
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
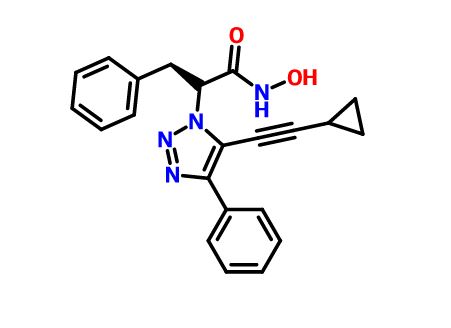

CAS 1620779-53-4
MF C22H20N4O2, MW 372.4
(S)-2-(5-(cyclopropylethynyl)-4-phenyl-1H-1,2,3-triazol-1-yl)-N-hydroxy-3-phenylpropanamide
1H-1,2,3-Triazole-1-acetamide, 5-(2-cyclopropylethynyl)-N-hydroxy-4-phenyl-α-(phenylmethyl)-, (αS)-
| Applicants: | TRUSTEES OF BOSTON UNIVERSITY DANA-FARBER CANCER INSTITUTE, INC. |
| Inventors: | Aaron Beaty BEELER John A. PORCO, JR. Oscar J. INGHAM James E. BRADNER |
|
As histone proteins bind DNA prior to transcription, their biochemical action plays a critical role in the regulation of gene expression and cellular differentiation. Histone deacetylases (HDACs) are an important family of proteins predominantly responsible for specific posttranslational modifications of histone proteins, the chief organizational component of chromatin. HDACs catalyze the removal of acetyl groups from histones and other cellular proteins. HDAC-mediated deacetylation of chromatin-bound histones regulates the expression of a variety of genes throughout the genome. Importantly, HDACs have been linked to cancer, as well as other health conditions. To date, eleven major HDAC isoforms have been described (HDACs 1-11). HDACs are categorized into two classes. Class I HDACs include HDAC1, HDAC2, HDAC3, HDAC8 and HDAC11. Class II HDACs include HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10. HDAC’s are validated targets for a number of disease states, including cancer, neurodegenerative diseases, sickle-cell anemia, muscular dystrophy, and HIV. There are currently two HDAC inhibitors on the market, Vorniostat and Romidepsin. Both are approved for treatment of T-cell lymphoma. However, they are both pan active inhibitors showing very little specificity of binding to HDAC subclasses. Because of this lack of specificity they have a number of side effects.
|
|
Non-selective HDAC inhibitors effect deacetylase activity of most, if not all, of the HDACs. The mechanisms of the anticancer effects of SAHA, a non-selective HDAC inhibitor, are not completely understood, and likely result from both altered gene expression and altered function of proteins regulating cell proliferation and cell death pathways. Non-selective HDAC inhibitors, such as SAHA, induce the accumulation of acetylated histone proteins and non histone proteins.
|
|
Small molecule HDAC inhibitors that are isoform-selective are useful as therapeutic agents with reduced toxicity and as tools for probing the biology of the HDAC isoforms. The present disclosure is related, in part to small molecules that are selective HDAC inhibitors.
|


1H NMR (500 MHz, d4-MeOD) 0.80 (2H, m), 0.98 (2H, m), 1.47 (1H, m), 3.51 (1H, dd, J = 11.2, 14.2 Hz), 3.71 (1H, dd, J = 3.9, 14.2 Hz), 5.49 (1H, dd, J = 3.9, 11.2 Hz), 6.96 (2H, m), 7.17-7.20 (3H, m), 7.37 (1H, t, J = 7.3 Hz), 7.43 (2H, t, J = 7.3 Hz), 7.99 (2H, d, J = 8.8 Hz);

13C NMR (100 MHz, d4-MeOD) 0.02, 8.55, 37.07, 60.83, 62.59, 109.09, 118.98, 125.9, 127.16, 128.55, 128.65, 128.71, 129.16, 130.07, 136.09, 147.10, 165.20;
HRMS calculated for C22H21N4O2 + (M+H): 373.1659, found: 373.1665.

PATENT
WO2014116962
https://www.google.com/patents/WO2014116962A1?cl=en
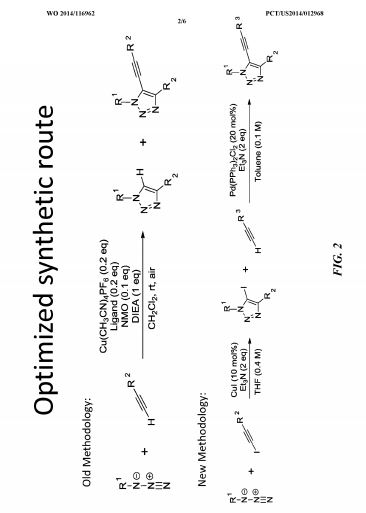
SAR. libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in Fig. 2.
In one study, compound 
was synthesized as outline in Scheme I.
Scheme I




PATENT
|
SAR libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in FIG. 2. In one study, compound
|
 was synthesized as outline in Scheme I.
was synthesized as outline in Scheme I.


|
The HDAC assays were carried out as described in Bowers A, West N, Taunton J, Schreiber S L, Bradner J E, Williams R M Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 11219-11222. Assay results revealed that among the analogues tested a cyclopropane analog was the most active at 0.4 nM (>1000 fold selectivity). These results demonstrated that a small aliphatic group in the 5-position on the triazole can increase potency. Also, compounds with an L-phenylalanine moiety at the 3-position showed significant potency. To expand our understanding of how the molecule interacts with the binding pocket of HDAC 8 and to understand our preliminary SAR, molecular modeling was carried out. The phenyl group from the original amino methyl ester fits snuggly into the Zn binding site and the alkynyl phenyl group sits flat in a hydrophobic groove. In summary, the inventors have developed a potent and highly selective small molecule which inhibits HDAC-8 at approximately 500 pM with over 1000-fold selectivity over HDAC-6 and significantly greater selectivity for all other HDACs. To inventors’ knowledge, to date there are no compounds with this level of potency and selectivity.
|
|
All patents and other publications identified in the specification and examples are expressly incorporated herein by reference for all purposes. These publications are provided solely for their disclosure prior to the filing date of the present application. Nothing in this regard should be construed as an admission that the inventors are not entitled to antedate such disclosure by virtue of prior invention or for any other reason. All statements as to the date or representation as to the contents of these documents is based on the information available to the applicants and does not constitute any admission as to the correctness of the dates or contents of these documents.
|
|
Although preferred embodiments have been depicted and described in detail herein, it will be apparent to those skilled in the relevant art that various modifications, additions, substitutions, and the like can be made without departing from the spirit of the invention and these are therefore considered to be within the scope of the invention as defined in the claims which follow. Further, to the extent not already indicated, it will be understood by those of ordinary skill in the art that any one of the various embodiments herein described and illustrated can be further modified to incorporate features shown in any of the other embodiments disclosed herein.
|
Paper

A novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8) has been discovered by the repurposing of a diverse compound collection. Medicinal chemistry optimization led to the identification of a highly potent (0.8 nM) and selective inhibitor of HDAC8.
Development of a Potent and Selective HDAC8 Inhibitor
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00239
file:///C:/Users/Inspiron/Downloads/ml6b00239_si_001.pdf
Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States

 Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.
Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.

Aaron Beeler
Aaron Beeler received his Ph.D. in 2002 from Professor John Rimoldi’s laboratory in the Department of Medicinal Chemistry at the University of Mississippi. He then joined the Porco group as a postodoctoral fellow and subsequently the Center for Chemical Methodology and Library Development at Boston University, now the Center for Molecular Discovery. He was promoted to Assistant Director of the CMLD-BU in January 2005. In 2012 Aaron joined the Department of Chemistry as a tenure-track professor in medicinal chemistry.
Degrees and Positions
- B.S. Belmont University, Biology,
- Ph.D. University of Mississippi, Medicinal Chemistry
Research
The Beeler Research Group is truly multidisciplinary, combining organic chemistry, engineering, and biology to solve problems in medicinal chemistry. All of these elements are combined and directed toward significant problems in human health. The Beeler Group is addressing focused disease areas (e.g., schizophrenia, Parkinson’s, cystic fibrosis), as well as project areas with broader impact potential (e.g., new methods for discovery of small molecules with anti-cancer properties).
- Medicinal Chemistry: The goals of medicinal chemistry projects are to optimize small molecules in order to: a) develop a probe that may be utilized as a tool in biological studies; b) develop a lead molecule to facilitate future therapeutics; and c) utilize small molecules to enhance understanding of biological targets that are important for human health. These projects provide students with training in organic chemistry, medicinal chemistry, and focused biology. Projects are selected based on their chemistry and/or biology significance and potential for addressing challenging questions.
- Technology: One of the core components of the research in the Beeler Group is development of technologies and paradigms that facilitate rapid modification of complex scaffolds. These technologies enable optimization of biologically active lead compounds and identification of small molecule leads in biological systems. The projects focus on utilizing automation, miniaturization, and microfluidics to carry out chemical transformations. These projects are highly interdisciplinary with both chemistry and engineering components.
- Photochemistry: This area focuses on photochemical transformations toward the synthesis of natural products, natural product scaffolds, and other complex chemotypes of interest to medicinal chemistry and chemical biology. The foundation of these projects is utilizing microfluidics to enable photochemical reaction development.
Techniques & Resources
Students in the Beeler Research Group will have opportunities to learn a number of exciting research disciplines. Organic synthesis will be at the heart of every project. This will include targeted synthesis, methodology development, and medicinal chemistry. Through collaborations with biological researchers and/or research projects carried out within the Beeler Group, students will learn methods for biological assays, pharmacology, and target identification. Many projects will also include aspects of engineering that will provide opportunities for learning techniques such as microfabrication and microfluidics.
Opportunities
It is becoming evident that successful and impactful science is realized in collaborative interdisciplinary environments. The Beeler Research Group’s multidisciplinary nature and collaborative projects provides opportunities to learn areas of research outside of traditional chemistry.
What’s Next for Graduates of the Beeler Group?
Members of the Beeler Research Group will be positioned for a wide range of future endeavors.
- Undergraduates will be prepared to enter into graduate school for organic chemistry, chemical biology, or chemical engineering or to start careers in industry;
- Graduate students will have the foundation required for postdoctoral studies in organic synthesis or chemical biology as well as an industrial career in biotech or pharma;
- Postdoctoral associates will gain training and experience critical for both academic and industrial careers.
Assistant Professor
Office: SCI 484C
Laboratory: SCI 484A
Phone: 617.358.3487
Fax: 617-358-2847
beelera@bu.edu
Office Hours: by Appointment
Beeler Group Homepage
Google Scholar Page
Oscar J. Ingham below

John A. PORCO, JR below


JAMES E. BRADNER, MD above
Dana-Farber Cancer Institute


 Ron Paranal
Ron Paranal
 Randolph A. Escobar
Randolph A. Escobar
 Han Yueh
Han Yueh
| US20090181943 * | Apr 9, 2008 | Jul 16, 2009 | Methylgene Inc. | Inhibitors of Histone Deacetylase |
| Reference | ||
|---|---|---|
| 1 | * | GERARD, B ET AL.: ‘Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal alkynes‘ TETRAHEDRON vol. 62, 12 May 2006, pages 6405 – 6411 |
| 2 | * | VANNINI, A ET AL.: ‘Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor.‘ PNAS, [Online] vol. 101, no. 42, 19 October 2004, pages 15064 – 15069 Retrieved from the Internet: <URL:http://www.pnas.org/content/101/42/15064> |
///////////epigenetic, HDAC, HDAC8, Histone deacetylase, histone deacetylase 8, triazole, PRECLINICAL, Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States, Oscar J. Ingham, Aaron Beeler
n1n(c(c(n1)c2ccccc2)C#CC3CC3)C(C(=O)NO)Cc4ccccc4

