
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


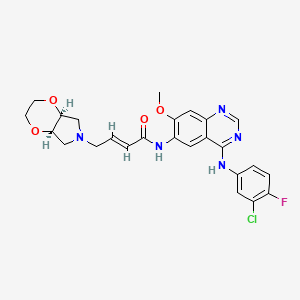
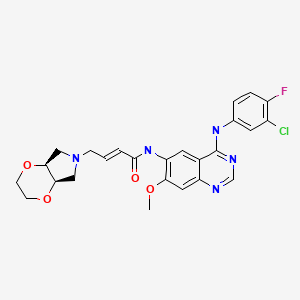

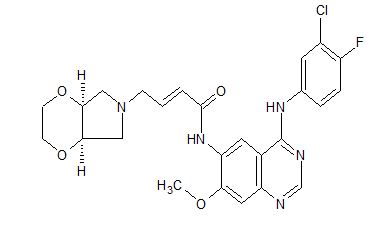
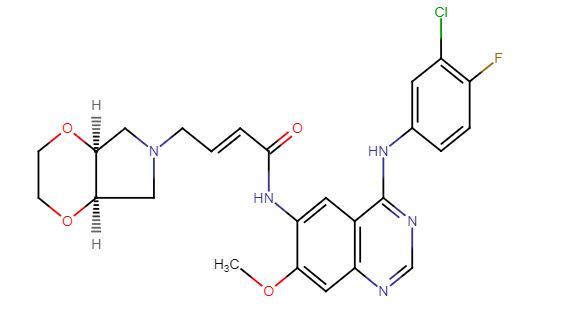
YINLITINIB
error EMAIL ME amcrasto@gmail.com
(E)-4-[(4aR,7aS)-2,3,4a,5,7,7a-hexahydro-[1,4]dioxino[2,3-c]pyrrol-6-yl]-N-[4-(3-chloro-4-fluoroanilino)-7-methoxyquinazolin-6-yl]but-2-enamide
(E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
- C25 H25 Cl F N5 O4, 513.95
DNT-04110 ; yinlitinib maleate , Guangdong Hec Pharmaceutical
Use for treating proliferative diseases, atherosclerosis and pulmonary fibrosis
Phase I CHINA
NOTE AND USE YOUR JUDGMENT ON DRUG SUBSTANCE, EMAIL ME amcrasto@gmail.com
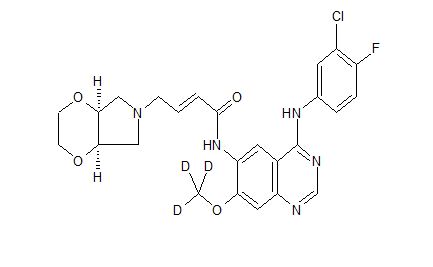
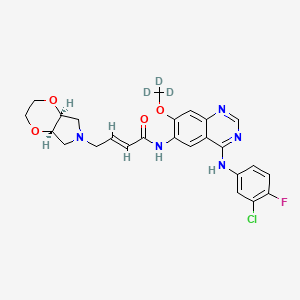
| Molecular Formula: | C25H25ClFN5O4 |
|---|---|
| Molecular Weight: | 516.973 g/mol |
Yinlitinib methoxy-d3
CAS 1637254-71-7
MAY BE DRUG COMD
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9556191 | AMINOQUINAZOLINE DERIVATIVES AND THEIR SALTS AND METHODS OF USE THEREOF |
2014-04-28
|
2016-02-11
|
In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained
Guangdong Hec Pharmaceutical is investigating an oral capsule formulation of yinlitinib maleate (DNT-04110), an irreversible pan-ErbB inhibitor, for the potential treatment of solid tumors . In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained . In December 2016, a phase I trial was planned in China
Protein kinases (PKs) represent a large family of proteins, which play an important role in the regulation of a wide variety of cellular processes and maintaining control over cellular functions. There are two classes of protein kinases (PKs): the protein tyrosine kinases (PTKs) and the serine-threonine kinases (STKs). The protein tyrosine kinase is an enzyme that catalytically transfers the phosphate group from ATP to the tyrosine residue located at the protein substrate, and has a play in the normal cell growth. Many growth factor receptor proteins operate via the tyrosine kinase, and influence the conduction of signal passage and further regulate the cell growth by this process. However, in some circumstances, these receptors become abnormal due to either mutation or overexpression, which cause the uncontrolled cell multiplication, cause the tumor growth, and finally initiate the well-known disease, i.e., cancer. The growth factor receptor protein tyrosine kinase inhibitor, via the inhibition of the above phosphorylation process, may treat cancers and other diseases characterized by the uncontrolled or abnormal cell growth.
Epidermal growth factor receptor (EGFR), a kind of receptor tyrosine kinases, is a multifunction glycoprotein that is widely distributed on the cell membranes of the tissues of the human body, and is an oncogene analog of avian erythroblastic leukemia viral (v-erb-b). Human EGFR/HER1/ErbB-1 and HER2 (human epidermal growth factor receptor-2)/ErbB-2/Teu/p185, HER3/ErbB-3, HER4/ErbB-4 and the like are grouped into the HER/ErbB family, and belong to protein tyrosine kinases (PTKs). They are single polypeptide chains, and each is encoded respectively by genes located on different chromosomes. EGFR and the like are expressed in the epithelia-derived tumors such as squamous cell carcinoma of head and neck, mammary cancer, rectal cancer, ovarian cancer, prostate carcinoma, non-small cell lung cancer, and the like, which are associated with cell proliferation, metastasis, and the like. Pan-HER tyrosine kinase inhibitor, via the competitive binding to the kinase catalytic sites in the intracellular region against ATP, blocks the autophosphorylation of intramolecular tyrosine, blocks the tyrosine kinase activation, inhibits HER-2 family activation, and therefore inhibits cell cycle progression, accelerates cell apoptosis, and exerts the therapeutic action.
EGFR, after binding to the ligand, forms a dimer with a subgroup of HER family, and then combines with ATP to activate the tyrosine kinase activity of the EGFR itself. Therefore, the autophosphorylation occurs in several tyrosine sites of the intracellular kinase region. Pan-HER tyrosine kinase inhibitor, via simultaneity acting on EGFR and HER2/4, inhibits the activation of HER family, and plays a good role in the tumor growth inhibition.
It is indicated in the study that Pan-HER tyrosine kinase irreversible inhibitor has an inhibition effect on HER2/4, besides it effectively inhibits EGFR. The pharmaceutical drugs of this kind, having an irreversible inhibition to both of HER/ErbB families, not only increase the drug activity, but also reduce the drug resistance, and have a substantial inhibition effect on H1975 cell lines which are resistant to erlotinib.
The pharmaceutical drugs that are now commercially available include selective EGFR tyrosine kinase inhibitor gefitinb (IRESSA®, ZD1839), erlotinib (TARCEVA®, OSI-774), double EGFR/HER2 inhibitor Lapatinib (TYKERB®, GW572016), and the like. These three drugs are all reversible EGF receptor tyrosine phosphorylation kinase inhibitor. It has been found in the study that they have good therapeutic response to some tumors initially. However, several months after the treatment, the disease progression appears again and therefore a natural or secondary drug resistance forms. For example, about half of the patients administered with gefitinib or erlotinib develop resistance to gefitinib or erlotinib, which can not lead to the desired therapeutic effect. And it has been indicated by study that the development of drug resistance to selective EGFR tyrosine kinase inhibitor relates to mutations in EGFR.
The mutations of EGFR gene mostly located in the tyrosing kinase coding domain (TK, exons 18-21) are mainly deletion mutation in exon 19 and point mutation in exon 21, both of which are drug-sensitive, and few are point mutation in exon 18 and insertion mutation in exon 20. T790M mutation recognized as one of the mechanism of drug resistance is a point mutation in exon 20 of EGFR. The presence of a second-site EGFR mutation leads to the substitution of methionine for threonine at position 790 (T790M) and changes in the structure of EGFR, which hinder the binding of EGFR inhibitors to EGFR or greatly increase the affinity between EGFR and ATP, so that ATP affinity back to the level of wild-type EGFR, thus resulting in drug resistance. Further studies shows that the pre-treatment tumor samples with mutations of EGFR contain T790M mutation, which indicates that T790M mutation is not just associated with drug resistance and it may have the carcinogenic potential itself.
Irreversible inhibitor can bind to EGFR tyrosine kinase by covalent bond. Thus, the drugs can act on the entire link of epidermal growth factor signal transduction pathway, and improve efficiency of drug blocking. Many clinical studies show that some irreversible inhibitors in current development can against T790M mutation, and overcome the drug resistance caused by T790M. Meanwhile, listed drug Afatinib (BIBW 2992) and some irreversible inhibitors in clinical development (e.g., Dacomitinib, PF00299804, etc.), can inhibit multiple members of EGFR receptor family, especially to the role of EGFR and HER-2, possibly by blocking collaborative signal pathway activated by homodimer and heterodimer to enhance inhibitory effect (Oncologist, 2009, 14 (11): 1116-1130).
Upon developing the drug having an excellent antineoplastic effect, being able to reduce the drug resistance and having a good tolerance, the present inventors discover a quinazoline derivatives as tyrosine kinase inhibitors having a Pan-HER irreversible inhibition function.
PATENT
https://patents.google.com/patent/US9556191
EXAMPLES Example 1 (E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70° C. and stirred for 4.0 hours. The reaction mixture was then cooled to 25° C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 349.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=9.4 Hz, 1H), 4.10 (s, 3H).
Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25° C. The reaction mixture was heated to 90° C. and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60±10° C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 319.1 [M+1]+.
Step 3) (E)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0° C. The reaction mixture was stirred at 0° C. for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
Step 4) (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0° C. The reaction mixture was then heated to 25° C. and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 465.1 [M+1]+.
Step 5) (E)-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N,N-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[1,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25° C., and the reaction mixture was then stirred at 25° C. for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH (v/v)=20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 514.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 10.60 (s, 1H), 9.35 (s, 1H), 8.90 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=8.4 Hz, 1H), 6.75-6.65 (m, 1H), 6.63 (d, J=16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J=6.2 Hz, 4H), 3.26 (t, J=4.4 Hz, 2H), 3.20 (dd, J1=7.8 Hz, J2=2.6 Hz, 2H), 2.20 (d, J=4.6 Hz, 4H).
PATENT
WO2017067447
DIFFERENT COMPD
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017067447
claiming novel crystalline polymorphic forms of a similar EGFR, useful for treating cancer. One of these two compounds is probably yinlitinib maleate , an irreversible pan-ErbB inhibitor, being developed by Guangdong Hec Pharmaceutical , another subsidiary of HEC Pharm , for treating solid tumors; in April 2017, yinlitinib maleate was reported to be in preclinical development
Chinese patent CN 103102344 A (publication number) have disclosed the structure of 4- [ (3-chloro-4-fluorophenyl) amino] -7-methoxy-6- [3- [ (1R, 6S) -2, 5-dioxa-8-azabicyclo [4.3.0] nonan-8-yl] propoxy] quinazoline in example 6 of specification, page 57, and the structure is shown as Formula (II) . The compound of Formula (II) has a high inhibition activity against EGFR, and can be used for treating proliferative disorders.
PATENT
https://patents.google.com/patent/WO2014177038A1/en
InventorYingjun ZhangBing LiuJinlei LiuJiancun ZhangChangchun Zheng
Original AssigneeSunshine Lake Pharma Co., Ltd.
PATENT
Inventor张英俊刘兵刘金雷张健存郑常春 Original Assignee广东东阳光药业有限公司
https://patents.google.com/patent/CN104119350B/en




Example 1
[0442] (E) -N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) -4- ((4aR, 7aS) – tetrahydro _2H_ [1,4] dioxin burning and [2,3_c] R ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0443]
[0444] Synthesis Step Shu: N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine
[0445] The N- (3- chloro-4-fluorophenyl) -7-fluoro-6-nitro-quinazolin-4-amine (10 • 0g, 29 • 8mmol) and sodium methoxide (2.80g, 51.8 mmol) was dissolved in methanol (150 mL), the reaction was warmed to 70 ° C 4. Oh. Was cooled to 25 ° C, the reaction mixture was poured into ice-water (500 mL), the precipitated yellow solid was filtered, the filter cake was dried in vacuo to give a yellow solid 9.00g, yield 86.9%.
[0446] MS (. ESI, pos ion) m / z: 349.1 [M + l] +;
[0447] bandit R (400MHz, DMS〇-d6) S: 11 • 60 (s, 1H), 9 • 55 (s, 1H), 8 • 08 (dd, Ji = 6 • 6Hz, J2 = 2.4Hz, lH), 7.90 (s, lH), 7.76-7.71 (m, lH), 7.58 (s, lH), 7.55 (t, J = 9.4Hz, 1H), 4.10 (s, 3H) square
[0448] Synthesis Step 2: n4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine
[0449] The N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine (9.00g, 25.9mmol) was dissolved in ethanol (100 mL), the was added reduced iron powder (14.5g, 259. Ommol) and concentrated hydrochloric acid (3mL) at 25 ° C, the reaction was warmed to 90 ° C 3.Oh. With 1M aqueous sodium hydroxide solution adjusted to pH 11, filtered hot to remove iron sludge, the mother liquor was concentrated and the residue was purified slurried with ethanol (50 mL), filtered, and the filter cake was dried in vacuo to a yellow solid 6.00g, yield 73.0%.
[0450] MS (ESI, pos ion.) M / z: 319.1 [M + l] + square
[0451] Synthesis Step 3: (E) -4- bromo-but-2-enoyl chloride
The [0452] square ° C Oxalyl chloride (4.19g, 33. Ommol) was slowly added dropwise to a solution containing 4-bromo crotonic acid (2.47g, 15. Ommol) and DMF (0.05mL) in dichloromethane (60 mL) solution of in 3. Oh reaction was stirred at 0 ° C. The reaction solution was concentrated, the residue was stored in a refrigerator until use.
[0453] Synthesis Step 4: (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) butan – 2_ dilute amide
[0454] The N4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine (4.00g, 12.6mmol) and triethylamine (6.0mL, 37.8mmol ) was dissolved in anhydrous tetrahydro-furan in Misaki (80 mL), cooled to 0 ° C, was slowly added (E) -4- bromo-2-dilute acid chloride (2.748,15.12 dirty 〇1), warmed to 25 ° ( : 2.011 reaction the reaction mixture was poured into water (1001 ^) and extracted with methylene chloride (50mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate filtered, concentrated and the residue with dichloromethane (30 mL). beating purified filtered, the filter cake was dried in vacuo 2.00g tan solid, yield 34.5%.
[0455] MS (ESI, pos ion.) M / z: 465.1 [M + l] + square
[0456] Synthesis Step 5: (E) -N- (4 _ ((3- chloro-4-fluorophenyl) amino) -7_ methoxy-quinazolin-6-yl) _4_ ((4aR, 7aS) – tetrahydro -2H- [1,4] dioxin burning and [2,3_c] P ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0457] The (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) but-2-ene amide (0.50g, 1.08mmol) and diisopropylethylamine (0.6mL, 3.24mmol) was dissolved in dimethylacetamide (10 mL) was added at 25 ° C (4aR, 7aS) – hexahydro–2H- [1,4] dioxane, and [2,3-c] pyrrole (0 • 42g, 3 • 24mmol) 5. Oh reaction was continued under stirring, 25 ° C. The reaction mixture was poured into water (70 mL) and extracted with methylene chloride (40mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate. Filtered, concentrated and the residue purified by column chromatography (CH2Cl2 / MeOH (V / v) = 20/1), to give 0.30g tan solid, yield 54.5%.
[0458] MS (. ESI, pos ion) m / z: 514.1 [M + l] +;
[0459] XH NMR (400MHz, DMS0-d6) 8: 10.60 (s, lH), 9.35 (s, lH), 8.90 (s, lH), 8.08 (dd, Ji = 6.6Hz, J2 = 2.4Hz, 1H ), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4Hz, 1H), 6.75-6.65 (m, lH), 6.63 (d, J = 16.2Hz, lH) , 4.10 (s, 3H), 3.78 (t, J = 6.2Hz, 4H), 3.26 (t, J = 4.4Hz, 2H), 3.20 (dd, Ji = 7.8Hz, J2 = 2.6Hz, 2H), 2.20 (d, J = 4.6Hz, 4H)
PATENT
Example 6
[00221] N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41dioxinor2,3-clpyrrol-6(3H -vn propoxy quinazolin-4-amine

[00222] Step Ubenzyl 3,4-dihvdroxypyrrolidine-l -carboxylate

To a solution of N- carbobenzoxy-3-pyrroline ( 1.00 g, 4.92 mmol, 1.0 eq) in acetone (20 mL) was added NMO ( 1.0 g, 7.38 mmol, 1.5 eq) followed by Os04 (cat. 10 mg in 1 mL ‘PrOH). The mixture was stirred for 3 h. To this, saturated NaHS03aqueous solution (5 mL) was added, and the mixture was stirred for another 0.5 h. The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mL x 3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (EtOAc) to give the compound as colorless oil (1.16 g, 100 %).
[00223] Step 2) benzyl tetrahvdro-2H-n.41dioxino[2.3-c1pyrrole-6(3H)- carboxylate

A mixture of NaOH aqueous solution (35 w/w %, 21 mL, aq.), C1CH2CH2C1 (21 mL), benzyl 3,4-dihydroxypyrrolidine-l -carboxylate (1.16 g, 4.9 mmol, 1.0 eq) and TBAB (0.31 g, 0.98 mmol, 0.2 eq) was heated at 55 °C for 48h in a round-bottom flask. The reaction mixture was cooled to room temperature and poured into water (50 mL), extracted with EtOAc (50 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified with a silica gel column chromatography ( 1 : 1 (v/v) PE/EtOAc) to give the product as colorless oil (0.50 g, 39 %).
[00224] Step 3) hexahvdro-2H-n.41dioxinor2.3-clpyrrole

To a solution of benzyl tetrahydro-2H-[l ,4]dioxino[2,3-c] pyrrole-6(3H)-carboxylate (0.46 g, 1 .94 mmol) in MeOH (20 mL) was added two drops of HC02H followed by 20 % Pd(OH)2 (50mg). The reaction mixture was stirred under H2 for 4h at rt and was filtered. The filtrate was concentrated in vacuo to give the crude product, which was used for the next step without further purification.
[00225] Step 4) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41 dioxinor2,3-clpyrrol-6(3H) -yl)propoxy)quinazolin-4-amine

A mixture of hexahydro-2H-[ l ,4]dioxino[2,3-c]pyrrole (1.0 eq), N-(3-chloro-4-fluorophenyI)-6- (3-chloropropoxy)-7-methoxyquinazolin-4-amine (710 mg, 1.8 mmol, 0.95 eq), 2C03 (524 mg, 3.8 mmol, 2.0 eq) and KI (16 mg, 0.095 mmol, 0.05 eq) in DMF (12 mL) was heated at 60 °C for 3 h and cooled to room temperature. The reaction mixture was quenched with water (10 mL) and diluted with EtOAc (20 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by a silica gel column chromatography (20: 1 (v/v) CH2Cl2/CH3OH) to give the crude product, which was recrystallized from CH2C12/PE to afford the title compound as a grayish-white solid (230 mg, 25.00 %), HPLC:99.1 1 % . The compound was characterized by the following spectroscopic data: MS (ESI, pos. ion) m/z: 489.9 (M+1 );’H NMR (400 MHz, CDC13) δ: 2.09 (2H, m), 2.74 (4H, m), 2.99 (2H, dd, = 3.3, 10.4 Hz), 3.56 (2H, m), 3.80 (2H, m), 3.99 (3H, s), 4.12 (2H, t, J = 3.5 Hz), 4.22 (2H, t, J = 6.8 Hz), 7.14 (1 H, t, J = 8.8 Hz), 7.23 (1 H, s), 7.29 ( 1 H, d, J = 15.8 Hz), 7.60 (1 H, m), 7.89 (1 H, dd, J = 2.5, 6.5 Hz), 8.63 (1 H, s) ppm.
PATENT
Example 1
[00192] (^-N 4 (3-Chloro -fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4 (4aR,7a5)-tetrahydro-2H-[ l,4]dioxino[2,3-c]pyrrol-6(3H)

[00193] Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70 °C and stirred for 4.0 hours. The reaction mixture was then cooled to 25 °C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 349.1 [M+l]+; and ‘H NMR (400 MHz, DMSO-<&) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 9.4 Hz, lH ), 4.10 (s, 3H).
[00194] Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25 °C. The reaction mixture was heated to 90 °C and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60 ± 10 °C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 319.1 [M+l]+.
[00195] Step 3) (£)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
[00196] Step 4) (ii)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0 °C. The reaction mixture was then heated to 25 °C and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL x 3). The combined organic phases were dried over anhydrous NaaSOzi, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 465.1 [M+l]+.
[00197] Step 5) (^-N 4 (3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl) (4aR,7aS)-tetrahydro-2H-[l,4]dioxino[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (iT)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N^V-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[l,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25 °C, and the reaction mixture was then stirred at 25 °C for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2 MeOH (v/v) = 20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 514.1 [M+l]+; and lH NMR (400 MHz, DMSO-t/tf) δ: 10.60 (s, 1H), 9.35 (s, 1H) , 8.90 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4 Hz, 1H ), 6.75-6.65 (m, 1H), 6.63(d, J = 16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J= 6.2 Hz, 4H), 3.26 (t, J = 4.4 Hz, 2H), 3.20 (dd, Jx = 7.8 Hz, J2 = 2.6 Hz, 2H), 2.20 (d, J= 4.6 Hz, 4H).
////////////DNT-04110, yinlitinib maleate , Guangdong Hec Pharmaceutical, PHASE 1, CHINA, yinlitinib
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC)c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC([2H])([2H])[2H])c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
SIMILAR COMPDS
|
1
|
MW: 485.9445 – |
|
|
2
|
MW: 558.8663 |
|
|
3
|
MW: 469.9455 |
|
|
4
|
MW: 485.9445 |
|
|
5
|
MW: 485.9445 |

