

| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-003807-37 | A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD) | Phase 3 | Ongoing | 2020-07-29 |
| 2015-001157-32 | An Exploratory Phase II Study to demonstrate the Safety and Efficacy of A4250 | Phase 2 | Completed | 2015-05-13 |
| 2014-004070-42 | An Exploratory, Phase IIa Cross-Over Study to Demonstrate the Efficacy | Phase 2 | Ongoing | 2014-12-09 |
| 2017-002325-38 | An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2) | Phase 3 | Ongoing | |
| 2017-002338-21 | A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1) | Phase 3 | Ongoing, Completed |
Home » Posts tagged 'Orphan Drug Status'
Tag Archives: Orphan Drug Status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Atumelnant
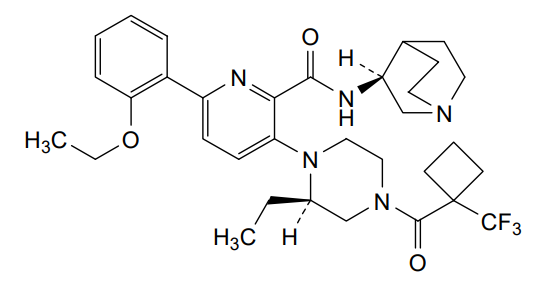
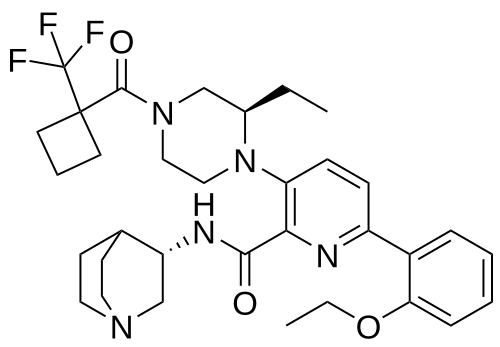
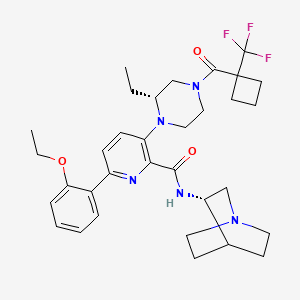
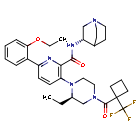
Atumelnant
CAS 2392970-97-5
MF C33H42F3N5O3 MW 613.7 g/mol
CRN04894, NR57FH6U1N
CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-{(2R)-2-ethyl-4-[1-(trifluoromethyl) cyclobutane-1-carbonyl]piperazin-1-yl}pyridine-2-carboxamide
Adrenocorticotropic hormone receptor antagonist
- OriginatorCrinetics Pharmaceuticals
- ClassAmides; Antineoplastics; Antisecretories; Benzene derivatives; Cyclobutanes; Ethers; Fluorocarbons; Ketones; Piperazines; Pyridines; Quinuclidines; Small molecules
- Mechanism of ActionMelanocortin type 2 receptor antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- Phase IICongenital adrenal hyperplasia; Cushing syndrome
- No development reportedEctopic ACTH syndrome
- 21 Aug 2025Atumelnant receives Orphan Drug status for Congenital adrenal hyperplasia in the US
- 07 Aug 2025Crinetics pharmaceuticals plans phase II/III clinical trial for Cushing’s disease in 1H 2026
- 08 May 2025Crinetics Pharmaceuticals plans the phase III CALM-CAH trial for Congenital adrenal hyperplasia (In adults) (PO), in the second half of 2025
Atumelnant (INNTooltip International Nonproprietary Name; developmental code name CRN04894) is an investigational new drug developed by Crinetics Pharmaceuticals for the treatment of adrenocorticotropic hormone (ACTH)-dependent endocrine disorders.[1] It is a selective antagonist of the melanocortin type 2 receptor (MC2R), also known as the ACTH receptor, which is primarily expressed in the adrenal glands.[1][2] The drug is orally active.[1] Atumelnant is being evaluated to treat conditions such as congenital adrenal hyperplasia (CAH) and ACTH-dependent Cushing’s syndrome caused for example by pituitary adenomas.[3]
Atumelnant is an orally bioavailable nonpeptide antagonist of the adrenocorticotropic hormone (ACTH) receptor (ACTHR; melanocortin receptor 2; MC2R), with potential steroid hormone production inhibitory activity. Upon oral administration, atumelnant competes with ACTH for receptor binding to MC2R in the adrenal cortex and inhibits ACTH signaling. This may inhibit the synthesis and secretion of steroid hormones. MC2R, a member of the melanocortin receptor subfamily of type 1 G protein-coupled receptors, plays a key role in adrenal steroidogenesis.
PAPER
Discovery of CRN04894: A Novel Potent Selective MC2R Antagonist
Publication Name: ACS Medicinal Chemistry Letters
Publication Date: 2024-03-19, PMCID: PMC11017392, PMID: 38628803
DOI: 10.1021/acsmedchemlett.3c00514
PATENTS
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B2Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024300920-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: JP-2024009837-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-102695210-B1Priority Date: 2018-06-05Grant Date: 2024-08-13
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024109866-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-BPriority Date: 2018-06-05Grant Date: 2024-10-29
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10981894-B2Priority Date: 2018-06-05Grant Date: 2021-04-20
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021002254-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021238164-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-11566015-B2Priority Date: 2018-06-05Grant Date: 2023-01-31
- Melanocortin subtype-2 receptor (MC2R) antagonists and their usesPublication Number: JP-7359783-B2Priority Date: 2018-06-05Grant Date: 2023-10-11
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020216415-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10766877-B2Priority Date: 2018-06-05Grant Date: 2020-09-08
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: EP-3802500-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-20210005995-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists for the treatment of diseasePublication Number: CN-117043146-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10562884-B2Priority Date: 2018-06-05Grant Date: 2020-02-18
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10604507-B2Priority Date: 2018-06-05Grant Date: 2020-03-31
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2019367481-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020010452-A1Priority Date: 2018-06-05
Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: US-2022313691-A1Priority Date: 2021-03-19 - Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: WO-2022197798-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: TW-202302108-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: AU-2022240609-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: EP-4308553-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of acth-dependent cushing’s syndromePublication Number: WO-2024211343-A1Priority Date: 2023-04-05
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: TW-202430167-APriority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: US-2024208963-A1Priority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: WO-2024130091-A1Priority Date: 2022-12-16
- Treatment of congenital adrenal hyperplasia and polycystic ovary syndromePublication Number: WO-2023163945-A1Priority Date: 2022-02-2
SYN
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US278278493&_cid=P22-MFXDN2-76849-1
Example 31: N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide (Compound 1-410)
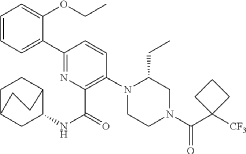
Step 31-1, Preparation of 6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxylic acid
Step 31-2, Preparation of N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Crinetics Pharmaceuticals”. AdisInsight. 21 January 2025. Retrieved 25 February 2025.
- “Atumelnant (CRN04894)”. crinetics.com. 14 August 2020.
- Varlamov EV, Gheorghiu ML, Fleseriu M (December 2024). “Pharmacological management of pituitary adenomas – what is new on the horizon?”. Expert Opinion on Pharmacotherapy. 26 (2): 119–125. doi:10.1080/14656566.2024.2446625. PMID 39718553.
| Clinical data | |
|---|---|
| Other names | CRN04894 |
| Routes of administration | Oral[1] |
| Drug class | Melanocortin MC2 receptor antagonist[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2392970-97-5 |
| PubChem CID | 146361282 |
| IUPHAR/BPS | 13339 |
| ChemSpider | 129750231 |
| UNII | NR57FH6U1N |
| KEGG | D13102 |
| Chemical and physical data | |
| Formula | C33H42F3N5O3 |
| Molar mass | 613.726 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Atumelnant, CRN04894, CRN 04894, NR57FH6U1N, CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia, PHASE 3
Asengeprast
Asengeprast
CAS 1001288-58-9
FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627,
Fast Track
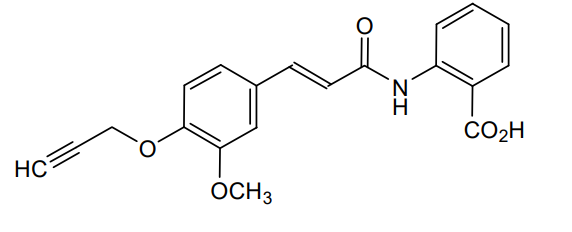
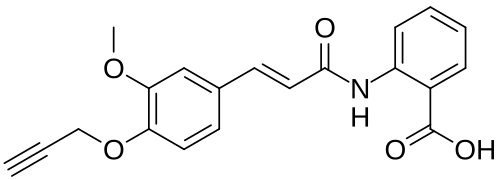
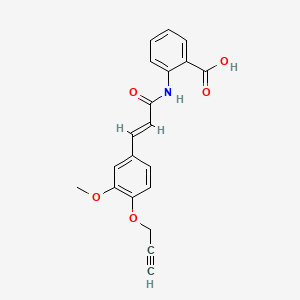
2-[[(E)-3-(3-methoxy-4-prop-2-ynoxyphenyl)prop-2-enoyl]amino]benzoic acid
2-[(2E)-3-{3-methoxy-4-[(prop-2-yn-1-yl)oxy]phenyl}prop-2-enamido]benzoic acid G protein-coupled receptor 68 (GPR68) antagonist,
anti-inflammatory
MF C20H17NO5 MW 351.4 g/mol. C6V7ZU2NPR
Asengeprast (development code FT011) is an experimental scleroderma drug candidate.[1] It is a small molecule inhibitor of the G-protein coupled receptor GPR68 with antifibrotic activity.[2] It is being developed by Certa Therapeutics.
The European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) has granted orphan drug status to FT011, for systemic sclerosis (SSc).[3]
Asengeprast has been reported to attenuate fibrosis and chronic heart failure in experimental diabetic cardiomyopathy.[4] Asengeprast can also inhibit kidney fibrosis and prevent kidney failure.[5] It was developed by structure-activity optimization of the antifibrotic activity of cinnamoyl anthranilates, by assessment of their ability to prevent TGF-beta-stimulated production of collagen.[6]
Effects of FT011 in Systemic Sclerosis, CTID: NCT04647890
Phase: Phase 2, Status: Completed, Date: 2023-12-20
SYN
Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates
Publication Name: Bioorganic & Medicinal Chemistry Letters
Publication Date: 2009-12-15
PMID: 19879136
DOI: 10.1016/j.bmcl.2009.09.120
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018144620&_cid=P21-MFTHV7-45829-1
PAT
Publication Number: WO-2008003141-A1
Priority Date: 2006-07-05
- Tranilast analogues (substituted cinnamoyl anthranilate compounds) for treatment of conditions associated with firbrosisPublication Number: NZ-574028-APriority Date: 2006-07-05
- Therapeutic CompoundsPublication Number: US-2010130497-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-2014357628-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-8765812-B2Priority Date: 2006-07-05Grant Date: 2014-07-01
- Therapeutic compoundsPublication Number: US-9561201-B2Priority Date: 2006-07-05Grant Date: 2017-02-07
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Asengeprast Ligand page”. IUPHAR/BPS Guide to PHARMACOLOGY.
- “Certa Therapeutics website”.
- Inácio P (23 July 2024). “Certa’s FT011 granted orphan drug status in Europe for SSc”. Scleroderma News.
- Zhang Y, Edgley AJ, Cox AJ, Powell AK, Wang B, Kompa AR, et al. (May 2012). “FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy”. European Journal of Heart Failure. 14 (5): 549–562. doi:10.1093/eurjhf/hfs011. PMID 22417655.
- Gilbert RE, Zhang Y, Williams SJ, Zammit SC, Stapleton DI, Cox AJ, et al. (2012). “A purpose-synthesised anti-fibrotic agent attenuates experimental kidney diseases in the rat”. PLOS ONE. 7 (10): e47160. Bibcode:2012PLoSO…747160G. doi:10.1371/journal.pone.0047160. PMC 3468513. PMID 23071743.
- Zammit SC, Cox AJ, Gow RM, Zhang Y, Gilbert RE, Krum H, et al. (December 2009). “Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates”. Bioorganic & Medicinal Chemistry Letters. 19 (24): 7003–7006. doi:10.1016/j.bmcl.2009.09.120. PMID 19879136.
| Chemical structure of asengeprast (FT011) | |
| Clinical data | |
|---|---|
| Other names | FT011 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1001288-58-9 |
| PubChem CID | 23648966 |
| ChemSpider | 24664633 |
| UNII | C6V7ZU2NPR |
| ChEMBL | ChEMBL1075834 |
| Chemical and physical data | |
| Formula | C20H17NO5 |
| Molar mass | 351.358 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- FT011, a Novel Cardiorenal Protective Drug, Reduces Inflammation, Gliosis and Vascular Injury in Rats with Diabetic RetinopathyPublication Name: PLOS ONEPublication Date: 2015-07-29PMCID: PMC4519240PMID: 26222724DOI: 10.1371/journal.pone.0134392
- A new anti-fibrotic drug attenuates cardiac remodeling and systolic dysfunction following experimental myocardial infarctionPublication Name: International Journal of CardiologyPublication Date: 2013-09-30PMID: 23219315DOI: 10.1016/j.ijcard.2012.11.067
- Attenuation of Armanni–Ebstein lesions in a rat model of diabetes by a new anti-fibrotic, anti-inflammatory agent, FT011Publication Name: DiabetologiaPublication Date: 2012-12-16PMID: 23242170DOI: 10.1007/s00125-012-2805-9
- A Purpose-Synthesised Anti-Fibrotic Agent Attenuates Experimental Kidney Diseases in the RatPublication Name: PLoS ONEPublication Date: 2012-10-10PMCID: PMC3468513PMID: 23071743DOI: 10.1371/journal.pone.0047160
- FT011, a new anti‐fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathyPublication Name: European Journal of Heart FailurePublication Date: 2012-05PMID: 22417655DOI: 10.1093/eurjhf/hfs011
///////////Asengeprast, FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627, C6V7ZU2NPR, Fast Track
Spesolimab
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYSFT SSWIHWVKQA PGQGLEWMGE INPGNVRTNY
NENFRNKVTM TVDTSISTAY MELSRLRSDD TAVYYCTVVF YGEPYFPYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPEAAGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSREEM
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
QIVLTQSPGT LSLSPGERAT MTCTASSSVS SSYFHWYQQK PGQAPRLWIY RTSRLASGVP
DRFSGSGSGT DFTLTISRLE PEDAATYYCH QFHRSPLTFG AGTKLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-H96, H146-H202, H222-L215, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’96, H’146-H’202, H’222-L’215, H’263-H’323, H’369-H’427, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Spesolimab
スペソリマブ (遺伝子組換え)
| Formula | C6480H9988N1736O2012S46 |
|---|---|
| cas | 2097104-58-8 |
| Mol weight | 145878.0547 |
| Antipsoriatic, Anti-IL-36 receptor antagonist |
fda approved 2022/9/1, spevigo
BI 655130; Spesolimab-sbzo
- OriginatorBoehringer Ingelheim
- ClassAnti-inflammatories; Antipsoriatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterleukin 36 receptor antagonists
- Orphan Drug StatusYes – Generalised pustular psoriasis
- RegisteredGeneralised pustular psoriasis
- Phase II/IIIUlcerative colitis
- Phase IICrohn’s disease; Hidradenitis suppurativa; Palmoplantar pustulosis
- DiscontinuedAtopic dermatitis
- 01 Sep 2022First global approval – Registered for Generalised pustular psoriasis in USA (IV)
- 01 Sep 2022Adverse events data from the Effisayil 1 phase II trial in Generalised pustular psoriasis released by Boehringer Ingelheim
- 03 Aug 2022Boehringer Ingelheim anticipates regulatory approval in Generalised pustular psoriasis by 2022
Spesolimab (BI 655130) is a humanised monoclonal antibody, being developed by Boehringer Ingelheim, for the treatment of generalised pustular psoriasis, Crohn’s disease, palmoplantar pustulosis, ulcerative colitis and hidradenitis suppurativa.
What causes Palmoplantar Pustulosis?
Researchers have found some possible causes including smoking, infections, certain medications and genetics. Smoking: Many patients who have PPP are smokers or have smoked in the past. Smoking may cause sweat glands to become inflamed, especially on the hands and feet, which causes pustules to form.
FDA approves the first treatment option for generalized pustular psoriasis flares in adults
- More than half of patients treated with SPEVIGO® (spesolimab-sbzo) injection, for intravenous use showed no visible pustules one week after receiving treatment
- Spesolimab is a monoclonal antibody that inhibits interleukin-36 (IL-36) signaling
Ridgefield, Conn., September 1, 2022 – Boehringer Ingelheim announced today the U.S. Food and Drug Administration has approved SPEVIGO, the first approved treatment option for generalized pustular psoriasis (GPP) flares in adults. SPEVIGO is a novel, selective antibody that blocks the activation of the interleukin-36 receptor (IL-36R), a key part of a signaling pathway within the immune system shown to be involved in the cause of GPP.
“GPP flares can greatly impact a patient’s life and lead to serious, life-threatening complications,” said Mark Lebwohl, M.D., lead investigator and publication author, and Dean for Clinical Therapeutics, Icahn School of Medicine at Mount Sinai, Kimberly and Eric J. Waldman Department of Dermatology, New York. “The approval of SPEVIGO is a turning point for dermatologists and clinicians. We now have an FDA-approved treatment that may help make a difference for our patients who, until now, have not had any approved options to help manage GPP flares.”
Distinct from plaque psoriasis, GPP is a rare and potentially life-threatening neutrophilic skin disease, which is characterized by flares (episodes of widespread eruptions of painful, sterile pustules). In the United States, it is estimated that 1 out of every 10,000 people has GPP. Given that it is so rare, recognizing the signs and symptoms can be challenging and consequently lead to delays in diagnosis.
“This important approval reflects our successful efforts to accelerate our research with the aim to bring innovative treatments faster to the people most in need,” said Carinne Brouillon, Member of the Board of Managing Directors, responsible for Human Pharma, Boehringer Ingelheim. “We recognize how devastating this rare skin disease can be for patients, their families and caregivers. GPP can be life-threatening and until today there have been no specific approved therapies for treating the devastating GPP flares. It makes me proud that with the approval of SPEVIGO we can now offer the first U.S. approved treatment option for those in need.”
In the 12-week pivotal Effisayil™ 1 clinical trial, patients experiencing a GPP flare (N=53) were treated with SPEVIGO or placebo. After one week, patients treated with SPEVIGO showed no visible pustules (54%) compared to placebo (6%).
In Effisayil™ 1, the most common adverse reactions (≥5%) in patients that received SPEVIGO were asthenia and fatigue, nausea and vomiting, headache, pruritus and prurigo, infusion site hematoma and bruising, and urinary tract infection.
“GPP can have an enormous impact on patients’ physical and emotional wellbeing. With the FDA approval of this new treatment, people living with GPP now have hope in knowing that there is an option to help treat their flares,” said Thomas Seck, M.D., Senior Vice President, Medicine and Regulatory Affairs, Boehringer Ingelheim. “SPEVIGO represents Boehringer Ingelheim’s commitment to delivering meaningful change for patients living with serious diseases with limited treatment options.”
About SPEVIGO
SPEVIGO is indicated for the treatment of GPP flares in adults. SPEVIGO is contraindicated in patients with severe or life-threatening hypersensitivity to spesolimab-sbzo or to any of the excipients in SPEVIGO. Reactions have included drug reaction with eosinophilia and systemic symptoms (DRESS).
What is SPEVIGO?
SPEVIGO is a prescription medicine used to treat generalized pustular psoriasis (GPP) flares in adults. It is not known if SPEVIGO is safe and effective in children.
U.S. FDA grants Priority Review for spesolimab for the treatment of flares in patients with generalized pustular psoriasis (GPP), a rare, life-threatening skin disease
December 15, 2021 – Boehringer Ingelheim today announced that the U.S. Food and Drug Administration (FDA) has accepted a Biologics License Application (BLA) and granted Priority Review for spesolimab for the treatment of generalized pustular psoriasis (GPP) flares.
FDA grants Priority Review to applications for medicines that, if approved, would offer significant improvement over available options in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions. The FDA has granted spesolimab Orphan Drug Designation for the treatment of GPP, and Breakthrough Therapy Designation for spesolimab for the treatment of GPP flares in adults.
“The FDA acceptance of our filing for spesolimab is a critical step in our efforts to bring this first-in-class treatment to people living with GPP,” said Matt Frankel, M.D., Vice President, Clinical Development and Medical Affairs, Specialty Care, Boehringer Ingelheim. “There is an urgent unmet need for an approved treatment option that can rapidly clear painful GPP flares.”
GPP is a rare, life-threatening neutrophilic skin disease, which is distinct from plaque psoriasis. It is characterized by episodes of widespread eruptions of painful, sterile pustules (blisters of non-infectious pus). There is a high unmet need for treatments that can rapidly and completely resolve the signs and symptoms of GPP flares. Flares greatly affect a person’s quality of life and can lead to hospitalization with serious complications, including heart failure, renal failure, sepsis, and death.
About spesolimab
Spesolimab is a novel, humanized, selective antibody that blocks the activation of the interleukin-36 receptor (IL-36R), a signaling pathway within the immune system shown to be involved in the pathogeneses of several autoimmune diseases, including GPP. Spesolimab is also under investigation for the prevention of GPP flares and for the treatment of other neutrophilic skin diseases, such as palmoplantar pustulosis (PPP) and hidradenitis suppurativa (HS).
About generalized pustular psoriasis (GPP)
GPP is a rare, heterogenous and potentially life-threatening neutrophilic skin disease, which is clinically distinct from plaque psoriasis. GPP is caused by neutrophils (a type of white blood cell) accumulating in the skin, resulting in painful, sterile pustules all over the body. The clinical course varies, with some patients having a relapsing disease with recurrent flares, and others having a persistent disease with intermittent flares. While the severity of GPP flares can vary, if left untreated they can be life-threatening due to complications such as sepsis and multisystem organ failure. This chronic, systemic disease has a substantial quality of life impact for patients and healthcare burden. GPP has a varied prevalence across different geographical regions and more women are affected than men.
Boehringer Ingelheim Immunology: Pioneering Science, Inspired By Patients
Living with fibrotic and inflammatory diseases greatly impacts patients’ lives emotionally and physically. These patients are our guides, partners and inspiration as we redefine treatment paradigms. As a family-owned company, we can plan long-term. Our goal is to discover and develop first-of-their-kind therapies. With a deep understanding of molecular pathways, we are pioneering scientific breakthroughs that target, repair and prevent many fibrotic and inflammatory diseases. By building on long-term external collaborations, we strive to bring treatment breakthroughs to patients in the shortest time. We won’t rest until we can give people the chance to live the lives they want.
Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.com.
MPR-US-101971
////////Spesolimab, monoclonal antibody, fda 2022, approvals 2022, Orphan Drug Status, Generalised pustular psoriasis, BI 655130, Spesolimab-sbzo, peptide, monoclonal antibody

NEW DRUG APPROVALS
ONE TIME
$10.00
MIRDAMETINIB

MIRDAMETINIB
391210-10-9
Chemical Formula: C16H14F3IN2O4
Molecular Weight: 482.19
PD0325901; PD 0325901; PD-325901; mirdametinib
FDA APPROVED 2/11/2025, Gomekli, To treat neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection
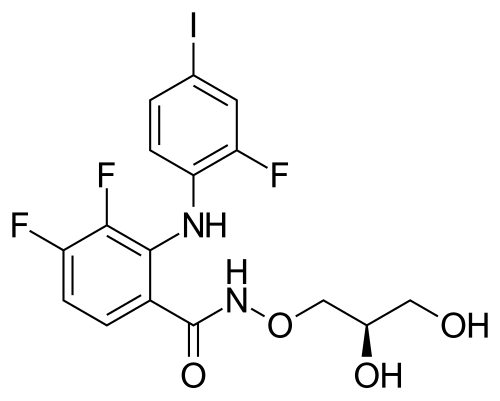
IUPAC/Chemical Name: (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino)benzamide
SpringWorks Therapeutics (a spin out of Pfizer ) is developing mirdametinib, a second-generation, non-ATP competitive, allosteric MEK1 and MEK2 inhibitor derived from CI-1040, for treating type 1 neurofibromatosis (NF1) and advanced solid tumors. In June 2021, a phase I/II trial was initiated in patients with low grade glioma.
- OriginatorPfizer
- DeveloperAstraZeneca; BeiGene; BIOENSIS; Pfizer; SpringWorks Therapeutics; St. Jude Childrens Research Hospital; University of Oxford
- ClassAniline compounds; Anti-inflammatories; Antineoplastics; Benzamides; Immunotherapies; Small molecules
- Mechanism of ActionMAP kinase kinase 1 inhibitors; MAP kinase kinase 2 inhibitors
- Orphan Drug StatusYes – Neurofibromatosis 1
- Phase IINeurofibromatosis 1
- Phase I/IIGlioma
- Phase ISolid tumours
- PreclinicalChronic obstructive pulmonary disease
- No development reportedCervical cancer
- DiscontinuedBreast cancer; Cancer; Colorectal cancer; Malignant melanoma; Non-small cell lung cancer
- 22 Jul 2021SpringWorks Therapeutics receives patent allowance for mirdametinib from the US Patent and Trademark Office for the treatment of Neurofibromatosis type 1-associated plexiform neurofibromas
- 16 Jun 2021SpringWorks Therapeutics and St. Jude Children’s Research Hospital agree to develop mirdametinib in USA for glioma
- 15 Jun 2021Efficacy and safety data from the phase IIb RENEU trial for Neurofibromatosis type 1-associated plexiform neurofibromas released by SpringWorks Therapeutics
Mirdametinib, sold under the brand name Gomekli, is a medication used for the treatment of people with neurofibromatosis type 1.[1] Mirdametinib is a kinase inhibitor.[1][2] It is taken by mouth.[1]
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib was approved for medical use in the United States in February 2025.[1][3]
SCHEME
SIDE CHAIN

MAIN
Medical uses
Mirdametinib is indicated for the treatment of people with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection.[1]
Adverse effects
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib can cause left ventricular dysfunction and ocular toxicity including retinal vein occlusion, retinal pigment epithelial detachment, and blurred vision.[3]
History
The efficacy of mirdametinib was evaluated in ReNeu (NCT03962543), a multicenter, single-arm trial in 114 participants aged two years of age and older (58 adults, 56 pediatric participants) with symptomatic, inoperable NF1-associated plexiform neurofibromas causing significant morbidity.[3] An inoperable plexiform neurofibromas was defined as a plexiform neurofibromas that could not be completely surgically removed without risk for substantial morbidity due to encasement or close proximity to vital structures, invasiveness, or high vascularity.[3]
The US Food and Drug Administration (FDA) granted the application for mirdametinib priority review, fast track, and orphan drug designations along with a priority review voucher.[3]
Society and culture
Legal status
Mirdametinib was approved for medical use in the United States in February 2025.[3][4][5]
PATENT
US-11066358
On July 20, 2021, SpringWorks Therapeutics announced that the United States Patent and Trademark Office (USPTO) has issued US11066358 , directed to mirdametinib , the Company’s product candidate in development for several oncology indications, including as a monotherapy for patients with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN) and was assigned to Warner-Lambert Company (a subsidiary of Pfizer ).This patent was granted on July 20, 2021, and expires on Feb 17, 2041. Novel crystalline forms of mirdametinib and compositions comprising them are claimed.
| N—((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (“mirdametinib”, or “PD-0325901”) is a small molecule drug which has been designed to inhibit mitogen-activated protein kinase kinase 1 (“MEK1”) and mitogen-activated protein kinase kinase 2 (“MEK2”). MEK1 and MEK2 are proteins that play key roles in the mitogen-activated protein kinase (“MAPK”) signaling pathway. The MAPK pathway is critical for cell survival and proliferation, and overactivation of this pathway has been shown to lead to tumor development and growth. Mirdametinib is a highly potent and specific allosteric non-ATP-competitive inhibitor of MEK1 and MEK2. By virtue of its mechanism of action, mirdametinib leads to significantly inhibited phosphorylation of the extracellular regulated MAP kinases ERK1 and ERK2, thereby leading to impaired growth of tumor cells both in vitro and in vivo. In addition, evidence indicates that inflammatory cytokine-induced increases in MEK/ERK activity contribute to the inflammation, pain, and tissue destruction associated with rheumatoid arthritis and other inflammatory diseases. |
Example 1: Production of Essentially Pure Form IV
Lab Scale Production of Essentially Pure Form IV
| All reactions were performed in toluene other than otherwise stated. Triflic anhydride gave the best yield. |
[TABLE-US-00002]TABLE 1 Coupling Agents for Step 1Entry No.Coupling AgentYieldNotes 1Mesyl Chloridedid not react 2Benzyl chloride27Had to heat 70° C. for 166 hr34-fluorobenzensulfonylchloride27Ran 93 hrs. at 70° C.44-chlorobenzensulfonylchloride35Complete after 68 hrs. 50° C.5Tosyl Chloride36Had to heat to 70° C. for 164 hrs6Benzyl chloride52study solvent effects: DMF, DMSO, NMP – all similar DMSO fastest all complete after 110 hrs., heated to 70° C. after 66 hrs.7Triflic anhydride91Cooled to −74° C. |
| [TABLE-US-00004]TABLE 3 Yields for base deprotection ReagentYield* Methyl hydrazine85-95% Anhydrous NH3 (sparged)78-90% Anhydrous NH3 (50 psi)80-92% Aqueous NH390-97% *from PD-0333760 |
Step 2: Fluoride Displacement
Pilot Plant Preparation of Essentially Pure Form IV
Step 1: Preparation of “Side Chain”, PD-0337792
Step 2: Preparation of PD-0315209
Step 3: Preparation of PD-0325901
Polymorph Transformation
| 21.4 kg PD-0315209, 9.7 kg CDI (1.05 equiv.), 91 kg solution of 9.7% PD-0337792 in Toluene (1.1 equiv.) were used and resulted in 12.74 kg of PD-0325901 (assay 99.4%, 100% Form IV, Yield 48%). |
PATENT
WO2006134469 , claiming methods of preparing MEK inhibitor, assigned to Warner-Lambert Co .
https://patents.google.com/patent/WO2006134469A1/enThe compound Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide represented by formula 1

i is a highly specific non-ATP-competitive inhibitor of MEK1 and MEK2. The compound of formula ± (Compound I) is also known as the compound PD 0325901. Compound I is disclosed in WO 02/06213; WO 04/045617; WO 2005/040098; EP 1262176; U.S. Patent Application Pub. No. 2003/0055095 A1 ; U.S. Patent Application Pub. No. 2004/0054172 A1; U.S. Patent Application Pub. No. 2004/0147478 A1 ; and U.S. Patent Application No. 10/969,681, the disclosures of which are incorporated herein by reference in their entireties.Numerous mitogen-activated protein kinase (MAPK) signaling cascades are involved in controlling cellular processes including proliferation, differentiation, apoptosis, and stress responses. Each MAPK module consists of 3 cytoplasmic kinases: a mitogen-activated protein kinase (MAPK), a mitogen-activated protein kinase kinase (MAPKK), and a mitogen-activated protein kinase kinase kinase (MAPKKK). MEK occupies a strategic downstream position in this intracellular signaling cascade catalyzing the phosphorylation of its MAP kinase substrates, ERK1 and ERK2. Anderson et al. “Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase.” Nature 1990, v.343, pp. 651-653. In the ERK pathway, MAPKK corresponds with MEK (MAP kinase ERK Kinase) and the MAPK corresponds with ERK (Extracellular Regulated Kinase). No substrates for MEK have been identified other than ERK1 and ERK2. Seger et al. “Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells.” J. Biol. Chem., 1992, v. 267, pp. 14373-14381. This tight selectivity in addition to the unique ability to act as a dual-specificity kinase is consistent with MEK’s central role in integration of signals into the MAPK pathway. The RAF-MEK-ERK pathway mediates proliferative and anti-apoptotic signaling from growth factors and oncogenic factors such as Ras and Raf mutant phenotypes that promote tumor growth, progression, and metastasis. By virtue of its central role in mediating the transmission of growth- promoting signals from multiple growth factor receptors, the Ras-MAP kinase cascade provides molecular targets with potentially broad therapeutic applications.One method of synthesizing Compound I is disclosed in the above-referenced WO 02/06213 andU.S. Patent Application Pub. No. 2004/0054172 A1. This method begins with the reaction of 2-fluoro-4- iodo-phenylamine and 2,3,4-trifluoro-benzoic acid in the presence of an organic base, such as lithium diisopropylamide, to form 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzoic acid, which is then reacted with (R)-0-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of a peptide coupling agent (e.g., diphenylphosphinic chloride) and a tertiary amine base (e.g., diisopropylethylamine). The resulting product is hydrolyzed under standard acidic hydrolysis conditions (e.g., p-TsOH in MeOH) to provide Compound 1. (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine is prepared by reaction of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol with N-hydroxyphthalimide in the presence of Ph3P and diethyl azodicarboxylate.Another method of synthesizing Compound I, which is disclosed in the above-referenced U.S.Patent Application No. 10/969,681, comprises reaction of 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzoic acid with (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of N1N1– carbonyldiimidazole. The resulting product is hydrolyzed with aqueous acid and crystallized to provide polymorphic form IV of Compound I.Although the described methods are effective synthetic routes for small-scale synthesis of Compound I, there remains a need in the art for new synthetic routes that are safe, efficient and cost effective when carried out on a commercial scale.The present invention provides a new synthetic route including Steps I through Step III to the MEK inhibitor Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I).Step I: Preparation of 0-{r(4RV2.2-dimethyl-1.3-dioxolan-4-ynmethyl}hydroxylanπine (6) The method of the present invention comprises a novel Step I of preparing of 0-{[(4R)-2,2- dimethyl-1 ,3-dioxolan-4-yl]methyl}hydroxylamine (6) from [(4S)-2,2-dimethyl-1 ,3-dioxoIan-4-yl]methanol (1) through the formation of [(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methyl trifluoromethanesulfonate (3) and its coupling with N-hydroxyphthalimide (4) to afford 2-{[(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methoxy}-1 H- isoindole-1 ,3(2H)-dione (5), which is subsequently de-protected to give 6 as shown in Scheme 1.Scheme 1



The reaction of compound (1) with trifluoromethanesulfonic anhydride (2) is carried out in the presence of a non-nucleophilic base, such as, for example, a tertiary organic amine, in an aprotic solvent at a temperature of from -5O0C to 50C, preferably, at a temperature less than -150C, to form triflate (3). A preferred tertiary organic amine is triethylamine, and a preferred solvent is toluene. Treatment of triflate (3) with N-hydroxyphthalimide (4) furnishes phthalimide (5), which can be isolated if desired. However, in order to minimize processing time and increase overall yield, 0-{[(4R)- 2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) can be prepared in a one-pot process with no phthalimide (S) isolation. Cleavage of the phthalimide function could be achieved by methods known in the art, for example, by hydrazinolysis. However, the use of less hazardous aqueous or anhydrous ammonia instead of methyl hydrazine (CH3NHNH2) is preferred.Step II: Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) As shown in Scheme 2, Step Il of the method of the present invention provides 3,4-difluoro-2-(2- fluoro-4-iodophenylamino)-benzoic acid (9).Scheme 2

Preparation of compound (9) can be carried out by reacting compound (7), wherein X is halogen, or O-SC^R^ or 0-P(3O)(OR^, wherein R^ is alkyl or aryl, with compound (8) optionally in a solvent, and in the presence of from about 1 mol equivalent to about 10 mol equivalents of at least one base, wherein the base is selected from: a Group I metal cation hydride or a Group 2 metal cation hydride, including lithium hydride, sodium hydride, potassium hydride, and calcium hydride, a Group I metal cation dialkylamide or a Group 2 metal cation dialkylamide, including lithium diisopropylamide, a Group I metal cation amide or a Group 2 metal cation amide, including lithium amide, sodium amide, potassium amide, a Group I metal cation alkoxide or a Group 2 metal cation alkoxide, including sodium ethoxide, potassium terf-butoxide, and magnesium ethoxide, and a Group I metal cation hexamethyldisilazide, including lithium hexamethyldisilazide; for a time, and at a temperature, sufficient to yield compound (9).Preferably, preparation of compound (9) is carried out by reacting compound (7), wherein X is halogen, more preferably, X is fluorine, in an aprotic solvent with compound (8) in the presence of from about 3 mol equivalents to about 5 mol equivalents of a Group I metal cation amide at a temperature of from 2O C to 55°C, more preferably, at a temperature from 45°C to 55°C. A catalytic amount of Group I metal cation dialkylamide can be added if necessary. A preferred Group I metal cation amide is lithium amide, a preferred Group I metal cation dialkylamide is lithium diisopropylamide, and a preferred solvent is tetrahydrofuran. Preferably, the reaction is performed by adding a small amount of compound (7) and compound (8) to lithium amide in tetrahydrofuran followed by slow continuous addition of the remaining portion. This procedure minimizes the risk of reactor over-pressurization due to gas side product (ammonia) generation.Step III: Preparation of N-((RV2.3-dihydroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I)Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using a carboxylic acid activating reagent such as, for example, COCI2, S(O)C^, S(O)2Cl2, P(O)Cl3, triphenylphosphine/diethylazodicarboxylate, diphenylphosphinic chloride, N, N’-dicyclohexylcarbodiimide, (benzotriazol-1 -yloxy)tripyrolidinophosphonium hexafluorophosphate, (benzotriazol-1 – yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, N-ethyl-N’-(3- dimethylaminopropyl)carbodiimide hydrochloride, or 1,1′-carbonyldiimidazole (CDI).A preferred carboxylic acid activating reagent is 1,1′-carbonyldimidazole (CDI) shown in Scheme 3. Preparation of the desirable polymorphic Form IV of Compound I using CDI is described in the above- referenced U.S. Patent Application No. 10/969,681.Scheme 3

10

10 11 Compound IIn according to the present invention, the method was modified to include the advantageous procedure for product purification and isolation, which procedure is performed in single-phase systems such as, for example, toluene/acetonitrile for the first isolation/crystallization and ethanol/toluene for the second recrystallization. Water addition, implemented in the previous procedure, was omitted to avoid the two-phase crystallization from the immiscible water-toluene system that caused inconsistent product purity. The one-phase procedure of the present invention provides consistent control and removal of un- reacted starting material and side products. Alternatively, Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using thionyl chloride (SOCI2) as shown in Scheme 4.Scheme 4


Compound IExamplesThe reagents and conditions of the reactions described herein are merely illustrative of the wide variety of starting materials, their amounts and conditions which may be suitably employed in the present invention as would be appreciated by those skilled in the art, and are not intended to be limiting in any way.HPLC (Conditions A): 10 μL injection volume onto Agilent Zorbax RX-C18 150 mm x 4.6 mm x 3.5 μm column at 30°C column temperature, 1.0 mL/min flow rate and detection at 246 nm. Mobile phase A (v/v): 25 mM Acetate Buffer, pH 6.0; Mobile phase B (v/v): Acetonitrile, and Linear Gradient Table:

Sample Preparation: Dilute 100 μL reaction mixture to 10 mL with acetonitrile. Mix in a vial 200 μL of this sample solution with 300 μL carbonate buffer pH 10.0 and 300 μL solution of 2-mercaptopyridine in acetonitrile (18 mM), heat the vial for 10 minutes at 500C and dilute to 1:1 ratio in mobile phase A.GC (Conditions B): 1 μL injection onto an RTX-5 column (30 m x 0.25 mm x 0.25 μm) with initial oven temperature of 120°C for 2 min. to final temperature of 250°C in 15°C/minute ramping and a final time of 2.33 min; Flow rate: 1 mL/min.HPLC (Conditions C): 5 μL injection onto Phenomenex Luna C18(2) 150 mm x 4.6 mm x 3μm column ; flow rate : 1.0 mL/min; detection at 225 nm; mobile phase A: 95/5 v/v Water/Acetonitrile with 0.1% Trifluoroacetic acid (TFA), mobile phase B: 5/95 v/v Water/Acetonitriie with 0.1% TFA; Linear Gradient Table:

Sample preparation: Dilute 1 ml_ reaction mixture to 100 mL with acetonitrile and dilute 1 mL of this solution to 10 mL with 50:50 Water/Acetonitrile.HPLC (Conditions D): 5 μL injection onto Waters SymmetryShield RP 18, 150 mm x 4.6 mm x 3.5 μm column; flow rate: 1.0 mL/min; detection at 235 nm; mobile phase A: 25 mM Acetate Buffer adjusted to pH 5.5, mobile phase B: Acetonitrile; Linear Gradient Table:

Sample preparation: Dilute 40 μL of reaction mixture in 20 mL acetonitrile.HPLC (Conditions E): 10 μL sample injection onto YMC ODS-AQ 5 μm, 250 mm x 4.6 mm column; flow rate: 1.0 ml_/min; detection at 280 nm; temperature 30°C; mobile phase : 75/25 v/v Acetonitrile/Water with 0.1% Formic acid.Sample preparation: Quench reaction mixture sample with dipropylamine and stir for about 5 minutes before further dilution with mobile phase.DSC measurement was performed using a Mettler-Toledo DSC 822, temperature range 25° to 150°C with 5°C/min heating rate in a 40 μL aluminum pan. Experimental Conditions for Powder X-Rav Diffraction (XRD):A Rigaku Miniflex+ X-ray diffractometer was used for the acquisition of the powder XRD patterns. The instrument operates using the Cu Ka1 emission with a nickel filter at 1.50451 units. The major instrumental parameters are set or fixed at:X-ray: Cu / 30 kV (fixed) / 15 mA (fixed)Divergence Slit: Variable Scattering Slit: 4.2° (fixed) Receiving Slit: 0.3 mm (fixed) Scan Mode: FT Preset Time: 2.0 s Scan Width: 0.050° Scan Axis: 2Theta/Theta Scan Range: 3.000° to 40.000°Jade Software Version: 5.0.36(SP1) 01/05/01 (Materials Data, Inc.) Rigaku Software: Rigaku Standard Measurement for Windows 3.1 Version 3.6(1994-1995) Example 1. Preparation of 0-ffl4R)-2.2-dimethyl-1.3-dioxolan-4-vπmethyl}hvdroxylamine (6)A solution containing [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol (1) (13.54 ml_, 0.109 mol) (DAISO Co., Ltd., CAS# 22323-82-6) and triethylamine (18.2 ml_, 0.131 mol) in 115 mL toluene was cooled to -15 C, then trifluoromethanesulfonic anhydride (2) (18.34 mL, 30.75 g, 0.109 mol) (Aldrich, Catalog # 17,617-6 ) was added drop wise while maintaining the temperature at less than -15°C. The mixture was then stirred for 2 hours, and transferred to a separate flask containing a mixture (slurry) of N- hydroxyphthalimide (4) (18.99 g, 0.116 mol) (Aldrich, Catalog # H5.370-4) and 18.2 mL (0.13 mol) triethylamine in 95 mL toluene. The resulting mixture was warmed to 20-25°C and stirred for at least 5 hours or until reaction completion (determined by HPLC (Conditions A)). Water (93 mL) was then added to quench the reaction mixture, the phases were separated, and the bottom aqueous layer was discarded. The water quench was repeated two more times resulting in a pale yellow organic layer. The organic layer was heated to 35 C and treated with 36.7 mL ammonium hydroxide solution (contains about 28-29% wt/wt ammonia). The mixture was stirred for at least 12 hours or until the reaction was deemed complete as determined by GC (Conditions B). The water was then removed under reduced pressure by co- distilling it with toluene to about half of the original volume at temperatures around 35-45 C. Toluene (170 mL) was added to the concentrated solution and the distillation was repeated. A sample was drawn for water content determination by Karl Fisher method (using EM Science Aquastar AQV-2000 Titrator with a sample injected to a pot containing methanol and salicylic acid). The distillation was repeated ifl water content was more than 0.1%. The concentrated solution was filtered to remove the white solid side product, and the filtrate was stored as 112mL (98 g) product solution containing 9.7% w/w compound 6 in toluene. This solution was ready for use in the final coupling step (Example 3). Overall chemical yield was 59%. A small sample was evaporated to yield a sample for NMR identification.1H NMR (400 MHz, CDCI3): δ 5.5 (bs, 2H), 4.35 (m, 1H), 4.07 (dd, 1H), 3.77 (m, 2H), 3.69 (dd, 1H), 1.44 (s, 3H), 1.37 (s, 3H).Example 2. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9)A solution of 2-fluoro-4-iodoaniline (8) (16.4 g, 0.069 mol) (Aldrich, Catalog # 30,660-6) and 2,3,4- trifluorobenzoic acid (7) (11.98 g, 0.068 mol) (Aldrich, Cat # 33,382-4) in 38 mL tetrahydrofuran (THF) was prepared and a portion (about 5%) of this solution was added to a stirring slurry of lithium amide (5 g, 0.22 mol) in 40 mL THF at 50-55 C. After about 15-30 min. an exotherm followed by gas release and color change are observed. The remaining portion of the (8) and (7) solution was added slowly over 1-2 hr while maintaining temperatures within 45-55°C. The mixture was stirred until the reaction was deemed complete (by HPLC (Conditions C). The final mixture was then cooled to 20-25°C and transferred to another reactor containing 6 N hydrochloric acid (47 mL) followed by 25 mL acetonitrile, stirred, and the bottom aqueous phase was discarded after treatment with 40 mL 50% sodium hydroxide solution. The organic phase was concentrated under reduced pressure and 57 mL acetone was added. The mixture was heated to 50°C, stirred, and added with 25 mL warm (40-50°C) water and cooled to 25-30°C to allow crystallization to occur (within 1-4 hours). Once the crystallization occurred, the mixture was further cooled to 0 to -5°C and stirred for about 2 hours. The solid product was filtered and the wet cake was dried in vacuum oven at about 55°C. Overall chemical yield was 21.4 g, 80%. 1H NMR (400 MHz, (CD3)2SO): δ 13.74 (bs, 1H), 9.15 (m, 1 H), 7.80 (dd, 1H), 7.62 (d, 1H), 7.41 (d, 1H), 7.10 (q, 1H), 6.81 (m, 1H).Example 2B. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) by the solid addition of lithium amide methodTo a stirring solution of 2,3,4-trifluorobenzoic acid (13) (5.0 g, 28.4 mmol) and 2-fluoro-4- iodoaniline (14) (6.73 g, 28.4 mmol) in MeCN (100 mL), under N2 atmosphere was added lithium amide (2.61 g, 113.6 mmol) in small portions. The reaction mixture was heated to reflux for 45 minutes, cooled to ambient temperature and quenched with 1 N HCI and then water. The yellowish white precipitate was filtered, washed with water. The solid was triturated in CH2CI2 (30 mL) for 1h, filtered and dried in a vacuum oven at 45°C for 14 hours to give 8.Og (72%) of compound (9) as an off-white solid, mp 201.5-203 °C.Example 3. Preparation of N-((R)-2.3-dihvdroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound \)3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (20 g, 0.051 mol) in 100 mL acetonitrile was treated with 1,1′-carbonyldiimidazole (CDI) (8.66 g, 0.053 mol) (Aldrich, Cat # 11,553-3) and stirred for about 2 hours at 20-25°C until the reaction was deemed complete by HPLC (Conditions D). 94 mL (84.9 g) of 9.7% w/w solution of O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) in toluene was then added and stirred for about 4 hours or until the reaction was deemed complete by HPLC (Conditions D). To this mixture was added 66 mL of 5.6 % hydrochloric acid solution, and after stirring, the bottom aqueous phase was discarded. Again 66 mL of 5.6 % hydrochloric acid solution was added to the organic phase and stirred at 20-25°C for 12-18 hours or until the reaction was deemed complete by HPLC (Conditions D). The bottom layer was then discarded and the remaining organic layer was concentrated under reduced pressure to remove about 10-20% solvent, and the volume was adjusted to about 9-11 mL/g with toluene (80 mL). Crude product was then crystallized at 10-15°C. The slurry was allowed to stir for about 2 hours and the crude solid product was filtered, and dried. The dried crude product was recharged to the reactor and dissolved into 150 mL of 5% v/v ethanol/toluene mixture at 55- 67°C. The solution was then clarified at this temperature through filter (line filter) to remove any remaining particulate matter. The solution was then cooled slowly to 5°C to crystallize and stirred for at least 2 h, filtered and dried. The dried solid product was redissolved in EtOH (60 mL) at 35°C, and product was precipitated out by adding water (300 mL) at 35°C followed by cooling to 200C. The slurry was stirred for at least 2 hours to transform the crystals to the desired polymorphic Form IV as determined by DSC and Powder X-ray Diffraction pattern (PXRD). The slurry was filtered and dried under vacuum oven at 70- 90°C to yield the final N-((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I) product. Overall chemical yield was 13 g, 53%. Melting point (DSC): 112+1° C. Appearance: White to off-white crystals.Shown in Figure 1, PXRD conforms to polymorphic crystal Form IV disclosed in the above mentioned U.S. Patent Application No. 10/969,681 1H NMR (400 MHz, (CD3)2SO): δ 11.89 (bs, 1H), 8.71 (bs, 1H), 7.57 (d, 1H), 7.37 (m, 2H), 7.20 (q, 1H), 6.67 (m, 1H), 4.84 (bs, 1H), 4.60 (m, 1H), 3.87 (m, 1 H), 3.7 (m, 2H), 3.34 (m, 2H).Example 4. Preparation of N-((R)-2.3-dihydroxypropoxyV3.4-difluoro-2-(2-fluoro-4-iodo-phenylanrιinoV benzamide (Compound \)To a stirring solution of 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (120 g, 0.30 mol) in a mixture of 1 mL N,N-dimethylformamide and 1000 mL toluene was added thionyl chloride (55 g, 0.462 mol). The mixture was heated to 50-65 C and stirred for 2 hours or until reaction completion as determined by HPLC (Conditions E). The final reaction mixture was then cooled and concentrated under reduced pressure to a slurry keeping the temperature below 35°C. Toluene (600 mL) was added to dissolve the slurry and vacuum distillation was repeated. Additional toluene (600 mL) was added to the slurry dissolving all solids and the solution was then cooled to 5° -10°C. The solution was then treated with O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) (63 g, 0.43 mol) solution in 207 mL toluene followed by potassium carbonate (65 g) and water (200 mL), stirred for at least 2 hours at 20- 25°C. The stirring was stopped to allow phase separation and the bottom phase was discarded. The remaining organic layer was treated with hydrochloric acid solution (7.4%, 240 mL) until pH was less than 1 and stirred for 2 hours. The final reaction mixture was slightly concentrated under vacuum collecting about 100 mL distillate and the resulting organic solution was cooled to 5°C to crystallize the product and filtered. The filter cake was washed with toluene (1000 mL) followed by water (100 mL) and the wet cake (crude product Compound I) was charged back to the flask. Toluene (100 mL), ethanol (100 mL) and water (100 mL) are then added, stirred at 30-35°C for about 15 min, and the bottom aqueous phase was discarded. Water (200 mL) was then added to the organic solution and the mixture was stirred at about 3O C to allow for crystallization. The stirring was continued for 2 hours after product crystallized, then it was further cooled to about 0°C and stirred for at least 2 hours. The slurry was filtered and wet cake was dried under reduced pressure at 55-85°C to yield the final product N-((R)-2,3-dihydroxypropoxy)-3,4- difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I) product. Overall chemical yield was 86 g, 58%.
PATENT
WO2002/006213 describes crystalline Forms I and II. U.S. Pat. No. 7,060,856 (“the ‘856 patent”)
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2002006213
| Clinical data | |
|---|---|
| Trade names | Gomekli |
| Other names | PD-0325901 |
| AHFS/Drugs.com | Gomekli |
| License data | US DailyMed: Mirdametinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EE05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 391210-10-9 |
| PubChem CID | 9826528 |
| IUPHAR/BPS | 7935 |
| DrugBank | DB07101 |
| ChemSpider | 10814340 |
| UNII | 86K0J5AK6M |
| KEGG | D11675 |
| ChEBI | CHEBI:9826528 |
| ChEMBL | ChEMBL507361 |
| PDB ligand | 4BM (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C16H14F3IN2O4 |
| Molar mass | 482.198 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Gomekli- mirdametinib capsule; Gomekli- mirdametinib tablet, for suspension”. DailyMed. 27 February 2025. Retrieved 2 April 2025.
- ^ Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ (June 2023). “Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas”. BMC Cancer. 23 (1): 553. doi:10.1186/s12885-023-10996-y. PMC 10273716. PMID 37328781.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection”. U.S. Food and Drug Administration (FDA). 11 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “UPDATE: SpringWorks Therapeutics Announces FDA Approval of Gomekli (mirdametinib) for the Treatment of Adult and Pediatric Patients with NF1-PN” (Press release). SpringWorks Therapeutics. 12 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025 – via GlobeNewswire News Room.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025.
External links
- “Mirdametinib (Code C52195)”. NCI Thesaurus.
- Clinical trial number NCT03962543 for “MEK Inhibitor Mirdametinib (PD-0325901) in Patients With Neurofibromatosis Type 1 Associated Plexiform Neurofibromas (ReNeu)” at ClinicalTrials.gov
- Moertel CL, Hirbe AC, Shuhaiber HH, Bielamowicz K, Sidhu A, Viskochil D, Weber MD, Lokku A, Smith LM, Foreman NK, Hajjar FM, McNall-Knapp RY, Weintraub L, Antony R, Franson AT, Meade J, Schiff D, Walbert T, Ambady P, Bota DA, Campen CJ, Kaur G, Klesse LJ, Maraka S, Moots PL, Nevel K, Bornhorst M, Aguilar-Bonilla A, Chagnon S, Dalvi N, Gupta P, Khatib Z, Metrock LK, Nghiemphu PL, Roberts RD, Robison NJ, Sadighi Z, Stapleton S, Babovic-Vuksanovic D, Gershon TR: ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025 Feb 20;43(6):716-729. doi: 10.1200/JCO.24.01034. Epub 2024 Nov 8. [Article]
- Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, Allen JC, Schorry E, Korf B, Robison NJ, Goldman S, Vinks AA, Emoto C, Fukuda T, Robinson CT, Cutter G, Edwards L, Dombi E, Ratner N, Packer R, Fisher MJ: NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol. 2021 Mar 1;39(7):797-806. doi: 10.1200/JCO.20.02220. Epub 2021 Jan 28. [Article]
- Ioannou M, Lalwani K, Ayanlaja AA, Chinnasamy V, Pratilas CA, Schreck KC: MEK Inhibition Enhances the Antitumor Effect of Radiotherapy in NF1-Deficient Glioblastoma. Mol Cancer Ther. 2024 Sep 4;23(9):1261-1272. doi: 10.1158/1535-7163.MCT-23-0510. [Article]
- FDA Approved Drug Products: GOMEKLI (mirdametinib) capsules and tablets for oral and oral suspension use (Feb 2024) [Link]
- FDA News: FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection [Link]
////////MIRDAMETINIB, Orphan Drug Status, Neurofibromatosis 1, PHASE 2, PD0325901, PD 0325901, PD-325901, FDA 2025, GOMEKLI, APPROVALS 2025
O=C(NOC[C@H](O)CO)C1=CC=C(F)C(F)=C1NC2=CC=C(I)C=C2F
Dasiglucagon




Dasiglucagon
Treatment of Hypoglycemia in Type 1 and Type 2 Diabetes Patients
| Formula | C152H222N38O50 |
|---|---|
| CAS | 1544300-84-6 |
| Mol weight | 3381.6137 |
FDA APPROVED, 2021/3/22, Zegalogue
Zealand Pharma A/S
UNIIAD4J2O47FQ
HypoPal rescue pen
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-2-methylpropanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-carboxy-1-[[(2S)-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-5-oxopentanoic acid
. [16-(2-methylalanine)(S>X),17-L-alanine(R>A),20-L-α-glutamyl(Q>E),21-L-αglutamyl(D>E),24-L-lysyl(Q>K),27-L-α-glutamyl(M>E),28-L-serine(N>S)]human glucagon
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L- phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L- lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L- arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L- valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-alpha-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-alpha-glutamyl-L-alphaC152 H222 N38 O50L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-Molecular Weight3381.61
Other Names
- L-Histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-L-threonine
- Developer Beta Bionics; Zealand Pharma
- ClassAntihyperglycaemics; Antihypoglycaemics; Peptides
- Mechanism of ActionGlucagon receptor agonists
- Orphan Drug StatusYes – Hypoglycaemia; Congenital hyperinsulinism
- RegisteredHypoglycaemia
- Phase IIICongenital hyperinsulinism
- Phase II/IIIType 1 diabetes mellitus
- 22 Mar 2021Registered for Hypoglycaemia (In children, In adolescents, In adults, In the elderly) in USA (SC) – First global approval
- 22 Mar 2021Zealand Pharma anticipates the launch of dasiglucagon in USA (SC, Injection) in June 2021
- 22 Mar 2021Pooled efficacy and safety data from three phase III trials in Hypoglycaemia released by Zealand Pharma
NEW DRUG APPROVALS
one time
$10.00
PATENTS
WO 2014016300
US 20150210744
PAPER
Pharmaceutical Research (2018), 35(12), 1-13
Dasiglucagon, sold under the brand name Zegalogue, is a medication used to treat severe hypoglycemia in people with diabetes.[1]
The most common side effects include nausea, vomiting, headache, diarrhea, and injection site pain.[1]
Dasiglucagon was approved for medical use in the United States in March 2021.[1][2][3] It was designated an orphan drug in August 2017.[4]
Dasiglucagon is under investigation in clinical trial NCT03735225 (Evaluation of the Safety, Tolerability and Bioavailability of Dasiglucagon Following Subcutaneous (SC) Compared to IV Administration).
Medical uses
Dasiglucagon is indicated for the treatment of severe hypoglycemia in people aged six years of age and older with diabetes.[1][2]
Contraindications
Dasiglucagon is contraindicated in people with pheochromocytoma or insulinoma.[1]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214231s000lbl.pdf
- ^ Jump up to:a b “Dasiglucagon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 March 2021.
- ^ “Zealand Pharma Announces FDA Approval of Zegalogue (dasiglucagon) injection, for the Treatment of Severe Hypoglycemia in People with Diabetes” (Press release). Zealand Pharma. 22 March 2021. Retrieved 22 March 2021 – via GlobeNewswire.
- ^ “Dasiglucagon Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 10 August 2017. Retrieved 22 March 2021.
External links
- “Dasiglucagon”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03378635 for “A Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Type 1 Diabetes Subjects” at ClinicalTrials.gov
- Clinical trial number NCT03688711 for “Trial to Confirm the Clinical Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Subjects With T1DM” at ClinicalTrials.gov
- Clinical trial number NCT03667053 for “Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in T1DM Children” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zegalogue |
| AHFS/Drugs.com | Zegalogue |
| License data | US DailyMed: Dasiglucagon |
| Routes of administration | Subcutaneous |
| Drug class | Glucagon receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1544300-84-6 |
| PubChem CID | 126961379 |
| DrugBank | DB15226 |
| UNII | AD4J2O47FQ |
| KEGG | D11359 |
| Chemical and physical data | |
| Formula | C152H222N38O50 |
| Molar mass | 3381.664 g·mol−1 |
| 3D model (JSmol) | Interactive image |
///////////Dasiglucagon, FDA 2021, APPROVALS 2021, Zegalogue, ダシグルカゴン, ZP 4207, ZP-GA-1, Hypoglycemia, Type 1, Type 2 , Diabetes Patients, Zealand Pharma A/S, Orphan Drug Status, Hypoglycaemia, Congenital hyperinsulinism, HypoPal rescue pen, DIABETES
#Dasiglucagon, #FDA 2021, #APPROVALS 2021, #Zegalogue, #ダシグルカゴン, #ZP 4207, ZP-GA-1, #Hypoglycemia, #Type 1, #Type 2 , #Diabetes Patients, #Zealand Pharma A/S, #Orphan Drug Status, #Hypoglycaemia, #Congenital hyperinsulinism, #HypoPal rescue pen, #DIABETESSMILES
- C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC3=CC=C(C=C3)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(=O)O)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC4=CC=CC=C4)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC5=CNC6=CC=CC=C65)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H](CC7=CNC=N7)N)O
Pemigatinib


Pemigatinib
INCB054828
| Formula | C24H27F2N5O4 |
|---|---|
| CAS | 1513857-77-62379919-96-5 HCL |
| Mol weight | 487.4991 |
2020/4/17FDA APPROVED, PEMAZYRE
佩米替尼 [Chinese] [INN]
3-(2,6-Difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholinomethyl)-1,3,4,6-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
2H-Pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one, 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-8-(4-morpholinylmethyl)-
3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- Originator Incyte Corporation
- Developer Incyte Corporation; Innovent Biologics
- ClassAntineoplastics; Ethers; Fluorobenzenes; Morpholines; Pyridines; Pyrimidinones; Pyrroles; Small molecules
- Mechanism of Action Type 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type 4 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Orphan Drug Status Yes – Myeloproliferative disorders; Lymphoma; Cholangiocarcinoma
- MarketedCholangiocarcinoma
- Phase IIBladder cancer; Lymphoma; Myeloproliferative disorders; Solid tumours; Urogenital cancer
- Phase I/IICancer
- 05 Nov 2020Preregistration for Cholangiocarcinoma (Late-stage disease, Metastatic disease, First line therapy, Inoperable/Unresectable) in Japan (PO) in November 2020
- 05 Nov 2020Incyte Corporation stops enrolment in the FIGHT-205 trial for Bladder cancer due to regulatory feedback
- 26 Oct 2020Preregistration for Cholangiocarcinoma (Second-line therapy or greater, Inoperable/Unresectable, Late-stage disease, Metastatic disease) in Canada (PO)
Pemigatinib, also known as INCB054828, is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3), with potential antineoplastic activity. FGFR inhibitor INCB054828 binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-related signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells.
Pemigatinib (INN),[2] sold under the brand name Pemazyre, is a medication for the treatment of adults with previously treated, unresectable locally advanced or metastatic bile duct cancer (cholangiocarcinoma) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[3][4] Pemigatinib works by blocking FGFR2 in tumor cells to prevent them from growing and spreading.[3]
Pemigatinib belongs to a group of medicines called protein kinase inhibitors.[5] It works by blocking enzymes known as protein kinases, particularly those that are part of receptors (targets) called fibroblast growth factor receptors (FGFRs).[5] FGFRs are found on the surface of cancer cells and are involved in the growth and spread of the cancer cells.[5] By blocking the tyrosine kinases in FGFRs, pemigatinib is expected to reduce the growth and spread of the cancer.[5]
The most common adverse reactions are hyperphosphatemia and hypophosphatemia (electrolyte disorders), alopecia (spot baldness), diarrhea, nail toxicity, fatigue, dysgeusia (taste distortion), nausea, constipation, stomatitis (sore or inflammation inside the mouth), dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain and dry skin.[3][4] Ocular (eye) toxicity is also a risk of pemigatinib.[3][4]
Medical uses
Cholangiocarcinoma is a rare form of cancer that forms in bile ducts, which are slender tubes that carry the digestive fluid bile from the liver to gallbladder and small intestine.[3] Pemigatinib is indicated for the treatment of adults with bile duct cancer (cholangiocarcinoma) that is locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) or metastatic (when cancer cells spread to other parts of the body) and who have tumors that have a fusion or other rearrangement of a gene called fibroblast growth factor receptor 2 (FGFR2).[3] It should be used in patients who have been previously treated with chemotherapy and whose cancer has a certain type of abnormality in the FGFR2 gene.[6]
History
Pemigatinib was approved for use in the United States in April 2020 along with the FoundationOne CDX (Foundation Medicine, Inc.) as a companion diagnostic for patient selection.[3][4][7]
The approval of pemigatinib in the United States was based on the results the FIGHT-202 (NCT02924376) multicenter open-label single-arm trial that enrolled 107 participants with locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement who had received prior treatment.[3][4][6] The trial was conducted at 67 sites in the United States, Europe, and Asia.[6] During the clinical trial, participants received pemigatinib once a day for 14 consecutive days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects.[3][4][6] To assess how well pemigatinib was working during the trial, participants were scanned every eight weeks.[3] The trial used established criteria to measure how many participants experienced a complete or partial shrinkage of their tumors during treatment (overall response rate).[3] The overall response rate was 36% (95% CI: 27%, 45%), with 2.8% of participants having a complete response and 33% having a partial response.[3] Among the 38 participants who had a response, 24 participants (63%) had a response lasting six months or longer and seven participants (18%) had a response lasting 12 months or longer.[3][4]
The U.S. Food and Drug Administration (FDA) granted the application for pemigatinib priority review, breakthrough therapy and orphan drug designations.[3][4][8][9] The FDA granted approval of Pemazyre to Incyte Corporation.[3]
On 24 August 2018, orphan designation (EU/3/18/2066) was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of biliary tract cancer.[5] On 17 October 2019, orphan designation EU/3/19/2216 was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK2.[10]
PATENT
US 20200281907
| The present disclosure is directed to, inter alia, methods of treating cancer in a patient in need thereof, comprising administering pemigatinib, which is 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one, having the structure shown below: |
Pemigatinib is described in U.S. Pat. No. 9,611,267, the entirety of which is incorporated herein by reference. Pemigatinib is further described in US Publication Nos.: 2019/0337948 and 2020/0002338, the entireties of which are incorporated herein by reference.
| Provided herein is a method of treating cancer comprising administering a therapy to a patient in need thereof, wherein the therapy comprises administering a therapeutically effective amount of pemigatinib to the patient while avoiding the concomitant administration of a CYP3A4 perpetrator. |
Example 1. Synthesis of Pemigatinib
Step 1: 4-(ethylamino)-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde
| A mixture of 4-chloro-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (CAS #958230-19-8, Lakestar Tech, Lot: 124-132-29: 3.0 g, 17 mmol) and ethylamine (10M in water, 8.3 mL, 83 mmol) in 2-methoxyethanol (20 mL, 200 mmol) was heated to 130° C. and stirred overnight. The mixture was cooled to room temperature then concentrated under reduced pressure. The residue was treated with 1N HCl (30 mL) and stirred at room temperature for 1 h then neutralized with saturated NaHCO 3 aqueous solution. The precipitate was collected via filtration then washed with water and dried to provide the desired product (2.9 g, 92%). LC-MS calculated for C 10H 12N 3O [M+H] + m/z: 190.1; found: 190.1. |
Step 2: 5-{[(2,6-difluoro-3,5-dimethoxyphenyl)amino]methyl}-N-ethyl-1H-pyrrolo[2,3-b]pyridin-4-amine
| A mixture of 4-(ethylamino)-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (7.0 g, 37 mmol), 2,6-difluoro-3,5-dimethoxyaniline (9.1 g, 48 mmol) and [(1S)-7,7-dimethyl-2-oxobicyclo[2.2.1]hept-1-yl]methanesulfonic acid (Aldrich, cat #21360: 2 g, 7 mmol) in xylenes (250 mL) was heated to reflux with azeotropic removal of water using Dean-Stark for 2 days at which time LC-MS showed the reaction was complete. The mixture was cooled to room temperature and the solvent was removed under reduced pressure. The residue was dissolved in tetrahydrofuran (500 mL) and then 2.0 M lithium tetrahydroaluminate in THF (37 mL, 74 mmol) was added slowly and the resulting mixture was stirred at 50° C. for 3 h then cooled to room temperature. The reaction was quenched by addition of water, 15% aqueous NaOH and water. The mixture was filtered and washed with THF. The filtrate was concentrated and the residue was washed with CH 2Cl 2 and then filtered to get the pure product (11 g, 82%). LC-MS calculated for C 18H 21F 2N 4O 2[M+H] + m/z: 363.2; found: 363.1. |
Step 3: 3-(2,6-Difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| A solution of triphosgene (5.5 g, 18 mmol) in tetrahydrofuran (30 mL) was added slowly to a mixture of 5-{[(2,6-difluoro-3,5-dimethoxyphenyl)amino]methyl}-N-ethyl-1H-pyrrolo[2,3-b]pyridin-4-amine (5.6 g, 15 mmol) in tetrahydrofuran (100 mL) at 0° C. and then the mixture was stirred at room temperature for 6 h. The mixture was cooled to 0° C. and then 1.0 M sodium hydroxide in water (100 mL, 100 mmol) was added slowly. The reaction mixture was stirred at room temperature overnight and the formed precipitate was collected via filtration, washed with water, and then dried to provide the first batch of the purified desired product. The organic layer in the filtrate was separated and the aqueous layer was extracted with methylene chloride. The combined organic layer was concentrated and the residue was triturated with methylene chloride then filtered and dried to provide another batch of the product (total 5.5 g, 92%). LC-MS calculated for C 19H 19F 2N 4O 3[M+H] + m/z: 389.1; found: 389.1. |
Step 4: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH 4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C 25H 23F 2N 4O 5S [M+H] + m/z: 529.1; found: 529.1. |
Step 5: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C 26H 23F 2N 4O 6S (M+H) + m/z: 557.1; found: 556.9. |
Step 6: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO 3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C 30H 32F 2N 5O 6S (M+H) + m/z: 628.2; found: 628.0. |
Step 7: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (pemigatinib)
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H 2O). LC-MS calculated for C 24H 28F 2N 5O 4 (M+H) + m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H). |
PATENT
WO 2019213506
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019213506
PATENT
WO 2019213544
The present disclosure is directed to, inter alia, solid forms, including crystalline forms and amorphous forms, of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)- 1 ,3,4,7 -tetrahydro-2H-pyrrolo [3 ‘,2’ : 5 ,6]pyrido [4,3 -d]pyrimidin-2-one
(Compound 1), and processes and intermediates for preparing the compound. The structure of Compound 1 is shown below.
Compound 1
Compound 1 is described in US Patent No. 9,611,267, the entirety of which is incorporated herein by reference.
Example 1
Synthesis of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l^, 4,7-tetrahydro-2H-pyrrolo[3f,2f:5,6]pyrido[4r3-d]pyrimidin-2-one (Compound 1) Scheme 1.
Step 1: Synthesis of 4-((4-chloro-5-(l, 3-dioxolan-2-yl)-l-(phenylsulfonyl)-lH-pyrrolo[2, 3-b ] pyridin-2-yl) methyl) morpholine
To a l-L flask was added 4-chloro-5-(l,3-dioxolan-2-yl)-l-(phenylsulfonyl)-lH-pyrrolo [2,3-b] pyridine (50.0 g, 137 mmol) (see, e.g., Example 2) and tetrahydrofuran (THF, 266 g, 300 mL) under N2. To this mixture at -70 °C was added 2.0 M lithium
diisopropylamide in THF/heptane/ethyl benzene (77.4 g, 95 mL, 190 mmol, 1.4 eq.). The mixture was stirred at -70 °C for 1 h. To the mixture was added /V- formyl morpholine (29.7 g, 258 mmol, 1.9 eq.) in THF (22. 2 g, 25 mL) dropwise. The reaction was done in 30 min after addition. LC/MS showed that the desired product, 4-chloro-5-(l, 3-dioxolan-2-yl)-l-(phenylsulfonyl)- 1 //-pyrrolo [2, 3-61 pyridine-2-carbaldehyde, was formed cleanly. The reaction was quenched with acetic acid (16.4 g, 15.6 mL, 274 mmol, 2.0 eq.) and the dry ice cooling was removed. To the mixture was added morpholine (33.7 g, 33.5 mL, 387 mmol, 2.83 eq.) followed by acetic acid (74.0 g, 70 mL, 1231 mmol, and 9.0 eq.) at 0 °C (internal temperature rose from 0 °C to 18 °C) and stirred overnight. Sodium triacetoxyborohydride (52.50 g, 247.7 mmol, 1.8 eq.) was added and the reaction mixture temperature rose from 20 °C to 32 °C. The mixture was stirred at room temperature for 30 min. HPLC & LC/MS indicated the reaction was complete. Water (100 g, 100 mL) was added followed by 2.0 M sodium carbonate (Na2C03) in water (236 g, 200 mL, 400 mmol, 2.9 eq.) slowly (off gas!). The mixture was stirred for about 30 min. The organic layer was separated and water (250 g, 250 mL) and heptane (308 g, 450 mL) were added. The resulting slurry was stirred for 1 h and the solid was collected by filtration. The wet cake was washed with heptane twice (75.00 mL x 2, 51.3 g x 2) before being dried in oven at 50 °C overnight to give the desired product, 4-((4-chloro-5-( 1 3-dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-2-yl)methyl)morpholine as a light brown solid (52.00 g, 81.8 % yield): LCMS calculated for C21H23CIN2O5S [M+H]+: 464.00; Found: 464.0; ftf NMR ^OO MHz, DMSO-de) d 8.48 (s, 1 H), 8.38 (m, 2H), 7.72 (m, 1H), 7.64 (m, 2H), 6.83 (s, 1H), 6.13 (s, 1H), 4.12 (m, 2H), 4.00 (m, 2H), 3.92 (s, 2H), 3.55 (m, 4H), 2.47 (m, 4H).
Step 2: Synthesis of 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo[2, 3-b] pyridine-5 -carbaldehyde
To a 2 L reactor with a thermocouple, an addition funnel, and a mechanical stirrer was charged 4-((4-chloro-5 -(1 ,3 -dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo [2,3 -6]pyridin-2-yl)methyl)morpholine (20.00 g, 43.1 mmol) and dichloromethane (265 g, 200 mL) at room temperature. The resulting mixture was stirred at room temperature (internal temperature
was 19.5 °C) to achieve a solution. To the resulting solution was added an aqueous hydrochloric acid solution (0.5 M, 240 g, 200.0 ml, 100 mmol, 2.32 eq.) at room temperature in 7 min. After over 23 h agitations at room temperature, the bilayer reaction mixture turned into a thick colorless suspension. When HPLC showed the reaction was complete, the slurry was cooled to 0-5 °C and aqueous sodium hydroxide solution (1 N, 104 g, 100 mL, 100 mmol, and 2.32 eq.) was added in about 10 min to adjust the pH of the reaction mixture to 10-11. «-Heptane (164 g, 240 mL) was added and the reaction mixture and the mixture were stirred at room temperature for 1 h. The solid was collected by filtration and the wet cake was washed with water (2 x 40 mL), heptane (2 x 40 ml) before being dried in oven at 50 °C under vacuum to afford the desired product, 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-/i |pyridine-5-carbaldehyde as a light brown solid (16.9 g, 93% yield): LCMS calculated for C19H19CIN3O4S [M+H]+: 420.00; Found: 420.0; ¾ NMR (400 MHz, DMSO-de) d 10.33 (s, 1H), 8.76 (s, 1 H), 8.42 (m, 2H), 7.74 (m, 1H), 7.65 (m, 2H), 6.98 (s, 1H), 3.96 (m, 2H), 3.564 (m, 4H), 2.51 (m, 4H).
Step 3: Synthesis ofN-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo [2, 3-h] pyridin-5-yl) methyl) -2, 6-difluoro-3,5-dimethoxyaniline
To a 2-L reactor equipped with a thermocouple, a nitrogen inlet and mechanical stirrer were charged AOV-dimethyl formamide (450 mL, 425 g), 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridine-5-carbaldehyde (30.0 g, 71.45 mmol) and 2,6-difluoro-3,5-dimethoxyanihne (14.2 g, 75.0 mmol). To this suspension (internal temperature 20 °C) was added chlorotrimethylsilane (19.4 g, 22. 7 mL, 179 mmol) dropwise in 10 min at room temperature (internal temperature 20-23 °C). The suspension changed into a solution in 5 min after the chlorotrimethylsilane addition. The solution was stirred at room temperature for 1.5 h before cooled to 0-5 °C with ice-bath. Borane-THF complex in THF (1.0 M, 71.4 mL, 71.4 mmol, 64.2 g, 1.0 eq.) was added dropwise via additional funnel over 30 min while maintaining temperature at 0-5 °C. After addition, the mixture was stirred for 4 h. Water (150 g, 150 mL) was added under ice-bath cooling in 20 min, followed by slow addition of ammonium hydroxide solution (28% N¾, 15.3 g, 17 ml, 252 mmol, 3.53 eq.) to pH 9-10 while maintaining the temperature below 10 °C. More water (250 mL, 250 g) was added through the additional funnel. The slurry was stirred for 30 min and the solids were collected by filtration. The wet cake was washed with water (90 g x 2, 90 ml x 2) and heptane (61.6 g x2, 90 ml x 2). The product w as suction dried overnight to give the desired product LG-((4-chloro-2-(morphohnomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-Z>]pyridin-5-yl)methyl)-2,6- difluoro-3,5-dimethoxyaniline (41.6 g, 96% yield): LCMS calculated for C27H28ClF2N405S[M+H]+: 593.10; Found: 593.1 ; ¾ NMR (400 MHz, DMSO-d6) 5 8.36 (m, 2H), 8.28 (s, 1H), 7.72 (m, 1H), 7.63 (m, 2H), 6.78 (s, 1H), 6.29 (m, 1H), 5.82 (m, 1H), 4.58 (m, 2H), 3.91 (s, 2H), 3.76 (s, 6H), 3.56 (m, 4H), 2.47 (m, 4H).
Step 4: Synthesis of l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo [2, 3-b ] pyridin-5-yl) methyl)-! -(2, 6-difluoro-3, 5-dimethoxyphenyl)-3-ethylurea
To a 2-L, 3-neck round bottom flask fitted with a thermocouple, a nitrogen bubbler inlet, and a magnetic stir were charged /V-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-b]pyridin-5-yl)methyl)-2,6-difluoro-3,5-dimethoxyaniline (67.0 g, 113 mmol) and acetonitrile (670 ml, 527 g). The suspension was cooled to 0-5 °C.
To the mixture was charged ethyl isocyanate (17.7 mL, 15.9 g, 224 mmol, 1.98 eq.) over 30 sec. The temperature stayed unchanged at 0.7 °C after the charge. Methanesulfonic acid (16.1 mL, 23.9 g, 248 mmol, 2.2 eq.) was charged dropwise over 35 min while maintaining the temperature below 2 °C. The mixture was warmed to room temperature and stirred overnight. At 24 h after addition showed that the product was 93.7%, unreacted SM was 0.73% and the major impurity (bis-isocyanate adduct) was 1.3%. The mixture was cooled with an ice-bath and quenched with sodium hydroxide (NaOH) solution (1.0M, 235 mL, 244 g, 235 mmol, 2.08 eq.) over 20 min and then saturated aqueous sodium bicarbonate
(NaHCCh) solution (1.07 M, 85 mL, 91 g, 0.091 mol, 0.80 eq.) over 10 min. Water (550 mL, 550 g) was added and the liquid became one phase. The mixture was stirred for 2 h and the solids were collected by filtration, washed with water (165 mL, 165 g) to give l-((4-chloro-2-(morpholinomethyl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo| 2.3-6 |p\ ri din-5 -y l (methy l )- 1 -(2,6-difluoro-3,5-dimethoxyphenyl)-3-ethylurea ( 70.3 g, 93.7% yield).
The crude l-((4-chloro-2-(morpholinomethyl)-l -(phenylsulfonyl)- li/-pyrrolo [2, 3-61 pyridin-5-yl) methyl)- 1 -(2, 6-difluoro-3, 5-dimethoxyphenyl)-3-ethylurea (68.5 g, 103 mmol) was added in to acetonitrile (616 mL, 485 g). The mixture was heated 60-65 °C and an amber colored thin suspension was obtained. The solid was filtered off with celite and the celite was washed with acetonitrile (68.5 mL, 53.8 g). To the pale yellow filtrate was added water (685 g, 685 ml) to form a slurry. The slurry was stirred overnight at room temperature and filtered. The solid was added to water (685 mL, 685 g) and stirred at 60 °C for 2 h. The solid was filtered and re-slurred in heptane (685 mL, 469 g) overnight. The product was dried in an oven at 50 °C under vacuum for 48 h to afford l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-difluoro-3.5-
dimethoxyphenyl)-3-ethylurea as a colorless solid (62.2 g, 90.8% yield, 99.9% purity by HPLC area%). KF was 0.028%. Acetonitrile (by ‘H NMR) was about 1.56%, DCM (by ‘H NMR) 2.0%: LCMS calculated for C30H33CIF2N5O6S [M+H]+: EM: 664.17; Found: 664.2; ¾ NMR (400 MHz, DMSO-de) d 8.33 (m, 2H), 8.31 (s, 1H), 7.72 (m, 1H), 7.64 (m, 1H), 6.96 (m, 2H), 6.73 (s, 1H), 6.43 (m, 1H), 4.87 (s, 2H), 3.90 (s, 2H), 3.77 (s, 6H), 3.54 (m, 4H),
3.03 (m, 2H), 2.46 (m, 4H), 0.95 (m, 3H).
Step 5: Synthesis of 3-(2, 6-difluoro-3, 5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-l, 3, 4, 7-tetrahydro-2H-pyrrolo[ 3 2’:5, 6 ]pyrido[ 4, 3-d]pyrimidin-2-one
To a 2000 mL flask equipped with a thermal couple, a nitrogen inlet, and a mechanical stirrer were charged dry l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-1 //-pyrrolo| 2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-dinuoro-3.5-dimetho\yphenyl)-3-ethylurea (30.0 g, 45.2 mmol, KF=0. l l%) and tetrahydrofuran (1200 mL, 1063 g). To this suspension at room temperature was charged 1.0 M lithium hexamethyldisilazide in THF (62.3 mL, 55.5 g, 62.3 mmol, 1.38 eq). The mixture turned into a solution after the base addition. The reaction mixture was stirred for 2 h and HPLC shows the starting material was not detectable. To this mixture was added 1.0 M hydrochloric acid (18.1 mL, -18.1 g. 18.1 mmol, 0.4 eq.). The solution was concentrated to 600 mL and water (1200 mL, 1200 g) was added. Slurry was formed after water addition. The slurry was stirred for 30 min at room temperature and the solid was collected by filtration. The wet cake was washed with water twice (60 mLx2,
60 gx2) and dried at 50 °C overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4, 3-d]pyrimidin-2-one as a light brown solid (26.58 g, as-is yield 93.7%): THF by ‘H NMR 0.32%, KF 5.26%, adjusted yield was 88.5%: LCMS calculated for C30H32F2N5O6S [M+H]+: EM: 628.20; Found: 628.2; ¾ NMR (400 MHz, DMSO-de) d 8.41 (m, 2H), 8.07 (s, 1H), 7.70 (m, 1H), 7.63 (m, 2H), 7.05 (m, 1H), 6.89 (s, 1H), 4.76 (s, 2H), 4.09 (m, 2H), 3.93 (s, 2H), 3.89 (s, 6H), 3.60 (m, 4H), 2.50 (m, 4H), 1.28 (m, 3H).
Step 6: Synthesis of 3-( 2, 6-difluoro-3, 5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-1,3, 4, 7 -tetrahydro-2H-pyrrolo [ 3 ‘, 2 5, 6 ]pyrido[ 4, 3-dJpyrimidin-2-one
To a stirring suspension of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholinomethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2i/-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (10.0 g, 15.93 mmol) in l,4-dioxane (100 ml, 103 g) in a 500 mL flask equipped with a nitrogen inlet, a condenser, a thermocouple and a heating mantle was added 1 M aqueous sodium hydroxide (63.7 ml, 66.3 g, 63.7 mmol). The reaction mixture was heated at 75 °C for 18 h. LCMS showed the reaction was complete. Water (100 mL, 100 g) was added to give a thick suspension. This slurry was stirred at room temperature for 1 h and filtered. The cake was washed with water (3 x 10 mL, 3 x 10 g) and heptane (2 x 10 mL, 2 x 6.84 g). The cake was dried overnight by pulling a vacuum through the filter cake and then dried in an oven at 50 °C under vacuum overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5, 6]pyrido[4,3-d]pyrimidin-2-one (6.8 g, 87.6% yield): LCMS calculated for C24H28F2N5O4 [M+H]+: 488.20; Found: 488.2.
PATENT
US 20130338134
https://patents.google.com/patent/US20130338134A1/en
Step 1: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0832]
- [0833]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (Example 49, Step 3: 900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C25H23F2N4O5S [M+H]+ m/z: 529.1; found: 529.1.
Step 2: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde
- [0834]
- [0835]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C26H23F2N4O6S (M+H)+ m/z: 557.1; found: 556.9.
Step 3: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0836]
- [0837]
To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C30H32F2N5O6S (M+H)+ m/z: 628.2; found: 628.0.
Step 4: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0838]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H2O). LC-MS calculated for C24H28F2N5O4 (M+H)+ m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H).
PATENTS
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2013338134-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-2017137424-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-2019127376-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-9611267-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2017-04-04 |
| WO-2014007951-A2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-6336665-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-06-06 |
| JP-6545863-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2019-07-17 |
| JP-6711946-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2020-06-17 |
| TW-201402574-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| US-10131667-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-11-20 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2015521600-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2017222709-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2018135377-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2019178156-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-6301321-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-03-28 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3176170-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| EP-3176170-B1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2018-11-14 |
| EP-3495367-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| ES-2704744-T3 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2019-03-19 |
| HU-E031916-T2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| DK-2861595-T5 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-01-15 |
| DK-3176170-T3 | Substituted tricyclic relations as fgfr inhibitors | 2012-06-13 | 2019-01-28 |
| EP-2861595-A2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| EP-2861595-B1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2016-12-21 |
| EP-2861595-B9 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2017-06-21 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019191707-A1 | Heterocyclic compounds as immunomodulators | 2018-03-30 | |
| AU-2013287176-A1 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| CA-2876689-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| CN-107383009-B | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2020-06-09 |
| DK-2861595-T3 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2017-02-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019213544-A2 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| WO-2019213544-A3 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| TW-202003511-A | Heterocyclic compounds as immunomodulators | 2018-03-30 | |
| US-10669271-B2 | Heterocyclic compounds as immunomodulators | 2018-03-30 | 2020-06-02 |
| US-2019300524-A1 | Heterocyclic compounds as immunomodulators | 2018-03-30 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| TW-201946630-A | Salts of an FGFR inhibitor | 2018-05-04 | |
| TW-202003516-A | Solid forms of an FGFR inhibitor and processes for preparing the same | 2018-05-04 | |
| US-2019337948-A1 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| US-2020002338-A1 | Salts of an fgfr inhibitor | 2018-05-04 | |
| WO-2019213506-A1 | Salts of an fgfr inhibitor | 2018-05-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019227007-A1 | Tricyclic heterocyclic compounds as sting activators | 2018-05-25 | |
| TW-201946626-A | Heterocyclic compounds as immunomodulators | 2018-05-11 | |
| US-10618916-B2 | Heterocyclic compounds as immunomodulators | 2018-05-11 | 2020-04-14 |
| US-2019345170-A1 | Heterocyclic compounds as immunomodulators | 2018-05-11 | |
| WO-2019217821-A1 | Tetrahydro-imidazo[4,5-c]pyridine derivatives as pd-l1 immunomodulators | 2018-05-11 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020040009-A1 | Tricyclic heteraryl compounds as sting activators | 2018-07-31 | |
| WO-2020028565-A1 | Tricyclic heteraryl compounds as sting activators | 2018-07-31 | |
| WO-2020028566-A1 | Heteroaryl amide compounds as sting activators | 2018-07-31 | |
| WO-2019238873-A1 | A method of precision cancer therapy | 2018-06-13 | |
| US-2019359608-A1 | Tricyclic heterocyclic compounds as sting activators | 2018-05-25 |
| Title | Priority Date | Grant Date | |
|---|---|---|---|
| WO-2020131627-A1 | Substituted pyrazolo[1,5-a]pyridine compounds as inhibitors of fgfr tyrosine kinases | 2018-12-19 | |
| WO-2020131674-A1 | 7-((3,5-dimethoxyphenyl)amino)quinoxaline derivatives as fgfr inhibitors for treating cancer | 2018-12-19 | |
| WO-2020081898-A1 | Non-invasive urinary biomarkers for the detection of urothelial carcinoma of the bladder | 2018-10-20 | |
| US-2020115378-A1 | Dihydropyrido[2,3-d]pyrimidinone compounds as cdk2 inhibitors | 2018-10-11 | |
| US-2020039994-A1 | Heteroaryl amide compounds as sting activators | 2018-07-31 |
References
- ^ “Pemigatinib (Pemazyre) Use During Pregnancy”. Drugs.com. 11 August 2020. Retrieved 24 September 2020.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3): 479. hdl:10665/330907.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves First Targeted Treatment for Patients with Cholangiocarcinoma, a Cancer of Bile Ducts”. U.S. Food and Drug Administration (FDA) (Press release). 17 April 2020. Retrieved 17 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to pemigatinib for cholangiocarcinoma”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e “EU/3/18/2066”. European Medicines Agency (EMA). 19 December 2018. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Drug Trials Snapshot: Pemazyre”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 5 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Pemazyre: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 21 April 2020.
- ^ “Pemigatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). Retrieved 19 April 2020.
- ^ “Pemigatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). Retrieved 19 April 2020.
- ^ “EU/3/19/2216”. European Medicines Agency (EMA). 23 January 2020. Retrieved 19 April 2020. This article incorporates text from this source, which is in the public domain.
Further reading
- Roskoski R (January 2020). “The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder”. Pharmacol. Res. 151: 104567. doi:10.1016/j.phrs.2019.104567. PMID 31770593.
External links
- “Pemigatinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Pemigatinib”. National Cancer Institute.
- Clinical trial number NCT02924376 for “Efficacy and Safety of Pemigatinib in Subjects With Advanced/Metastatic or Surgically Unresectable Cholangiocarcinoma Who Failed Previous Therapy – (FIGHT-202)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pemazyre |
| Other names | INCB054828 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620028 |
| License data | US DailyMed: Pemigatinib |
| Pregnancy category | US: N (Not classified yet)[1] |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1513857-77-6 |
| PubChem CID | 86705695 |
| DrugBank | DB15102 |
| ChemSpider | 68007304 |
| UNII | Y6BX7BL23K |
| KEGG | D11417 |
| ChEMBL | ChEMBL4297522 |
| Chemical and physical data | |
| Formula | C24H27F2N5O4 |
| Molar mass | 487.508 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCN1C2=C3C=C(NC3=NC=C2CN(C1=O)C4=C(C(=CC(=C4F)OC)OC)F)CN5CCOCC5 | |
| InChI[hide]InChI=1S/C24H27F2N5O4/c1-4-30-21-14(11-27-23-16(21)9-15(28-23)13-29-5-7-35-8-6-29)12-31(24(30)32)22-19(25)17(33-2)10-18(34-3)20(22)26/h9-11H,4-8,12-13H2,1-3H3,(H,27,28)Key:HCDMJFOHIXMBOV-UHFFFAOYSA-N |
/////////Pemigatinib, 佩米替尼 , PEMAZYRE, FDA 2020, 2020 APPROVALS, INCB054828, INCB 054828, Orphan Drug Status, Myeloproliferative disorders, Lymphoma, Cholangiocarcinoma, INCYTE
O=C1N(CC)C2=C3C(NC(CN4CCOCC4)=C3)=NC=C2CN1C5=C(F)C(OC)=CC(OC)=C5F.[H]Cl
Odevixibat

Odevixibat
A-4250, AR-H 064974
CAS 501692-44-0
BUTANOIC ACID, 2-(((2R)-2-((2-((3,3-DIBUTYL-2,3,4,5-TETRAHYDRO-7-(METHYLTHIO)-1,1-DIOXIDO-5-PHENYL-1,2,5-BENZOTHIADIAZEPIN-8-YL)OXY)ACETYL)AMINO)-2-(4-HYDROXYPHENYL)ACETYL)AMINO)-, (2S)-
(2S)-2-[[(2R)-2-[[2-[(3,3-dibutyl-7-methylsulfanyl-1,1-dioxo-5-phenyl-2,4-dihydro-1λ6,2,5-benzothiadiazepin-8-yl)oxy]acetyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]butanoic acid
| Molecular Formula | C37H48N4O8S2 |
| Molecular Weight | 740.929 |
-
-
-
- UPDATE 7/20/2021FDA APPROVED, To treat pruritus,
-
- New Drug Application (NDA): 215498
Company: ALBIREO PHARMA INC
-
- AZD8294WHO 10706AR-H064974HY-109120CS-0078340D11716US9694018, 5Originator Albireo AB
- Developer Albireo AB; Albireo Pharma
- ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
- Mechanism of Action Sodium-bile acid cotransporter inhibitors
- Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
- New Molecular Entity Yes
- Phase III Biliary atresia; Intrahepatic cholestasis
- Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
- No development reported Non-alcoholic steatohepatitis
- 22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
Odevixibat, sold under the trade name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis (PFIC).[1]
The most common side effects include diarrhea, abdominal pain, hemorrhagic diarrhea, soft feces, and hepatomegaly (enlarged liver).[1]
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).[1][2]
In May 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for odevixibat for the treatment of PFIC in people aged six months or older.[1][3]
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
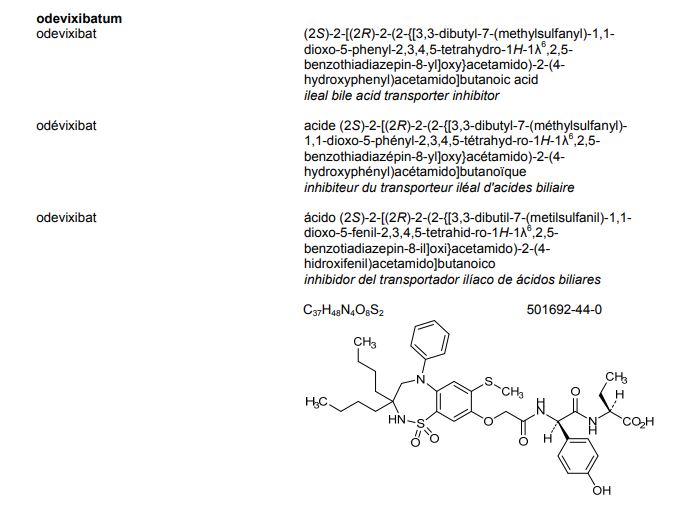
![]()
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
For more information about the PEDFIC 2 or BOLD studies, please visit ClinicalTrials.gov or contact medinfo@albireopharma.com.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.

PATENT
https://patents.google.com/patent/US9694018B1/en
Example 5
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N—{(R)-α-[N—((S)-1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine, Mw. 740.94.
This compound is prepared as described in Example 29 of WO3022286.
PATENT
https://patents.google.com/patent/WO2003022286A1/sv
Example 29
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N-((R)-α-[N-((S)- 1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
PATENT
https://patents.google.com/patent/US20140323412A1/en
PATENT
https://patents.google.com/patent/WO2013063526A1/e
PATENT
https://patents.google.com/patent/WO2019245448A1/en
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.

As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
References
- ^ Jump up to:a b c d “First treatment for rare liver disease”. European Medicines Agency (EMA) (Press release). 21 May 2021. Retrieved 21 May 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Odevixibat”. Albireo Pharma. Retrieved 21 May 2021.
- ^ “Bylvay: Pending EC decision”. European Medicines Agency (EMA). 19 May 2021. Retrieved 21 May 2021.
External links
- “Odevixibat”. Drug Information Portal. U.S. National Library of Medicine.
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04336722 | Efficacy and Safety of Odevixibat in Children With Biliary Atresia Who Have Undergone a Kasai HPE (BOLD) | Phase 3 | Recruiting | 2020-09-02 |
| NCT04483531 | Odevixibat for the Treatment of Progressive Familial Intrahepatic Cholestasis | Available | 2020-08-25 | |
| NCT03566238 | This Study Will Investigate the Efficacy and Safety of A4250 in Children With PFIC 1 or 2 | Phase 3 | Active, not recruiting | 2020-03-05 |
| NCT03659916 | Long Term Safety & Efficacy Study Evaluating The Effect of A4250 in Children With PFIC | Phase 3 | Recruiting | 2020-01-21 |
| NCT03608319 | Study of A4250 in Healthy Volunteers Under Fasting, Fed and Sprinkled Conditions | Phase 1 | Completed | 2018-09-19 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02630875 | A4250, an IBAT Inhibitor in Pediatric Cholestasis | Phase 2 | Completed | 2018-03-29 |
| NCT02360852 | IBAT Inhibitor A4250 for Cholestatic Pruritus | Phase 2 | Terminated | 2017-02-23 |
| NCT02963077 | A Safety and Pharmakokinetic Study of A4250 Alone or in Combination With A3384 | Phase 1 | Completed | 2016-11-16 |
EU Clinical Trials Register
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Bylvay |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C37H48N4O8S2 |
| Molar mass | 740.93 g·mol−1 |
| 3D model (JSmol) | |
////////////odevixibat, Orphan Drug Status, phase 3, Albireo, A-4250, A 4250, AR-H 064974
CCCCC1(CN(C2=CC(=C(C=C2S(=O)(=O)N1)OCC(=O)NC(C3=CC=C(C=C3)O)C(=O)NC(CC)C(=O)O)SC)C4=CC=CC=C4)CCCC
| publicationnumber | |||||||
| US-2020046635-A1 | |||||||
| US-2020046636-A1 | US-2020046757-A1 | US-2020046758-A1 | US-2020002299-A1 | WO-2019245448-A1 | WO-2019245449-A1 | US-2019046451-A1 | US-2019070217-A1 |
| US-10441605-B2 | |||||||
| US-2017224720-A1 | |||||||
| US-2017224721-A1 | |||||||
| US-2018264029-A1 | |||||||
| US-2018360869-A1 | |||||||
| US-2018360870-A1 | |||||||
| US-2018360871-A1 | |||||||
| WO-2017133517-A1 | |||||||
| US-2017143738-A1 | |||||||
| US-2017143783-A1 | |||||||
| EP-2968230-A2 | |||||||
| EP-2968262-A1 | |||||||
| US-2014271734-A1 | |||||||
| US-2014275090-A1 | |||||||
| WO-2014144485-A1 | |||||||
| WO-2014144485-A9 | |||||||
| WO-2014144650-A2 | |||||||
| EP-2770990-A1 | |||||||
| EP-2771003-A1 | |||||||
| EP-2771003-B1 | |||||||
| EP-3266457-A1 | |||||||
| EP-3278796-A1 | |||||||
| US-10512657-B2 | |||||||
| US-2013108573-A1 | |||||||
| US-2013109671-A1 | |||||||
| US-2013338093-A1 | |||||||
| US-2014243281-A1 | |||||||
| US-2014323412-A1 | |||||||
| US-2016310518-A1 | |||||||
| US-2017368085-A1 | |||||||
| US-2019169217-A1 | |||||||
| US-2020069715-A1 | |||||||
| WO-2013063512-A1 | |||||||
| WO-2013063526-A1 | |||||||
| EP-2739286-A2 | |||||||
| WO-2013020108-A2 | |||||||
| EP-2637646-B1 | |||||||
| EP-2637668-B1 | |||||||
| EP-3023102-A1 | |||||||
| EP-3023102-B1 | |||||||
| EP-3400944-A1 | |||||||
| US-10000528-B2 | |||||||
| US-10011633-B2 | |||||||
| US-10093697-B2 | |||||||
| US-10221212-B2 | |||||||
| US-2012114588-A1 | |||||||
| US-2013225511-A1 | |||||||
| US-2013236541-A1 | |||||||
| US-2015031636-A1 | |||||||
| US-2015031637-A1 | |||||||
| US-2016193277-A1 | |||||||
| US-2016194353-A1 | |||||||
| US-2017182059-A1 | |||||||
| US-2017182115-A1 | |||||||
| US-2018022776-A1 | |||||||
| US-2018030088-A1 | |||||||
| US-2018030089-A1 | |||||||
| US-2018162904-A1 | |||||||
| US-2018362577-A1 | |||||||
| US-9688720-B2 | |||||||
| US-9694018-B1 | |||||||
| US-10555950-B2 | |||||||
| US-2016220577-A1 | |||||||
| US-9339480-B2 | |||||||
| WO-2008039829-A2 | |||||||
| US-2009069285-A1 | |||||||
| US-7842684-B2 | |||||||
| WO-2007051995-A2 | |||||||
| EP-1896408-A1 | |||||||
| EP-1896409-A1 | |||||||
| EP-1896457-A1 | |||||||
| US-2010048529-A1 | |||||||
| US-2010048530-A1 | |||||||
| US-2010137273-A1 | |||||||
| US-2010152156-A1 | |||||||
| US-2010168039-A1 | |||||||
| US-2010168075-A1 | |||||||
| US-7893048-B2 | |||||||
| US-7906502-B2 | |||||||
| WO-2006137792-A1 | |||||||
| WO-2006137794-A1 | |||||||
| WO-2006137795-A1 | |||||||
| US-2010216759-A1 | |||||||
| US-7863265-B2 | |||||||
| US-2008194494-A1 | |||||||
| US-2009186834-A1 | |||||||
| WO-2006102674-A2 | |||||||
| US-2009005321-A1 | |||||||
| EP-1831151-A1 | |||||||
| US-2008114064-A1 | |||||||
| WO-2006065214-A1 | |||||||
| EP-1699759-A1 | |||||||
| US-2007142304-A1 | |||||||
| US-2008064676-A1 | |||||||
| US-2010099657-A2 | |||||||
| US-7871998-B2 | |||||||
| WO-2005061452-A1 | |||||||
| EP-1638922-A1 | |||||||
| EP-1638926-A1 | |||||||
| EP-1638930-A1 | |||||||
| EP-1675820-A2 | |||||||
| EP-1676833-A1 | |||||||
| US-2005148656-A1 | |||||||
| US-2006142389-A1 | |||||||
| US-2006178432-A1 | |||||||
| US-2006194879-A1 | |||||||
| US-2006258866-A1 | |||||||
| US-2007099928-A1 | |||||||
| US-2007099997-A1 | |||||||
| US-2007244198-A1 | |||||||
| US-7309720-B2 | |||||||
| WO-2004110984-A1 | |||||||
| WO-2004113270-A2 | |||||||
| WO-2004113276-A1 | |||||||
| WO-2004113283-A1 | |||||||
| EP-1610770-A1 | |||||||
| EP-1610770-B1 | |||||||
| EP-1894564-A2 | |||||||
| US-2006199797-A1 | |||||||
| US-7514421-B2 | |||||||
| WO-2004089350-A1 | |||||||
| EP-1572626-A1 | |||||||
| US-2005131068-A1 | |||||||
| WO-2004056748-A1 | |||||||
| EP-1539120-A1 | |||||||
| US-2006083790-A1 | |||||||
| WO-2004006899-A1 | |||||||
| EP-1521742-A1 | |||||||
| US-2005239766-A1 | |||||||
| US-7470678-B2 | |||||||
| WO-2004005247-A1 | |||||||
| EP-1517679-A1 | |||||||
| EP-1517679-B1 | |||||||
| EP-1517883-A1 | |||||||
| EP-1517883-B1 | |||||||
| EP-1517883-B8 | |||||||
| US-2005222261-A1 | |||||||
| US-2005256198-A1 | |||||||
| US-2005267149-A1 | |||||||
| US-7351858-B2 | |||||||
| US-7355069-B2 | |||||||
| US-7521461-B2 | |||||||
| WO-2004000294-A1 | |||||||
| WO-2004000790-A1 | |||||||
| EP-1478368-A1 | |||||||
| US-2005124557-A1 | |||||||
| WO-03061663-A1 | |||||||
| EP-1458672-A1 | |||||||
| EP-1458672-B1 | |||||||
| EP-1458673-A1 | |||||||
| EP-1458673-B1 | |||||||
| EP-1458677-A1 | |||||||
| EP-1458677-B1 | |||||||
| US-2005113362-A1 | |||||||
| US-2005171204-A1 | |||||||
| US-2005215630-A1 | |||||||
| US-2005282822-A1 | |||||||
| US-7256307-B2 | |||||||
| US-7276539-B2 | |||||||
| US-7488844-B2 | |||||||
| US-7514471-B2 | |||||||
| WO-03051821-A1 | |||||||
| WO-03051822-A1 | |||||||
| WO-03051826-A1 | |||||||
| EP-1427423-B1 | |||||||
| EP-1427423-B9 | |||||||
| US-2005038009-A1 | |||||||
| US-7132416-B2 |
TILDACERFONT
TILDACERFONT
| Synonyms: |
Tildacerfont 1014983-00-6 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(1-ethyl-propyl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine 7-(1-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine |
|---|---|
| MW/ MF | 420 g/mol/ C20H26ClN5OS |
- Originator Spruce Biosciences
- Class2 ring heterocyclic compounds; Morpholines; Pyrazoles; Pyrimidines; Small molecules; Thiazoles
- Mechanism of Action Corticotropin receptor antagonists
- Orphan Drug Status Yes – Congenital adrenal hyperplasia
- New Molecular Entity Yes
- Phase II Congenital adrenal hyperplasia
- 09 Jul 2020 Spruce Biosciences initiates a phase II trial in Congenital adrenal hyperplasia in USA (PO) (NCT04457336)
- 24 Sep 2019 Spruce Biosciences completes a phase II trial in Congenital adrenal hyperplasia in USA (NCT03687242)
- 19 Sep 2019 Updated safety and efficacy data from a phase II trial in Congenital adrenal hyperplasia release by Spruce Biosciences
Deuterated pyrazolo[1,5-a]pyrimidine derivatives, particularly tildacerfont (SPR-001), useful as CRF antagonists for treating congenital adrenal hyperplasia. Spruce Bioscience is developing tildacerfont under license from Lilly as an oral capsule formulation for the treatment of congenital adrenal hyperplasia; in July 2017, a phase II trial for CAH was initiated.
Corticotropin releasing factor (CRF) is a 41 amino acid peptide that is the primary physiological regulator of proopiomelanocortin (POMC) derived peptide secretion from the anterior pituitary gland. In addition to its endocrine role at the pituitary gland, immunohistochemical localization of CRF has demonstrated that the hormone has a broad extrahypothalamic distribution in the central nervous system and produces a wide spectrum of autonomic, electrophysiological and behavioral effects consistent with a neurotransmitter or neuromodulator role in the brain. There is also evidence that CRF plays a significant role in integrating the response in the immune system to physiological, psychological, and immunological stressors.
PATENT
Product case, WO2008036579 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008036579
Example 16
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo [ 1 ,5 -α]pyrimidine
Under a nitrogen atmosphere dissolve 3-(4-bromo-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (116 mg, 0.25 mmol) in THF (1.5 mL) and chill to -78 0C. Add n-butyl lithium (0.1 mL. 2.5 M in hexane, 0.25 mmol) and stir at -78 0C for 30 min. Add N-chlorosuccinimide (33.4 mg, 0.25 mmol) and stir for another 30 min, slowly warming to room temperature. After stirring overnight, quench the reaction by adding a solution of saturated ammonia chloride and extract with ethyl acetate. Wash the organic layer with brine, dry over sodium sulfate, filter, and concentrate to a residue. Purify the crude material by flash chromatography, eluting with hexanes:dichloromethane: ethyl acetate (5:5:2) to provide the title compound (54 mg). MS (APCI) m/z (35Cl) 420.6 (M+l)+; 1H NMR (400 MHz, CDCl3): 6.44 (s, IH), 3.79 (t, 4H, J=4.8 Hz), 3.63-3.56 (m, IH), 3.47 (t, 4H, J=4.8 Hz), 2.55 (s, 3H), 2.45 (s, 3H), 1.88-1.75 (m, 4H), 0.87 (t, 6H, J=7.5 Hz).
Alternate Preparation from Preparation 6:
Combine 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine, (9 g,
26.2 mmol) and 4-chloro-2-morpholino-thiazole (7.5 g, 36.7 mmol) in
dimethylformamide (90 mL) previously degassed with nitrogen. Add cesium carbonate (17.8 g, 55 mmol), copper iodide (250 mg, 1.31 mmol), triphenylphosphine (550 mg, 2.09 mmol) and palladium acetate (117 mg, 0.52 mmol). Heat the mixture to 125 0C for 16 h and then cool to 22 0C. Add water (900 mL) and extract with methyl-?-butyl ether (3 x 200 mL). Combine the organic portions and evaporate the solvent. Purify by silica gel chromatography eluting with hexanes/ethyl acetate (4/1) to afford the title compound (6.4 g, 62%). ES/MS m/z (35Cl) 420 (M+l)+.
Example 16a
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo[l,5-α]pyrimidine, hydrochloride
Dissolve 3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (1.40 g, 3.33 mmol) in acetone (10 mL) at 50 0C and cool to room temperature. Add hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and stir well in a sonicator. Concentrate the solution a little and add a minimal amount of diethyl ether to crystallize the HCl salt. Cool the mixture in a refrigerator overnight. Add additional hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and cool in a refrigerator. Filter the crystalline material and dry to obtain the title compound (1.15 g, 75%). ES/MS m/z (35Cl) 420 (M+l)+; 1H NMR(CDCO): 9.18 (br, IH), 6.86 (s, IH), 3.72 ( m, 4H), 3.49(m, IH), 3.39 (m, 4H), 2.48 (s, 3H), 2.38(s, 3H), 1.79 (m, 4H), 0.79 (m, 6H).
PATENT
US-20200255436
PATENT
WO2019210266
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019210266
claiming the use of CRF-1 antagonists (eg tildacerfont).
PATENT
WO 2010039678
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010039678
EXAMPLES
Example 1 : 7-(l-ethyl-propyl)-3-(‘2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine nthroline
Charge 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.03 g, 3.00 mmoles), K3PO4 (1.95 g, 9.00 mmoles), 2,4-dichlorothiazole (0.58 g, 3.75 mmoles), 1,10 phenanthroline (0.05 g, 0.30 mmoles) and anhydrous DMAC (5 mL) to a round bottom flask equipped with a magnetic stir bar, thermal couple and N2 inlet. Degas the yellow heterogeneous reaction mixture with N2 (gas) for 30 min. and then add CuI (0.06 g, 0.30 mmoles) in one portion followed by additional 30 min. degassing with N2 (gas). Stir the reaction mixture at 120 0C for about 6 hr. Cool the reaction mixture to room temperature overnight, add toluene (10 mL) and stir for 1 hr. Purify the mixture through silica gel eluting with toluene (10ml). Extract with 1 M HCl (10 mL), water (10 mL), brine (10 mL) and concentrate under reduced pressure to give a yellow solid. Recrystallize the solid from methanol (5ml) to yield the title compound as a yellow crystalline solid. (0.78 g, 70% yield, >99% pure by LC) MS(ES) = 369 (M+ 1). 1H NMR (CDCl3)= 6.5 (IH, s); 3.6 (IH, m); 2.6 (3H, s); 2.5 (3H, s); 1.9 (4H, m); 0.9 (6H, t).
Example 2: 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolol“! ,5-aipyrimidine
Charge 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (0.37 g, 1.00 mmoles), K2CO3 (0.28 g, 2.00 mmoles) and anhydrous morpholine (3 mL) to a round bottom flask equipped with a magnetic stir bar and N2 inlet. Stir the yellow mixture at 100 0C for about 4 hr., during which time the reaction becomes homogeneous. Cool the reaction mixture to room temperature, add H2O (10 mL) and stir the heterogeneous reaction mixture overnight at room temperature. Collected the yellow solid by filtration, wash with H2O and allowed to air dry overnight to give the crude title compound (391mg). Recrystallize from isopropyl alcohol (3 mL) to yield the title compound as a light yellow crystalline solid (380 mg, 90.6% yield, >99% by LC). MS(ES) = 420 (M+l). 1H NMR (CDCl3)= 6.45 (IH, s); 3.81 (m, 4H); 3.62 (IH, m); 3.50 (m, 4H); 2.6 (3H, s); 2.45 (3 H, s); 1.85 (4H, m); 0.9 (6H, t).
Example 3 :
The reactions of Example 1 are run with various other catalysts, ligands, bases and solvents, which are found to have the following effects on yield of 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. (See Tables 1 – 4).
Table 1 : Evaluation of different li ands
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole, 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.5 mmol CuI, 0.5 mmol ligand and 2.1 mmol Cs2CO3 in 4 mL DMAC. The reactions are degassed under N2 for 30 min. and then heated at between 80 and
1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak. Longer reaction times are shown in parenthesis) Table 2: Evaluation of various solvents
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.25 mmol CuI, 0.25 mmol 1,10-phenanthroline and 2.1 mmol Cs2CO3 in 3 mL specified solvent. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 3 : Evaluation of different copper sources
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.05 mmol CuX, 0.01 mmol 1,10-phenanthroline and 3 equivalents K3PO4 in 3 mL DMAC. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 4: Evaluation of various inorganic bases
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.1 mmol CuI, 0.1 mmol 1,10-phenanthroline and 2.1 mmol base and degassed for 30 minutes prior to the addition of 3 mL DMAC. The reactions are degassed under N2 for 10 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Example 4. Use of morpholine both as a reactant and base in 2-MeTHF as solvent.
solvent
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (15.2 g, 41.16 mmoles) is charged into a 250 mL 3-necked round bottomed flask, followed by addition of 2-MeTHF (61 mL, 4.0 volumes), the yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (19 g, 218.18 mmoles) is added over 2-5 minutes. Contents are heated to reflux and maintained at reflux for 12 hr. The slurry is cooled to 25 0C, followed by addition of 2-MeTHF (53 mL, 3.5 volumes) and water ( 38 mL 2.5 volumes). The reaction mixture is warmed to 40 0C, where upon a homogenous solution with two distinct layers formed. The layers are separated, the organic layer is filtered and concentrated to ~3 volumes at atmospheric pressure. Four volumes 2-propanol (61 mL) are added. The solution is concentrated to ~3 volumes followed by addition of 4 volumes 2-propanol (61 mL), re-concentrated to ~3 volumes, followed by addition of another 6 volumes 2-propanol (91 mL), and refluxed for 15 min. The clear solution is gradually cooled to 75 0C, seeded with 0.45 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 2 mL 2-propanol, rinsed with an additional 2 mL 2-propanol and transferred to a crystallization flask. The slurry is cooled to between 0-5 0C, maintained for 1 hr, filtered and the product rinsed with 2-propanol (30 mL, 2 volumes). The solid is dried at 60 0C in a vacuum oven to afford 16.92 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. Purity of product by HPLC assay is 100.00 %. XRPD and DSC data of product is consistant with reference sample. MS(ES) = 420 (M+ 1).
Example 5. Use of morpholine as both reactant and base in 2-propanol as solvent.
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (11.64 mmoles) is charged into a 100 mL 3 -necked round bottomed flask followed by addition of 2-propanol ( 16 mL, 3.72 volumes). The yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (3.3 g, 37.84 mmoles) is added over 2-5 minutes. Contents are refluxed for 6 hr. The slurry is cooled to 25 0C. 2-Propanol ( 32 mL, 7.44 volumes) and water ( 8.6 mL, 2.0 volumes) are added and the mixture warmed to 70-75 0C, filtered and concentrated to ~ 9 volumes at atmospheric pressure. The clear solution is gradually cooled to 55 0C, seeded with 0.06 g of crystalline 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 0.5 mL 2-propanol, rinsed with additional 0.5 mL 2-propanol and added to crystallization flask. The slurry is cooled to 0-5 0C, maintained for 1 hr., filtered and the product rinsed with 2-propanol ( 9 mL, 2.1 volumes). Suctioned dried under vacuum at 60 0C to afford 4.6 g of dry 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (88.8 % yield, purity by HPLC assay is 99.88 % ). MS(ES) = 420 (M+ 1).
Example 6: 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine
7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (10 g, 29.17 mmoles), 2, 4-dichlorothiazole (5.2 g , 33.76 mmoles), cesium carbonate(19.9g, 61.07 mmoles) and 1,10-phenanthroline (1 g, 5.5 mmoles) are charged into a 250 mL 3-necked round bottomed flask, followed by 2-MeTHF (36 mL, 3.6 volumes). The reaction mixture is degassed with nitrogen and then evacuated. Cuprous chloride (0.57 g, 5.7 mmoles), DMAC (10 mL, 1 volume) and 2-MeTHF (4 mL, 0.4 volumes) are added in succession. The reaction mixture is degassed with nitrogen and then evacuated. The contents are refluxed for 20 hr. The reaction mixture is cooled to -70 0C and 2-MeTHF (100 mL, 10 volumes) is added. The contents are filtered at ~70 0C and the residual cake is washed with 2-MeTHF (80 mL, 8 volumes) at about 65-72°C. The filtrate is transferred into a separatory funnel and extracted with water. The organic layer is separated and washed with dilute HCl. The resulting organic layer is treated with Darco G60, filtered hot (600C). The filtrate is concentrated at atmospheric pressure to -2.8 volumes. 25 mL 2-propanol is added, followed by re-concentration to -2.8 volumes. An additional 25 mL 2-propanol is added, followed again by re-concentration to -2.8 volumes. Finally, 48 mL 2-propanol is added. The contents are cooled to -7 0C, maintained at -7 0C for 1 hr., filtered and rinsed with 20 mL chilled 2-propanol. Product is suction dried and then vacuum dried at 60 0C to afford 9.41 g 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (purity of product by HPLC assay is 95.88 %). MS(ES) = 369 (M+ 1).
Example 7. Synthesis of 7-(l-ethyl-propyl)-3-(2, 4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori,5-a1pyrimidine using 1,4-Dioxane solvent and CuCl catalyst
Add dioxane (9.06X), Cs2CO3 (2.00X), 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.0 equivalent), 2,4-dichlorothiazole (0.54 equivalent) to a reactor under N2. Purge the reactor with N2 three times, degas with N2 for 0.5-1 hr., and then add 1,10-phenanthroline (0.3 eq) and CuCl (0.3eq) under N2 , degassing with N2 for 0.5-1 hr. Heat the reactor to 1000C -1100C under N2 . Stir the mixture for 22-24 hr. at 100 0C -1100C. Cool to 10~20°C and add water (10V) and CH3OH (5V), stir the mixture for 1-1.5 hr. at 10~20°C. Filter the suspension, resuspend the wet cake in water, stirr for 1-1.5 hr. at 10~20°C, and filter the suspension again. Charge the wet cake to n-heptane (16V) and EtOAc (2V) under N2. Heat the reactor to 40 °C~500C under N2.
Active carbon (0. IX) is added at 40 °C~500C. The reactor is heated to 55°C~650C under N2 and stirred at 55 °C~650C for 1-1.5 hr. The suspension is filtered at 40~55°C through diatomite (0.4 X). The cake is washed with n-heptane (2.5V). The filtrate is transferred to another reactor. EtOAc (10V) is added and the the organic layer washed with 2 N HCl (10V) three times, followed by washing two times with water (10X, 10V). The organic layer is concentrated to 3-4V below 500C. The mixture is heated to 80-90 0C. The mixture is stirred at this temperature for 40-60 min. The mixture is cooled to 0~5°C, stirred for 1-1.5 hr. at 0~5°C and filtered. The cake is washed with n-heptane (IV) and vacuum dried at 45-500C for 8-10 hr. The crude product is dissolved in 2-propanol (7.5V) under N2, and re-crystallized with 2-propanol. The cake is dried in a vacuum oven at 45°C~50°C for 10-12 hr. (55-80% yield). 1H NMR56.537 (s, IH) 3.591-3.659 (m, IH, J=6.8Hz), 2.593 (s, 3H), 2.512 (s, 3H), 1.793-1.921(m, 4H), 0.885-0.903 (m, 6H).
REFERENCES
1: Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol. 2014 Apr;35(2):234-44. doi: 10.1016/j.yfrne.2014.01.001. Epub 2014 Jan 20. Review. PubMed PMID: 24456850; PubMed Central PMCID: PMC4213066.
/////////////tildacerfont, SPR 001, Orphan Drug Status, Congenital adrenal hyperplasia, SPRUCE BIOSCIENCES, PHASE 2
CCC(CC)C1=CC(=NC2=C(C(=NN12)C)C3=C(N=C(S3)N4CCOCC4)Cl)C
Blarcamesine, ブラルカメシン ,

Blarcamesine
ブラルカメシン;
[(2,2-diphenyloxolan-3-yl)methyl]dimethylamine
- Anavex 2-73
- Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanemethanamine
- THD-DP-FM
- AE-37 / AE37 / ANAVEX 2-73 FREE BASE
- UNII 9T210MMZ3F
| Formula |
C19H23NO
|
|---|---|
| Cas |
195615-83-9
195615-84-0 HCL
|
| Mol weight |
281.392
|
Treatment of Rett syndrome, Investigated for use/treatment in breast cancer.
Anti-amnesic, Muscarinic/sigma receptor agonist
- Originator Anavex Life Sciences
- Developer ABX-CRO; Anavex Life Sciences; The Michael J. Fox Foundation for Parkinsons Research
- Class Antidementias; Antidepressants; Antiepileptic drugs; Antiparkinsonians; Anxiolytics; Behavioural disorder therapies; Dimethylamines; Furans; Neuroprotectants; Neuropsychotherapeutics; Nootropics; Small molecules
- Mechanism of Action Muscarinic receptor modulators; Sigma-1 receptor agonists
- Orphan Drug Status Yes – Epilepsy; Rett syndrome
- Phase II/III Alzheimer’s disease
- Phase II Parkinson’s disease; Rett syndrome
- Preclinical Amyotrophic lateral sclerosis; Angelman syndrome; Anxiety disorders; Autistic disorder; Fragile X syndrome; Multiple sclerosis
- No development reported Cognition disorders; Epilepsy; Stroke
- 28 Oct 2019 No recent reports of development identified for phase-I development in Cognition-disorders in USA
- 09 Oct 2019 Anavex Life Sciences initiates enrolment in the long term extension ATTENTION-AD trial for Alzheimer’s disease in (country/ies)
- 02 Oct 2019 Anavex Life Sciences has patent protection covering compositions of matter and methods of treating Alzheimer’s disease for blarcamesine in USA
- Anavex Life Sciences is developing ANAVEX-2-73 and its active metabolite ANAVEX-19-144, for treating Alzheimer’s disease, epilepsy, stroke and Rett syndrome.
ANAVEX2-73 is an experimental drug is in Phase II trials for Alzheimer’s disease, phase I trials for epilepsy, and in preclinical trials for amyotrophic lateral sclerosis, Parkinson’s disease, Rett syndrome, stroke.[1][2] ANAVEX2-73 acts as a muscarinic receptor and a moderate sigma1 receptor agonist.[1] ANAVEX2-73 may function as a pro-drug for ANAVEX19-144 as well as a drug itself. ANAVEX19-144 is the active metabolite of ANAVEX 1-41, which is similar to ANAVEX2-73 but it is not as selective for sigma receptor.[2]
Properties and uses
ANAVEX2-73 has an inhibitory constant (ki) lower than 500 nM for all M1–M4 muscarinic acetylcholine receptor subtypes, demonstrating that it acts as a powerful antimuscarinic compound.[2] ANAVEX2-73 was originally tested in mice against the effect of the muscarinic receptor antagonist scopolamine, which induces learning impairment.[1] M1 receptor agonists are known to reverse the amnesia caused by scopolamine.[3] Scopolamine is used in the treatment of Parkinson’s disease and motion sickness by reducing the secretions of the stomach and intestines and can also decreases nerve signals to the stomach.[3] This is via competitive inhibition of muscarinic receptors.[3] Muscarinic receptors are involved in the formation of both short term and long term memories.[1] Experiments in mice have found that M1 and M3 receptor agonists inhibit the formation of amyloid-beta and target GSK-3B.[clarification needed]Furthermore, stimulation of the M1 receptor activates AF267B, which in turn blocks β-secretase, which cleaves the amyloid precursor protein to produce the amyloid-beta peptide. These amyloid-beta peptides aggregate together to form plaques. This enzyme[clarification needed] is involved in the formation of Tau plaques, which are common in Alzheimer’s disease.[clarification needed][4]Therefore. M1 receptor activation appears to decreases tau hyperphosphorylation and amyloid-beta accumulation.[4]
Sigma1 activation appears to be only involved in long-term memory processes. This partly explains why ANAVEX2-73 seems to be more effective in reversing scopolamine-induced long-term memory problems compared to short-term memory deficits.[1] The sigma-1 receptor is located on mitochondria-associated endoplasmic reticulum membranes and modulates the ER stress response and local calcium exchanges with the mitochondria. ANAVEX2-73 prevented Aβ25-35-induced increases in lipid peroxidation levels, Bax/Bcl-2ratio and cytochrome c release into the cytosol, which are indicative of elevated toxicity.[clarification needed] ANAVEX2-73 inhibits mitochondrial respiratory dysfunction and therefore prevents against oxidative stress and apoptosis. This drug prevented the appearance of oxidative stress. ANAVEX2-73 also exhibits anti-apoptotic and anti-oxidant activity. This is due in part because sigma-1 agonists stimulate the anti-apoptoic factor Bcl-2 due to reactive oxygen species dependent transcriptional activation of nuclear factor kB.[5] Results from Marice (2016) demonstrate that sigma1 compounds offer a protective potential, both alone and possibly with other agents like donepezil, an acetylcholinesterase inhibitor, or the memantine, a NMDA receptor antagonist.[6]
PATENT
WO9730983
PATENT
Novel crystalline forms of A2-73 (blarcamesine hydrochloride, ANAVEX2-73, AV2-73), a mixed muscarinic receptor ligand and Sig-1 R agonist useful for treating Alzheimer’s disease.
PATENT
WO2017013498
SYN
By Foscolos, George B. et alFrom Farmaco, 51(1), 19-26; 1996

References
- ^ Jump up to:a b c “ANAVEX 2-73 – AdisInsight”. Adisinsight.springer.com. Retrieved 2016-05-25.
- ^ Jump up to:a b c Malviya, M; Kumar, YC; Asha, D; Chandra, JN; Subhash, MN; Rangappa, KS (2008). “Muscarinic receptor 1 agonist activity of novel N-arylthioureas substituted 3-morpholino arecoline derivatives in Alzheimer’s presenile dementia models”. Bioorg Med Chem. 16: 7095–7101. doi:10.1016/j.bmc.2008.06.053.
- ^ Jump up to:a b Leal, NS; Schreiner, B; Pinho, CM; Filadi, R; Wiehager, B; Karlström, H; Pizzo, P; Ankarcrona, M (2016). “Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production”. J Cell Mol Med. 20: 1686–1695. doi:10.1111/jcmm.12863. PMC 4988279. PMID 27203684.
- ^ Lahmy, V; Long, R; Morin, D; Villard, V; Maurice, T (2015-09-28). “Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model”. Front Cell Neurosci. 8: 463. doi:10.3389/fncel.2014.00463. PMC 4299448. PMID 25653589.
- ^ Maurice, T (2015-09-28). “Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments”. Behav. Brain Res. 296: 270–8. doi:10.1016/j.bbr.2015.09.020. PMID 26386305.
//////////Blarcamesine, ブラルカメシン , Orphan Drug Status, PHASE 2
CN(C)CC1CCOC1(C1=CC=CC=C1)C1=CC=CC=C1
Reldesemtiv
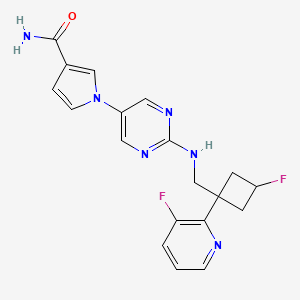
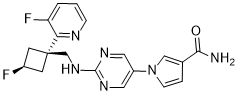
Reldesemtiv
CK-2127107
CAS 1345410-31-2
UNII-4S0HBYW6QE, 4S0HBYW6QE
MW 384.4 g/mol, MF C19H18F2N6O
1-[2-({[trans-3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methyl}amino)pyrimidin-5-yl]-1H-pyrrole-3- carboxamide
1-[2-[[3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methylamino]pyrimidin-5-yl]pyrrole-3-carboxamide
Reldesemtiv, also known as CK-2127107, is a skeletal muscle troponin activator (FSTA) and is a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue such as SMA, COPD, and ALS.
Cytokinetics , in collaboration with Astellas , is developing reldesemtiv, the lead from a program of selective fast skeletal muscle troponin activators, in an oral suspension formulation, for the treatment of indications associated with neuromuscular dysfunction, including spinal muscular atrophy and amyotrophic lateral sclerosis.
- Originator Cytokinetics
- Developer Astellas Pharma; Cytokinetics
- Class Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Troponin stimulants
- Orphan Drug Status Yes – Spinal muscular atrophy
- Phase II Amyotrophic lateral sclerosis; Chronic obstructive pulmonary disease; Spinal muscular atrophy
- Suspended Muscle fatigue
- No development reported Muscular atrophy
- 05 May 2019 Safety and efficacy data from the phase II FORTITUDE-ALS trial in Amyotrophic lateral sclerosis presented at the American Academy of Neurology Annual Meeting (AAN-2019)
- 07 Mar 2019 Cytokinetics completes the phase III FORTITUDE-ALS trial for Amyotrophic lateral sclerosis in USA, Australia, Canada, Spain, Ireland and Netherlands (PO) (NCT03160898)
- 22 Jan 2019 Cytokinetics plans a phase I trial in Healthy volunteers in the first quarter of 2019
Reldesemtiv, a next-generation, orally-available, highly specific small-molecule is being developed by Cytokinetics, in collaboration with Astellas Pharma, for the improvement of skeletal muscle function associated with neuromuscular dysfunction, muscle weakness and/or muscle fatigue in spinal muscular atrophy (SMA), chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS). The drug candidate is a fast skeletal muscle troponin activator (FSTA) or troponin stimulant intended to slow the rate of calcium release from the regulatory troponin complex of fast skeletal muscle fibers. Clinical development for ALS, COPD and SMA is underway in the US, Australia, Canada, Ireland, Netherlands and Spain. No recent reports of development had been identified for phase I development for muscular atrophy in the US. Due to lack of of efficacy determined at interim analysis Cytokinetics suspended phase I trial in muscle fatigue in the elderly.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
[0003] Of the thirteen distinct classes of myosin in human cells, the myosin-II class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-II forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere’s thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-binding of the globular head from the actin filament (Ca2+ regulated) coupled with return of the catalytic domain and light chain to their starting conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin-tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the
Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
U.S. Patent No. 8962632 discloses l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide, a next-generation fast skeletal muscle troponin activator (FSTA) as a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue.
PATENT
WO 2011133888

PATENT
WO2016039367 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016039367&tab=FULLTEXT
claiming the use of a similar compound for treating stress urinary incontinence.
Compound A is 1- [2-({[trans-3-fluoro-1- (3-fluoropyridin-2-yl) cyclobutyl] methyl} amino) pyrimidin-5-yl] -1H Pyrrole-3-carboxamide, which is the compound described in Example 14 of the aforementioned US Pat. The chemical structure is as shown below.
[Chemical formula 1]

PATENT
WO-2019133605
Process for preparing reldesemtiv , a myosin, actin, tropomyosin, troponin C, troponin I, troponin T modulator, useful for treating neuromuscular disorders, muscle wasting, claudication and metabolic syndrome.
Scheme 1
[0091] Scheme 1 illustrates a scheme of synthesizing the compound of Formula (1C).
Scheme 2
[0092] Scheme 2 illustrates an alternative scheme of synthesizing the compound of Formula (1C).
M
TFAA DS, toluene
Et
to
2
HCI, H20
50°C
Scheme 3
[0093] Scheme 3 illustrates a scheme of converting the compound of Formula (1C) to the compound of Formula (II).
H2
Ni Raney
NH3
Scheme 4
[0094] Scheme 4 illustrates a scheme of converting the compound of Formula (II) to the compound of Formula (1).
Examples
[0095] To a flask was added N-methylpyrrolidone (30 mL), tert-butyl cyanoacetate (8.08 g) at room temperature. To a resulting solution was added potassium tert-butoxide (7.71 g), l,3-dibromo-2,2-dimethoxy propane (5.00 g) at 0 °C. To another flask, potassium iodide (158 mg), 2,6-di-tert-butyl-p-cresol (42 mg), N-methylpyrrolidone (25 mL) were added at room temperature and then resulting solution was heated to 165 °C. To this solution, previously prepared mixture was added dropwise at 140-165 °C, then stirred for 2 hours at 165 °C. To the reaction mixture, water (65 mL) was added. A resulting solution was extracted with toluene (40 mL, three times) and then combined organic layer was washed with water (20 mL, three times) and 1N NaOH aq. (20 mL). A resulting organic layer was concentrated below 50 °C under reduced pressure to give 3, 3 -dimethoxy cyclobutane- l-carbonitrile (66% yield,
GC assay) as toluene solution. 1H MR (CDCl3, 400 MHz) d 3.17 (s, 3H), 3.15 (s, 3H), 2.93-2.84 (m, 1H), 2.63-2.57 (m, 2H), 2.52-2.45 (m, 2H).
Example 2 Synthesis of methyl 3,3-dimethoxycyclobutane-l-carboxylate
[0096] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. MeOH (339.00 kg), 3-oxocyclobutanecarboxylic acid (85.19 kg, 746.6 mol, 1.0 eq.), Amberlyst-l5 ion exchange resin (8.90 kg, 10% w/w), and
trimethoxymethane (196.00 kg, 1847.3 mol, 2.5 eq.) were charged into the reactor and the resulting mixture was heated to 55±5°C and reacted for 6 hours to give methyl 3,3-dimethoxycyclobutane-l-carboxylate solution in MeOH. 1H NMR (CDCl3, 400 MHz) d 3.70 (s, 3H), 3.17 (s, 3H), 3.15 (s, 3H), 2.94-2.85 (m, 1H), 2.47-2.36 (m, 4H).
Example 3 Synthesis of 3, 3-dimethoxycyclobutane-l -carboxamide
[0097] The methyl 3, 3 -dimethoxy cyclobutane- l-carboxylate solution in MeOH prepared as described in Example 2 was cooled to below 25°C and centrifuged. The filter cake was washed with MeOH(7.00 kg) and the filtrate was pumped to the reactor. The solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH
(139.40 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH (130.00 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. Half of the resulting solution was diluted with MeOH (435.00 kg) and cooled to below 30°C. NH3 gas (133.80 kg) was injected into the reactor below 35°C for
24 hours. The mixture was stirred at 40±5°C for 72 hours. The resulting solution was
concentrated under vacuum below 50°C until the system had no more than 2 volumes.
MTBE(l8l.OO kg) was charged into the reactor. The resulting solution was concentrated under vacuum below 50°C until the system had no more than 2 volumes. PE (318.00 kg) was charged into the reactor. The resulting mixture was cooled to 5±5°C, stirred for 4 hours at 5±5°C, and centrifuged. The filter cake was washed with PE (42.00 kg) and the wet filter cake was put into a vacuum oven. The filter cake was dried at 30±5°C for at least 8 hours to give 3,3-dimethoxycyclobutane-l-carboxamide as off-white solid (112.63 kg, 94.7% yield). 1H NMR (CDCf, 400 MHz) d 5.76 (bs, 1H), 5.64 (bs, 1H), 3.18 (s, 3H), 3.17 (s, 3H), 2.84-2.76 (m, 1H), 2.45-2.38 (m, 4H).
[0098] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Toluene (500.00 kg), 3,3-dimethoxycyclobutane-l-carboxamide (112.54kg, 706.9 mol, 1.0 eq.), and TEA (158.00 kg, 1561.3 mol, 2.20 eq) were charged into the reactor and the resulting mixture was cooled to 0+ 5°C. TFAA (164.00 kg, 781 mol, 1.10 eq.) was added dropwise at 0±5°C. The resulting mixture was stirred for 10 hours at 20±5°C and cooled below 5±5°C. H20 (110.00 kg) was charged into the reactor at below 15 °C. The resulting mixture was stirred for 30 minutes and the water phase was separated. The aqueous phase was extracted with toluene (190.00 kg) twice. The organic phases were combined and washed with H20 (111.00 kg). H20 was removed by azeotrope until the water content was no more than 0.03%. The resulting solution was cooled to below 20°C to give 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene (492.00 kg with 17.83% assay content, 87.9% yield).
Example 5 Synthesis of l-(3-fluoropyridin-2-yl)-3,3-dimethoxycyclobutane-l-carbonitrile
[0099] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene prepared as described in Example 4 (246.00 kg of a 17.8% solution of 3,3-dimethoxycyclobutane-l-carbonitrile in toluene, 1.05 eq.) and 2-chloro-3-fluoropyridine (39.17 kg, 297.9 mol, 1.00 eq.) were charged into the reactor. The reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The mixture was slowly cooled to -20±5°C. NaHDMS (2M in THF) (165.71 kg, 1.20 eq) was added
dropwise at -20±5°C. The resulting mixture was stirred at -l5±5°C for 1 hour. The mixture was stirred until the content of 2-chloro-3-fluoropyridine is no more than 2% as measured by HPLC. Soft water (16.00 kg) was added dropwise at below 0°C while maintaining the reactor temperature. The resulting solution was transferred to another reactor. Aq. NH4Cl (10% w/w, 88.60 Kg) was added dropwise at below 0°C while maintaining the reactor temperature. Soft water (112.00 kg) was charged into the reactor and the aqueous phase was separated and collected. The aqueous phase was extracted with ethyl acetate (70.00 kg) and an organic phase was collected. The organic phase was washed with sat. NaCl (106.00 kg) and collected. The above steps were repeated to obtain another batch of organic phase. The two batches of organic phase were concentrated under vacuum below 70°C until the system had no more than 2 volumes. The resulting solution was cooled to below 30°C to give a l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution. 1H NMR (CDC13, 400 MHz) d 8.42-8.38 (m, 1H), 7.50-7.45 (m, 1H), 7.38-7.33 (m, 1H), 3.28 (s, 3 H), 3.13 (s, 3H), 3.09-3.05 (m, 4H).
Example 6 Synthesis of I-(3-fluoropyridin-2-yl)-3-oxocyclohutanecarhonitrile
[0100] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Water (603.00 kg) was added to the reactor and was stirred.
Concentrated HC1 (157.30 kg) was charged into the reactor at below 35°C. The l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution prepared as described in Example 5 (206.00 kg) was charged into the reactor and the resulting mixture was heated to 50±5°C and reacted for 3 hours at 50±5°C. The mixture was reacted until the content of 1-(3 -fluoropyridin-2-yl)-3, 3 -dimethoxycyclobutane- l-carbonitrile was no more than 2.0% as measured by HPLC. The reaction mixture was cooled to below 30°C and extracted with ethyl acetate (771.00 kg). An aqueous phase was collected and extracted with ethyl acetate (770.00 kg). The organic phases were combined and the combined organic phase was washed with soft water (290.00 kg) and brine (385.30 kg). The organic phase was concentrated under vacuum at below 60°C until the system had no more than 2 volumes. Propan-2-ol (218.00 kg) was charged into the reactor. The organic phase was concentrated under vacuum at below
60°C until the system had no more than 1 volume. PE (191.00 kg) was charged into the reactor at 40±5 °C and the resulting mixture was heated to 60±5 °C and stirred for 1 hour at 60±5 °C. The mixture was then slowly cooled to 5±5 °C and stirred for 5 hours at 5±5 °C. The mixture was centrifuged and the filter cake was washed with PE (48.00 kg) and the wet filter cake was collected. Water (80.00 kg), concentrated HC1 (2.20 kg), propan-2-ol (65.00 kg), and the wet filter cake were charged in this order into a drum. The resulting mixture was stirred for 10 minutes at 20±5 °C. The mixture was centrifuged and the filter cake was washed with a mixture solution containing 18.00 kg of propan-2-ol, 22.50 kg of soft water, and 0.60 kg of concentrated HC1. The filter cake was put into a vacuum oven and dried at 30±5°C for at least 10 hours. The filter cake was dried until the weight did not change to give l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile as off-white solid (77.15 kg, 68.0% yield). 1H NMR (CDCl3, 400 MHz) d 8.45-8.42 (m, 1H), 7.60-7.54 (m, 1H), 7.47-7.41 (m, 1H), 4.18-4.09 (m, 2H), 4.02-3.94 (m, 2H).
Example 7 Synthesis of I-(3-fhtoropyridin-2-yl)-3-hydroxycyclobulanecarbonilrile
[0101] To a solution of l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g,
1.22 mol) in a mixture ofDCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78° C. The reaction mixture was stirred at -78°C. for 1 hour and quenched with a mixture of methanol and water (1 : 1). The organic layer was washed with water (500 mL><3), dried over Na2S04, and concentrated. The residue was purified on silica gel (50% EtO Ac/hexanes) to provide the title compound as an amber oil (185.8 g, 77.5%). Low Resolution Mass
Spectrometry (LRMS) (M+H) m/z 193.2.
Example 8 Synthesis of (ls,3s)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutane-l-carbonitrile
[0102] To a solution of 1 -(3 -fluoropyridin-2-yl)-3 -hydroxy cyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 hours. The reaction was cooled to rt and poured onto sat. NaHCCf solution. The mixture was separated and the organic layer was washed with water, dried over Na2S04, and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (116g) in a 8: 1 transxis mixture. The above brown oil (107 g) was dissolved in toluene (110 mL) and hexanes (330mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g). LRMS (M+H) m/z 195.1.
Example 9 Synthesis of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine
[0103] A mixture of ( 1.v,3.v)-3-fluoro- 1 -(3-fluoropyridin-2-yl)cyclobutane- 1 -carbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g, 97.6%). LRMS (M+H) m/z 199.2.
Example 10 Synthesis of t-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate
[0104] A mixture of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine (37.6 g, 190 mmol), 5-bromo-2-fluoropyrimidine (32.0 g, 181 mmol), DIPEA (71 mL, 407 mmol), and NMP (200 mL) was stirred at rt overnight. The reaction mixture was then diluted with EtOAc (1500 mL) and washed with saturated sodium bicarbonate (500 mL). The
organic layer was separated, dried over Na2S04, and concentrated. The resultant solid was dissolved in THF (600 mL), followed by the slow addition of DMAP (14 g, 90 mmol) and Boc20 (117.3 g, 542 mmol). The reaction was heated to 60° C. and stirred for 3 h. The reaction mixture was then concentrated and purified by silica gel chromatography
(EtO Ac/hex) to give 59.7 g oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a white solid.
Example 11 Synthesis of t-butyl 5-(3-cyano- 1 H -pyrrol- 1 -yl)pyrimidin-2-yl(((lrans)-3-fhtoro-l-(3-fluoropyridin-2-yl)cyclohutyl)methyl)carhamate
[0105] To a solution oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate (1.0 g, 2.8 mmol) in 15 mL of toluene (degassed with nitrogen) was added copper iodide (100 mg, 0.6 mmol), potassium phosphate (1.31 g, 6.2 mmol), trans-N,N’-dimethylcyclohexane-l, 2-diamine (320 mg, 2.2 mmol), and 3-cyanopyrrole (310 mg, 3.6 mmol). The reaction was heated to 100 °C and stirred for 2 h. The reaction was then concentrated and purified by silica gel chromatography (EtOAc/hexanes) to afford 1.1 g of t-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a clear oil.
Example 12 Synthesis of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide
[0106] To a solution oft-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate (1.1 g, 3.1 mmol) in DMSO (10 mL) was added potassium carbonate (1.3 g, 9.3 mmol). The mixture was cooled to 0 °C and hydrogen peroxide (3 mL) was slowly added. The reaction was warmed to rt and stirred for 90 min. The reaction was diluted with EtO Ac (75 mL) and washed three times with brine (50 mL). The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% MeOH/CH2Cl2) to afford 1.07 g of a white solid compound. This compound was dissolved in 25% TFA/CH2CI2 and stirred for 1 hour. The reaction was then concentrated, dissolved in ethyl acetate (75 mL), and washed three times with saturated potassium carbonate solution. The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was triturated with 75% ethyl acetate/hexanes. The resultant slurry was sonicated and filtered to give 500 mg of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3 -carboxamide as a white solid. LRMS (M+H=385).
REFERENCES
1: Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018 May;57(5):729-734. doi: 10.1002/mus.26017. Epub 2017 Dec 11. PubMed PMID: 29150952.
2: Gross N. The COPD Pipeline XXXII. Chronic Obstr Pulm Dis. 2016 Jul 14;3(3):688-692. doi: 10.15326/jcopdf.3.3.2016.0150. PubMed PMID: 28848893; PubMed Central PMCID: PMC5556764.
//////////////CK-2127107, CK 2127107, CK2127107, Reldesemtiv, Cytokinetics, Astellas, neuromuscular disorders, muscle wasting, claudication, metabolic syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, Orphan Drug Status, Spinal muscular atrophy, Phase II
C1C(CC1(CNC2=NC=C(C=N2)N3C=CC(=C3)C(=O)N)C4=C(C=CC=N4)F)F

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}