

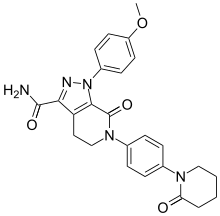
Apixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]


Ruth R. Wexler, executive director of cardiovascular diseases chemistry at Bristol-Myers Squibb, who led the group that designed and synthesized Eliquis (apixaban) to reduce the risk of stroke in patients with an abnormal heart rhythm called atrial fibrillation, recalls hearing about the drug’s success in late-stage clinical trials for the first time.
“I was at the European Society of Cardiology meeting when the results of ARISTOTLE, our large Phase 3 trial, were announced,” she says. “I was sitting in the audience, and it was just amazing to see the data released for the first time. It blew my mind that the data was that spectacular.”
In the trial, which compared apixaban with the workhorse anticoagulant Coumadin (warfarin), apixaban reduced the risk of stroke in patients with atrial fibrillation by 21%, major bleeding by 31%, and mortality by 11%. Unlike Coumadin, apixaban doesn’t require regular monitoring of the blood.
Medical uses
Apixaban is indicated for the following:[5]
- To lower the risk of stroke and embolism in patients with nonvalvular atrial fibrillation.
- Deep vein thrombosis (DVT) prophylaxis. DVT’s may lead to pulmonary embolism (PE) in knee or hip replacement surgery patients.
- Treatment of both DVT and PE.
- To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]
FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.




Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html


![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://i0.wp.com/pic8.molbase.net/molpic/ae/b8/543077.png) APIXABAN
APIXABAN
PREDICTIONS
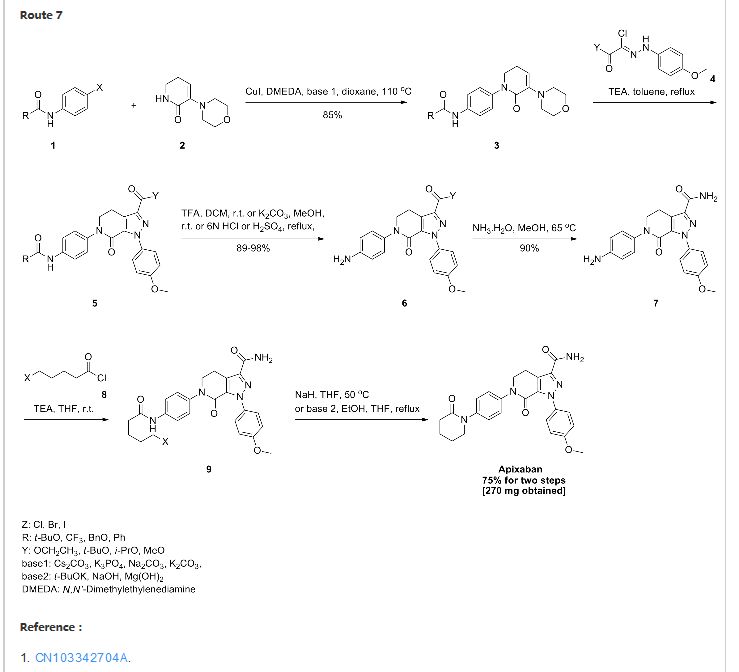
1H NMR

13C NMR

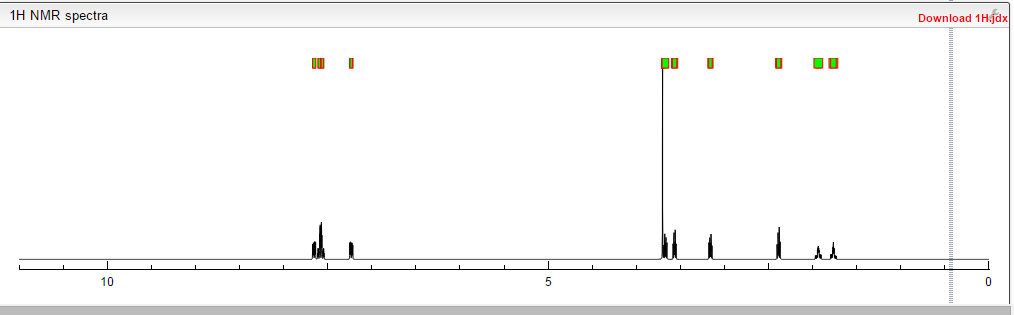
COSY
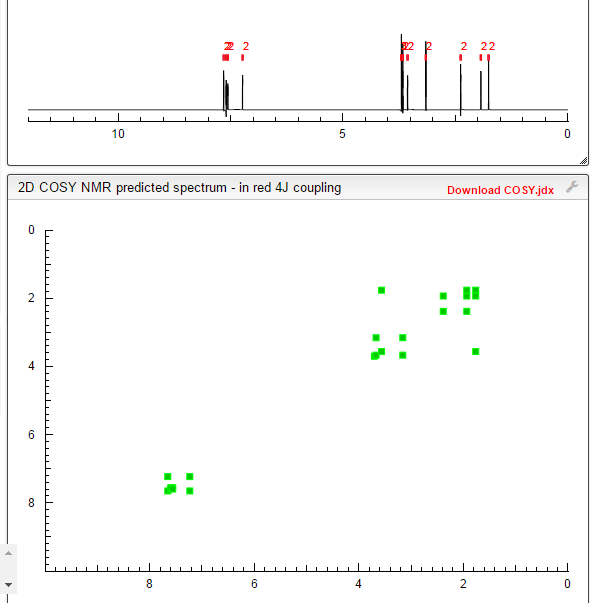
1H NMR PREDICT


13 C NMR PREDICT


|
|
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-1h.png)
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-13c.png)
|
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.
 (I)
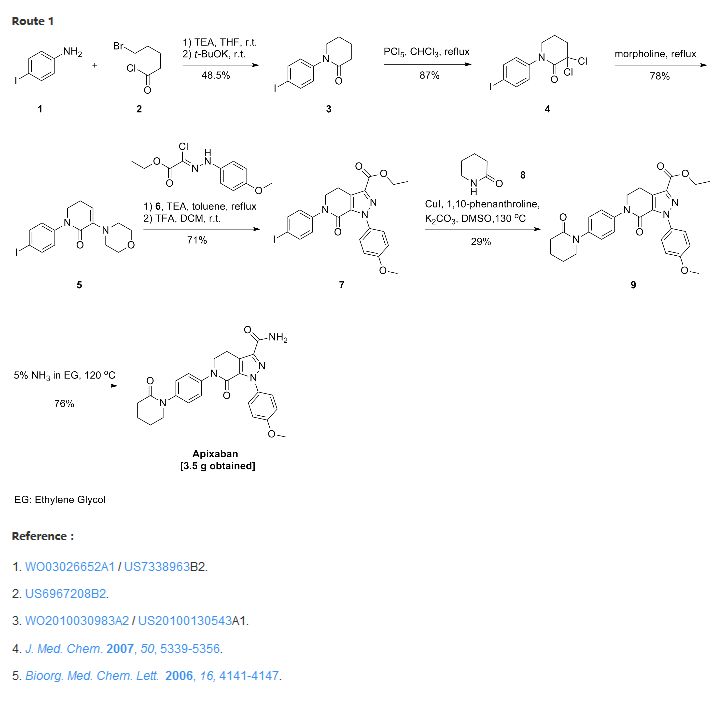
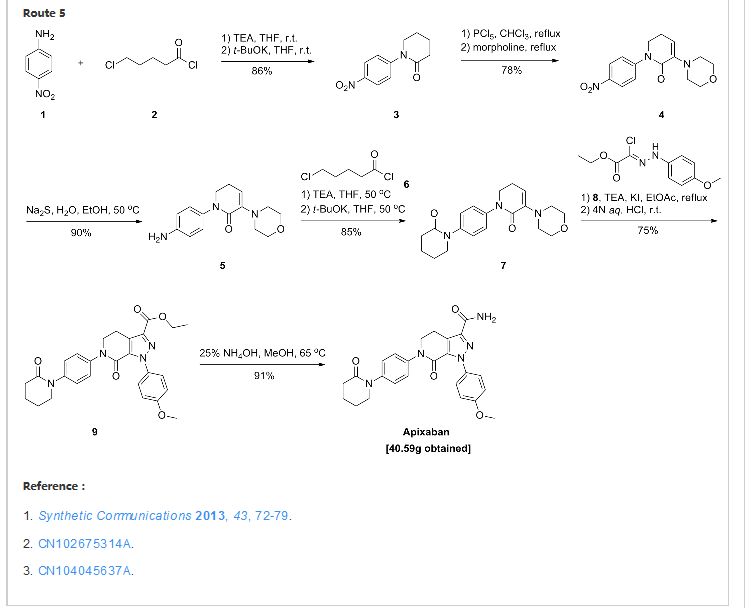
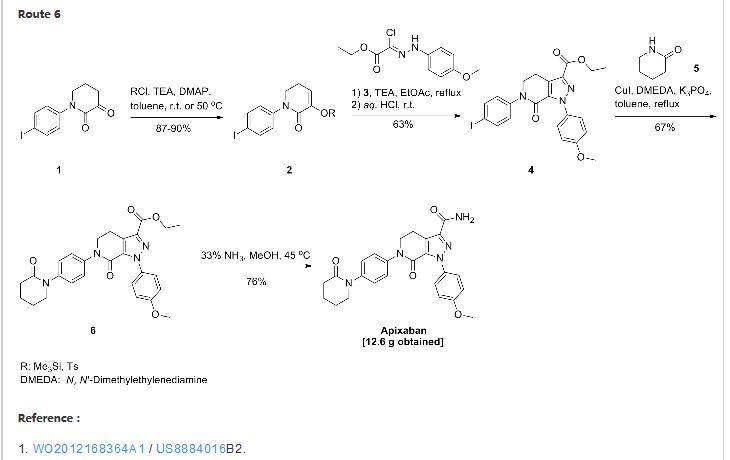
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban
 B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
 The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%
 .
1H NMR (300 MHz, CDC13): DELTA
7.47 (2H, dd, J0=8.7 Hz, Ar-H),
7.31(2H, dd, J0=8.7 Hz, Ar-H),
7.23 (2H, dd, J0=8.7 Hz, Ar-H),
6.93 (2H, dd, J0=8.7 Hz, Ar-H),
6.83 (1H, s, N-H),
5.53 (1H, s, N-H),
4.1 1 (2H, t, J=6.6 Hz, CH2CH2N),
3.81 (3H, s, Ar-OCH3),
3.59 (2H, m, NCH2CH2CH2CH2CO)
3.37 (2H, t, J=6.6 Hz, CH2CH2N),
2.55 (2H, m, NCH2CH2CH2CH2CO),
1.93 (4H, m, NCH2CH^CH2CH2CO).
SEE
NMR For apixaban (in CDCl3) 1  Zhou , J. C. ; Oh , L. M. ; Ma , P. ; Li , H. Y. Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones. WO Patent 2003/0 49681, June 19 , 2003 .
  J. Med. Chem. 2007 , 50 , 5339 – 5356 .
1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (1)[ref ]J. Med. Chem. 2007 , 50 , 5339 – 5356 .To the advanced intermediate 2 (2.44 g, 5.0 mmol) was added 25% ammonia water (1.5 mL, 20 mmol) in methanol (20 mL), and the mixture was heated to 65 °C for 5 h in an autoclave of 50 mL. The resulting mixture was cooled to room temperature, poured into water (30 mL), and crystalized below 0°C. The precipitate was filtrated and dried in vacuo at 50°C to afford the desired product 1 as a pale white solid. Yield: 2.09 g, 91%; mp 171–173 °C; IR (KBr, cm−1): 3448 and 3298 (N-H stretching), 2940 (C-H aliphatic), 1669 (C˭N stretching), 1614 (C˭O stretching), 1544 (aliphatic C˭C), 1513, 1463 and 1441 (aromatic C˭C), 1334, 1300 and 1254 (C-N stretching), 1146, 1111, 1090 and 1024 (C-O stretching), 835, 816, 794 and 758 (Ar-H aromatic bending); 1H NMR (500 MHz, CDCl3, ppm), δ: 7.48 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 5.66 (brs, 2H), 4.12 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.55–3.65 (m, 2H), 3.39 (t, J = 5.6 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H), 1.91–2.01 (m, 4H); 13C NMR (125 MHz, CDCl3, ppm), δ: 170.9, 164.4, 160.5, 158.0, 142.1, 140.6 (2C), 134.0, 133.2, 127.4 (4C), 126.9 (2C), 126.5, 114.4 (2C), 56.2, 52.3, 51.8, 33.5, 24.2, 22.1, 21.9; MS/EI m/z = 459.2 (M+).
SEE
http://www.google.com/patents/WO2014056434A1?cl=en ………………….. http://www.google.com/patents/WO2014108919A2?cl=en HPLC method of Analysis:
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.
 methyl esterImpurity Chloro Impurity Dehydro Impurity
Scheme-I:
Apixaban
 Scheme-II:
 Pure Apixaban Formula-1 [Apixaban]
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
Particle size distribution: D(0.1): 9.183 μπι; D(0.5): 25.991 um; D(0.9): 60.749 μιη; D[4,3]: 31.066 μπι.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
/…………………
|
References
- “ELIQUIS® (apixaban) Approved In Europe For Preventing Venous Thromboembolism After Elective Hip Or Knee Replacement” (Press release). Pfizer. April 20, 2012. Retrieved 2012-05-29.
- http://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_eliquis_apixaban_for_the_treatment_of_deep_vein_thrombosis_dvt_and_pulmonary_embolism_pe_and_for_the_reduction_in_the_risk_of_recurrent_dvt_and_pe_following_initial_therapy
- “Bristol-Myers Squibb News Release 26 April 2007”. Archived from the original on 11 September 2007. Retrieved 2007-09-15.
- Nainggolan, Lisa. “Apixaban better than European enoxaparin regimen for preventing VTE”. Retrieved 2011-04-01.
- “Eliquis (apixaban) [prescribing information]” (PDF). Princeton, NJ: Bristol-Myers Squibb. March 2014. Retrieved 2014-10-29.
- “www.nice.org.uk”.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. PMID 24455237.
- “ELIQUIS® (apixaban) tablets Factor Xa Inhibitor” (PDF). FDA. August 2014. Retrieved 2014-11-02.
- Enriquez A, Lip GY, Baranchuk A (2015). “Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants”. Europace. doi:10.1093/europace/euv030. PMID 25816811.
- Frost C, Wang J, Nepal S; et al. (February 2013). “Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects”. Br J Clin Pharmacol 75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID 22759198.
- Granger, M.D. et. al., Christopher (September 15, 2011). “Apixaban versus Warfarin in Patients with Atrial Fibrillation”. New England Journal of Medicine (365): 981–992. doi:10.1056/NEJMoa1107039. Retrieved 17 September 2015.
- FDA approves Eliquis to reduce the risk of stroke, blood clots in patients with non-valvular atrial fibrillation
Neale, Todd (March 14, 2014). “FDA OKs Apixaban for DVT Prevention”. MedPage Today. Retrieved 17 September 2015.
//////////
SEE ABAN SERIES……http://organicsynthesisinternational.blogspot.in/p/aban-series.html
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
 COCK WILL TEACH YOU
COCK WILL TEACH YOU
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com

