PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
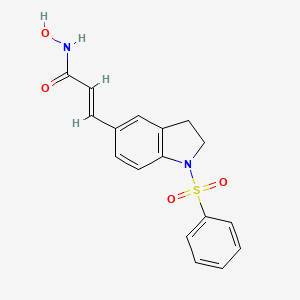
Imofinostat (also known as ABT-301 or MPT0E028) is an orally bioavailable, small-molecule histone deacetylase (HDAC) inhibitor primarily being developed as an innovative precision oncology treatment. Developed by companies like AnBogen Therapeutics and Formosa Pharmaceuticals, it is designed to reactivate tumor suppressor genes that cancer cells have silenced, thereby triggering cancer cell death (apoptosis) and stopping tumor growth.
Mechanism of Action
Imofinostat works through a distinct multi-modality approach to fight cancer cells:
HDAC Inhibition: It acts as a potent inhibitor of human pan-histone deacetylase enzymes, showing preferential selectivity for Class I HDACs (especially HDAC3). This blocks the deacetylation of histone proteins, causing chromatin to remodel and forcing cancer cells to express tumor-suppressor genes.
Akt Pathway Targeting: Independent of its epigenetic effects, it can directly target and reduce the activation (phosphorylation) of the Akt protein kinase, a major pathway that cancer cells use to survive and multiply.
Microenvironment Modulation: Preclinical data shows it alters the tumor microenvironment by converting “cold tumors” (invisible to the immune system) into “hot tumors” by promoting the infiltration of CD8+ cytotoxic T cells.
Current Clinical Status & Indications
Imofinostat is actively moving through clinical trial pipelines, focusing heavily on combination therapies to overcome treatment resistance:
Colorectal Cancer (CRC): It is currently being evaluated in a global Phase 1/2 clinical trial (NCT07244705). It is combined with the immune checkpoint inhibitor tislelizumab (Tevimbra®) and the anti-angiogenic drug bevacizumab to treat advanced, metastatic colorectal cancer.
Pancreatic Cancer: Recent data presented at the 2026 American Association for Cancer Research (AACR) Annual Meeting demonstrates that imofinostat disrupts the HDAC3-NRF2 pathway. This action breaks down chemotherapy resistance in highly aggressive KRAS-mutant pancreatic ductal adenocarcinoma, making tumors much more sensitive to treatments like gemcitabine.
Other Solid Tumors: Phase 1 monotherapy trials have confirmed that the drug possesses a highly competitive safety profile across a broad variety of advanced solid tumors.
Imofinostat is an orally bioavailable N-hydroxyacrylamide-derived inhibitor of both human pan-histone deacetylase (HDAC) enzymes and the serine/threonine protein kinase Akt (protein kinase B), with potential antineoplastic activity. Upon administration, imofinostat selectively binds to and inhibits HDACs, which inhibits deacetylation of histone proteins and leads to the accumulation of highly acetylated histones. This may result in both an induction of chromatin remodeling, and the selective transcription of tumor suppressor genes. This prevents cell division and induces both cell cycle arrest and apoptosis, which may inhibit the proliferation of susceptible tumor cells. In addition, imofinostat inhibits the phosphorylation and activation of Akt, which prevents the activation of downstream signaling pathways, independent of its HDAC inhibitory activity. HDACs, upregulated in many tumor cell types, are a family of enzymes that deacetylate histone proteins. Akt, overexpressed in many tumor cell types, plays a key role in tumor cell proliferation and survival.
Dose-Seeking Study of MPT0E028 in Subjects With Advanced Solid Malignancies Without Standard Treatment
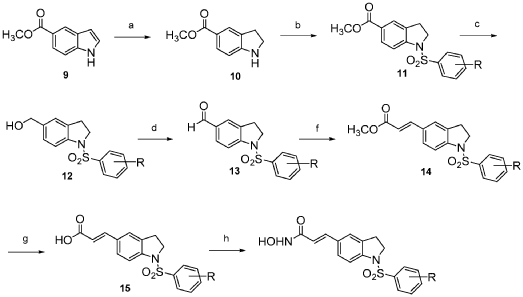
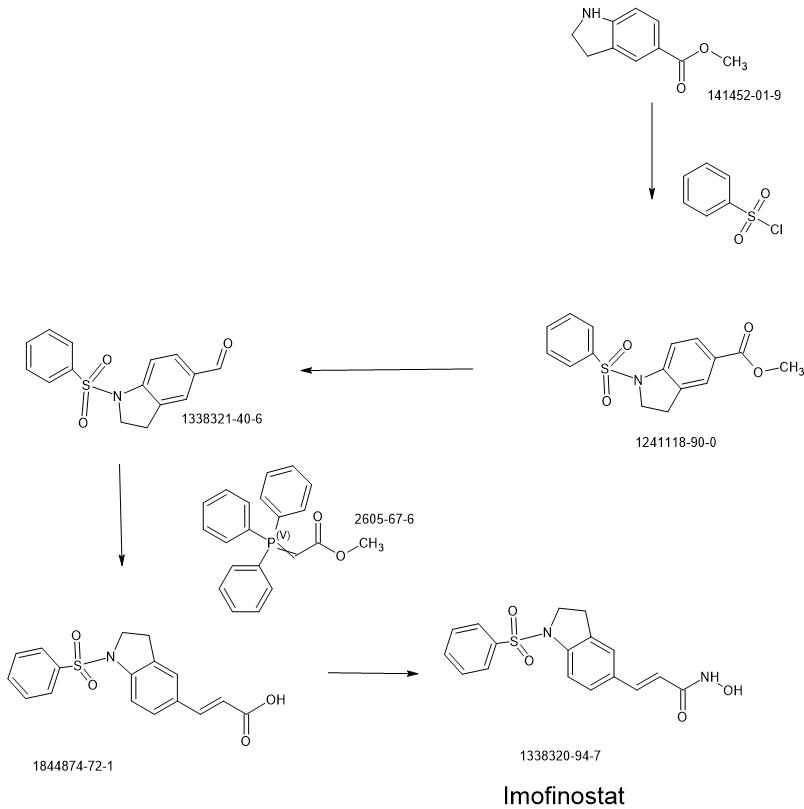
Compound 12 was synthesized via the route as shown in Scheme 3 above (reagents and conditions: (a) NaBH3CN, AcOH; (b) Benzenesulfonyl chloride, 4-methoxybenzenesulfonyl chloride, 3,4-dimethoxybenzenesulfonyl chloride, 4-fluorobenzenesulfonyl chloride, or 4-nitrobenzenesulfonyl chloride, pyridine; (c) L1AIH4, THF; (d) PDC, MS, CH2C12; f) Ph3P = CH-COOCH3, CH2C12; (g) 1M LiOH(aq), dioxane; (h) (i) NH2OTHP, PyBOP, NEt3, DMF; (ii) TFA, MeOH; (i) Fe, NH4C1, Isopropanol, H20).
2,3-Dihydro-lH-indole-5-carboxylic acid methyl ester (10): sodium cyanoborohydride (0.16 g, 2.57 mmol) was added to a solution of methyl indole-5-carboxylate (9) (0.30 g, 1.71 mmol) in AcOH (2 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water at 0 °C. Concentrated NaOH was added to reach pH=10. The aqueous layer was extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 10 (0.28 g). 1H NMR (500MHz, CDC13): δ 3.06 (t, J= 8.5 Hz, 2H), 3.65 (t, J= 8.5 Hz, 2H), 3.84 (s, 3H), 6.53-6.55 (m, 1H), 7.75-7.76 (m, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carboxylic acid methyl ester (11): To a solution of 10 (0.28 g, 1.58 mmol) in pyridine (2 mL), benzenesulfonyl chloride (0.40 ml, 3.16 mmol) was added. The reaction mixture was refluxed overnight. The mixture was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 11 (0.40 g). 1H NMR (500MHz, CDCI3): δ 2.99 (t, J= 8.6 Hz, 2H), 3.87 (s, 3H), 3.97 (t, J= 8.6 Hz, 2H), 7.45-7.48 (m, 2H), 7.56-7.59 (m, 1H), 7.66 (d, J= 8.5 Hz, 1H), 7.75 (s, 1H), 7.82 (d, J= 7.7 Hz, 2H), 7.90 (d, J= 7.9 Hz, 1H).
(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-methanol (12): LAH (0.10 g, 2.52 mmol) was added to a solution of 11 (0.40 g, 1.26 mmol) in THF (10 mL) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h before it was quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The reaction mixture was purified by silica gel chromatography (EtOAc: n-hexane = 1 : 1) to afford 12 (0.24 g). 1H NMR (500MHz, CDC13): δ 2.83 (t, J= 8.4 Hz, 2H), 3.92 (t, J= 8.5 Hz, 2H), 4.49 (s, 2H), 7.09 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 7.46-7.49 (m, 2H), 7.53 (d, J= 8.2 Hz, 1H), 7.60 (t, J= 7.5 Hz, 1H), 7.76 (d, J= 7.7 Hz, 2H).
l-Benzenesulfonyl-2,3-dihydro-lH-indole-5-carbaldehyde (13): molecular sieves (0.63g) were added to a solution of 12 (0.24 g, 0.83 mmol) in CH2C12 (10 mL), PDC (0.63 g, 1.66 mmol). The mixture was stirred at room temperature overnight before it was filtered through celite. The organic layer was concentrated under reduced pressure then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 2) to afford 13 (0.19 g). 1H NMR (500MHz, CDC13): δ 3.05 (t, J= 8.6 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 7.46-7,49 (m, 2H), 7.58-7.62 (m, 2H), 7.71 (d, J= 8.3 Hz, 1H), 7.75 (d, J= 8.3 Hz, 1H), 7.84 (d, J= 7.8 Hz, 2H), 9.85 (s, 1H).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid methyl ester (14): Methyl (triphenylphosphoranylidene) acetate (0.27 g, 0.79 mmol) was added to a solution of 13 (0.19g,
0.66 mmol) in CH2CI2 (10 mL). The mixture was stirred at room temperature for 3h before it was
quenched with water and then extracted with CH2CI2 (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure to give a yellow residue, which was then purified by silica gel chromatography (EtOAc: n-hexane = 1 : 3) to afford 14
(0.20 g).
3-(l-Benzenesulfonyl-2,3-dihydro-lH-indol-5-yl)-acrylic acid (15): 1M LiOH aqueous solution (1.16 ml, 1.16 mmol) was added to a solution of 14 (0.20g, 0.58 mmol) in dioxane
(15 mL). The reaction mixture was stirred at 40 °C overnight before it was concentrated under reduced pressure. The residue was dissolved in water and concentrated HCl was added up to acidic pH to give the precipitation, which was dried by vacuum to afford 15 (0.16 g). 1H NMR (500MHz, CD3OD): δ 2.92 (t, J= 8.5 Hz, 2H), 3.96 (t, J= 8.5 Hz, 2H), 6.33 (d, J= 15.9 Hz, 1H), 7.38 (s, 1H), 7.41 (d, J= 8.5 Hz, 1H), 7.50-7.53 (m, 2H), 7.55 (d, J= 16.1 Hz, 1H), 7.58-7.64 (m, 2H), 7.82 (d, J = 7.6 Hz, 2H).
(Compound 12): NH2OTHP (0.05 g, 0.44 mmol) was added to a solution of 15 (0.12 g, 0.37 mmol), PyBOP (0.20 g, 0.39 mmol), triethylamine (0.12 ml, 0.88 mmol) in DMF (1.5 mL). The reaction mixture was stirred at room temperature for 1 h before it was quenched with water, followed by extraction with EtOAc (15 mL x 3). The combined organic layer was dried over anhydrous MgS04 and concentrated under reduced pressure. The residue was purified by silica gel chromatography (CH2C12: CH3OH = 30 : 1 : l%NH3(aq)) to give a white solid, which was treated with TFA (1.13 ml, 15.21 mmol) in the presence of CH3OH (25 mL) and stirred overnight at room temperature. The reaction mixture was concentrated under reduced pressure to give a white residue, which was recrystallized by CH3OH to afford Compound 12 (0.12 g). 1H NMR (500MHz,
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
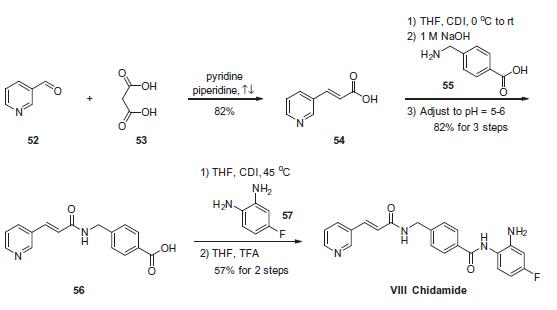
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID23541946.
^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID25829268. S2CID36758974.
Dacinostat, also known as LAQ824, is a hydroxamate histone deacetylase inhibitor with potential anticancer activity. LAQ824 sensitized nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. LAQ824 reduced clonogenic survival of the H23 and H460 cell lines five-fold compared with controls and four-fold compared with either agent alone (P<0.001). In phase I trials, LAQ824 was well tolerated at doses that induced accumulation of histone acetylation, with higher doses inducing changes consistent with HSP90 inhibition.
NVP-LAQ824 inhibits histone deacetylase enzymatic activities in vitro and transcriptionally activated the p21 promoter in reporter gene assays. When tested on a variety of solid tumour cell lines, NVP-LAQ824 exhibited selective anti-proliferative effects, inducing cell growth inhibition in some, while inducing cell death in others. To induce cell death, a minimum of 16 h exposure to NVP-LAQ824 is required. Flow cytometry studies revealed that both tumour cell lines and normal diploid fibroblasts arrested in the G2/M phase of the cell cycle after compound treatment. However, an increased sub-G1 population at 48 h (reminiscent of apoptotic cells) was only observed in the cancer cell lines.
Annexin V staining data confirmed that NVP-LAQ824 induced apoptosis in tumour cells, but not in normal cells. To relate HDAC inhibition to the anti-proliferative effects of NVP-LAQ824, expression of HDAC 1 was inhibited using antisense and this was sufficient to activate p21 expression, hypophosphorylate Rb and inhibit cell growth. Furthermore, tumour cells treated with NVP-LAQ824 caused acetylation of HSP90 and degradation of its cargo oncoproteins. Finally, NVP-LAQ824 exhibited antitumour effects in a xenograft animal model.
To determine if NVP-LAQ824 inhibited histone deacetylases in vivo, tumours treated with the drug were immunoblotted with an antibody specific for acetylated histones H3 and H4 and the results indicated increased histone H3 and 114 acetylation levels in NVP-LAQ824 treated cancer cells. Together, our data indicated that the activity of NVP-LAQ824 was consistent with its intended mechanism of action. This novel HDAC inhibitor is currently in clinical trials as an anticancer agent. see: http://www.ncbi.nlm.nih.gov/pubmed/15171259.
Reversible acetylation of histones is a major regulator of gene expression that acts by altering accessibility of transcription factors to DNA. In normal cells, histone deacetylase (HDA) and histone acetyltrasferase together control the level of acetylation of histones to maintain a balance. Inhibition of HDA results in the accumulation of hyperacetylated histones, which results in a variety of cellular responses.
Inhibitors of HDA have been studied for their therapeutic effects on cancer cells. For example, butyric acid and its derivatives, including sodium phenylbutyrate, have been reported to induce apoptosis in vitro in human colon carcinoma, leukemia and retinoblastoma cell lines. However, butyric acid and its derivatives are not useful pharmacological agents because they tend to be metabolized rapidly and have a very short half-life in vivo. Other inhibitors of HDA that have been widely studied for their anti-cancer activities are trichostatin A and trapoxin. Trichostatin A is an antifungal and antibiotic and is a reversible inhibitor of mammalian HDA. Trapoxin is a cyclic tetrapeptide, which is an irreversible inhibitor of mammalian HDA.
Although trichostatin and trapoxin have been studied for their anti-cancer activities, the in vivo instability of the compounds makes them less suitable as anti-cancer drugs. There remains a need for an active compound that is suitable for treating tumors, including cancerous tumors, that is highly efficacious and stable
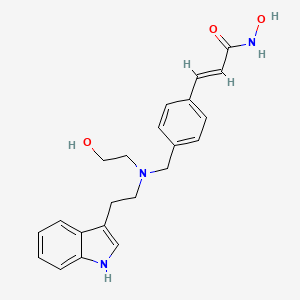
The esterification of 4-formylcinnamic acid (I) with methanol and HCl gives the methyl ester (II), which can be obtained by Heck coupling of 4-bromobenzaldehyde (III) with methyl acrylate (IV). The reductocondensation of (II) with tryptamine (V) by means of NaBH(OAc)3 in dichloroethane yields the secondary amine (VI), which is alkylated with 2-(tert-butyldimethylsilyloxy)ethyl bromide (VII) by means of DIEA in DMSO to afford the tertiary amine (VIII). The reaction of the methyl ester group of (VIII) with KOH and hydroxylamine in methanol provides the silylated hydroxamic acid (IX), which is finally deprotected with TFA in water.
1: Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011 Apr 1;186(7):3986-96. doi: 10.4049/jimmunol.1001101. Epub 2011 Mar 2. PubMed PMID: 21368229.
2: Schwarz K, Romanski A, Puccetti E, Wietbrauk S, Vogel A, Keller M, Scott JW, Serve H, Bug G. The deacetylase inhibitor LAQ824 induces notch signalling in haematopoietic progenitor cells. Leuk Res. 2011 Jan;35(1):119-25. doi: 10.1016/j.leukres.2010.06.024. Epub 2010 Jul 31. PubMed PMID: 20674020.
3: Cho YS, Whitehead L, Li J, Chen CH, Jiang L, Vögtle M, Francotte E, Richert P, Wagner T, Traebert M, Lu Q, Cao X, Dumotier B, Fejzo J, Rajan S, Wang P, Yan-Neale Y, Shao W, Atadja P, Shultz M. Conformational refinement of hydroxamate-based histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824). J Med Chem. 2010 Apr 8;53(7):2952-63. doi: 10.1021/jm100007m. PubMed PMID: 20205394.
4: Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, Craft N, Economou JS, Marincola FM, Wang E, Ribas A. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009 Nov 15;69(22):8693-9. doi: 10.1158/0008-5472.CAN-09-1456. Epub 2009 Oct 27. PubMed PMID: 19861533; PubMed Central PMCID: PMC2779578.
5: Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, Johnstone RW. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009 Jul 9;114(2):380-93. doi: 10.1182/blood-2008-10-182758. Epub 2009 Apr 21. PubMed PMID: 19383971.
6: de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, Rowinsky EK, Aherne W, Atadja P, Scott JW, Patnaik A. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008 Oct 15;14(20):6663-73. doi: 10.1158/1078-0432.CCR-08-0376. PubMed PMID: 18927309.
7: Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, Atadja P, Griffiths JR, Judson IR, Workman P, Leach MO, Beloueche-Babari M. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia. 2008 Apr;10(4):303-13. PubMed PMID: 18392140; PubMed Central PMCID: PMC2288545.
8: Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs. 2007 Aug;18(7):793-800. PubMed PMID: 17581301.
9: Kato Y, Salumbides BC, Wang XF, Qian DZ, Williams S, Wei Y, Sanni TB, Atadja P, Pili R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007 Jan;6(1):70-81. PubMed PMID: 17237267.
10: Leyton J, Alao JP, Da Costa M, Stavropoulou AV, Latigo JR, Perumal M, Pillai R, He Q, Atadja P, Lam EW, Workman P, Vigushin DM, Aboagye EO. In vivo biological activity of the histone deacetylase inhibitor LAQ824 is detectable with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Cancer Res. 2006 Aug 1;66(15):7621-9. PubMed PMID: 16885362.
FDA Approves Farydak (panobinostat) for Multiple Myeloma
February 23, 2015 — The U.S. Food and Drug Administration today approved Farydak (panobinostat) for the treatment of patients with multiple myeloma.
Multiple myeloma is a form of blood cancer that arises from plasma cells, a type of white blood cell, found in bone marrow. According to the National Cancer Institute, approximately 21,700 Americans are diagnosed with multiple myeloma and 10,710 die from the disease annually
SAN DIEGO,Aug. 11, 2014/PRNewswire/ — Mirati Therapeutics, Inc. (NASDAQ:MRTX) today announced that the U.S. FDA has granted Orphan Drug Designation to mocetinostat, a spectrum selective HDAC inhibitor, for diffuse large B-cell lymphoma (DLBCL). In June, mocetinostat was granted Orphan Drug Designation as a treatment for myelodysplastic syndrome (MDS). Orphan drug designation is also being sought for bladder cancer patients with specific genetic alterations.
Mocetinostat is an orally-bioavailable, spectrum-selective HDAC inhibitor. Mocetinostat is enrolling patients in a Phase 2 dose confirmation study in combination with Vidaza as treatment for intermediate and high-risk MDS. Mirati also plans to initiate Phase 2 studies of mocetinostat as a single agent in patients with mutations in histone acetyl transferases in bladder cancer and DLBCL. Initial data from the Phase 2 studies is expected by the end of 2014. In addition to the ongoing Phase 2 clinical trials, mocetinostat has completed 13 clinical trials in more than 400 patients with a variety of hematologic malignancies and solid tumors.
About Mirati Therapeutics
Mirati Therapeutics is a targeted oncology company developing an advanced pipeline of breakthrough medicines for precisely defined patient populations. Mirati’s approach combines the three most important factors in oncology drug development – drug candidates with complementary and compelling targets, creative and agile clinical development, and a highly accomplished precision medicine leadership team. The Mirati team is using a proven blueprint for developing targeted oncology medicines to advance and maximize the value of its pipeline of drug candidates, including MGCD265 and MGCD516, which are orally bioavailable, multi-targeted kinase inhibitors with distinct target profiles, and mocetinostat, an orally bioavailable, spectrum-selective histone deacetylase inhibitor. More information is available atwww.mirati.com.
In eukaryotic cells, nuclear DNA associates with histones to form a compact complex called chromatin. The histones constitute a family of basic proteins which are generally highly conserved across eukaryotic species. The core histones, termed H2A, H2B, H3, and H4, associate to form a protein core. DNA winds around this protein core, with the basic amino acids of the histones interacting with the negatively charged phosphate groups of the DNA. Approximately 146 base pairs of DNA wrap around a histone core to make up a nucleosome particle, the repeating structural motif of chromatin.
Csordas, Biochem. J., 286: 23-38 (1990) teaches that histones are subject to posttranslational acetylation of the α,ε-amino groups of N-terminal lysine residues, a reaction that is catalyzed by histone acetyl transferase (HAT1). Acetylation neutralizes the positive charge of the lysine side chain, and is thought to impact chromatin structure. Indeed, Taunton et al., Science, 272: 408-411 (1996), teaches that access of transcription factors to chromatin templates is enhanced by histone hyperacetylation. Taunton et al. further teaches that an enrichment in underacetylated histone H4 has been found in transcriptionally silent regions of the genome.
Histone acetylation is a reversible modification, with deacetylation being catalyzed by a family of enzymes termed histone deacetylases (HDACs). Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999), teaches that HDACs are divided into two classes, the first represented by yeast Rpd3-like proteins, and the second represented by yeast Hda1-like proteins. Grozinger et al. also teaches that the human HDAC1, HDAC2, and HDAC3 proteins are members of the first class of HDACs, and discloses new proteins, named HDAC4, HDAC5, and HDAC6, which are members of the second class of HDACs. Kao et al., Genes & Dev., 14: 55-66 (2000), discloses HDAC7, a new member of the second class of HDACs. More recently, Hu et al. J. Bio. Chem. 275:15254-13264 (2000) and Van den Wyngaert, FEBS, 478: 77-83 (2000) disclose HDAC8, a new member of the first class of HDACs.
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998), discloses that HDAC activity is inhibited by trichostatin A (TSA), a natural product isolated from Streptomyces hygroscopicus, and by a synthetic compound, suberoylanilide hydroxamic acid (SAHA). Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988), teaches that TSA causes arrest of rat fibroblasts at the G1 and G2 phases of the cell cycle, implicating HDAC in cell cycle regulation. Indeed, Finnin et al., Nature, 401: 188-193 (1999), teaches that TSA and SAHA inhibit cell growth, induce terminal differentiation, and prevent the formation of tumors in mice. Suzuki et al., U.S. Pat. No. 6,174,905, EP 0847992, JP 258863/96, and Japanese Application No. 10138957, disclose benzamide derivatives that induce cell differentiation and inhibit HDAC. Delorme et al., WO 01/38322 and PCT/IB01/00683, disclose additional compounds that serve as HDAC inhibitors.
The molecular cloning of gene sequences encoding proteins with HDAC activity has established the existence of a set of discrete HDAC enzyme isoforms. Some isoforms have been shown to possess specific functions, for example, it has been shown that HDAC-6 is involved in modulation of microtubule activity. However, the role of the other individual HDAC enzymes has remained unclear.
These findings suggest that inhibition of HDAC activity represents a novel approach for intervening in cell cycle regulation and that HDAC inhibitors have great therapeutic potential in the treatment of cell proliferative diseases or conditions. To date, few inhibitors of histone deacetylase are known in the art.
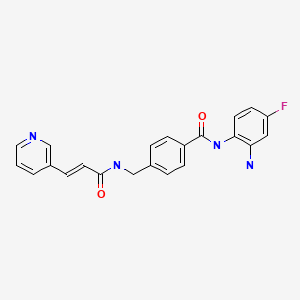
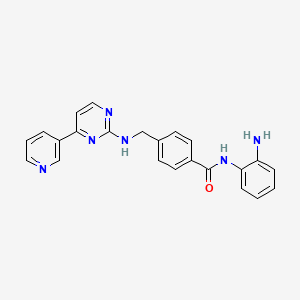
Example 426Synthesis of N-(2-Amino-phenyl)-4-[(4-pyridin-3-pyrimidin-2-ylamino)-methyl]-benzamide
Step 1: Synthesis of 4-Guanidinomethyl-benzoic acid methyl ester Intermediate 1
The mixture of 4-Aminomethyl-benzoic acid methyl ester HCl (15.7 g, 77.8 mmol) in DMF (85.6 mL) and DIPEA (29.5 mL, 171.2 mmol) was stirred at rt for 10 min. Pyrazole-1-carboxamidine HCl (12.55 g, 85.6 mmol) was added to the reaction mixture and then stirred at rt for 4 h to give clear solution. The reaction mixture was evaporated to dryness under vacuum. Saturated NaHCO3solution (35 mL) was added to give nice suspension. The suspension was filtered and the filter cake was washed with cold water. The mother liquid was evaporated to dryness and then filtered. The two solids were combined and re-suspended over distilled H2O (50 ml). The filter cake was then washed with minimum quantities of cold H2O and ether to give 12.32 g white crystalline solid intermediate 1 (77% yield, M+1: 208 on MS).
Step 2: Synthesis of 3-Dimethylamino-1-pyridin-3-yl-propenone Intermediate 2
3-Acetyl-pyridine (30.0 g, 247.6 mmol) and DMF dimethyl acetal (65.8 mL, 495.2 mmol) were mixed together and then heated to reflux for 4 h. The reaction mixture was evaporated to dryness and then 50 mL diethyl ether was added to give brown suspension. The suspension was filtered to give 36.97 g orange color crystalline product (85% yield, M+1: 177 on MS).
Step 3: Synthesis of 4-[(4Pyridin-3-pyrimidin-2-ylamino)-methyl]benzoic acid methyl ester Intermediate 3
Intermediate 1 (0.394 g, 1.9 mmol) and intermediate 2 (0.402 g, 2.3 mmol) and molecular sieves (0.2 g, 4A, powder, >5 micron) were mixed with isopropyl alcohol (3.8 mL). The reaction mixture was heated to reflux for 5 h. MeOH (50 mL) was added and then heated to reflux. The cloudy solution was filtrated over a pad of celite. The mother liquid was evaporated to dryness and the residue was triturated with 3 mL EtOAc. The suspension was filtrated to give 0.317 g white crystalline solid Intermediate 3 (52%, M+1: 321 on MS).
Step 4: Synthesis of N-(2-Amino-phenyl)-4-[(4-pyrymidin-2-ylamino)-methyl]-benzamide
Intermediate 3 (3.68 g, 11.5 mmol) was mixed with THF (23 mL), MeOH (23 mL) and H2O (11.5 mL) at rt. LiOH (1.06 g, 25.3 mmol) was added to reaction mixture. The resulting reaction mixture was warmed up to 40° C. overnight. HCl solution (12.8 mL, 2N) was added to adjust pH=3 when the mixture was cooled down to rt. The mixture was evaporated to dryness and then the solid was washed with minimum quantity of H2O upon filtration. The filter cake was dried over freeze dryer to give 3.44 g acid of the title compound (95%, M+1: 307 on MS).
Acid (3.39 g, 11.1 mmol) of the title compound, BOP (5.679 g, 12.84 mmol) and o-Ph(NH2)2(2.314 g, 21.4 mmol) were dissolved in the mixture of DMF (107 mL) and Et3N (2.98 mL, 21.4 mmol). The reaction mixture was stirred at rt for 5 h and then evaporated to dryness. The residue was purified by flash column (pure EtOAc to 5% MeOH/EtOAc) and then interested fractions were concentrated. The final product was triturated with EtOAc to give 2.80 g of title product
Pfefferli, Catherine; Müller, Fritz; Ja¿wi¿ska, Anna; Wicky, Chantal (2014). “Specific NuRD components are required for fin regeneration in zebrafish”.BMC Biol.12(30).doi:10.1186/1741-7007-12-30.PMID24779377.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....































{kind=link}
{kind=link}