
Home » Posts tagged 'sNDA'
Tag Archives: sNDA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
(sNDA)…..FDA okays Shire ADHD drug Vyvanse (lisdexamfetamine dimesylate) for binge eating


Yet more good news for Shire has come with the US Food and Drug Administration approving its attention-deficit hyperactivity disorder blockbuster Vyvanse for binge-eating disorder, the first medicine approved by the agency to treat this condition.

The agency has expanded approval on Vyvanse (lisdexamfetamine dimesylate) for adults with BED based on two Phase III studies which showed that it was statistically superior to placebo in terms of number of binge days per week. BED affects around 2.8 million US adults and is more prevalent than anorexia nervosa and bulimia nervosa combined.
Read more at: http://www.pharmatimes.com/Article/15-01-30/FDA_okays_Shire_ADHD_drug_Vyvanse_for_binge_eating.aspx

Originally discovered and developed by New River Pharmaceuticals, the company entered into a collaborative agreement with Shire Pharmaceuticals in 2005 for global commercialization of the drug candidate. After Shire’s acquisition of New River Pharmaceuticals in April 2007, lisdexamfetamine entered the product portfolio of Shire.

In 2009, the compound was licensed to GlaxoSmithKline by Shire in the U.S. for comarketing for the treatment of attention deficit/hyperactivity disorder (ADHD). In 2010, this license agreement was terminated. The product was licensed to Shionogi by Shire in Japan for co-development, co-commercialization, and co-promotion for the treatment of attention deficit/hyperactivity disorder (ADHD).

Lisdexamfetamine (NRP-104), a conditionally bioreversible derivative of amphetamine, was launched in the U.S. in 2007 for the treatment of attention deficit hyperactivity disorder (ADHD) in children aged 6-12 years old. In 2008, the product was approved for use in adults, and in 2009 it was approved in Canada, followed by commercialization in 2010. In 2010, FDA approval was obtained for use in treatment of ADHD in adolescents aged 13 to 17 years and launch took place the same year. Approval for the treatment of adolescents was assigned in Canada in 2011.

In 2012, Shire filed a regulatory application in Europe via the decentralized procedure with the U.K. acting as the reference member state, for the treatment of ADHD in children and adolescent patients aged 6 to 17 years. This indication was approved in 2013. Also, in 2012 FDA approval was granted for the maintenance treatment for adults with ADHD. U.K., DK and SE are awaiting approval for the same indication in a decentralized procedure initiated in 2014 with the U.K. acting as the reference member state. In 2014, the company filed with priority review a supplemental New Drug Application (sNDA) in the U.S. for the treatment binge eating in adults.
cas 608137-33-3
(2S)-2,6-Diamino-N-[(1S)-1-methyl-2-phenylethyl]hexanamide dimethanesulfonate

Binge eating,,,,,, express their stress as temper tantrums or by indulging in compulsive eating spree

In terms of clinical development, phase III clinical trials are ongoing at Shionogi in Japan for the treatment of ADHD.. The National Institute on Drug Abuse (NIDA) is evaluating the compound in early clinical studies for the treatment of methamphetamine dependence. Phase III trials were underway as an adjunctive treatment of major depressive disorder; however, they were discontinued due to lack of efficacy. A phase II clinical trial for the treatment of excessive daytime sleepiness (EDS) has been completed. Shire had been evaluating the compound in clinical studies for the treatment of chronic fatigue syndrome.
In 2013, Shire cancelled its phase III program evaluating the product for the negative symptoms of schizophrenia based on a review and prioritization of the company’s development portfolio.
http://www.google.co.in/patents/US7662787



RIVER PHARMA

Patent and Exclusivity Search Results from query on Appl No 021977 Product 003 in the OB_Rx list.
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N021977 | 003 | 7105486 | Jun 29, 2023 | U – 727 | |||
| N021977 | 003 | 7223735 | Jun 29, 2023 | Y | |||
| N021977 | 003 | 7655630 | Feb 24, 2023 | Y | |||
| N021977 | 003 | 7659253 | Feb 24, 2023 | Y | Y | U – 727 | |
| N021977 | 003 | 7659254 | Feb 24, 2023 | U – 1034 | |||
| N021977 | 003 | 7662787 | Feb 24, 2023 | Y | |||
| N021977 | 003 | 7662788 | Feb 24, 2023 | U – 727 | |||
| N021977 | 003 | 7671030 | Feb 24, 2023 | Y | U – 727 | ||
| N021977 | 003 | 7671031 | Feb 28, 2023 | U – 727 | |||
| N021977 | 003 | 7674774 | Mar 18, 2023 | Y | U – 842 | ||
| N021977 | 003 | 7678770 | Mar 25, 2023 | U – 842 | |||
| N021977 | 003 | 7678771 | Mar 25, 2023 | Y | U – 842 | ||
| N021977 | 003 | 7687466 | Feb 24, 2023 | Y | |||
| N021977 | 003 | 7687467 | Apr 8, 2023 | Y | U – 842 | ||
| N021977 | 003 | 7700561 | Jun 29, 2023 | Y | |||
| N021977 | 003 | 7713936 | Feb 24, 2023 | U – 727 | |||
| N021977 | 003 | 7718619 | Feb 24, 2023 | Y | U – 842 | ||
| N021977 | 003 | 7723305 | Feb 24, 2023 | Y | U – 842 |
Exclusivity Data
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N021977 | 003 | I – 645 | Jan 31, 2015 |
Eliquis, Apixaban for the Treatment of Deep Vein Thrombosis and Pulmonary Embolism
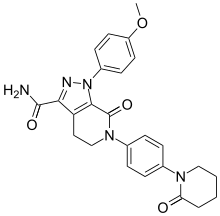
Apixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]


INVENTIVE
Donald Pinto (left) and Michael Orwat (right) work on developing new products for BMS.
Credit: Bristol-Myers Squibb
Ruth R. Wexler, executive director of cardiovascular diseases chemistry at Bristol-Myers Squibb, who led the group that designed and synthesized Eliquis (apixaban) to reduce the risk of stroke in patients with an abnormal heart rhythm called atrial fibrillation, recalls hearing about the drug’s success in late-stage clinical trials for the first time.
“I was at the European Society of Cardiology meeting when the results of ARISTOTLE, our large Phase 3 trial, were announced,” she says. “I was sitting in the audience, and it was just amazing to see the data released for the first time. It blew my mind that the data was that spectacular.”
In the trial, which compared apixaban with the workhorse anticoagulant Coumadin (warfarin), apixaban reduced the risk of stroke in patients with atrial fibrillation by 21%, major bleeding by 31%, and mortality by 11%. Unlike Coumadin, apixaban doesn’t require regular monitoring of the blood.
Medical uses
Apixaban is indicated for the following:[5]
- To lower the risk of stroke and embolism in patients with nonvalvular atrial fibrillation.
- Deep vein thrombosis (DVT) prophylaxis. DVT’s may lead to pulmonary embolism (PE) in knee or hip replacement surgery patients.
- Treatment of both DVT and PE.
- To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]
FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.
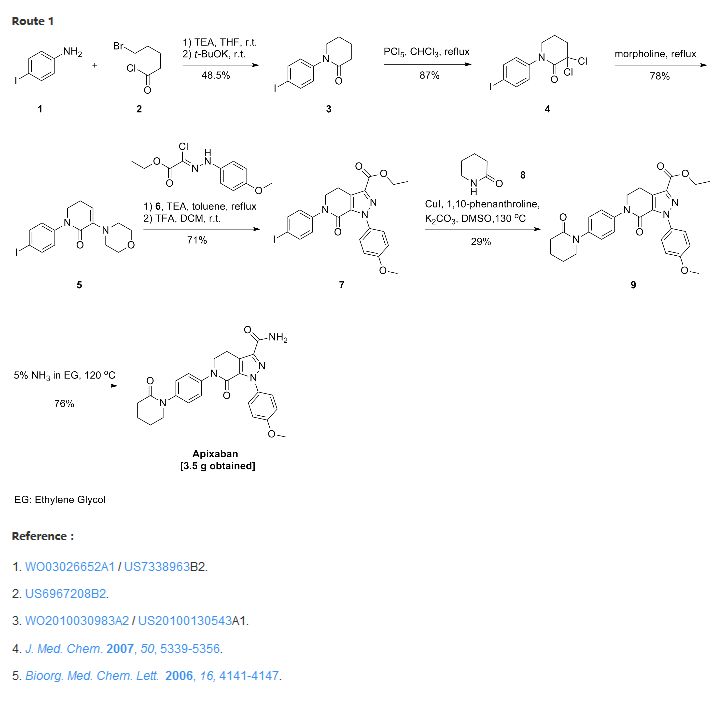



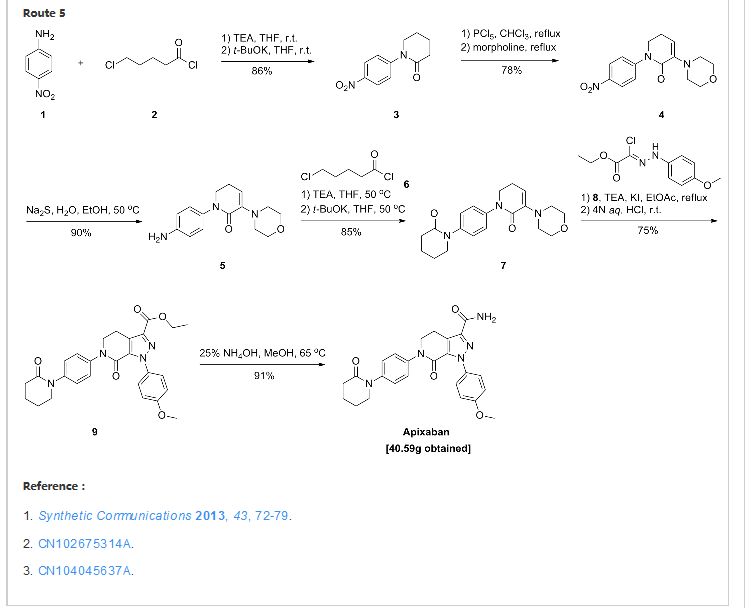
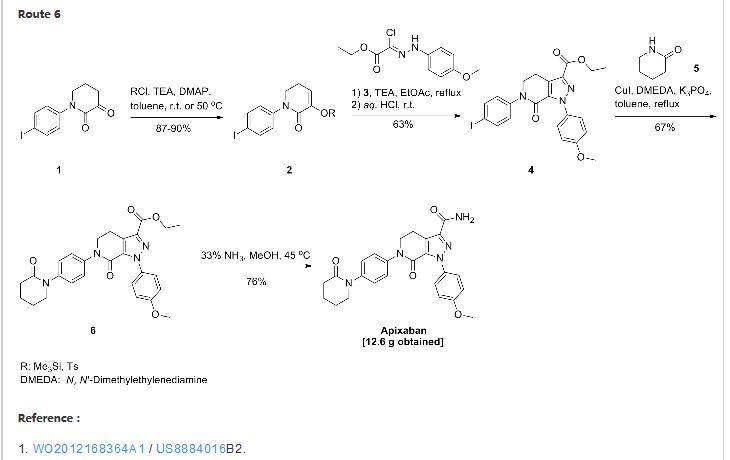

Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html


![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://i0.wp.com/pic8.molbase.net/molpic/ae/b8/543077.png) APIXABAN
APIXABAN
PREDICTIONS
1H NMR

13C NMR

COSY
1H NMR PREDICT


13 C NMR PREDICT


|
|
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-1h.png)
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-13c.png)
C-NMR spectral analysis
|
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.
 (I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban
 B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
 The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%
 .
1H NMR (300 MHz, CDC13): DELTA
7.47 (2H, dd, J0=8.7 Hz, Ar-H),
7.31(2H, dd, J0=8.7 Hz, Ar-H),
7.23 (2H, dd, J0=8.7 Hz, Ar-H),
6.93 (2H, dd, J0=8.7 Hz, Ar-H),
6.83 (1H, s, N-H),
5.53 (1H, s, N-H),
4.1 1 (2H, t, J=6.6 Hz, CH2CH2N),
3.81 (3H, s, Ar-OCH3),
3.59 (2H, m, NCH2CH2CH2CH2CO)
3.37 (2H, t, J=6.6 Hz, CH2CH2N),
2.55 (2H, m, NCH2CH2CH2CH2CO),
1.93 (4H, m, NCH2CH^CH2CH2CO).
SEE
NMR For apixaban (in CDCl3) 1  Zhou , J. C. ; Oh , L. M. ; Ma , P. ; Li , H. Y. Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones. WO Patent 2003/0 49681, June 19 , 2003 .
  J. Med. Chem. 2007 , 50 , 5339 – 5356 .
1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (1)[ref ]J. Med. Chem. 2007 , 50 , 5339 – 5356 .To the advanced intermediate 2 (2.44 g, 5.0 mmol) was added 25% ammonia water (1.5 mL, 20 mmol) in methanol (20 mL), and the mixture was heated to 65 °C for 5 h in an autoclave of 50 mL. The resulting mixture was cooled to room temperature, poured into water (30 mL), and crystalized below 0°C. The precipitate was filtrated and dried in vacuo at 50°C to afford the desired product 1 as a pale white solid. Yield: 2.09 g, 91%; mp 171–173 °C; IR (KBr, cm−1): 3448 and 3298 (N-H stretching), 2940 (C-H aliphatic), 1669 (C˭N stretching), 1614 (C˭O stretching), 1544 (aliphatic C˭C), 1513, 1463 and 1441 (aromatic C˭C), 1334, 1300 and 1254 (C-N stretching), 1146, 1111, 1090 and 1024 (C-O stretching), 835, 816, 794 and 758 (Ar-H aromatic bending); 1H NMR (500 MHz, CDCl3, ppm), δ: 7.48 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 5.66 (brs, 2H), 4.12 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.55–3.65 (m, 2H), 3.39 (t, J = 5.6 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H), 1.91–2.01 (m, 4H); 13C NMR (125 MHz, CDCl3, ppm), δ: 170.9, 164.4, 160.5, 158.0, 142.1, 140.6 (2C), 134.0, 133.2, 127.4 (4C), 126.9 (2C), 126.5, 114.4 (2C), 56.2, 52.3, 51.8, 33.5, 24.2, 22.1, 21.9; MS/EI m/z = 459.2 (M+).
SEE
http://www.google.com/patents/WO2014056434A1?cl=en ………………….. http://www.google.com/patents/WO2014108919A2?cl=en HPLC method of Analysis:
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.
 methyl esterImpurity Chloro Impurity Dehydro Impurity
Scheme-I:
Apixaban
 Scheme-II:
 Pure Apixaban Formula-1 [Apixaban]
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
Particle size distribution: D(0.1): 9.183 μπι; D(0.5): 25.991 um; D(0.9): 60.749 μιη; D[4,3]: 31.066 μπι.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
/…………………
|
References
- “ELIQUIS® (apixaban) Approved In Europe For Preventing Venous Thromboembolism After Elective Hip Or Knee Replacement” (Press release). Pfizer. April 20, 2012. Retrieved 2012-05-29.
- http://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_eliquis_apixaban_for_the_treatment_of_deep_vein_thrombosis_dvt_and_pulmonary_embolism_pe_and_for_the_reduction_in_the_risk_of_recurrent_dvt_and_pe_following_initial_therapy
- “Bristol-Myers Squibb News Release 26 April 2007”. Archived from the original on 11 September 2007. Retrieved 2007-09-15.
- Nainggolan, Lisa. “Apixaban better than European enoxaparin regimen for preventing VTE”. Retrieved 2011-04-01.
- “Eliquis (apixaban) [prescribing information]” (PDF). Princeton, NJ: Bristol-Myers Squibb. March 2014. Retrieved 2014-10-29.
- “www.nice.org.uk”.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. PMID 24455237.
- “ELIQUIS® (apixaban) tablets Factor Xa Inhibitor” (PDF). FDA. August 2014. Retrieved 2014-11-02.
- Enriquez A, Lip GY, Baranchuk A (2015). “Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants”. Europace. doi:10.1093/europace/euv030. PMID 25816811.
- Frost C, Wang J, Nepal S; et al. (February 2013). “Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects”. Br J Clin Pharmacol 75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID 22759198.
- Granger, M.D. et. al., Christopher (September 15, 2011). “Apixaban versus Warfarin in Patients with Atrial Fibrillation”. New England Journal of Medicine (365): 981–992. doi:10.1056/NEJMoa1107039. Retrieved 17 September 2015.
- FDA approves Eliquis to reduce the risk of stroke, blood clots in patients with non-valvular atrial fibrillation
Neale, Todd (March 14, 2014). “FDA OKs Apixaban for DVT Prevention”. MedPage Today. Retrieved 17 September 2015.
//////////
SEE ABAN SERIES……http://organicsynthesisinternational.blogspot.in/p/aban-series.html
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
 COCK WILL TEACH YOU
COCK WILL TEACH YOU
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
The USFDA has approved Navidea Biopharmaceuticals’ Supplemental New Drug Application (sNDA) for the expanded use of Lymphoseek (technetium Tc 99m tilmanocept) Injection
 [99mTc]-DTPA-mannosyl-dextran [99mTc]-DTPA-mannosyl-dextran |
|||
| Systematic (IUPAC) name | |||
|---|---|---|---|
| Dextran 3-[(2-aminoethyl)thio]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D-mannopyranosylthio)ethyl]amino]ethyl]thio]propyl ether technetium-99m complexes…………………………………………………..………………..OTHER NAME ………………Dextran 3-[(2-aminoethyl)thio]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D-mannopyranosylthio)ethyl]amino]ethyl]thio]propyl ether technetium-99Tc complexes (1-6)-alpha-D-pyranoglucan partially etherified by 3-[(2-aminoethyl)sulfanyl]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadecyl and 3-[(2-{[2-(L-mannopyranosylsulfanyl)acetimidoyl]amino}ethyl)sulfanyl]propyl [99mTc]technetium coordination compound [99mTc]-DTPA-mannosyl-dextran composed of a dextran backbone linked to multiple units of mannose and DTPA (diethylenetriamine pentaacetic acid) with an average molecular weight of 35800………………..LAUNCHED………….Launched – 2013 |
|||
| Clinical data | |||
| Trade names | Lymphoseek | ||
| AHFS/Drugs.com | entry | ||
| Pregnancy cat. | C (US) | ||
| Legal status | ℞-only (US) | ||
| Routes | Intradermal, subcutaneous | ||
| Pharmacokinetic data | |||
| Half-life | 1.75 to 3.05 hours at injection site | ||
| Identifiers | |||
| ATC code | V09IA09 | ||
| Chemical data | |||
| Formula | (C6H10O5)n(C19H28N4O9S99mTc)3–8(C13H24N2O5S2)12–20(C5H11NS)0–17 | ||
| Mol. mass 15,281–23,454 g/mol[1]……………………..CODES1600 NEO3-06 TcDTPAmanDx Tilmanocepthttp://chem.sis.nlm.nih.gov/chemidplus/rn/1262984-82-6NDA N202207 APPROVED
|
|||
PATENT US 6409990, EXPMay 12, 2020
商品名:Lymphoseek 通用名:Technetium Tc 99m tilmanocept 中文名:未知
药企:Navidea Biopharmaceuticals, Inc.
FDA approves Navidea’s Lymphoseek for expanded use in head and neck cancer patients
The US Food and Drug Administration (FDA) has approved Navidea Biopharmaceuticals’ Supplemental New Drug Application (sNDA) for the expanded use of Lymphoseek (technetium Tc 99m tilmanocept) Injection indicated for guiding sentinel lymph node (SLN) biopsy in head and neck cancer patients with squamous cell carcinoma of the oral cavity.
NCI: 99mTc-DTPA-mannosyl-dextran A radiolabeled macromolecule consisting of the chelating agent diethylenetriamine pentaacetic acid (DTPA) and mannose each attached to a dextran backbone and labeled with metastable technetiumTc-99 (Tc-99m), with mannose binding and radioisotopic activities. Upon injection, the mannose moiety of 99mTc-DTPA-mannosyl-dextran binds to mannose-binding protein (MBP). As MBPs reside on the surface of dendritic cells and macrophages, this gamma-emitting macromolecule tends to accumulate in lymphatic tissue where it may be imaged using gamma scintigraphy. This agent exhibits rapid clearance from the injection site, rapid uptake and high retention within the first draining lymph node, and low uptake by the remaining lymph nodes. MBP is a C-type lectin that binds mannose or fucose carbohydrate residues, such as those found on the surfaces of many pathiogens, and once bound activates the complement system.
The active ingredient in technetium Tc 99m tilmanocept is technetium Tc 99m tilmanocept. The active ingredient is formed when Technetium Tc 99m pertechnetate, sodium injection is added to the tilmanocept powder vial.
Technetium Tc 99m binds to the diethylenetriaminepentaacetic acid (DTPA) moieties of the tilmanocept molecule.
Chemically, technetium Tc 99m tilmanocept consists of technetium Tc 99m, dextran 3-[(2- aminoethyl)thio]propyl 17-carboxy-10,13,16- tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16- tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D- mannopyranosylthio) ethyl]amino]ethyl]thio]propyl ether complexes. Technetium Tc 99m tilmanocept has the following structural formula:

Empirical formula: [C6H10O5]n.(C19H28N4O9S99mTc)b.(C13H24N2O5S2)c.(C5H11NS)a
Calculated average molecular weight: 15,281 to 23,454 g/mol
It contains 3-8 conjugated DTPA (diethylenetriamine pentaacetic acid) molecules (b); 12-20 conjugated mannose molecules (c) with 0-17 amine side chains (a) remaining free.
The tilmanocept powder vial contains a sterile, non-pyrogenic, white to off-white powder that consists of a mixture of 250 mcg tilmanocept, 20 mg trehalose dihydrate, 0.5 mg glycine, 0.5 mg sodium ascorbate, and 0.075 mg stannous chloride dihydrate. The contents of the vial are lyophilized and are under nitrogen.
Technetium Tc 99m tilmanocept injection is supplied as a Kit. The Kit includes tilmanocept powder vials which contain the necessary non-radioactive ingredients needed to produce technetium Tc 99m tilmanocept. The Kit also contains DILUENT for technetium Tc 99m tilmanocept. The diluent contains a preservative and is specifically formulated for technetium Tc 99m tilmanocept. No other diluent should be used.
The DILUENT for technetium Tc 99m tilmanocept contains 4.5 mL sterile buffered saline consisting of 0.04% (w/v) potassium phosphate, 0.11% (w/v) sodium phosphate (heptahydrate), 0.5% (w/v) sodium chloride, and 0.4% (w/v) phenol. The pH is 6.8 – 7.2.http://www.druginformation.com/RxDrugs/T/Technetium%20Tc%2099m%20Tilmanocept%20Injection.html

Lymphoseek(TM) is a lymphatic tissue-targeting agent which was first launched in 2013 in the U.S. by Navidea Biopharmaceuticals (formerly known as Neoprobe) for lymphatic mapping with a hand-held gamma counter to assist in the localization of lymph nodes draining a primary tumor site in patients with breast cancer or melanoma. In 2014, a supplemental NDA was approved in the U.S. for its use as a sentinel lymph node tracing agent in patients with head and neck squamous cell carcinoma of the oral cavity. Although several tracing agents exist that are used in “off-label” capacities, Lymphoseek is the first tracing agent specifically labeled for lymph node detection.
In 2012, an MAA was filed in the E.U. for the detection of lymphatic tissue in patients with solid tumors, and in 2013, a supplemental MAA was filed in the E.U. for sentinel lymph node detection in patients with head and neck cancer. The products is also awaiting registration to support broader and more flexible use in imaging and lymphatic mapping procedures, including lymphoscintigraphy and other optimization capabilities.
Navidea holds an exclusive worldwide license of Lymphoseek(TM) through the University of California at San Diego (UCSD), and, in 2007, Lymphoseek(TM) was licensed to Cardinal Health by Navidea for marketing and distribution in the U.S.
Lymphoseek(TM), also known as [99mTc]DTPA-mannosyl-dextran, is a receptor-binding radiopharmaceutical designed specifically for the mapping of sentinel lymph nodes in connection with gamma detection devices in a surgical procedure known as intraoperative lymphatic mapping (ILM). It is made up of multiple DTPA and mannose units, each attached by a 5-carbon thioether spacer to a dextran backbone. The compound features subnanomolar affinity for the mannose binding protein receptor, and consequently shows low distal node accumulation. Additionally, its small molecular diameter of 7 nanometers allows for enhanced diffusion into lymphatic channels and capillaries.

1600
99mTc-tilmanocept
Tc-DTPA-mannosyl-dextran
Technetium Tc 99m Tilmanocept
Tilmanocept
UNII-8IHI69PQTC
Chemical structure of [99mTc]tilmanocept. [99mTc]Tilmanocept is composed of a dextran backbone (black) to which are attached multiple units of mannose (green) and DTPA (blue). The mannose units provide a molecular mechanism by which [99mTc]tilmanocept avidly binds to a receptor specific to reticuloendothelial cells (CD206), and the DTPA units provide a highly stable means to radiolabel tilmanocept with 99mtechnetium (red). The molecular weight of [99mTc]tilmanocept is approximately 19,000 g/mol; the molecular diameter is 7.1 nm
[(99m)Tc]Tilmanocept is a CD206 receptor-targeted radiopharmaceutical designed for sentinel lymph node (SLN) identification. Two nearly identical nonrandomized phase III trials compared [(99m)Tc]tilmanocept to vital blue dye.
Technetium (99mTc) tilmanocept, trade name Lymphoseek, is a radiopharmaceutical diagnostic imaging agent approved by the U.S. Food and Drug Administration (FDA) for the imaging of lymph nodes.[1][2] It is used to locate those lymph nodes which may be draining from tumors, and assist doctors in locating those lymph nodes for removal during surgery.[3]

http://blog.sina.com.cn/u/1242475203
…………………….
WO 2000069473
http://www.google.com/patents/EP1178838A2?cl=en
…………………………………………..
US 6409990
http://www.google.co.in/patents/US6409990
References
- FDA Professional Drug Information
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm343525.htm
- Marcinow, A. M.; Hall, N.; Byrum, E.; Teknos, T. N.; Old, M. O.; Agrawal, A. (2013). “Use of a novel receptor-targeted (CD206) radiotracer, 99mTc-tilmanocept, and SPECT/CT for sentinel lymph node detection in oral cavity squamous cell carcinoma: Initial institutional report in an ongoing phase 3 study”. JAMA otolaryngology– head & neck surgery 139 (9): 895–902. doi:10.1001/jamaoto.2013.4239. PMID 24051744.
http://www.google.com/patents/US8247538
Radiopharmaceuticals for use in therapy employ radionuclides which are generally longer in half-life and weaker in penetration capability, but emit stronger radiation, sufficient to kill cells, in relation to that for use in diagnosis. Alpha ray-emitting radionuclides are excluded from radiopharmaceuticals for the reason that they are highly radioactive and difficult to purchase and to attach to other compounds. All of the radionuclides currently used in pharmaceuticals are species that emit beta rays.
As mentioned above, radiopharmaceuticals, whether for use in therapy or diagnosis, are prepared by labeling pharmaceuticals with specific radionuclides. Technetium-99m (99mTc) is known as the radioisotope most widely used to label radiopharmaceuticals. Technetium-99m has a half life of as short as 6 hours and emits gamma rays at 140 KeV, and thus it is not so toxic to the body. In addition, gamma radiation from the radioisotope is highly penetrative enough to obtain images. Thanks to these advantages, technetium-99m finds a broad spectrum of therapeutic and diagnostic applications in the nuclear medicine field (Sivia, S. J., John, D. L., Potential technetium small molecule radiopharmaceuticals. Chem. Rev. 99, 2205-2218, 1999; Shuang, L., Edwards, D. S., 99mTc-Labeled small peptides as diagnostic radiopharmaceuticals. Chem. Rev. 99, 2235-2268, 1999).
Methods of labeling 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid are well known in the art (Callery, P. S., Faith, W. C., et al., 1976. Tissue distribution of technetium-99m and carbon-labeled N-(2,6)-dimetylphenylcarbamoylmethyl iminodiacetic acid. J. Med. Chem. 19, 962-964; Motter, M. and Kloss, G., 1981. Properties of various IDA derivatives. J. Label. Compounds Padiopharm. 18, 56-58; Cao, Y. and Suresh, M. R. 1998. A Simple And Efficient Method For Radiolabeling Of Preformed Liposomes. J Pharm Pharmaceut Sci. 1 (1), 31-37).
Basically, the conventional methods are based on the following reaction formula. In practice, a solution of SnCl2.2H2O, serving as a reducing agent of technetium-99m, in 0.1 N HCl and 0.1 ml (10 mCi) of sodium pertechnetium were added to lyophilized 2,6-diisopropylacetanilidoiminodiacetic acid in a vial, followed by stirring at room temperature for 30 min to prepare 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid. The preparation of 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid may be realized according to the following reaction formula.

Such conventional processes of preparing radiopharmaceuticals labeled with technetium-99m can be divided into reactions between the radioisotope and a physiologically active material to be labeled and the separation of labeled compounds from unlabeled compounds.
| M. Molter, et al., Properties of Various IDA Derivatives, J. Label. Compounds Padiopharm., vol. 18, pp. 56-58, 1981. | ||
| 2 | Patrick S. Callery, et al., Tissue Distribution of Technetium-99m and Carbon . . . , J. Med. Chem., vol. 19, pp. 962-964, 1976. | |
| 3 | * | Sang Hyun Park et al. Synthesis and Radiochemical Labeling of N-(2,6-diisopropylacetanilido)-Iminodiacetic acid and it s analogues under microwave irradiation: A hepatobiliary imaging agent, QSAR Comb. Sci. 2004, 23, 868-874. |
| 4 | Shuang Liu, et al., 99mTc-Labeled Small Peptides as Diagnostic . . . , Chem. Rev., vol. 99, pp. 2235-2268, 1999. | |
| 5 | Shuang Liu, et al., 99mTc—Labeled Small Peptides as Diagnostic . . . , Chem. Rev., vol. 99, pp. 2235-2268, 1999. | |
| 6 | Silvia S. Jurisson, et al., Potential Technetium Small Molecule . . . , Chem. Rev., vol. 99, pp. 2205-2218, 1999. | |
| 7 | Y. Cao, et al., A Simple and Efficient Method for Radiolabeling . . . , J. Phar,. Pharmaceut. Sci., pp. 31-37, 1998. |
|
Diagnostic radiopharmaceuticals (V09)
|
|
|---|---|
| Central nervous system | |
| Skeletal system | |
| Renal | |
| Hepatic/reticuloendothelial | |
| Respiratory system | |
| Cardiovascular system | |
| Inflammation/infection |
|
| Tumor | |
| Adrenal cortex | |
| Radionuclides (including tracers) |
|
FDA Accepts Eliquis sNDA
FDA Accepts For Review ELIQUIS® (apixaban) Supplemental New Drug Application for the Treatment of Deep Vein Thrombosis (DVT) and Pulmonary Embolism (PE), and for the Reduction in the Risk of Recurrent DVT and PE
PRINCETON, N.J. & NEW YORK, December 19, 2013–(BUSINESS WIRE)–Bristol-Myers Squibb Company (NYSE:BMY) and Pfizer Inc. (NYSE:PFE) today announced that the U.S. Food and Drug Administration (FDA) has accepted for review a Supplemental New Drug Application (sNDA) for ELIQUIS® (apixaban) for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the reduction in the risk of recurrent DVT and PE. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by the FDA is August 25, 2014.
sNDA – FDA accepts AMAG Feraheme (Ferumoxytol) sNDA for review

Feraheme (ferumoxytol)
Iron(II,III) oxide
Fe3O4
CUT PASTE OF INFO….
7 MAR 2013
The US Food and Drug Administration (FDA) has accepted for review AMAG Pharmaceuticals’ supplemental new drug application (sNDA) for Feraheme (ferumoxytol) injection for Intravenous (IV) use.
The sNDA filed is to expand the indication for ferumoxytol for the treatment of iron deficiency anemia (IDA) in adult patients with chronic kidney disease (CKD), who have failed or could not take oral iron treatment.
Ferumoxytol is currently indicated for oral use for the treatment of IDA in adult patients with CKD, according to the company.
The sNDA included the data from a global phase III program, which included two phase III clinical trials such as as IDA-301 (placebo comparator) and IDA-302 (active comparator).
The trials, which enrolled 1,400 patients, evaluated the use of ferumoxytol in a broad range of adult IDA patients, all of whom had failed or could not take oral iron treatment.
Both studies achieved the primary efficacy endpoints with statistically significant improvements in hemoglobin from baseline to the 35-day.
The studies, which also included patient-reported outcomes data as pre-specified secondary and exploratory endpoints, found no new safety signals, outside of those described in the current Feraheme (ferumoxytol) label, were observed with ferumoxytol treatment in these studies, claims the company.
In response to the application, the FDA said it will complete the review of Feraheme sNDA by 21 October 2013.
Feraheme, an iron replacement product, is a non-stoichiometric magnetite (superparamagnetic iron oxide) coated with polyglucose sorbitol carboxymethylether. The overall colloidal particle size is 17-31 nm in diameter. The chemical formula of Feraheme is Fe5874O8752-C11719H18682O9933Na414 with an apparent molecular weight of 750 kDa.
Feraheme injection is an aqueous colloidal product that is formulated with mannitol. It is a black to reddish brown liquid, and is provided in single use vials containing 510 mg of elemental iron. Each mL of the sterile colloidal solution of Feraheme injection contains 30 mg of elemental iron and 44 mg of mannitol, and has low bleomycin-detectable iron. The formulation is isotonic with an osmolality of 270-330 mOsm/kg. The product contains no preservatives, and has a pH of 6 to 8.
Ferumoxytol
STRUCTURE SOURCE http://chem.sis.nlm.nih.gov/chemidplus/rn/1309-38-2
Molecular Formulas
-
Fe.O
-
Fe3-O4
Molecular Weight
- 231.531
Ferumoxytol [USAN]
RN: 1309-38-2
Polyglucose sorbitol carboxymethyl ether-coated non-stoichiometric magnetite. Ferumoxytol is a superparamagnetic iron oxide that is coated with a low molecular weight semi-synthetic carbohydrate, polyglucose sorbitol carboxymethyl ether. The iron oxide is a superparamagnetic form of non-stoichiometric magnetite with crystal size of 6.2 to 7.3 nm. In solution, the colloidal particle of ferumoxytol has a Stokes diameter of 18-20 nm. Molecular weight is approximately 308,000
Iron oxide (Fe3O4). It is a black ore of IRON that forms opaque crystals and exerts strong magnetism. The NANOPARTICLES; and MICROSPHERES of its mineral form, magnetite, have many biomedical applications.
Ferumoxytol is the generic ingredient in one branded drug marketed by Amag Pharms Inc and is included in one NDA. There are six patents protecting this compound and one Paragraph IV challenge. Additional information is available in the individual branded drug profile pages.
This ingredient has eleven patent family members in ten countries.
There is one drug master file entry for ferumoxytol. One supplier is listed for this compound.
Phase II
Cas 722492-56-0
Launched – 2009, Anemia, iron deficiency
7228
AMI-7228
Code-7228
A superparamagnetic iron oxide (non-stoichiometric magnetite) coated with a low molecular weight semi-synthetic carbohydrate polyglucose carboxymethyl ether; USAN (OO-74) (Advanced Magnetics, Cambridge, MA, USA)
Other Names
- C 7228
- Code 7228
- Cytogen
- Feraheme
- Rienso
Superparamagnetic iron oxide coated with a low molecular weight semi-synthetic carbohydrate polyglucose sorbitol carboxymethyl ether. The iron oxide is a superparamagnetic form of non-stoichiometric magnetite with crystal size of 6.2 to 7.3 nm. In solution, the colloidal particle has a Stokes diameter of 18-20 nm
CLICK ON IMAGE

CLICK O IMAGE

Feraheme, an iron replacement product, is a non-stoichiometric magnetite (superparamagnetic iron oxide) coated with polyglucose sorbitol carboxymethylether. The overall colloidal particle size is 17-31 nm in diameter. The chemical formula of Feraheme is Fe5874O8752C11719H18682O9933Na414 with an apparent molecular weight of 750 kDa.
Feraheme Injection is an aqueous colloidal product that is formulated with mannitol. It is a black to reddish brown liquid, and is provided in single use vials containing 510 mg of elemental iron. Each mL of the sterile colloidal solution of Feraheme Injection contains 30 mg of elemental iron and 44 mg of mannitol, and has low bleomycin-detectable iron. The formulation is isotonic with an osmolality of 270-330 mOsm/kg. The product contains no preservatives, and has a pH of 6 to 8.
Ferumoxytol is AMAG Pharmaceuticals’ lead investigational compound. In 2007, the company filed a regulatory application seeking approval in the U.S. for use as an intravenous iron replacement therapeutic in patients who may be on dialysis and are suffering from anemic chronic kidney disease (CKD). In 2009, FDA approval was assigned and the product became available on the market. A regulatory application was filed in the E.U. in 2010 for this indication and a positive opinion was received in 2012. Final E.U. approval was obtained in June 2012. In 2012, AMAG Pharmaceuticals submitted a supplemental NDA to the FDA for the treatment of patients with iron-deficiency anemia (IDA) who are not candidates for oral iron, for which they received a complete response letter in January 2014. In 2013, Takeda filed for approval for this indication in the E.U. This application was withdrawn in 2015 due to safety concerns.
In terms of clinical studies, phase II trials are underway at AMAG and at Oregon Health and Science University for use in magnetic resonance angiography (MRA). The National Cancer Institute is also conducting phase II trials for the imaging of primary high-grade brain tumors and/or cerebral metastases from lung or breast cancer. Phase I clinical trials are ongoing at Dana-Farber Cancer Institute for use in magnetic resonance imaging in pediatric and adult patients with malignant sarcoma.
The drug consists of intravenously administered bioavailable iron which allows for more efficient replenishment of the body’s iron stores than oral iron supplements, without their associated common side effects. Ferumoxytol is a blood pool agent, a true intravascular contrast agent that remains in the blood stream for an extended period of time. Based on this quality, the product may be useful as a contrast agent in a wide range of applications in MRI.
In 2008, fast track designation was received in the U.S. as a diagnostic agent for vascular-enhanced magnetic resonance imaging (VE-MRI) to improve the assessment of peripheral arterial disease in patients with known or suspected chronic kidney disease. In 2010, a license, development and commercialization agreement was established between Takeda and AMAG Pharmaceuticals in Asia Pacific countries (excluding Japan, China and Taiwan), Canada, Europe, the Commonwealth of Independent States and Turkey. However, in December 2014, both companies announced the termination of this license agreement. In 2011, orphan drug designation was assigned by the FDA for use in magnetic resonance imaging in brain metastases. This designation was assigned in 2012 for use in magnetic resonance imaging to assess, and monitor treatment of solid tumor malignancies previously diagnosed in pediatric patients (age 16 years and younger).
SFDA

As announced in May 2008, we entered into a development and commercialization agreement with AMAG Pharmaceuticals, Inc. (“AMAG”) (NASDAQ:AMAG), a US biopharmaceutical company, for ferumoxytol, an intravenous iron replacement therapeutic agent being developed to treat iron deficiency anemia in CKD patients and in patients requiring hemodialysis.
Under the terms of the agreement, AMAG granted us exclusive rights to develop and commercialize ferumoxytol in the PRC, initially for CKD, and with an option to expand into additional indications. We will be responsible for the clinical development, registration, and commercialization of ferumoxytol in the PRC. We and AMAG will form a joint steering committee, with equal representation from both parties, to oversee and guide the development and commercialization of ferumoxytol in China. The agreement has an initial duration of 13 years and will be automatically renewed for a set term if minimum sales thresholds are achieved. AMAG will retain all manufacturing rights for ferumoxytol and will provide, under a separate agreement, commercial supply to us at a predetermined supply price.
Ferumoxytol was approved in June 2009 by the U.S. Food and Drug Administration to treat iron deficiency anemia in CKD patients and launched commercially in the U.S. by AMAG in July 2009. Ferumoxytol received marketing approval in Canada in December 2011 and a positive recommendation for approval from the Committee for Medicinal Products for Human Use of the European Medicines Agency in April 2012.
We have submitted the application for a registrational clinical trial for ferumoxytol to SFDA, as announced in January 2010. Once approved by the SFDA, we will commence a multi-center randomized efficacy and safety study in China with approximately 200 CKD patients, measuring the mean change in hemoglobin from baseline at Day 35 after first dose.
https://www.google.com/patents/US20100266644
Ferumoxytol is a newer parenteral iron formulation but limited information is available as to its efficacy and administration. See e.g., Landry et al. (2005) Am J Nephrol 25, 400-410, 408; and Spinowitz et al. (2005) Kidney Intl 68, 1801-1807; U.S. Pat. No. 6,599,498.
Another example of a preferred iron carbohydrate complex for use in the methods described herein is a carboxyalkylated reduced polysaccharide iron oxide complex (e.g., ferumoxytol, described in U.S. Pat. No. 6,599,498).
Another preferred iron carbohydrate complex for use in the methods described herein is a polyglucose sorbitol carboxymethyl ether-coated non-stoichiometric magnetite (e.g., “ferumoxytol”). Ferumoxytol is known in the art to be effective for treating anemia (at single unit doses lower than described herein). See e.g., Spinowitz et al. (2005) Kidney Intl 68, 1801-1807. Ferumoxytol is a superparamagnetic iron oxide that is coated with a low molecular weight semi-synthetic carbohydrate, polyglucose sorbitol carboxymethyl ether. Ferumoxytol and its synthesis are described in U.S. Pat. No. 6,599,498, incorporated herein by reference. Safety, efficacy, and pharmacokinetics of ferumoxytol are as described, for example, in Landry et al. (2005) Am J Nephrol 25, 400-410, 408; and Spinowitz et al. (2005) Kidney Intl 68, 1801-1807.
The iron oxide of ferumoxytol is a superparamagnetic form of non-stoichiometric magnetite with a crystal size of 6.2 to 7.3 nm. Average colloidal particle size can be about 30 nm, as determined by light scattering. Molecular weight is approximately 750 kD. The osmolarity of ferumoxytol is isotonic at 297 mOsm/kg and the pH is neutral. The blood half-life of ferumoxytol is approximately 10-14 hours. It has been previously reported that ferumoxytol can be given by direct intravenous push over 1-5 minutes in doses up to 1,800 mg elemental iron per minute, with maximal total dose up to 420 mg per injection. Landry et al. (2005) Am J Nephrol 25, 400-410, 408.
About Feraheme® (ferumoxytol)/Rienso
In the United States, Feraheme (ferumoxytol) Injection for Intravenous (IV) use is indicated for the treatment of iron deficiency anemia (IDA) in adult patients who have failed oral iron therapy. Feraheme received marketing approval from the FDA on June 30, 2009 for the treatment of IDA in adult chronic kidney disease (CKD) patients and was commercially launched by AMAG in the U.S. shortly thereafter.
Ferumoxytol is protected in the U.S. by five issued patents covering the composition and dosage form of the product. Each issued patent is listed in the FDA’s Orange Book. These patents are set to expire in March 2020; a request for patent term extension has been filed, which, if granted, may extend the patent term to June 2023 for one of the patents.
Ferumoxytol received marketing approval in Canada in December 2011, where it is marketed by Takeda as Feraheme, and in the European Union in June 2012 and Switzerland in August 2012, where it is marketed by Takeda as Rienso.
For additional U.S. product information, including full prescribing information, please visit www.feraheme.com.
AMAG now has five Orange Book-listed patents for ferumoxytol, with patent protection through March 2020, without patent term extension. AMAG has applied for a patent term extension for an Orange Book-listed ferumoxytol patent, which would lengthen that patent term through June 2023.
//////////Ferumoxytol, AMAG Pharmaceuticals, Phase II, 722492-56-0, Launched, 2009, Anemia, iron deficiency, 7228 , AMI-7228 , Code-7228
[Fe](O[Fe]=O)O[Fe]=O
