
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Mocetinostat
SAN DIEGO, Aug. 11, 2014 /PRNewswire/ — Mirati Therapeutics, Inc. (NASDAQ: MRTX) today announced that the U.S. FDA has granted Orphan Drug Designation to mocetinostat, a spectrum selective HDAC inhibitor, for diffuse large B-cell lymphoma (DLBCL). In June, mocetinostat was granted Orphan Drug Designation as a treatment for myelodysplastic syndrome (MDS). Orphan drug designation is also being sought for bladder cancer patients with specific genetic alterations.
| Identifiers | |
|---|---|
| CAS number | 726169-73-9 |
| PubChem | 9865515 |
| ChemSpider | 8041206 |
| ChEMBL | CHEMBL272980 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C23H20N6O |
| Molar mass | 396.44 g mol−1 |
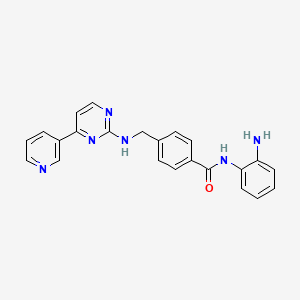
Mocetinostat (MGCD0103) is a benzamide histone deacetylase inhibitor undergoing clinical trials for treatment of various cancers including follicular lymphoma, Hodgkin’s lymphoma and acute myelogenous leukemia.[1][2][3]
One clinical trial (for refractory follicular lymphoma) was temporarily put on hold due to cardiac problems but resumed recruiting in 2009.[4]
In 2010 favourable results were announced from the phase II trial for Hodgkin’s lymphoma.[5]
MGCD0103 has also been used as a research reagent where blockage of members of the HDAC-family of histone deacetylases is required.[6]
Mechanism of action
It works by inhibiting mainly histone deacetylase 1 (HDAC1), but also HDAC2, HDAC3, and HDAC11.[7]
About Mocetinostat
Mocetinostat is an orally-bioavailable, spectrum-selective HDAC inhibitor. Mocetinostat is enrolling patients in a Phase 2 dose confirmation study in combination with Vidaza as treatment for intermediate and high-risk MDS. Mirati also plans to initiate Phase 2 studies of mocetinostat as a single agent in patients with mutations in histone acetyl transferases in bladder cancer and DLBCL. Initial data from the Phase 2 studies is expected by the end of 2014. In addition to the ongoing Phase 2 clinical trials, mocetinostat has completed 13 clinical trials in more than 400 patients with a variety of hematologic malignancies and solid tumors.
About Mirati Therapeutics
Mirati Therapeutics is a targeted oncology company developing an advanced pipeline of breakthrough medicines for precisely defined patient populations. Mirati’s approach combines the three most important factors in oncology drug development – drug candidates with complementary and compelling targets, creative and agile clinical development, and a highly accomplished precision medicine leadership team. The Mirati team is using a proven blueprint for developing targeted oncology medicines to advance and maximize the value of its pipeline of drug candidates, including MGCD265 and MGCD516, which are orally bioavailable, multi-targeted kinase inhibitors with distinct target profiles, and mocetinostat, an orally bioavailable, spectrum-selective histone deacetylase inhibitor. More information is available at www.mirati.com.
In eukaryotic cells, nuclear DNA associates with histones to form a compact complex called chromatin. The histones constitute a family of basic proteins which are generally highly conserved across eukaryotic species. The core histones, termed H2A, H2B, H3, and H4, associate to form a protein core. DNA winds around this protein core, with the basic amino acids of the histones interacting with the negatively charged phosphate groups of the DNA. Approximately 146 base pairs of DNA wrap around a histone core to make up a nucleosome particle, the repeating structural motif of chromatin.
Csordas, Biochem. J., 286: 23-38 (1990) teaches that histones are subject to posttranslational acetylation of the α,ε-amino groups of N-terminal lysine residues, a reaction that is catalyzed by histone acetyl transferase (HAT1). Acetylation neutralizes the positive charge of the lysine side chain, and is thought to impact chromatin structure. Indeed, Taunton et al., Science, 272: 408-411 (1996), teaches that access of transcription factors to chromatin templates is enhanced by histone hyperacetylation. Taunton et al. further teaches that an enrichment in underacetylated histone H4 has been found in transcriptionally silent regions of the genome.
Histone acetylation is a reversible modification, with deacetylation being catalyzed by a family of enzymes termed histone deacetylases (HDACs). Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999), teaches that HDACs are divided into two classes, the first represented by yeast Rpd3-like proteins, and the second represented by yeast Hda1-like proteins. Grozinger et al. also teaches that the human HDAC1, HDAC2, and HDAC3 proteins are members of the first class of HDACs, and discloses new proteins, named HDAC4, HDAC5, and HDAC6, which are members of the second class of HDACs. Kao et al., Genes & Dev., 14: 55-66 (2000), discloses HDAC7, a new member of the second class of HDACs. More recently, Hu et al. J. Bio. Chem. 275:15254-13264 (2000) and Van den Wyngaert, FEBS, 478: 77-83 (2000) disclose HDAC8, a new member of the first class of HDACs.
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998), discloses that HDAC activity is inhibited by trichostatin A (TSA), a natural product isolated from Streptomyces hygroscopicus, and by a synthetic compound, suberoylanilide hydroxamic acid (SAHA). Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988), teaches that TSA causes arrest of rat fibroblasts at the G1 and G2 phases of the cell cycle, implicating HDAC in cell cycle regulation. Indeed, Finnin et al., Nature, 401: 188-193 (1999), teaches that TSA and SAHA inhibit cell growth, induce terminal differentiation, and prevent the formation of tumors in mice. Suzuki et al., U.S. Pat. No. 6,174,905, EP 0847992, JP 258863/96, and Japanese Application No. 10138957, disclose benzamide derivatives that induce cell differentiation and inhibit HDAC. Delorme et al., WO 01/38322 and PCT/IB01/00683, disclose additional compounds that serve as HDAC inhibitors.
The molecular cloning of gene sequences encoding proteins with HDAC activity has established the existence of a set of discrete HDAC enzyme isoforms. Some isoforms have been shown to possess specific functions, for example, it has been shown that HDAC-6 is involved in modulation of microtubule activity. However, the role of the other individual HDAC enzymes has remained unclear.
These findings suggest that inhibition of HDAC activity represents a novel approach for intervening in cell cycle regulation and that HDAC inhibitors have great therapeutic potential in the treatment of cell proliferative diseases or conditions. To date, few inhibitors of histone deacetylase are known in the art.
………………..
http://www.google.com/patents/WO2011112623A1?cl=en

Mocetinostat (MGCD-0103)
N-(2-aminophenyl)-4-[[(4-pyridin-3-ylpyrimidin-2-yl)amino]methyl^^
…………………………
http://www.google.co.in/patents/US6897220
Example 426 Synthesis of N-(2-Amino-phenyl)-4-[(4-pyridin-3-pyrimidin-2-ylamino)-methyl]-benzamide

Step 1: Synthesis of 4-Guanidinomethyl-benzoic acid methyl ester Intermediate 1
The mixture of 4-Aminomethyl-benzoic acid methyl ester HCl (15.7 g, 77.8 mmol) in DMF (85.6 mL) and DIPEA (29.5 mL, 171.2 mmol) was stirred at rt for 10 min. Pyrazole-1-carboxamidine HCl (12.55 g, 85.6 mmol) was added to the reaction mixture and then stirred at rt for 4 h to give clear solution. The reaction mixture was evaporated to dryness under vacuum. Saturated NaHCO3 solution (35 mL) was added to give nice suspension. The suspension was filtered and the filter cake was washed with cold water. The mother liquid was evaporated to dryness and then filtered. The two solids were combined and re-suspended over distilled H2O (50 ml). The filter cake was then washed with minimum quantities of cold H2O and ether to give 12.32 g white crystalline solid intermediate 1 (77% yield, M+1: 208 on MS).
Step 2: Synthesis of 3-Dimethylamino-1-pyridin-3-yl-propenone Intermediate 2
3-Acetyl-pyridine (30.0 g, 247.6 mmol) and DMF dimethyl acetal (65.8 mL, 495.2 mmol) were mixed together and then heated to reflux for 4 h. The reaction mixture was evaporated to dryness and then 50 mL diethyl ether was added to give brown suspension. The suspension was filtered to give 36.97 g orange color crystalline product (85% yield, M+1: 177 on MS).
Step 3: Synthesis of 4-[(4Pyridin-3-pyrimidin-2-ylamino)-methyl]benzoic acid methyl ester Intermediate 3
Intermediate 1 (0.394 g, 1.9 mmol) and intermediate 2 (0.402 g, 2.3 mmol) and molecular sieves (0.2 g, 4A, powder, >5 micron) were mixed with isopropyl alcohol (3.8 mL). The reaction mixture was heated to reflux for 5 h. MeOH (50 mL) was added and then heated to reflux. The cloudy solution was filtrated over a pad of celite. The mother liquid was evaporated to dryness and the residue was triturated with 3 mL EtOAc. The suspension was filtrated to give 0.317 g white crystalline solid Intermediate 3 (52%, M+1: 321 on MS).
Step 4: Synthesis of N-(2-Amino-phenyl)-4-[(4-pyrymidin-2-ylamino)-methyl]-benzamide
Intermediate 3 (3.68 g, 11.5 mmol) was mixed with THF (23 mL), MeOH (23 mL) and H2O (11.5 mL) at rt. LiOH (1.06 g, 25.3 mmol) was added to reaction mixture. The resulting reaction mixture was warmed up to 40° C. overnight. HCl solution (12.8 mL, 2N) was added to adjust pH=3 when the mixture was cooled down to rt. The mixture was evaporated to dryness and then the solid was washed with minimum quantity of H2O upon filtration. The filter cake was dried over freeze dryer to give 3.44 g acid of the title compound (95%, M+1: 307 on MS).
Acid (3.39 g, 11.1 mmol) of the title compound, BOP (5.679 g, 12.84 mmol) and o-Ph(NH2)2 (2.314 g, 21.4 mmol) were dissolved in the mixture of DMF (107 mL) and Et3N (2.98 mL, 21.4 mmol). The reaction mixture was stirred at rt for 5 h and then evaporated to dryness. The residue was purified by flash column (pure EtOAc to 5% MeOH/EtOAc) and then interested fractions were concentrated. The final product was triturated with EtOAc to give 2.80 g of title product
(66%, MS+1: 397 on MS).
1H NMR (400 MHz, DMSO-D6) δ (ppm): 9.57 (s, 1H), 9.22 (s, 1H), 8.66 (d, J=3.5 Hz, 1H), 8.39 (d, J=5.1 Hz, 2H), 8.00 (t, J=6.5 Hz, 1H), 7.90 (d, J=8.2 Hz, 2H), 7.50 (m, 3H), 7.25 (d, J=5.1 Hz, 1H), 7.12 (d, J=7.4 Hz, 1H), 6.94 (dd, J=7.0, 7.8 Hz, 1H), 6.75 (d, J=8.2 Hz, 1H), 6.57 (dd, J=7.0, 7.8 Hz, 1H), 4.86 (s, 2H), 4.64 (d, J=5.9 Hz, 2H).
References
- “Pharmion Corporation (PHRM) Release: Clinical Data On Oncology HDAC Inhibitor MGCD0103, Presented At The American Society of Clinical Oncology 42nd Annual Meeting” (Press release). Colorado, United States: BioSpace. June 6, 2006.
- Gelmon, K.; Tolcher, A.; Carducci, M.; Reid, G. K.; Li, Z.; Kalita, A.; Callejas, V.; Longstreth, J. et al. (2005). “Phase I trials of the oral histone deacetylase (HDAC) inhibitor MGCD0103 given either daily or 3x weekly for 14 days every 3 weeks in patients (pts) with advanced solid tumors”. J. Clin. Oncol. 2005 ASCO Annual Meeting. 23 (16S). 3147.
- MethylGene to Resume Development of its HDAC Inhibitor, MGCD0103 (Mocetinostat), Sept 2009
- “METHYLGENE TO RESUME DEVELOPMENT OF ITS HDAC INHIBITOR, MGCD0103 (MOCETINOSTAT)”. 21 Sep 2009.
- “Final Phase 2 Clinical Data for Mocetinostat (MGCD0103) in Relapsed/Refractory Hodgkin Lymphoma Patients”. 6 Dec 2010.
- Pfefferli, Catherine; Müller, Fritz; Ja¿wi¿ska, Anna; Wicky, Chantal (2014). “Specific NuRD components are required for fin regeneration in zebrafish”. BMC Biol. 12 (30). doi:10.1186/1741-7007-12-30. PMID 24779377.

- MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo
|
3-20-2009
|
THERAPEUTIC COMBINATIONS AND METHODS FOR CARDIOVASCULAR IMPROVEMENT AND TREATING CARDIOVASCULAR DISEASE
|
|
|
10-3-2008
|
COMBINATION OF ERa+ LIGANDS AND HISTONE DEACETYLASE INHIBITORS FOR THE TREATMENT OF CANCER
|
|
|
12-21-2007
|
Assay for efficacy of histone deacetylase inhibitors
|
|
|
5-25-2005
|
Inhibitors of histone deacetylase
|
|
2-8-2012
|
HDAC INHIBITORS AND HORMONE TARGETED DRUGS FOR THE TREATMENT OF CANCER
|
|
|
6-3-2011
|
Sequential Administration of Chemotherapeutic Agents for Treatment of Cancer
|
|
|
5-6-2011
|
METHODS FOR TREATING OR PREVENTING COLORECTAL CANCER
|
|
|
1-12-2011
|
Inhibitors of histone deacetylase
|
|
|
1-12-2011
|
Inhibitors of Histone Deacetylase
|
|
|
11-24-2010
|
Inhibitors of histone deacetylase
|
|
|
3-5-2010
|
INTRAOCULAR PRESSURE-LOWERING AGENT COMPRISING COMPOUND HAVING HISTONE DEACETYLASE INHIBITOR EFFECT AS ACTIVE INGREDIENT
|
|
|
6-12-2009
|
Administration of an Inhibitor of HDAC and an mTOR Inhibitor
|
|
|
5-22-2009
|
Combinations of HDAC Inhibitors and Proteasome Inhibitors
|
|
|
5-15-2009
|
Combination Therapy
|
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html

