
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
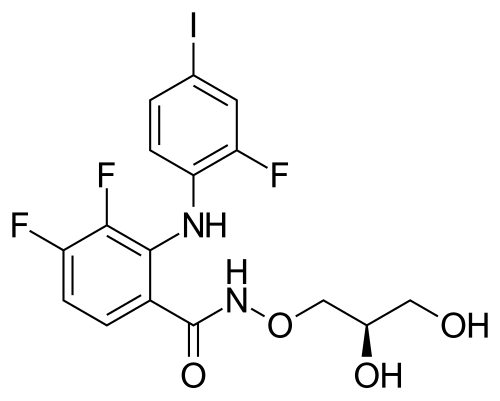

MIRDAMETINIB
391210-10-9
Chemical Formula: C16H14F3IN2O4
Molecular Weight: 482.19
PD0325901; PD 0325901; PD-325901; mirdametinib
FDA APPROVED 2/11/2025, Gomekli, To treat neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection
IUPAC/Chemical Name: (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino)benzamide
SpringWorks Therapeutics (a spin out of Pfizer ) is developing mirdametinib, a second-generation, non-ATP competitive, allosteric MEK1 and MEK2 inhibitor derived from CI-1040, for treating type 1 neurofibromatosis (NF1) and advanced solid tumors. In June 2021, a phase I/II trial was initiated in patients with low grade glioma.
- OriginatorPfizer
- DeveloperAstraZeneca; BeiGene; BIOENSIS; Pfizer; SpringWorks Therapeutics; St. Jude Childrens Research Hospital; University of Oxford
- ClassAniline compounds; Anti-inflammatories; Antineoplastics; Benzamides; Immunotherapies; Small molecules
- Mechanism of ActionMAP kinase kinase 1 inhibitors; MAP kinase kinase 2 inhibitors
- Orphan Drug StatusYes – Neurofibromatosis 1
- Phase IINeurofibromatosis 1
- Phase I/IIGlioma
- Phase ISolid tumours
- PreclinicalChronic obstructive pulmonary disease
- No development reportedCervical cancer
- DiscontinuedBreast cancer; Cancer; Colorectal cancer; Malignant melanoma; Non-small cell lung cancer
- 22 Jul 2021SpringWorks Therapeutics receives patent allowance for mirdametinib from the US Patent and Trademark Office for the treatment of Neurofibromatosis type 1-associated plexiform neurofibromas
- 16 Jun 2021SpringWorks Therapeutics and St. Jude Children’s Research Hospital agree to develop mirdametinib in USA for glioma
- 15 Jun 2021Efficacy and safety data from the phase IIb RENEU trial for Neurofibromatosis type 1-associated plexiform neurofibromas released by SpringWorks Therapeutics
Mirdametinib, sold under the brand name Gomekli, is a medication used for the treatment of people with neurofibromatosis type 1.[1] Mirdametinib is a kinase inhibitor.[1][2] It is taken by mouth.[1]
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib was approved for medical use in the United States in February 2025.[1][3]
SCHEME
SIDE CHAIN

MAIN
Medical uses
Mirdametinib is indicated for the treatment of people with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection.[1]
Adverse effects
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib can cause left ventricular dysfunction and ocular toxicity including retinal vein occlusion, retinal pigment epithelial detachment, and blurred vision.[3]
History
The efficacy of mirdametinib was evaluated in ReNeu (NCT03962543), a multicenter, single-arm trial in 114 participants aged two years of age and older (58 adults, 56 pediatric participants) with symptomatic, inoperable NF1-associated plexiform neurofibromas causing significant morbidity.[3] An inoperable plexiform neurofibromas was defined as a plexiform neurofibromas that could not be completely surgically removed without risk for substantial morbidity due to encasement or close proximity to vital structures, invasiveness, or high vascularity.[3]
The US Food and Drug Administration (FDA) granted the application for mirdametinib priority review, fast track, and orphan drug designations along with a priority review voucher.[3]
Society and culture
Legal status
Mirdametinib was approved for medical use in the United States in February 2025.[3][4][5]
PATENT
US-11066358
On July 20, 2021, SpringWorks Therapeutics announced that the United States Patent and Trademark Office (USPTO) has issued US11066358 , directed to mirdametinib , the Company’s product candidate in development for several oncology indications, including as a monotherapy for patients with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN) and was assigned to Warner-Lambert Company (a subsidiary of Pfizer ).This patent was granted on July 20, 2021, and expires on Feb 17, 2041. Novel crystalline forms of mirdametinib and compositions comprising them are claimed.
| N—((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (“mirdametinib”, or “PD-0325901”) is a small molecule drug which has been designed to inhibit mitogen-activated protein kinase kinase 1 (“MEK1”) and mitogen-activated protein kinase kinase 2 (“MEK2”). MEK1 and MEK2 are proteins that play key roles in the mitogen-activated protein kinase (“MAPK”) signaling pathway. The MAPK pathway is critical for cell survival and proliferation, and overactivation of this pathway has been shown to lead to tumor development and growth. Mirdametinib is a highly potent and specific allosteric non-ATP-competitive inhibitor of MEK1 and MEK2. By virtue of its mechanism of action, mirdametinib leads to significantly inhibited phosphorylation of the extracellular regulated MAP kinases ERK1 and ERK2, thereby leading to impaired growth of tumor cells both in vitro and in vivo. In addition, evidence indicates that inflammatory cytokine-induced increases in MEK/ERK activity contribute to the inflammation, pain, and tissue destruction associated with rheumatoid arthritis and other inflammatory diseases. |
Example 1: Production of Essentially Pure Form IV
Lab Scale Production of Essentially Pure Form IV
| All reactions were performed in toluene other than otherwise stated. Triflic anhydride gave the best yield. |
[TABLE-US-00002]TABLE 1 Coupling Agents for Step 1Entry No.Coupling AgentYieldNotes 1Mesyl Chloridedid not react 2Benzyl chloride27Had to heat 70° C. for 166 hr34-fluorobenzensulfonylchloride27Ran 93 hrs. at 70° C.44-chlorobenzensulfonylchloride35Complete after 68 hrs. 50° C.5Tosyl Chloride36Had to heat to 70° C. for 164 hrs6Benzyl chloride52study solvent effects: DMF, DMSO, NMP – all similar DMSO fastest all complete after 110 hrs., heated to 70° C. after 66 hrs.7Triflic anhydride91Cooled to −74° C. |
| [TABLE-US-00004]TABLE 3 Yields for base deprotection ReagentYield* Methyl hydrazine85-95% Anhydrous NH3 (sparged)78-90% Anhydrous NH3 (50 psi)80-92% Aqueous NH390-97% *from PD-0333760 |
Step 2: Fluoride Displacement
Pilot Plant Preparation of Essentially Pure Form IV
Step 1: Preparation of “Side Chain”, PD-0337792
Step 2: Preparation of PD-0315209
Step 3: Preparation of PD-0325901
Polymorph Transformation
| 21.4 kg PD-0315209, 9.7 kg CDI (1.05 equiv.), 91 kg solution of 9.7% PD-0337792 in Toluene (1.1 equiv.) were used and resulted in 12.74 kg of PD-0325901 (assay 99.4%, 100% Form IV, Yield 48%). |
PATENT
WO2006134469 , claiming methods of preparing MEK inhibitor, assigned to Warner-Lambert Co .
https://patents.google.com/patent/WO2006134469A1/enThe compound Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide represented by formula 1

i is a highly specific non-ATP-competitive inhibitor of MEK1 and MEK2. The compound of formula ± (Compound I) is also known as the compound PD 0325901. Compound I is disclosed in WO 02/06213; WO 04/045617; WO 2005/040098; EP 1262176; U.S. Patent Application Pub. No. 2003/0055095 A1 ; U.S. Patent Application Pub. No. 2004/0054172 A1; U.S. Patent Application Pub. No. 2004/0147478 A1 ; and U.S. Patent Application No. 10/969,681, the disclosures of which are incorporated herein by reference in their entireties.Numerous mitogen-activated protein kinase (MAPK) signaling cascades are involved in controlling cellular processes including proliferation, differentiation, apoptosis, and stress responses. Each MAPK module consists of 3 cytoplasmic kinases: a mitogen-activated protein kinase (MAPK), a mitogen-activated protein kinase kinase (MAPKK), and a mitogen-activated protein kinase kinase kinase (MAPKKK). MEK occupies a strategic downstream position in this intracellular signaling cascade catalyzing the phosphorylation of its MAP kinase substrates, ERK1 and ERK2. Anderson et al. “Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase.” Nature 1990, v.343, pp. 651-653. In the ERK pathway, MAPKK corresponds with MEK (MAP kinase ERK Kinase) and the MAPK corresponds with ERK (Extracellular Regulated Kinase). No substrates for MEK have been identified other than ERK1 and ERK2. Seger et al. “Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells.” J. Biol. Chem., 1992, v. 267, pp. 14373-14381. This tight selectivity in addition to the unique ability to act as a dual-specificity kinase is consistent with MEK’s central role in integration of signals into the MAPK pathway. The RAF-MEK-ERK pathway mediates proliferative and anti-apoptotic signaling from growth factors and oncogenic factors such as Ras and Raf mutant phenotypes that promote tumor growth, progression, and metastasis. By virtue of its central role in mediating the transmission of growth- promoting signals from multiple growth factor receptors, the Ras-MAP kinase cascade provides molecular targets with potentially broad therapeutic applications.One method of synthesizing Compound I is disclosed in the above-referenced WO 02/06213 andU.S. Patent Application Pub. No. 2004/0054172 A1. This method begins with the reaction of 2-fluoro-4- iodo-phenylamine and 2,3,4-trifluoro-benzoic acid in the presence of an organic base, such as lithium diisopropylamide, to form 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzoic acid, which is then reacted with (R)-0-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of a peptide coupling agent (e.g., diphenylphosphinic chloride) and a tertiary amine base (e.g., diisopropylethylamine). The resulting product is hydrolyzed under standard acidic hydrolysis conditions (e.g., p-TsOH in MeOH) to provide Compound 1. (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine is prepared by reaction of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol with N-hydroxyphthalimide in the presence of Ph3P and diethyl azodicarboxylate.Another method of synthesizing Compound I, which is disclosed in the above-referenced U.S.Patent Application No. 10/969,681, comprises reaction of 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzoic acid with (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of N1N1– carbonyldiimidazole. The resulting product is hydrolyzed with aqueous acid and crystallized to provide polymorphic form IV of Compound I.Although the described methods are effective synthetic routes for small-scale synthesis of Compound I, there remains a need in the art for new synthetic routes that are safe, efficient and cost effective when carried out on a commercial scale.The present invention provides a new synthetic route including Steps I through Step III to the MEK inhibitor Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I).Step I: Preparation of 0-{r(4RV2.2-dimethyl-1.3-dioxolan-4-ynmethyl}hydroxylanπine (6) The method of the present invention comprises a novel Step I of preparing of 0-{[(4R)-2,2- dimethyl-1 ,3-dioxolan-4-yl]methyl}hydroxylamine (6) from [(4S)-2,2-dimethyl-1 ,3-dioxoIan-4-yl]methanol (1) through the formation of [(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methyl trifluoromethanesulfonate (3) and its coupling with N-hydroxyphthalimide (4) to afford 2-{[(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methoxy}-1 H- isoindole-1 ,3(2H)-dione (5), which is subsequently de-protected to give 6 as shown in Scheme 1.Scheme 1



The reaction of compound (1) with trifluoromethanesulfonic anhydride (2) is carried out in the presence of a non-nucleophilic base, such as, for example, a tertiary organic amine, in an aprotic solvent at a temperature of from -5O0C to 50C, preferably, at a temperature less than -150C, to form triflate (3). A preferred tertiary organic amine is triethylamine, and a preferred solvent is toluene. Treatment of triflate (3) with N-hydroxyphthalimide (4) furnishes phthalimide (5), which can be isolated if desired. However, in order to minimize processing time and increase overall yield, 0-{[(4R)- 2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) can be prepared in a one-pot process with no phthalimide (S) isolation. Cleavage of the phthalimide function could be achieved by methods known in the art, for example, by hydrazinolysis. However, the use of less hazardous aqueous or anhydrous ammonia instead of methyl hydrazine (CH3NHNH2) is preferred.Step II: Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) As shown in Scheme 2, Step Il of the method of the present invention provides 3,4-difluoro-2-(2- fluoro-4-iodophenylamino)-benzoic acid (9).Scheme 2

Preparation of compound (9) can be carried out by reacting compound (7), wherein X is halogen, or O-SC^R^ or 0-P(3O)(OR^, wherein R^ is alkyl or aryl, with compound (8) optionally in a solvent, and in the presence of from about 1 mol equivalent to about 10 mol equivalents of at least one base, wherein the base is selected from: a Group I metal cation hydride or a Group 2 metal cation hydride, including lithium hydride, sodium hydride, potassium hydride, and calcium hydride, a Group I metal cation dialkylamide or a Group 2 metal cation dialkylamide, including lithium diisopropylamide, a Group I metal cation amide or a Group 2 metal cation amide, including lithium amide, sodium amide, potassium amide, a Group I metal cation alkoxide or a Group 2 metal cation alkoxide, including sodium ethoxide, potassium terf-butoxide, and magnesium ethoxide, and a Group I metal cation hexamethyldisilazide, including lithium hexamethyldisilazide; for a time, and at a temperature, sufficient to yield compound (9).Preferably, preparation of compound (9) is carried out by reacting compound (7), wherein X is halogen, more preferably, X is fluorine, in an aprotic solvent with compound (8) in the presence of from about 3 mol equivalents to about 5 mol equivalents of a Group I metal cation amide at a temperature of from 2O C to 55°C, more preferably, at a temperature from 45°C to 55°C. A catalytic amount of Group I metal cation dialkylamide can be added if necessary. A preferred Group I metal cation amide is lithium amide, a preferred Group I metal cation dialkylamide is lithium diisopropylamide, and a preferred solvent is tetrahydrofuran. Preferably, the reaction is performed by adding a small amount of compound (7) and compound (8) to lithium amide in tetrahydrofuran followed by slow continuous addition of the remaining portion. This procedure minimizes the risk of reactor over-pressurization due to gas side product (ammonia) generation.Step III: Preparation of N-((RV2.3-dihydroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I)Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using a carboxylic acid activating reagent such as, for example, COCI2, S(O)C^, S(O)2Cl2, P(O)Cl3, triphenylphosphine/diethylazodicarboxylate, diphenylphosphinic chloride, N, N’-dicyclohexylcarbodiimide, (benzotriazol-1 -yloxy)tripyrolidinophosphonium hexafluorophosphate, (benzotriazol-1 – yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, N-ethyl-N’-(3- dimethylaminopropyl)carbodiimide hydrochloride, or 1,1′-carbonyldiimidazole (CDI).A preferred carboxylic acid activating reagent is 1,1′-carbonyldimidazole (CDI) shown in Scheme 3. Preparation of the desirable polymorphic Form IV of Compound I using CDI is described in the above- referenced U.S. Patent Application No. 10/969,681.Scheme 3

10

10 11 Compound IIn according to the present invention, the method was modified to include the advantageous procedure for product purification and isolation, which procedure is performed in single-phase systems such as, for example, toluene/acetonitrile for the first isolation/crystallization and ethanol/toluene for the second recrystallization. Water addition, implemented in the previous procedure, was omitted to avoid the two-phase crystallization from the immiscible water-toluene system that caused inconsistent product purity. The one-phase procedure of the present invention provides consistent control and removal of un- reacted starting material and side products. Alternatively, Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using thionyl chloride (SOCI2) as shown in Scheme 4.Scheme 4


Compound IExamplesThe reagents and conditions of the reactions described herein are merely illustrative of the wide variety of starting materials, their amounts and conditions which may be suitably employed in the present invention as would be appreciated by those skilled in the art, and are not intended to be limiting in any way.HPLC (Conditions A): 10 μL injection volume onto Agilent Zorbax RX-C18 150 mm x 4.6 mm x 3.5 μm column at 30°C column temperature, 1.0 mL/min flow rate and detection at 246 nm. Mobile phase A (v/v): 25 mM Acetate Buffer, pH 6.0; Mobile phase B (v/v): Acetonitrile, and Linear Gradient Table:

Sample Preparation: Dilute 100 μL reaction mixture to 10 mL with acetonitrile. Mix in a vial 200 μL of this sample solution with 300 μL carbonate buffer pH 10.0 and 300 μL solution of 2-mercaptopyridine in acetonitrile (18 mM), heat the vial for 10 minutes at 500C and dilute to 1:1 ratio in mobile phase A.GC (Conditions B): 1 μL injection onto an RTX-5 column (30 m x 0.25 mm x 0.25 μm) with initial oven temperature of 120°C for 2 min. to final temperature of 250°C in 15°C/minute ramping and a final time of 2.33 min; Flow rate: 1 mL/min.HPLC (Conditions C): 5 μL injection onto Phenomenex Luna C18(2) 150 mm x 4.6 mm x 3μm column ; flow rate : 1.0 mL/min; detection at 225 nm; mobile phase A: 95/5 v/v Water/Acetonitrile with 0.1% Trifluoroacetic acid (TFA), mobile phase B: 5/95 v/v Water/Acetonitriie with 0.1% TFA; Linear Gradient Table:

Sample preparation: Dilute 1 ml_ reaction mixture to 100 mL with acetonitrile and dilute 1 mL of this solution to 10 mL with 50:50 Water/Acetonitrile.HPLC (Conditions D): 5 μL injection onto Waters SymmetryShield RP 18, 150 mm x 4.6 mm x 3.5 μm column; flow rate: 1.0 mL/min; detection at 235 nm; mobile phase A: 25 mM Acetate Buffer adjusted to pH 5.5, mobile phase B: Acetonitrile; Linear Gradient Table:

Sample preparation: Dilute 40 μL of reaction mixture in 20 mL acetonitrile.HPLC (Conditions E): 10 μL sample injection onto YMC ODS-AQ 5 μm, 250 mm x 4.6 mm column; flow rate: 1.0 ml_/min; detection at 280 nm; temperature 30°C; mobile phase : 75/25 v/v Acetonitrile/Water with 0.1% Formic acid.Sample preparation: Quench reaction mixture sample with dipropylamine and stir for about 5 minutes before further dilution with mobile phase.DSC measurement was performed using a Mettler-Toledo DSC 822, temperature range 25° to 150°C with 5°C/min heating rate in a 40 μL aluminum pan. Experimental Conditions for Powder X-Rav Diffraction (XRD):A Rigaku Miniflex+ X-ray diffractometer was used for the acquisition of the powder XRD patterns. The instrument operates using the Cu Ka1 emission with a nickel filter at 1.50451 units. The major instrumental parameters are set or fixed at:X-ray: Cu / 30 kV (fixed) / 15 mA (fixed)Divergence Slit: Variable Scattering Slit: 4.2° (fixed) Receiving Slit: 0.3 mm (fixed) Scan Mode: FT Preset Time: 2.0 s Scan Width: 0.050° Scan Axis: 2Theta/Theta Scan Range: 3.000° to 40.000°Jade Software Version: 5.0.36(SP1) 01/05/01 (Materials Data, Inc.) Rigaku Software: Rigaku Standard Measurement for Windows 3.1 Version 3.6(1994-1995) Example 1. Preparation of 0-ffl4R)-2.2-dimethyl-1.3-dioxolan-4-vπmethyl}hvdroxylamine (6)A solution containing [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol (1) (13.54 ml_, 0.109 mol) (DAISO Co., Ltd., CAS# 22323-82-6) and triethylamine (18.2 ml_, 0.131 mol) in 115 mL toluene was cooled to -15 C, then trifluoromethanesulfonic anhydride (2) (18.34 mL, 30.75 g, 0.109 mol) (Aldrich, Catalog # 17,617-6 ) was added drop wise while maintaining the temperature at less than -15°C. The mixture was then stirred for 2 hours, and transferred to a separate flask containing a mixture (slurry) of N- hydroxyphthalimide (4) (18.99 g, 0.116 mol) (Aldrich, Catalog # H5.370-4) and 18.2 mL (0.13 mol) triethylamine in 95 mL toluene. The resulting mixture was warmed to 20-25°C and stirred for at least 5 hours or until reaction completion (determined by HPLC (Conditions A)). Water (93 mL) was then added to quench the reaction mixture, the phases were separated, and the bottom aqueous layer was discarded. The water quench was repeated two more times resulting in a pale yellow organic layer. The organic layer was heated to 35 C and treated with 36.7 mL ammonium hydroxide solution (contains about 28-29% wt/wt ammonia). The mixture was stirred for at least 12 hours or until the reaction was deemed complete as determined by GC (Conditions B). The water was then removed under reduced pressure by co- distilling it with toluene to about half of the original volume at temperatures around 35-45 C. Toluene (170 mL) was added to the concentrated solution and the distillation was repeated. A sample was drawn for water content determination by Karl Fisher method (using EM Science Aquastar AQV-2000 Titrator with a sample injected to a pot containing methanol and salicylic acid). The distillation was repeated ifl water content was more than 0.1%. The concentrated solution was filtered to remove the white solid side product, and the filtrate was stored as 112mL (98 g) product solution containing 9.7% w/w compound 6 in toluene. This solution was ready for use in the final coupling step (Example 3). Overall chemical yield was 59%. A small sample was evaporated to yield a sample for NMR identification.1H NMR (400 MHz, CDCI3): δ 5.5 (bs, 2H), 4.35 (m, 1H), 4.07 (dd, 1H), 3.77 (m, 2H), 3.69 (dd, 1H), 1.44 (s, 3H), 1.37 (s, 3H).Example 2. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9)A solution of 2-fluoro-4-iodoaniline (8) (16.4 g, 0.069 mol) (Aldrich, Catalog # 30,660-6) and 2,3,4- trifluorobenzoic acid (7) (11.98 g, 0.068 mol) (Aldrich, Cat # 33,382-4) in 38 mL tetrahydrofuran (THF) was prepared and a portion (about 5%) of this solution was added to a stirring slurry of lithium amide (5 g, 0.22 mol) in 40 mL THF at 50-55 C. After about 15-30 min. an exotherm followed by gas release and color change are observed. The remaining portion of the (8) and (7) solution was added slowly over 1-2 hr while maintaining temperatures within 45-55°C. The mixture was stirred until the reaction was deemed complete (by HPLC (Conditions C). The final mixture was then cooled to 20-25°C and transferred to another reactor containing 6 N hydrochloric acid (47 mL) followed by 25 mL acetonitrile, stirred, and the bottom aqueous phase was discarded after treatment with 40 mL 50% sodium hydroxide solution. The organic phase was concentrated under reduced pressure and 57 mL acetone was added. The mixture was heated to 50°C, stirred, and added with 25 mL warm (40-50°C) water and cooled to 25-30°C to allow crystallization to occur (within 1-4 hours). Once the crystallization occurred, the mixture was further cooled to 0 to -5°C and stirred for about 2 hours. The solid product was filtered and the wet cake was dried in vacuum oven at about 55°C. Overall chemical yield was 21.4 g, 80%. 1H NMR (400 MHz, (CD3)2SO): δ 13.74 (bs, 1H), 9.15 (m, 1 H), 7.80 (dd, 1H), 7.62 (d, 1H), 7.41 (d, 1H), 7.10 (q, 1H), 6.81 (m, 1H).Example 2B. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) by the solid addition of lithium amide methodTo a stirring solution of 2,3,4-trifluorobenzoic acid (13) (5.0 g, 28.4 mmol) and 2-fluoro-4- iodoaniline (14) (6.73 g, 28.4 mmol) in MeCN (100 mL), under N2 atmosphere was added lithium amide (2.61 g, 113.6 mmol) in small portions. The reaction mixture was heated to reflux for 45 minutes, cooled to ambient temperature and quenched with 1 N HCI and then water. The yellowish white precipitate was filtered, washed with water. The solid was triturated in CH2CI2 (30 mL) for 1h, filtered and dried in a vacuum oven at 45°C for 14 hours to give 8.Og (72%) of compound (9) as an off-white solid, mp 201.5-203 °C.Example 3. Preparation of N-((R)-2.3-dihvdroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound \)3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (20 g, 0.051 mol) in 100 mL acetonitrile was treated with 1,1′-carbonyldiimidazole (CDI) (8.66 g, 0.053 mol) (Aldrich, Cat # 11,553-3) and stirred for about 2 hours at 20-25°C until the reaction was deemed complete by HPLC (Conditions D). 94 mL (84.9 g) of 9.7% w/w solution of O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) in toluene was then added and stirred for about 4 hours or until the reaction was deemed complete by HPLC (Conditions D). To this mixture was added 66 mL of 5.6 % hydrochloric acid solution, and after stirring, the bottom aqueous phase was discarded. Again 66 mL of 5.6 % hydrochloric acid solution was added to the organic phase and stirred at 20-25°C for 12-18 hours or until the reaction was deemed complete by HPLC (Conditions D). The bottom layer was then discarded and the remaining organic layer was concentrated under reduced pressure to remove about 10-20% solvent, and the volume was adjusted to about 9-11 mL/g with toluene (80 mL). Crude product was then crystallized at 10-15°C. The slurry was allowed to stir for about 2 hours and the crude solid product was filtered, and dried. The dried crude product was recharged to the reactor and dissolved into 150 mL of 5% v/v ethanol/toluene mixture at 55- 67°C. The solution was then clarified at this temperature through filter (line filter) to remove any remaining particulate matter. The solution was then cooled slowly to 5°C to crystallize and stirred for at least 2 h, filtered and dried. The dried solid product was redissolved in EtOH (60 mL) at 35°C, and product was precipitated out by adding water (300 mL) at 35°C followed by cooling to 200C. The slurry was stirred for at least 2 hours to transform the crystals to the desired polymorphic Form IV as determined by DSC and Powder X-ray Diffraction pattern (PXRD). The slurry was filtered and dried under vacuum oven at 70- 90°C to yield the final N-((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I) product. Overall chemical yield was 13 g, 53%. Melting point (DSC): 112+1° C. Appearance: White to off-white crystals.Shown in Figure 1, PXRD conforms to polymorphic crystal Form IV disclosed in the above mentioned U.S. Patent Application No. 10/969,681 1H NMR (400 MHz, (CD3)2SO): δ 11.89 (bs, 1H), 8.71 (bs, 1H), 7.57 (d, 1H), 7.37 (m, 2H), 7.20 (q, 1H), 6.67 (m, 1H), 4.84 (bs, 1H), 4.60 (m, 1H), 3.87 (m, 1 H), 3.7 (m, 2H), 3.34 (m, 2H).Example 4. Preparation of N-((R)-2.3-dihydroxypropoxyV3.4-difluoro-2-(2-fluoro-4-iodo-phenylanrιinoV benzamide (Compound \)To a stirring solution of 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (120 g, 0.30 mol) in a mixture of 1 mL N,N-dimethylformamide and 1000 mL toluene was added thionyl chloride (55 g, 0.462 mol). The mixture was heated to 50-65 C and stirred for 2 hours or until reaction completion as determined by HPLC (Conditions E). The final reaction mixture was then cooled and concentrated under reduced pressure to a slurry keeping the temperature below 35°C. Toluene (600 mL) was added to dissolve the slurry and vacuum distillation was repeated. Additional toluene (600 mL) was added to the slurry dissolving all solids and the solution was then cooled to 5° -10°C. The solution was then treated with O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) (63 g, 0.43 mol) solution in 207 mL toluene followed by potassium carbonate (65 g) and water (200 mL), stirred for at least 2 hours at 20- 25°C. The stirring was stopped to allow phase separation and the bottom phase was discarded. The remaining organic layer was treated with hydrochloric acid solution (7.4%, 240 mL) until pH was less than 1 and stirred for 2 hours. The final reaction mixture was slightly concentrated under vacuum collecting about 100 mL distillate and the resulting organic solution was cooled to 5°C to crystallize the product and filtered. The filter cake was washed with toluene (1000 mL) followed by water (100 mL) and the wet cake (crude product Compound I) was charged back to the flask. Toluene (100 mL), ethanol (100 mL) and water (100 mL) are then added, stirred at 30-35°C for about 15 min, and the bottom aqueous phase was discarded. Water (200 mL) was then added to the organic solution and the mixture was stirred at about 3O C to allow for crystallization. The stirring was continued for 2 hours after product crystallized, then it was further cooled to about 0°C and stirred for at least 2 hours. The slurry was filtered and wet cake was dried under reduced pressure at 55-85°C to yield the final product N-((R)-2,3-dihydroxypropoxy)-3,4- difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I) product. Overall chemical yield was 86 g, 58%.
PATENT
WO2002/006213 describes crystalline Forms I and II. U.S. Pat. No. 7,060,856 (“the ‘856 patent”)
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2002006213
| Clinical data | |
|---|---|
| Trade names | Gomekli |
| Other names | PD-0325901 |
| AHFS/Drugs.com | Gomekli |
| License data | US DailyMed: Mirdametinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EE05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 391210-10-9 |
| PubChem CID | 9826528 |
| IUPHAR/BPS | 7935 |
| DrugBank | DB07101 |
| ChemSpider | 10814340 |
| UNII | 86K0J5AK6M |
| KEGG | D11675 |
| ChEBI | CHEBI:9826528 |
| ChEMBL | ChEMBL507361 |
| PDB ligand | 4BM (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C16H14F3IN2O4 |
| Molar mass | 482.198 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Gomekli- mirdametinib capsule; Gomekli- mirdametinib tablet, for suspension”. DailyMed. 27 February 2025. Retrieved 2 April 2025.
- ^ Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ (June 2023). “Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas”. BMC Cancer. 23 (1): 553. doi:10.1186/s12885-023-10996-y. PMC 10273716. PMID 37328781.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection”. U.S. Food and Drug Administration (FDA). 11 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “UPDATE: SpringWorks Therapeutics Announces FDA Approval of Gomekli (mirdametinib) for the Treatment of Adult and Pediatric Patients with NF1-PN” (Press release). SpringWorks Therapeutics. 12 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025 – via GlobeNewswire News Room.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025.
External links
- “Mirdametinib (Code C52195)”. NCI Thesaurus.
- Clinical trial number NCT03962543 for “MEK Inhibitor Mirdametinib (PD-0325901) in Patients With Neurofibromatosis Type 1 Associated Plexiform Neurofibromas (ReNeu)” at ClinicalTrials.gov
- Moertel CL, Hirbe AC, Shuhaiber HH, Bielamowicz K, Sidhu A, Viskochil D, Weber MD, Lokku A, Smith LM, Foreman NK, Hajjar FM, McNall-Knapp RY, Weintraub L, Antony R, Franson AT, Meade J, Schiff D, Walbert T, Ambady P, Bota DA, Campen CJ, Kaur G, Klesse LJ, Maraka S, Moots PL, Nevel K, Bornhorst M, Aguilar-Bonilla A, Chagnon S, Dalvi N, Gupta P, Khatib Z, Metrock LK, Nghiemphu PL, Roberts RD, Robison NJ, Sadighi Z, Stapleton S, Babovic-Vuksanovic D, Gershon TR: ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025 Feb 20;43(6):716-729. doi: 10.1200/JCO.24.01034. Epub 2024 Nov 8. [Article]
- Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, Allen JC, Schorry E, Korf B, Robison NJ, Goldman S, Vinks AA, Emoto C, Fukuda T, Robinson CT, Cutter G, Edwards L, Dombi E, Ratner N, Packer R, Fisher MJ: NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol. 2021 Mar 1;39(7):797-806. doi: 10.1200/JCO.20.02220. Epub 2021 Jan 28. [Article]
- Ioannou M, Lalwani K, Ayanlaja AA, Chinnasamy V, Pratilas CA, Schreck KC: MEK Inhibition Enhances the Antitumor Effect of Radiotherapy in NF1-Deficient Glioblastoma. Mol Cancer Ther. 2024 Sep 4;23(9):1261-1272. doi: 10.1158/1535-7163.MCT-23-0510. [Article]
- FDA Approved Drug Products: GOMEKLI (mirdametinib) capsules and tablets for oral and oral suspension use (Feb 2024) [Link]
- FDA News: FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection [Link]
////////MIRDAMETINIB, Orphan Drug Status, Neurofibromatosis 1, PHASE 2, PD0325901, PD 0325901, PD-325901, FDA 2025, GOMEKLI, APPROVALS 2025
O=C(NOC[C@H](O)CO)C1=CC=C(F)C(F)=C1NC2=CC=C(I)C=C2F


{kind=link}