
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Mekinist (trametinib)
JTP-74057, GSK212, GSK1120212
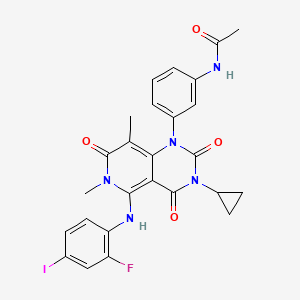
N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4, 3-d] pyrimidin-1-yl] phenyl}acetamide
| Molecular Weight | 615.39 |
| Formula | C26H23FIN5O4 |
| CAS Number | 871700-17-3 |

Trametinib (GSK1120212) is experimental cancer drug. It is a MEK inhibitor drug with anti-cancer activity.[1]
It inhibits MEK1 and MEK2.[1]
Trametinib had good results for V600E mutated metastatic melanoma in a phase III clinical trial.[2]
- Trametinib, NCI Drug Dictionary
- METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM).
GSK1120212 (JTP-74057) is a potent and selective allosteric inhibitor of the MEK1 and MEK2 (MEK1/2) enzymes with promising antitumor activity in a phase I clinical trial (ASCO 2010). GSK1120212 (JTP-74057) inhibits MEK1/2 kinase activity and prevents Raf-dependent MEK phosphorylation (S217 for MEK1), producing prolonged p-ERK1/2 inhibition. Potent cell growth inhibition was evident in most tumor lines with mutant BRAF or Ras. In xenografted tumor models, GSK1120212 orally dosed once daily had a long circulating half-life and sustained suppression of p-ERK1/2 for more than 24 hours; GSK1120212 also reduced tumor Ki67, increased p27 (Kip1/CDKN1B), and caused tumor growth inhibition in multiple tumor models.
May 29, 2013 —
GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Mekinist (trametinib) as a single-agent oral treatment for unresectable or metastatic melanoma in adult patients with BRAF V600E or V600K mutations. Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Mekinist is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma. It is also approved for patients with the V600K mutation, which makes up approximately 10 percent of all BRAF V600 mutations in metastatic melanoma.
Melanoma is the most serious and deadly form of skin cancer.[iii] According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States.[iv] When melanoma spreads in the body, the disease is called metastatic melanoma.[v] Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.2 One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis,while in the U.S., the five-year survival rate was 16 percent (2003-2009). The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Mekinist (trametinib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E and V600K mutations as detected by an FDA-approved test. Limitation of use: Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy.
(WO2005121142A1). Aniline a reaction with CDI was added cyclopropylamine get two , two and malonic acid cyclization get 3 . 3 chlorination with phosphorus oxychloride reaction with methylamine 4 , as well as byproducts 5 (ratio of 2:1). Mixture 4 + 5 and acid 6 crystals obtained after cyclization compound 7 (pure substance). 7 with activated trifluoromethanesulfonyl chloride to the amide 8 SNAr reaction occurs 9 , 9 in alkaline conditions rearrangement trimetazidine imatinib.

…………………
http://www.google.com/patents/WO2014039375A1?cl=en
The term “trametinib” as used herein means the MEK inhibitor represented by the structure of formula (I):

or a pharmaceutically acceptable salt or solvate thereof. Trametinib is preferably administered as a solvate in the form of N-{3-[3- cyclopropyl-5-(2-fluoro-4-iodo-phenylamino)6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydro- 2H-pyrido[4,3-d]pyrimidin-1 -yl]phenyl}acetamide dimethyl sulfoxide (solvate).
Depending on naming convention, the compound of formula (I) may also properly be referred to as N-{3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7- trioxo-3,4,6,7-tetrahydropyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl}acetamide.
Trametinib is disclosed and claimed, along with pharmaceutically acceptable salts thereof, and also as solvates thereof, as being useful as an inhibitor of MEK activity, particularly in treatment of cancer, in WO 2005/121 142. Trametinib can be prepared as described in WO 2005/121 142.
Suitably, trametinib is in the form of a dimethyl sulfoxide solvate. Suitably, trametinib is in the form of a sodium salt. Suitably, trametinib is in the form of a solvate selected from: hydrate, acetic acid, ethanol, nitromethane, chlorobenzene, 1 -pentancol, isopropyl alcohol, ethylene glycol and 3-methyl-1 -butanol. These solvates and salt forms can be prepared by one of skill in the art from the description in WO2005/121 142.
…………………….
http://www.google.com/patents/WO2005121142A1?cl=en
Example 3-10 By treating N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4 , 3-d]pyrimidin-1-yl] phenyl Jmethanesulfonamide 46 according to conventional methods, sodium salt and potassium salt thereof were obtained.
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide sodium salt:
^-NMR (DMSO-de, 300 MHz) δ 0.47 (brs, 2H) , 0.70-0.90 (m, 2H) , 1.23(s, 3H) , 2.35(brs, IH) , 2.82(s, 3H) , 3.22(s, 3H) , 6.69(t, J=8.8Hz, IH) , 6.81 (d, J=8.1Hz , IH) , 6.98 (s, IH) , 7.02 (d, J=8.8Hz , IH) , 7.10-7.30 (m, 2H) , 7.38(d, J=9.2Hz , IH) , 10.22(brs, IH) . MS (ESI) m/z 652 [MH]+.
N-{3-[3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide potassium salt: Example 4-1
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 ,8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1-yl] – phenyl}-acetamide Step 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) rea

47 48 Under a nitrogen atmosphere, to N,N-carbonyldiimidazole (39.9 g) were added N,N-dimethylformamide (200 ml) and triethylamine (34.3 ml) and a solution of 2-fluoro-4-iodoaniline 47 (48.5 g) in N,N-dimethylformamide (50 ml) was added dropwise with stirring under ice-cooling. After the completion of the dropwise addition, the mixture was stirred at room temperature for 18 hrs. The reaction mixture was ice-cooled, and cyclopropylamine (21.3 ml) was added dropwise. The reaction mixture was stirred at room temperature for 1 hr and added dropwise to water-toluene [2:1 (volume ratio), 750 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4-iodophenyl) urea 48 (61.3 g, yield 93.4%) as colorless crystals. Step 2 Synthesis of l-cyclopropyl-3- (2-fluoro-4- iodophenyl) pyrimidine-2 , 4 , 6-trione

To l-cyclopropyl-3- (2-fluoro-4-iodophenyl) urea 48 (61.0 g) obtained in Step 1 and malonic acid 4 (19.9 g) were added acetic anhydride (300 ml) and acetyl chloride (27.2 ml), and the mixture was stirred under a nitrogen atmosphere at 60°C for 3 hrs. After allowing to cool to room temperature, the reaction mixture was added dropwise to water-toluene [2:1 (volume ratio), 900 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4- iodophenyl)pyrimidine-2,4, 6-trione 49 (60.9 g, yield 82%) as pale-yellow crystals.
Step 3 Synthesis of 6-chloro-3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -lH-pyrimidine-2 , -dione

49 50 51 To l-cyclopropyl-3- (2-fluoro-4-iodophenyl) -pyrimidine-
2, 4, 6-trione 49 (59.0 g) obtained in Step 2 were added phosphorus oxychloride (85.0 ml) and dimethylaniline (29.0 ml), and water (8.3 ml) was added dropwise to the mixture at room temperature with stirring. After the completion of the dropwise addition, the mixture was stirred with heating at 110°C for 1 hr. After allowing to cool to room temperature, the reaction mixture was added dropwise to ice water-toluene [2:1 (volume ratio), 900 ml] with stirring. The mixture was stirred at room temperature for 1 hr. The organic layer was separated, and washed successively with water (300 ml) and brine (300 ml) . Anhydrous magnesium sulfate and activated carbon were added and the mixture was stirred. Anhydrous magnesium sulfate and activated carbon were filtered off, and the filtrate was concentrated under reduced pressure to give a 1:2 mixture (62.9 g) of 6-chloro-3-cyclopropyl-l- (2- fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 50 and 6-chloro-l- cyclopropyl-3- (2-fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 51 as a yellow foamy oil, which was used for the next step without purification.
Step 4 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6- methylamino-lH-pyrimidine-2 , 4-dione

To a 1:2 mixture (62.9 g) of 6-chloro-3-cyclopropyl-l- (2- fluoro-4-iodophenyl) -lH-pyrimidine-2 ,4-dione 50 and 6-chloro-l- cyclopropyl-3- (2-fluoro-4-iodophenyl) -lH-pyrimidine-2 , -dione 51 obtained in Step 3 were added methanol (189 ml) and a solution (126 ml) of 40% methylamine in methanol, and the mixture was stirred at room temperature for 2 hrs . The precipitated crystals were filtered off and the filtrate was concentrated under reduced pressure. The residue was extracted with chloroform (200 ml) and water (200 ml) , and the organic layer was washed with brine (200 ml) and dried over anhydrous magnesium sulfate. Anhydrous magnesium sulfate was filtered off and the filtrate was concentrated under reduced pressure to give a 2:1 mixture (34.55 g) of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6-methylamino-lH- pyrimidine-2 ,4-dione 52 and l-cyclopropyl-3- (2-fluoro-4- iodophenyl) -6-methylamino-lH-pyrimidine-2,4 ,-dione 53 as yellow crystals, which were used for the next step without purification. Step 5 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -5- hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 , 3-d] pyrimidine-2 , 4 , 7-trione

To a 2:1 mixture (34.6 g) of 3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6-methylamino-lH-pyrimidine-2, 4-dione 52 and 1- cyclopropyl-3- (2-fluoro-4-iodo-phenyl) 6-methylamino-lH- pyrimidine-2,4,-dione 53 obtained in Step 4, and 2-methylmalonic acid 54 (10.2 g) was added acetic anhydride (173 ml) , and the mixture was stirred at 100°C for 2 hrs. After allowing to cool to room temperature, the reaction mixture was concentrated under reduced pressure. Acetone (104 ml) was added to the residue, and the mixture was stirred with heating under reflux for 30 min. After allowing to cool to room temperature, the precipitated crystals were collected by filtration and dried to give 3- cyclopropyl-1- (2-fluoro-4-iodophenyl) -5-hydroxy-6 , 8-dimethyl- lH,8H-pyrido [2, 3-d] pyrimidine-2, 4, 7-trione 55 (15.1 g, yield from 48, 21%) as colorless crystals.
Step 6 Synthesis of trifluoromethanesulfonic acid 3-cyclopropyl- 1- (2-fluoro-4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

55 56 43 Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro-
4-iodophenyl) -5-hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2,3- d] pyrimidine-2 ,4 ,7-trione 55 (33.0 g) obtained in Step 5 were added chloroform (165 ml) and 2 , 6-lutidine (10.4 ml), and trifluoromethanesulfonic anhydride 56 (14.4 ml) was added dropwise under ice-cooling with stirring. After the completion of the dropwise addition, the mixture was stirred at same temperature for 30 min and at room temperature for 2 hrs. The reaction mixture was washed successively with aqueous sodium hydrogen carbonate (165 ml) , IN hydrochloric acid (165 ml) and brine (165 ml) and dried over anhydrous magnesium sulfate. Anhydrous magnesium sulfate was filtered off and the filtrate was concentrated under reduced pressure. 2-Propanol (198 ml) was added to the residue, and the mixture was stirred with heating under reflux, and allowed to return to room temperature. The crystals were collected by filtration and dried to give trifluoromethanesulfonic acid 3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (31.9 g, yield 93%) as colorless crystals.
Step 7 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 ,4 , 7-trioxo-l ,2,3,4,7 , 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-ylamino] phenyl } acetamide

To trifluoromethanesulfonic acid 3-cyclopropyl-l- (2-fluoro-
4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (25.0 g) obtained in Step 6 and 3 ‘-aminoacetanilide 57 (7.33 g) were added N,N- dimethylacetamide (50.0 ml) and 2,6-lutidine (5.68 ml), and the mixture was stirred at 130°C for 5 hrs. After allowing to cool to room temperature, methanol-water [1:2 (volume ratio), 150 ml] was added with stirring. The crystals were collected by filtration and dried to give N- {3- [3-cyclopropyl-l- (2-fluoro-4-iodophenyl) – 6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro-pyrido [2,3- d]pyrimidin-5-ylamino] phenyl}acetamide 58 (24.8 g, yield 99%) as colorless crystals. Step 8 Synthesis of N- { 3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4″, 7-trioxo-3 , 4 , 6 , 7-tetrahydro-2H- pyrido [4 , 3 -d]pyrimidin-l-yl] phenyl} acetamide

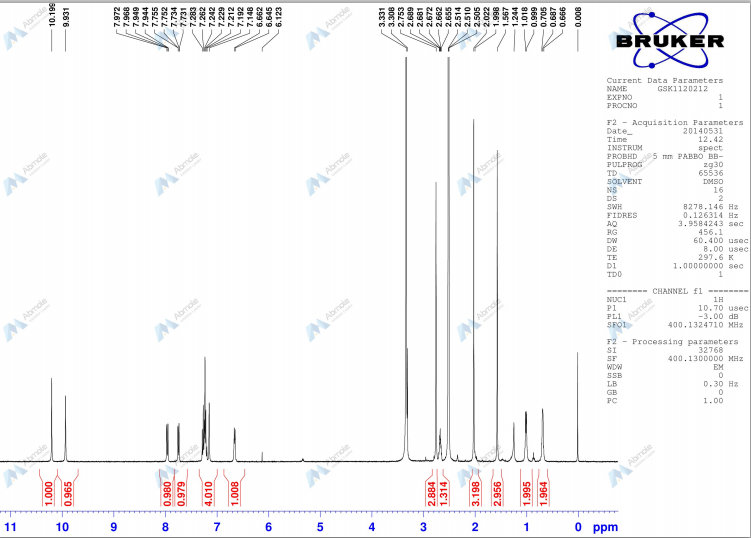
Under a nitrogen atmosphere, to a solution (1.57 g) of 28% sodium methoxide in methanol was added tetrahydrofuran (40 ml) , N- {3- [3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro-pyrido [2 , 3-d] pyrimidin-5- ylamino]phenyl}acetamide 58 (5.00 g) obtained in Step 7 was added, and the mixture was stirred at room temperature for 4 hrs. Acetic acid (0.56 ml) was added, and the mixture was stirred at room temperature for 30 min. Water (40 ml) was added and the mixture was further stirred for 1 hr. The crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4, 3-d] pyrimidin-1-yl] phenyl}acetamide 59 (4.75 g, yield 95%) as colorless crystals. MS ESI m/e: 616 (M+H) , 614 (M-H) .
1H-NMR(DMSO-d6, 400MHz) δ 0.63-0.70 (m, 2H) , 0.91-1.00 (m, 2H) , 1.25(s, 3H) , 2.04(s, 3H) , 2.58-2.66(m, IH) , 3.07(s, 3H) , 6.92(t,
J=8.8Hz, IH) , 7.00-7.05 (m, IH) , 7.36 (t, J=8.2Hz , IH) , 7.52-7.63 (m,
3H) , 7.79(dd, J=2.0 , 10.4Hz, IH) , 10.10(s, IH) , 11.08(s, IH) .
Example 4-1 (alternative method)
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d] pyrimidin-1-yl] – phenyl } -acetamide
Step 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -urea

48 47 Under a nitrogen atmosphere, to N,N-carbonyldiimidazole (82.1 g) were added N, N-dimethylformamide (400 ml) and triethylamine (70.5 ml) , and a solution of 2-fluoro-4-iodoaniline 47 (100 g) in N, N-dimethylformamide (100 ml) was added dropwise under ice-cooling. After the completion of the dropwise addition, the mixture was stirred at room temperature for 5 hrs . The reaction mixture was ice-cooled, and cyclopropylamine (44.0 ml) was added dropwise. The mixture was stirred at room temperature for 1 hr, and the reaction mixture was added dropwise to water- toluene [2:1 (volume ratio) , 1500 ml] with stirring. The precipitated crystals were collected by filtration and dried to give l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -urea 48 (129 g, yield 95.5%) as colorless crystals. Step 2 Synthesis of 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro- 4-iodo-phenyl) -urea

48 73 74 Under a nitrogen atmosphere, to l-cyclopropyl-3- (2-fluoro- 4-iodo-phenyl) -urea 48 (167 g) and cyanoacetic acid 73 (80.0 g) , was added N, N-dimethylformamide (836 ml) , and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at room temperature. The mixture was stirred at room temperature for 4 hrs. The reaction mixture was cooled with water, and water- isopropanol [2:1 (volume ratio) , 1670 ml] was added dropwise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) – urea 74 (192 g) .
Step 3 Synthesis of 6-amino-3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -lH-pyrimidine-2 , 4-dione

74 75 To 1- (2-cyano-acetyl) -l-cyclopropyl-3- (2-fluoro-4-iodo- phenyl)-urea 74 (192 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml) , and the mixture was stirred with heating at 80°C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -1H- pyrimidine-2, 4-dione 75 (178g, yield from 48, 88%) as pale-yellow crystals . Step 4 Synthesis of N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) – 2 , 6-dioxo-l ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N ,N-dimethyl- formamidine

75 76 Under a nitrogen atmosphere, to 6-amino-3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -lH-pyrimidine-2 , 4-dione 75 (178 g) were added N,N-dimethylformamide (356 ml) and N,N-dimethylformamide dimethylacetal (178 ml) , and the mixture was stirred at room temperature for 2 hrs. Isopropanol (178 ml) was added with stirring at room temperature, and water (1068 ml) was added dropwise. The mixture was stirred at room temperature for 2 hrs, and the precipitated crystals were collected by filtration and dried to give N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -2,6- dioxo-1 ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N,N-dimethyl-formamidine 76 (188 g, yield 92%) as yellow crystals.
Step 5 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6- methylamino- lH-pyrimidine-2 ,4-dione

76 52 Under a nitrogen atmosphere, to t-butanol-ethanol [2:1 (volume ratio) , 250 ml] was added sodium borohydride (6.41 g) , and the mixture was stirred at room temperature for 1 hr. Under water-cooling, N’- [l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -2 ,6- dioxo-1 ,2,3, 6-tetrahydro-pyrimidin-4-yl] -N,N-dimethy1-formamidine 76 (50.0 g) was added, and the mixture was stirred for 2.5 hrs. Under water-cooling, water (225 ml) and 10% aqueous citric acid solution (175 ml) were successively added dropwise, and the mixture was stirred for 3 hrs . The precipitated crystals were collected by filtration and dried to give crude crystals (34.5 g, LC purity 91%) of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6- methylamino- lH-pyrimidine-2 ,4-dione 52, which were used for the next reaction without purification. Step 6 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -5- hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 , 3-d] pyrimidine-2 ,4 ,7-trione

Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro- 4-iodo-phenyl) -6-methylamino-lH-pyrimidine-2, -dione 52 (34.4 g) and 2-methyl-malonic acid 54 (15.2 g) was added acetic anhydride (34.4 ml) , and the mixture was stirred with heating at 100°C for 3 hrs. After allowing to cool to 50°C, acetone (68.8 ml) was added dropwise, and the mixture was stirred as it was for 30 min. Water (172 ml) was further added dropwise, and the mixture was stirred for 1 hr. After allowing to cool to room temperature with stirring, the precipitated crystals were collected by filtration and dried to give crude crystals (37.7 g, LC purity 91%) of 3- cyclopropyl-1- (2-fluoro-4-iodo-phenyl) -5-hydroxy-6 , 8-dimethyl- lH,8H-pyrido [2 ,3-d] pyrimidine-2 ,4 ,7-trione 55. Isopropanol (92.0 ml) was added to the obtained crude crystals (30.7 g) , and the mixture was stirred at room temperature for 4 hrs . The crystals were collected by filtration and dried to give 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -5-hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2 ,3- d] pyrimidine-2 ,4 ,7-trione 55 (25.9 g, yield from 76, 58%) as pale-yellow crystals.
Step 7 Synthesis of p-toluenesulfonic acid 3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 , , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

55 11 77 Under a nitrogen atmosphere, to 3-cyclopropyl-l- (2-fluoro- 4-iodo-phenyl) -5-hydroxy-6 ,8-dimethyl-lH,8H-pyrido [2,3- d] pyrimidine-2 ,4,7-trione 55 (23.9 g) was added acetonitrile (167 ml) , and the mixture was stirred under ice-cooling. Triethylamine (11.0 ml) and trimethylamine hydrochloride (2.37 g) were added, and a solution of p-toluenesulfonyl chloride 11 (12.3 g) in acetonitrile (72.0 ml) was added dropwise. The mixture was stirred under ice-cooling for 1 hr, and stirred at room temperature for 3 hrs. Methanol (239 ml) was added, and the mixture was stirred at room temperature for 1 hr. The crystals were collected by filtration and dried to give p-toluenesulfonic acid 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro-pyrido [2 ,3-d]pyrimidin-5-yl ester 77 (28.7 g, yield 91%) as colorless crystals.
Step 8 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 ,3-d]pyrimidin-5-ylamino] -phenyl}-acetamide

To p-toluenesulfonic acid 3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -6 , 8-dimethyl-2 ,4,7- trioxo-1 ,2,3,4,7, 8-hexahydro- pyrido [2,3-d]pyrimidin-5-yl ester 77 (28.0 g) and 3′- aminoacetanilide 57 (13.2 g) were added N,N-dimethylacetamide (84.0 ml) and 2,6-lutidine (15.3 ml), and the mixture was stirred at 130°C for 4 hrs. After allowing to cool with stirring, methanol (196 ml) was added dropwise, and the mixture was stirred at room temperature. The crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) – 6 , 8-dimethyl-2 , , 7-trioxo-l ,2,3,4,7, 8-hexahydro-pyrido [2,3- d]pyrimidin-5-ylamino] -phenyl}-acetamide 58 (25.2 g, yield 93%) as colorless crystals. Step 9 Synthesis of N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodo- phenylamino) -6 , 8-dimethyl-2 ,4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4 ,3-d] pyrimidin-1-yl] -phenyl}-acetamide

58 59 Under a nitrogen atmosphere, to N-{3- [3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 ,4 , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 ,3-d]pyrimidin-5-ylamino] -phenyl }-acetamide 58 (45.7 g) was added tetrahydrofuran (366 ml), and a solution (15.7 g) of 28% sodium methoxide in methanol was added dropwise with stirring at room temperature and the mixture was stirred at room temperature for 4 hrs. Acetic acid (5.61 ml) was added, and the mixture was stirred at room temperature for 30 min. With stirring at 70°C in an oil bath, water (366 ml) was added dropwise, and the mixture was stirred for 1 hr. After allowing to cool with stirring, the crystals were collected by filtration and dried to give crystal 1 (46.0 g) of N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodo-phenylamino) -6 , 8-dimethyl-2 ,4 , 7-trioxo-3 ,4,6, 7-tetrahydro- 2H-pyrido [4 , 3-d]pyrimidin-1-yl] -phenyl}-acetamide 59. N,N-Dimethylacetamide (184 ml) was added to crystal 1 (46.0 g) , and the mixture was stirred with heating at 130°C. After complete dissolution, the solution was filtered by suction using with paper (5B) , and washed with N,N-dimethylacetamide (92.0 ml).
The filtrate was stirred under heating at 130°C, 1-butanol (138 ml) and water (96.0 ml) were successively added dropwise, and the mixture was stirred for 30 min. Water (46.0 ml) was further added dropwise, and the mixture was stirred for 30 min allowed to cool with stirring. The crystals were collected by filtration and dried to give crystal 2 (41.7 g) of N-{3- [3-cyclopropyl-5- (2- fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [ ,3-d]pyrimidin-l-yl] -phenyl}-acetamide 59 as colorless crystals. To crystal 2 (41.5 g) was added 1-butanol-water [19:1 (volume ratio) , 415 ml] , and the mixture was stirred at 130°C for 18 hrs. After allowing to cool with stirring, the crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [4 ,3-d]pyrimidin-1-yl] -phenyl}-acetamide 59 (40.7 g, yield 89%) as colorless crystals
Combination Small Molecule MEK and PI3K Inhibition Enhances Uveal Melanoma Cell Death in a Mutant GNAQ- and GNA11-Dependent Manner.
Khalili JS et al. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. PMID: 22733540.
Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212.
Jing J et al. Mol Cancer Ther. 2012 Mar;11(3):720-9. PMID: 22169769.
Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo.
Yamaguchi T et al. Int J Oncol. 2011 Jul;39(1):23-31. PMID: 21523318.
GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition.
Gilmartin AG et al. Clin Cancer Res. 2011 Mar 1;17(5):989-1000. PMID: 21245089.
//////


