
Home » Posts tagged 'histone deacetylase.'
Tag Archives: histone deacetylase.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8)
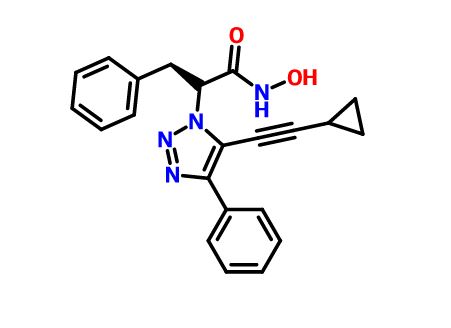

CAS 1620779-53-4
MF C22H20N4O2, MW 372.4
(S)-2-(5-(cyclopropylethynyl)-4-phenyl-1H-1,2,3-triazol-1-yl)-N-hydroxy-3-phenylpropanamide
1H-1,2,3-Triazole-1-acetamide, 5-(2-cyclopropylethynyl)-N-hydroxy-4-phenyl-α-(phenylmethyl)-, (αS)-
| Applicants: | TRUSTEES OF BOSTON UNIVERSITY DANA-FARBER CANCER INSTITUTE, INC. |
| Inventors: | Aaron Beaty BEELER John A. PORCO, JR. Oscar J. INGHAM James E. BRADNER |
|
As histone proteins bind DNA prior to transcription, their biochemical action plays a critical role in the regulation of gene expression and cellular differentiation. Histone deacetylases (HDACs) are an important family of proteins predominantly responsible for specific posttranslational modifications of histone proteins, the chief organizational component of chromatin. HDACs catalyze the removal of acetyl groups from histones and other cellular proteins. HDAC-mediated deacetylation of chromatin-bound histones regulates the expression of a variety of genes throughout the genome. Importantly, HDACs have been linked to cancer, as well as other health conditions. To date, eleven major HDAC isoforms have been described (HDACs 1-11). HDACs are categorized into two classes. Class I HDACs include HDAC1, HDAC2, HDAC3, HDAC8 and HDAC11. Class II HDACs include HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10. HDAC’s are validated targets for a number of disease states, including cancer, neurodegenerative diseases, sickle-cell anemia, muscular dystrophy, and HIV. There are currently two HDAC inhibitors on the market, Vorniostat and Romidepsin. Both are approved for treatment of T-cell lymphoma. However, they are both pan active inhibitors showing very little specificity of binding to HDAC subclasses. Because of this lack of specificity they have a number of side effects.
|
|
Non-selective HDAC inhibitors effect deacetylase activity of most, if not all, of the HDACs. The mechanisms of the anticancer effects of SAHA, a non-selective HDAC inhibitor, are not completely understood, and likely result from both altered gene expression and altered function of proteins regulating cell proliferation and cell death pathways. Non-selective HDAC inhibitors, such as SAHA, induce the accumulation of acetylated histone proteins and non histone proteins.
|
|
Small molecule HDAC inhibitors that are isoform-selective are useful as therapeutic agents with reduced toxicity and as tools for probing the biology of the HDAC isoforms. The present disclosure is related, in part to small molecules that are selective HDAC inhibitors.
|

1H NMR (500 MHz, d4-MeOD) 0.80 (2H, m), 0.98 (2H, m), 1.47 (1H, m), 3.51 (1H, dd, J = 11.2, 14.2 Hz), 3.71 (1H, dd, J = 3.9, 14.2 Hz), 5.49 (1H, dd, J = 3.9, 11.2 Hz), 6.96 (2H, m), 7.17-7.20 (3H, m), 7.37 (1H, t, J = 7.3 Hz), 7.43 (2H, t, J = 7.3 Hz), 7.99 (2H, d, J = 8.8 Hz);

13C NMR (100 MHz, d4-MeOD) 0.02, 8.55, 37.07, 60.83, 62.59, 109.09, 118.98, 125.9, 127.16, 128.55, 128.65, 128.71, 129.16, 130.07, 136.09, 147.10, 165.20;
HRMS calculated for C22H21N4O2 + (M+H): 373.1659, found: 373.1665.

PATENT
WO2014116962
https://www.google.com/patents/WO2014116962A1?cl=en
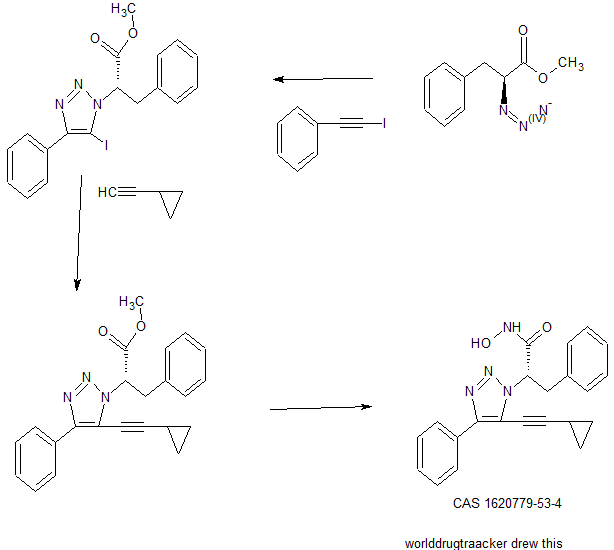
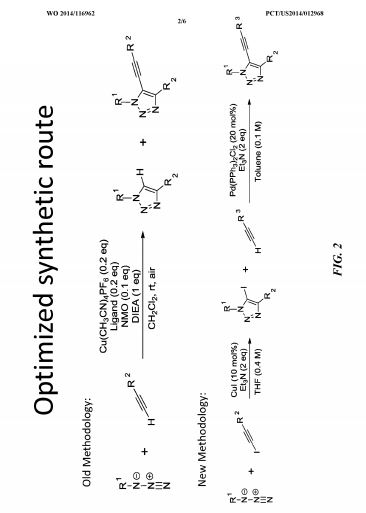
SAR. libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in Fig. 2.
In one study, compound 
was synthesized as outline in Scheme I.
Scheme I




PATENT
|
SAR libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in FIG. 2. In one study, compound
|
 was synthesized as outline in Scheme I.
was synthesized as outline in Scheme I.


|
The HDAC assays were carried out as described in Bowers A, West N, Taunton J, Schreiber S L, Bradner J E, Williams R M Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 11219-11222. Assay results revealed that among the analogues tested a cyclopropane analog was the most active at 0.4 nM (>1000 fold selectivity). These results demonstrated that a small aliphatic group in the 5-position on the triazole can increase potency. Also, compounds with an L-phenylalanine moiety at the 3-position showed significant potency. To expand our understanding of how the molecule interacts with the binding pocket of HDAC 8 and to understand our preliminary SAR, molecular modeling was carried out. The phenyl group from the original amino methyl ester fits snuggly into the Zn binding site and the alkynyl phenyl group sits flat in a hydrophobic groove. In summary, the inventors have developed a potent and highly selective small molecule which inhibits HDAC-8 at approximately 500 pM with over 1000-fold selectivity over HDAC-6 and significantly greater selectivity for all other HDACs. To inventors’ knowledge, to date there are no compounds with this level of potency and selectivity.
|
|
All patents and other publications identified in the specification and examples are expressly incorporated herein by reference for all purposes. These publications are provided solely for their disclosure prior to the filing date of the present application. Nothing in this regard should be construed as an admission that the inventors are not entitled to antedate such disclosure by virtue of prior invention or for any other reason. All statements as to the date or representation as to the contents of these documents is based on the information available to the applicants and does not constitute any admission as to the correctness of the dates or contents of these documents.
|
|
Although preferred embodiments have been depicted and described in detail herein, it will be apparent to those skilled in the relevant art that various modifications, additions, substitutions, and the like can be made without departing from the spirit of the invention and these are therefore considered to be within the scope of the invention as defined in the claims which follow. Further, to the extent not already indicated, it will be understood by those of ordinary skill in the art that any one of the various embodiments herein described and illustrated can be further modified to incorporate features shown in any of the other embodiments disclosed herein.
|
Paper

A novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8) has been discovered by the repurposing of a diverse compound collection. Medicinal chemistry optimization led to the identification of a highly potent (0.8 nM) and selective inhibitor of HDAC8.
Development of a Potent and Selective HDAC8 Inhibitor
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00239
file:///C:/Users/Inspiron/Downloads/ml6b00239_si_001.pdf
Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States

 Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.
Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.

Aaron Beeler
Aaron Beeler received his Ph.D. in 2002 from Professor John Rimoldi’s laboratory in the Department of Medicinal Chemistry at the University of Mississippi. He then joined the Porco group as a postodoctoral fellow and subsequently the Center for Chemical Methodology and Library Development at Boston University, now the Center for Molecular Discovery. He was promoted to Assistant Director of the CMLD-BU in January 2005. In 2012 Aaron joined the Department of Chemistry as a tenure-track professor in medicinal chemistry.
Degrees and Positions
- B.S. Belmont University, Biology,
- Ph.D. University of Mississippi, Medicinal Chemistry
Research
The Beeler Research Group is truly multidisciplinary, combining organic chemistry, engineering, and biology to solve problems in medicinal chemistry. All of these elements are combined and directed toward significant problems in human health. The Beeler Group is addressing focused disease areas (e.g., schizophrenia, Parkinson’s, cystic fibrosis), as well as project areas with broader impact potential (e.g., new methods for discovery of small molecules with anti-cancer properties).
- Medicinal Chemistry: The goals of medicinal chemistry projects are to optimize small molecules in order to: a) develop a probe that may be utilized as a tool in biological studies; b) develop a lead molecule to facilitate future therapeutics; and c) utilize small molecules to enhance understanding of biological targets that are important for human health. These projects provide students with training in organic chemistry, medicinal chemistry, and focused biology. Projects are selected based on their chemistry and/or biology significance and potential for addressing challenging questions.
- Technology: One of the core components of the research in the Beeler Group is development of technologies and paradigms that facilitate rapid modification of complex scaffolds. These technologies enable optimization of biologically active lead compounds and identification of small molecule leads in biological systems. The projects focus on utilizing automation, miniaturization, and microfluidics to carry out chemical transformations. These projects are highly interdisciplinary with both chemistry and engineering components.
- Photochemistry: This area focuses on photochemical transformations toward the synthesis of natural products, natural product scaffolds, and other complex chemotypes of interest to medicinal chemistry and chemical biology. The foundation of these projects is utilizing microfluidics to enable photochemical reaction development.
Techniques & Resources
Students in the Beeler Research Group will have opportunities to learn a number of exciting research disciplines. Organic synthesis will be at the heart of every project. This will include targeted synthesis, methodology development, and medicinal chemistry. Through collaborations with biological researchers and/or research projects carried out within the Beeler Group, students will learn methods for biological assays, pharmacology, and target identification. Many projects will also include aspects of engineering that will provide opportunities for learning techniques such as microfabrication and microfluidics.
Opportunities
It is becoming evident that successful and impactful science is realized in collaborative interdisciplinary environments. The Beeler Research Group’s multidisciplinary nature and collaborative projects provides opportunities to learn areas of research outside of traditional chemistry.
What’s Next for Graduates of the Beeler Group?
Members of the Beeler Research Group will be positioned for a wide range of future endeavors.
- Undergraduates will be prepared to enter into graduate school for organic chemistry, chemical biology, or chemical engineering or to start careers in industry;
- Graduate students will have the foundation required for postdoctoral studies in organic synthesis or chemical biology as well as an industrial career in biotech or pharma;
- Postdoctoral associates will gain training and experience critical for both academic and industrial careers.
Assistant Professor
Office: SCI 484C
Laboratory: SCI 484A
Phone: 617.358.3487
Fax: 617-358-2847
beelera@bu.edu
Office Hours: by Appointment
Beeler Group Homepage
Google Scholar Page
Oscar J. Ingham below

John A. PORCO, JR below


JAMES E. BRADNER, MD above
Dana-Farber Cancer Institute


 Ron Paranal
Ron Paranal
 Randolph A. Escobar
Randolph A. Escobar
 Han Yueh
Han Yueh
| US20090181943 * | Apr 9, 2008 | Jul 16, 2009 | Methylgene Inc. | Inhibitors of Histone Deacetylase |
| Reference | ||
|---|---|---|
| 1 | * | GERARD, B ET AL.: ‘Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal alkynes‘ TETRAHEDRON vol. 62, 12 May 2006, pages 6405 – 6411 |
| 2 | * | VANNINI, A ET AL.: ‘Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor.‘ PNAS, [Online] vol. 101, no. 42, 19 October 2004, pages 15064 – 15069 Retrieved from the Internet: <URL:http://www.pnas.org/content/101/42/15064> |
///////////epigenetic, HDAC, HDAC8, Histone deacetylase, histone deacetylase 8, triazole, PRECLINICAL, Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States, Oscar J. Ingham, Aaron Beeler
n1n(c(c(n1)c2ccccc2)C#CC3CC3)C(C(=O)NO)Cc4ccccc4
Mirati Therapeutics Receives Orphan Designation from U.S. FDA for Mocetinostat in Diffuse Large B-Cell Lymphoma

Mocetinostat
SAN DIEGO, Aug. 11, 2014 /PRNewswire/ — Mirati Therapeutics, Inc. (NASDAQ: MRTX) today announced that the U.S. FDA has granted Orphan Drug Designation to mocetinostat, a spectrum selective HDAC inhibitor, for diffuse large B-cell lymphoma (DLBCL). In June, mocetinostat was granted Orphan Drug Designation as a treatment for myelodysplastic syndrome (MDS). Orphan drug designation is also being sought for bladder cancer patients with specific genetic alterations.
| Identifiers | |
|---|---|
| CAS number | 726169-73-9 |
| PubChem | 9865515 |
| ChemSpider | 8041206 |
| ChEMBL | CHEMBL272980 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C23H20N6O |
| Molar mass | 396.44 g mol−1 |
Mocetinostat (MGCD0103) is a benzamide histone deacetylase inhibitor undergoing clinical trials for treatment of various cancers including follicular lymphoma, Hodgkin’s lymphoma and acute myelogenous leukemia.[1][2][3]
One clinical trial (for refractory follicular lymphoma) was temporarily put on hold due to cardiac problems but resumed recruiting in 2009.[4]
In 2010 favourable results were announced from the phase II trial for Hodgkin’s lymphoma.[5]
MGCD0103 has also been used as a research reagent where blockage of members of the HDAC-family of histone deacetylases is required.[6]
Mechanism of action
It works by inhibiting mainly histone deacetylase 1 (HDAC1), but also HDAC2, HDAC3, and HDAC11.[7]
About Mocetinostat
Mocetinostat is an orally-bioavailable, spectrum-selective HDAC inhibitor. Mocetinostat is enrolling patients in a Phase 2 dose confirmation study in combination with Vidaza as treatment for intermediate and high-risk MDS. Mirati also plans to initiate Phase 2 studies of mocetinostat as a single agent in patients with mutations in histone acetyl transferases in bladder cancer and DLBCL. Initial data from the Phase 2 studies is expected by the end of 2014. In addition to the ongoing Phase 2 clinical trials, mocetinostat has completed 13 clinical trials in more than 400 patients with a variety of hematologic malignancies and solid tumors.
About Mirati Therapeutics
Mirati Therapeutics is a targeted oncology company developing an advanced pipeline of breakthrough medicines for precisely defined patient populations. Mirati’s approach combines the three most important factors in oncology drug development – drug candidates with complementary and compelling targets, creative and agile clinical development, and a highly accomplished precision medicine leadership team. The Mirati team is using a proven blueprint for developing targeted oncology medicines to advance and maximize the value of its pipeline of drug candidates, including MGCD265 and MGCD516, which are orally bioavailable, multi-targeted kinase inhibitors with distinct target profiles, and mocetinostat, an orally bioavailable, spectrum-selective histone deacetylase inhibitor. More information is available at www.mirati.com.
In eukaryotic cells, nuclear DNA associates with histones to form a compact complex called chromatin. The histones constitute a family of basic proteins which are generally highly conserved across eukaryotic species. The core histones, termed H2A, H2B, H3, and H4, associate to form a protein core. DNA winds around this protein core, with the basic amino acids of the histones interacting with the negatively charged phosphate groups of the DNA. Approximately 146 base pairs of DNA wrap around a histone core to make up a nucleosome particle, the repeating structural motif of chromatin.
Csordas, Biochem. J., 286: 23-38 (1990) teaches that histones are subject to posttranslational acetylation of the α,ε-amino groups of N-terminal lysine residues, a reaction that is catalyzed by histone acetyl transferase (HAT1). Acetylation neutralizes the positive charge of the lysine side chain, and is thought to impact chromatin structure. Indeed, Taunton et al., Science, 272: 408-411 (1996), teaches that access of transcription factors to chromatin templates is enhanced by histone hyperacetylation. Taunton et al. further teaches that an enrichment in underacetylated histone H4 has been found in transcriptionally silent regions of the genome.
Histone acetylation is a reversible modification, with deacetylation being catalyzed by a family of enzymes termed histone deacetylases (HDACs). Grozinger et al., Proc. Natl. Acad. Sci. USA, 96: 4868-4873 (1999), teaches that HDACs are divided into two classes, the first represented by yeast Rpd3-like proteins, and the second represented by yeast Hda1-like proteins. Grozinger et al. also teaches that the human HDAC1, HDAC2, and HDAC3 proteins are members of the first class of HDACs, and discloses new proteins, named HDAC4, HDAC5, and HDAC6, which are members of the second class of HDACs. Kao et al., Genes & Dev., 14: 55-66 (2000), discloses HDAC7, a new member of the second class of HDACs. More recently, Hu et al. J. Bio. Chem. 275:15254-13264 (2000) and Van den Wyngaert, FEBS, 478: 77-83 (2000) disclose HDAC8, a new member of the first class of HDACs.
Richon et al., Proc. Natl. Acad. Sci. USA, 95: 3003-3007 (1998), discloses that HDAC activity is inhibited by trichostatin A (TSA), a natural product isolated from Streptomyces hygroscopicus, and by a synthetic compound, suberoylanilide hydroxamic acid (SAHA). Yoshida and Beppu, Exper. Cell Res., 177: 122-131 (1988), teaches that TSA causes arrest of rat fibroblasts at the G1 and G2 phases of the cell cycle, implicating HDAC in cell cycle regulation. Indeed, Finnin et al., Nature, 401: 188-193 (1999), teaches that TSA and SAHA inhibit cell growth, induce terminal differentiation, and prevent the formation of tumors in mice. Suzuki et al., U.S. Pat. No. 6,174,905, EP 0847992, JP 258863/96, and Japanese Application No. 10138957, disclose benzamide derivatives that induce cell differentiation and inhibit HDAC. Delorme et al., WO 01/38322 and PCT/IB01/00683, disclose additional compounds that serve as HDAC inhibitors.
The molecular cloning of gene sequences encoding proteins with HDAC activity has established the existence of a set of discrete HDAC enzyme isoforms. Some isoforms have been shown to possess specific functions, for example, it has been shown that HDAC-6 is involved in modulation of microtubule activity. However, the role of the other individual HDAC enzymes has remained unclear.
These findings suggest that inhibition of HDAC activity represents a novel approach for intervening in cell cycle regulation and that HDAC inhibitors have great therapeutic potential in the treatment of cell proliferative diseases or conditions. To date, few inhibitors of histone deacetylase are known in the art.
………………..
http://www.google.com/patents/WO2011112623A1?cl=en

Mocetinostat (MGCD-0103)
N-(2-aminophenyl)-4-[[(4-pyridin-3-ylpyrimidin-2-yl)amino]methyl^^
…………………………
http://www.google.co.in/patents/US6897220
Example 426 Synthesis of N-(2-Amino-phenyl)-4-[(4-pyridin-3-pyrimidin-2-ylamino)-methyl]-benzamide

Step 1: Synthesis of 4-Guanidinomethyl-benzoic acid methyl ester Intermediate 1
The mixture of 4-Aminomethyl-benzoic acid methyl ester HCl (15.7 g, 77.8 mmol) in DMF (85.6 mL) and DIPEA (29.5 mL, 171.2 mmol) was stirred at rt for 10 min. Pyrazole-1-carboxamidine HCl (12.55 g, 85.6 mmol) was added to the reaction mixture and then stirred at rt for 4 h to give clear solution. The reaction mixture was evaporated to dryness under vacuum. Saturated NaHCO3 solution (35 mL) was added to give nice suspension. The suspension was filtered and the filter cake was washed with cold water. The mother liquid was evaporated to dryness and then filtered. The two solids were combined and re-suspended over distilled H2O (50 ml). The filter cake was then washed with minimum quantities of cold H2O and ether to give 12.32 g white crystalline solid intermediate 1 (77% yield, M+1: 208 on MS).
Step 2: Synthesis of 3-Dimethylamino-1-pyridin-3-yl-propenone Intermediate 2
3-Acetyl-pyridine (30.0 g, 247.6 mmol) and DMF dimethyl acetal (65.8 mL, 495.2 mmol) were mixed together and then heated to reflux for 4 h. The reaction mixture was evaporated to dryness and then 50 mL diethyl ether was added to give brown suspension. The suspension was filtered to give 36.97 g orange color crystalline product (85% yield, M+1: 177 on MS).
Step 3: Synthesis of 4-[(4Pyridin-3-pyrimidin-2-ylamino)-methyl]benzoic acid methyl ester Intermediate 3
Intermediate 1 (0.394 g, 1.9 mmol) and intermediate 2 (0.402 g, 2.3 mmol) and molecular sieves (0.2 g, 4A, powder, >5 micron) were mixed with isopropyl alcohol (3.8 mL). The reaction mixture was heated to reflux for 5 h. MeOH (50 mL) was added and then heated to reflux. The cloudy solution was filtrated over a pad of celite. The mother liquid was evaporated to dryness and the residue was triturated with 3 mL EtOAc. The suspension was filtrated to give 0.317 g white crystalline solid Intermediate 3 (52%, M+1: 321 on MS).
Step 4: Synthesis of N-(2-Amino-phenyl)-4-[(4-pyrymidin-2-ylamino)-methyl]-benzamide
Intermediate 3 (3.68 g, 11.5 mmol) was mixed with THF (23 mL), MeOH (23 mL) and H2O (11.5 mL) at rt. LiOH (1.06 g, 25.3 mmol) was added to reaction mixture. The resulting reaction mixture was warmed up to 40° C. overnight. HCl solution (12.8 mL, 2N) was added to adjust pH=3 when the mixture was cooled down to rt. The mixture was evaporated to dryness and then the solid was washed with minimum quantity of H2O upon filtration. The filter cake was dried over freeze dryer to give 3.44 g acid of the title compound (95%, M+1: 307 on MS).
Acid (3.39 g, 11.1 mmol) of the title compound, BOP (5.679 g, 12.84 mmol) and o-Ph(NH2)2 (2.314 g, 21.4 mmol) were dissolved in the mixture of DMF (107 mL) and Et3N (2.98 mL, 21.4 mmol). The reaction mixture was stirred at rt for 5 h and then evaporated to dryness. The residue was purified by flash column (pure EtOAc to 5% MeOH/EtOAc) and then interested fractions were concentrated. The final product was triturated with EtOAc to give 2.80 g of title product
(66%, MS+1: 397 on MS).
1H NMR (400 MHz, DMSO-D6) δ (ppm): 9.57 (s, 1H), 9.22 (s, 1H), 8.66 (d, J=3.5 Hz, 1H), 8.39 (d, J=5.1 Hz, 2H), 8.00 (t, J=6.5 Hz, 1H), 7.90 (d, J=8.2 Hz, 2H), 7.50 (m, 3H), 7.25 (d, J=5.1 Hz, 1H), 7.12 (d, J=7.4 Hz, 1H), 6.94 (dd, J=7.0, 7.8 Hz, 1H), 6.75 (d, J=8.2 Hz, 1H), 6.57 (dd, J=7.0, 7.8 Hz, 1H), 4.86 (s, 2H), 4.64 (d, J=5.9 Hz, 2H).
References
- “Pharmion Corporation (PHRM) Release: Clinical Data On Oncology HDAC Inhibitor MGCD0103, Presented At The American Society of Clinical Oncology 42nd Annual Meeting” (Press release). Colorado, United States: BioSpace. June 6, 2006.
- Gelmon, K.; Tolcher, A.; Carducci, M.; Reid, G. K.; Li, Z.; Kalita, A.; Callejas, V.; Longstreth, J. et al. (2005). “Phase I trials of the oral histone deacetylase (HDAC) inhibitor MGCD0103 given either daily or 3x weekly for 14 days every 3 weeks in patients (pts) with advanced solid tumors”. J. Clin. Oncol. 2005 ASCO Annual Meeting. 23 (16S). 3147.
- MethylGene to Resume Development of its HDAC Inhibitor, MGCD0103 (Mocetinostat), Sept 2009
- “METHYLGENE TO RESUME DEVELOPMENT OF ITS HDAC INHIBITOR, MGCD0103 (MOCETINOSTAT)”. 21 Sep 2009.
- “Final Phase 2 Clinical Data for Mocetinostat (MGCD0103) in Relapsed/Refractory Hodgkin Lymphoma Patients”. 6 Dec 2010.
- Pfefferli, Catherine; Müller, Fritz; Ja¿wi¿ska, Anna; Wicky, Chantal (2014). “Specific NuRD components are required for fin regeneration in zebrafish”. BMC Biol. 12 (30). doi:10.1186/1741-7007-12-30. PMID 24779377.

- MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo
|
3-20-2009
|
THERAPEUTIC COMBINATIONS AND METHODS FOR CARDIOVASCULAR IMPROVEMENT AND TREATING CARDIOVASCULAR DISEASE
|
|
|
10-3-2008
|
COMBINATION OF ERa+ LIGANDS AND HISTONE DEACETYLASE INHIBITORS FOR THE TREATMENT OF CANCER
|
|
|
12-21-2007
|
Assay for efficacy of histone deacetylase inhibitors
|
|
|
5-25-2005
|
Inhibitors of histone deacetylase
|
|
2-8-2012
|
HDAC INHIBITORS AND HORMONE TARGETED DRUGS FOR THE TREATMENT OF CANCER
|
|
|
6-3-2011
|
Sequential Administration of Chemotherapeutic Agents for Treatment of Cancer
|
|
|
5-6-2011
|
METHODS FOR TREATING OR PREVENTING COLORECTAL CANCER
|
|
|
1-12-2011
|
Inhibitors of histone deacetylase
|
|
|
1-12-2011
|
Inhibitors of Histone Deacetylase
|
|
|
11-24-2010
|
Inhibitors of histone deacetylase
|
|
|
3-5-2010
|
INTRAOCULAR PRESSURE-LOWERING AGENT COMPRISING COMPOUND HAVING HISTONE DEACETYLASE INHIBITOR EFFECT AS ACTIVE INGREDIENT
|
|
|
6-12-2009
|
Administration of an Inhibitor of HDAC and an mTOR Inhibitor
|
|
|
5-22-2009
|
Combinations of HDAC Inhibitors and Proteasome Inhibitors
|
|
|
5-15-2009
|
Combination Therapy
|
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
BELINOSTAT, FAST TRACK, ORPHAN DRUG, A hydroxamate-type inhibitor of histone deacetylase.

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
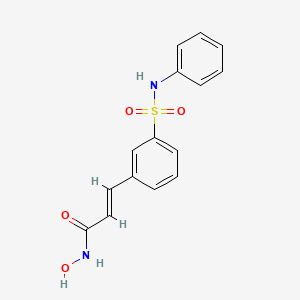
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


