparing aripiprazole pharmaceutical formulation, comprising the steps of: an acidic solution containing aripiprazole is dissolved in the acidulant, to obtain an acidic solution containing the drug; Thereafter, the resulting drug-containing acidic solution alkalizing agents and materials prepared by wet granulation or suspension to give aripiprazole pharmaceutical formulation; said excipients include antioxidants)
Zheng Si Ji; Tan wave; Fulin Yong; Liu Xiaoyi; Yuanshao Qing; Cao Zhihui; aripiprazole Ⅰ type microcrystalline, aripiprazole solid preparation and preparation methods; application number: 201110180032.0; Publication Number: CN102850268A; Publication Date: 2013.01.02
Cai Fu Bo, Qin Xinrong, Du Xiaochun, Li Ling; kind of aripiprazole improved method of synthesis; Chengdu Nakasone Pharmaceutical Group Co., Ltd.; Application Number: 200910058148.X; Publication Number: CN101781246A; Publication date: 2010.07.21 (the invention provides a method of synthesis of aripiprazole improved method according to the modified method of the present invention, aripiprazole into the etherification reaction and condensation reaction of two-step synthesis, by an etherification reaction in the quinolone compound and at least 6-fold molar equivalents of 1,4 – dihalo-butane reacted with a non-polar solvent ether aripiprazole precipitate, and recovering 1,4 – dihalo-butane recycling; azeotropic condensation reaction of a ketone to be / water mixture as solvent, aripiprazole etherified with a piperazine compound or a salt thereof in the presence of a base under reflux and alkaline metal iodide compound conditions, the amount of water added to the end of the reaction, cooling crystallization, filtration, and dried to give aripiprazole. improved high yield synthesis of high purity, step simple, low cost, suitable for industrial production.)
GUPTA, Vijay Shankar, KUMAR, Pramod, VIR, Dharam; Process for producing aripiprazole in anhydrous type i crystals; JUBILANT LIFE SCIENCES LIMITED; WO 2012131451 A1
SRIVASTAVA JAYANT GUPTA Vijay Shankar;. Improved process for the preparation of 7 (4-bromobutoxy) 3,4-dihydrocarbostyril, a precursor of aripiprazole; wo2011030213 A1
No Generic Abilify in the US until April 2015
On May 7, 2012, The US Court of Appeals for the Federal Circuit ruled in favor of Otsuka Pharmaceutical Co., Ltd. In its patent litigation against several companies including Israel-based Teva and Weston, Ontario-based Apotex seeking FDA approval to market generic copies of Abilify ®.. The Federal Circuit Affirmed a Decision of the U.S. District Court for the District of New Jersey Holding that the asserted claims ofU.S. Patent No. 5,006,528 Covering aripiprazole, the active Ingredient in Abilify ®, are Valid, THUS Maintaining Patent and Regulatory Protection for Abilify ® in the U.S. until at least April 20, 2015 . The Case is Otsuka Pharma Co.. V. sand Inc.., 2011-1126 and 2011-1127, US Court of Appeals for the Federal Circuit (Washington). The lower court case is Otsuka Pharmaceutical Co. v. Sandoz Inc., 07cv1000, US District Court for the District of New Jersey (Trenton).
Chemical Name for Aripiprazole (Abilify for active Ingredient): 7 – {4 – [4 – (2,3-Dichlorophenyl) piperazin-1-yl] butoxy} 3 ,4-dihydroquinolin-2 (1H)-One
CAS Number 129722 -12-9
aripiprazole chemical name 7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro-2 ( 1H) – quinolinone
Aripiprazole (, Aripiprazole, Abilify) is an atypical antipsychotic medication for the quinoline derivatives, aripiprazole is a dopamine system stabilizer first, positive and schizophrenia negative symptoms have a significant effect. For the treatment of schizophrenia, the development of Otsuka Pharmaceutical Co., Ltd., in November 15, 2002 by the U.S. Food and Drug Administration (FDA) approval in the U.S., domestic aripiprazole has (Booz clear (brisking, manufacturers : Chengdu Nakasone Pharmaceutical), Austrian (Manufacturer: Shanghai Pharmaceutical Co., Ltd. and Western)) have been approved by the listing in China. On sale in the United States where the law by Bristol-Myers Squibb is responsible. An law where the main patent protection in the United States, and more than three-quarters of its sales from the U.S., patent will expire in April 2015.
Aripiprazole synthetic route
7 – hydroxy-3 ,4. Dihydro -2 (1H) – quinolinone as a starting material, 1,4. Dibromobutane ether to give 7 – (4 – Bromo-butoxy) -3,4 – dihydro – 2 (1H) quinolinone, and then with 1 – (2,3 – dichlorophenyl) piperazine acid condensation aripiprazole (7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro -2 (1H) – quinolinone)
Aripiprazole preparation method
7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone
A reaction flask was added 7 – hydroxy – 3,4 – dihydro -2 (1H) – quinolone 32.6 g (0.2mol), 1,4 – dibromo butane 129.5g (0.6mol), 11.2% KOH solution 250ml (0.5mol) and DMF975ml, was heated to 60 º C for 2h diluted with 1L water, the aqueous layer with ethyl acetate. acetate (300ml × 2) and the combined organic layers were washed with water, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to recover the solvent, the residue was recrystallized from isopropanol, to give 7 – (4 – Bromo-butoxy) – 3,4 – dihydro -2 (1H) – quinolone 38.7g, yield 68%, mp108 ~ 110 º C.
Synthesis of aripiprazole
in the reaction flask was added 7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone, 29.8g (0.1mol), KI25g (0.15mol) 95% Ethanol 596ml, stirred and heated to 60 º C, was added N-2 30min after 3 – dichlorophenyl piperazine 23.1g (0.1mol) and triethylamine 20ml (0.15mol), stirred for 8h at 60 º C the mixture is filtered. crystallization filtrate was cooled, filtered and the filter cake was recrystallized twice from ethanol and dried to obtain aripiprazole 25.6g, yield 57%, mp138.9 ~ 139.6 º C.
emea
Aripiprazole is a new antipsychotic belonging to the class of atypical antipsychotic drugs. It has been proposed that aripiprazole antipsychotic action could be mediated through a combination of partial agonist action at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors
Aripiprazole is a quinolinone derivative with the chemical name 7-[4-[4-(2,3-dichlorophenyl)-1- piperazinyl]butoxy]- 3,4-dihydro-2(1H)-quinolinone.
The active substance does not contain any chiral centres and does not exhibit any optical isomerism.
Aripiprazole active substance is a white crystalline powder and is practically insoluble in water and its solubility is pH dependent. Therefore, a particle size effect on dissolution of the tablets can be expected. In order to ensure batch-to-batch consistency of the product, and to ensure adequate bioavailability, aripiprazole is subject to milling.
Aripiprazole can exist in several crystalline forms, Form I was chosen for the development and commercialisation.. Aripiprazole is synthesised by a 2-step process. In the first step, 7-hydroxy-3,4-dihydro-2(H)- quinolinone is transformed into an intermediate, which is reacted with 1-(2,3-dichlorophenyl) piperazine hydrochloride to obtain aripiprazole. The process, specifications and control methods are adequately described in the restricted section of the EDMF.
Schizophrenia is a major psychotic disorder. Its essential features consist of a mixture of characteristic
signs and symptoms that have been present for a significant length of time during a 1-month period (or
for a shorter time if successfully treated), with some signs of the disorder persisting for at least 6
months.
The characteristic symptoms of schizophrenia have often been conceptualized as falling into two
broad categories positive and negative (or deficit) symptoms with a third category, disorganized,
recently added because statistical analyses have revealed that it is a dimension independent of the
positive symptom category, where it was previously included. The positive symptoms include
delusions and hallucinations. Disorganized symptoms include disorganized speech (thought disorder)
and disorganized behaviour and poor attention. Negative symptoms include restricted range and
intensity of emotional expression (affective flattening), reduced thought and speech productivity
(alogia), anhedonia, and decreased initiation of goal-directed behaviour (avolition).
The onset of schizophrenia typically occurs during adolescence or early adulthood. It affects men and
women with equal frequency. The peak age at onset for males, however, is the early 20s, and for
women it is the late 20s and early 30s. The majority of patients alternate between acute psychotic
episodes and stable phases with full or partial remission. Inter-episode residual symptoms are
common. This often-chronic illness can be characterized by three phases that merge into one another
without absolute, clear boundaries between them. These phases, which form the structure for
integrating treatment approaches, are described below:
Acute phase. During this florid psychotic phase, patients exhibit severe psychotic symptoms, such as
delusions and/or hallucinations and severely disorganized thinking, and are usually unable to care for
themselves appropriately. Negative symptoms often become more severe as well.
Stabilization phase. During this phase, acute psychotic symptoms decrease in severity. This phase may
last for 6 or more months after the onset of an acute episode.
Stable phase. Symptoms are relatively stable and, if present at all, are almost always less severe than
in the acute phase. Patients can be asymptomatic; others may manifest non-psychotic symptoms, such
as tension, anxiety, depression, or insomnia. When negative (deficit) symptoms and/or positive
symptoms, such as delusions, hallucinations, or thought disorder, persist, they are often present in
attenuated, non-psychotic forms (e.g., circumstantiality rather than looseness, illusions rather than hallucinations, overvalued ideas rather than delusions).
There are a number of antipsychotics in use but none is ideal in particular because their safety profile is complex. The in vitro affinity profile of aripiprazole for dopamine and serotonin receptors is similar to the one of so-called atypical antipsychotics. It is postulated that aripiprazole’s mechanism of action is novel as it involves a combination of partial agonist action (agonist/antagonism) at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors.
Aripiprazole is a new antipsychotic belonging to the class of atypical antipsychotic drugs. It has been
proposed that aripiprazole antipsychotic action could be mediated through a combination of partial
agonist at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors.
The non-clinical characterization of aripiprazole as a dopamine D2 receptor partial agonist at pituitary
lactotrophs predicts a low potential to induce hyperprolactinemia in humans but it may be dose related.
Studies on the sedative liability of aripiprazole suggest a reduced sedative potential compared to
typical antipsychotics. The safety pharmacology profile of aripiprazole r

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











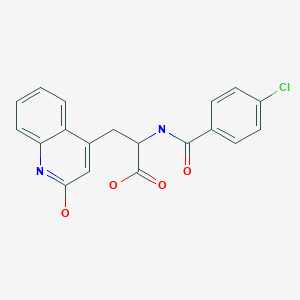






 3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.