
Home » Posts tagged 'china'
Tag Archives: china
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Convidicea (Ad5-nCoV)
| A vial of Convidecia vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Viral vector |
| Clinical data | |
| Trade names | Convidecia |
| Routes of administration | Intramuscular, Intranasal |
| ATC code | None |
| Legal status | |
| Legal status | Full and Emergency authorizations |
| Identifiers | |
| DrugBank | DB15655 |
Convidicea (Ad5-nCoV)
Recombinant vaccine (adenovirus type 5 vector)
Recombinant Novel Coronavirus Vaccine (Adenovirus Type 5 Vector)
CanSino Biologics, china
see https://covid19.trackvaccines.org/vaccines/2/
AD5-nCOV, trade-named Convidecia, is a single-dose[1] viral vector vaccine for COVID-19 developed by CanSino Biologics. It conducted its Phase III trials in Argentina,[2] Chile,[3] Mexico,[4] Pakistan,[5] Russia,[6] and Saudi Arabia[7] with 40,000 participants.
In February 2021, global data from Phase III trials and 101 COVID cases showed that the vaccine had a 65.7% efficacy in preventing moderate symptoms of COVID-19, and 91% efficacy in preventing severe disease.[8] It has similar efficacy to Johnson & Johnson’s Ad26.COV2.S, another one-shot adenovirus vector vaccine with 66% efficacy in a global trial.[9][1] Convidecia is similar to other viral vector vaccines like AZD1222, Gam-COVID-Vac, and Ad26.COV2.S.[10] Its single-dose regimen and normal refrigerator storage requirement (2°to 8 °C) could make it a favorable vaccine option for many countries.[9]
Convidecia is approved for use by some countries in Asia,[11][12][13] Europe,[14][15] and Latin America.[16][17][18] Production capacity for Ad5-NCov should reach 500 million doses in 2021. Manufacturing will take place in China,[19] Malaysia,[13] Mexico,[20] and Pakistan.[21]
Ad5-nCoV is a recombinant adenovirus type-5 vector (Ad5) vaccine currently being investigated for prophylaxis against SARS-CoV-2.1,2 It is being developed by CanSino Biologics Inc., in partnership with the Beijing Institute of Biotechnology, who in March 2020 announced the approval of a phase I clinical trial (ChiCTR2000030906)1 with an expected completion in December 2020. The study will evaluate antibody response in healthy patients between the ages of 18 and 60 who will receive one of three study doses, with follow-up taking place at weeks 2 and 4 and months 3 and 6 post-vaccination.2
- Chinese Clinical Trial Register: A phase I clinical trial for recombinant novel coronavirus (2019-COV) vaccine (adenoviral vector) [Link]
- Antibody Society: COVID-19 Archives [Link]
Technology
Convidecia is a viral vector vaccine similar to AstraZeneca‘s AZD1222 and Gamaleya‘s Gam-COVID-Vac.[10] Ad5-nCOV can be stored in less extreme cold conditions compared to mRNA vaccines.[22][9]
Efficacy
In February 2021, data released from an interim analysis of Phase III trials with 30,000 participants and 101 COVID cases showed that globally, the vaccine had an efficacy of 65.7% at preventing moderate cases of COVID-19 and 90.98% efficacy at preventing severe cases. In the Pakistan trial subset, the vaccine had an efficacy of 74.8% at preventing symptomatic cases 100% for preventing severe disease.[8]
While the efficacy rates were lower than the Pfizer–BioNTech and Moderna vaccines, its single-dose regimen and normal refrigerator storage requirement (2 to 8 °C) could make it a favorable option for many countries. It has similar efficacy to Johnson & Johnson’s Ad26.COV2.S, another one-shot adenovirus vaccine found to be 66% effective in a global trial.[9][1]
Clinical trials
Phase I-II
In early 2020, Chen Wei led a joint team of the Institute of Biotechnology, the Academy of Military Medical Sciences and CanSino Biologics to develop AD5-nCOV. According to the Chinese state media, the team registered an experimental COVID-19 vaccine for Phase I trial in China on 17 March 2020 to test its safety. The trial was conducted on 108 healthy adults aged 18 to 60 in two medical facilities in Wuhan, Hubei province.[23]
In April, Ad5-nCoV became the first COVID-19 vaccine candidate in the world to begin Phase II trials.[24] The Phase II trial results were published in the peer-reviewed journal The Lancet in August 2020, and noted neutralizing antibody and T cell responses based on statistical analyses of data involving 508 eligible participants.[25] In September, Zeng Guang, chief scientist of the Chinese Center for Disease Control and Prevention said the amount of COVID-19 antibodies in subjects from the Phase I trials remained high six months after the first shot. Zeng said the high levels of antibodies suggested the shots may provide immunity for an extended period of time, although Phase III results were still required.[26] On September 24, CanSino began Phase IIb trials on 481 participants to evaluate the safety and immunogenicity of Ad5-nCoV for children ages 6–17 and elderly individuals ages 56 and above.[27]
In August, China’s National Intellectual Property Administration issued the country’s first COVID-19 vaccine patent to CanSino.[28]
On 16 May 2020, Canadian Prime Minister Justin Trudeau announced Health Canada had approved Phase II trials to be conducted by the Canadian Center for Vaccinology (CCfV) on the COVID-19 vaccine produced by CanSino. Scott Halperin, director of the CCfV said the vaccine would not be the only one going into clinical trials in Canada, and any potential vaccine would not be publicly available until after Phase 3 is complete.[29][30] If the vaccine trials were successful, then the National Research Council would work with CanSino to produce and distribute the vaccine in Canada.[30] In August 2020, the National Research Council disclosed the vaccine had not been approved by Chinese customs to ship to Canada, after which the collaboration between CanSino and the Canadian Center for Vaccinology was abandoned.[31]
Nasal spray trials
In September, CanSino began a Phase I trial in China with 144 adults to determine the safety and immunogenicity of the vaccine to be administered as a nasal spray, in contrast with most COVID-19 vaccine candidates which require intramuscular injection.[32] On June 3, 2021, Chen Wei announced the expansion of clinical trials was approved by the NMPA, in the meantime, they are applying for Emergency Use Listing for the nasal spray.[33]
Phase III
In August, Saudi Arabia confirmed it would begin Phase III trials on 5,000 people for Ad5-nCoV in the cities of Riyadh, Dammam, and Mecca.[7]
In October, Mexico began Phase III trials on 15,000 volunteers.[34][4]
In September, Russia began Phase III trials on 500 volunteers,[35] which Petrovax later received approval from the government to expand to 8,000 more volunteers.[36][6]
In September, Pakistan began Phase III trials on 40,000 volunteers as part of a global multi-center study.[5] As of December, about 13,000 volunteers have participated in trials of Ad5-nCoV.[22]
In November, Chile began Phase III trials on 5,200 volunteers to be managed by University of La Frontera.[37][3]
In December, Argentina’s Fundación Huésped began Phase III trials in 11 health centers in the metropolitan area of Buenos Aires and Mar del Plata.[2]
Combination trials
In April 2021, a new trial was registered in Jiangsu involving one dose of Convidecia followed by a dose of ZF2001 28 or 56 days later using different technologies as a way to further boost efficacy.[38]
Manufacturing
In February, Chen Wei who lead the development of the vaccine, said annual production capacity for Ad5-NCov could reach 500 million doses in 2021.[19]
In February, Mexico received the first batch of active ingredients for Convidecia, which is being packaged in Querétaro by Drugmex.[20]
In Malaysia, final filling and packaging of the vaccine for distribution would be completed by Solution Biologics.[13]
In May, Pakistan began filling and finishing 3 million doses a month at the National Institute of Health, which would be branded as PakVac for domestic distribution.[39]
If the vaccine is approved in Russia, Petrovax said it would produce 10 million doses per month in 2021.[40]
Marketing and deployment
See also: List of COVID-19 vaccine authorizations § Convidecia
Asia
On 25 June 2020, China approved the vaccine for limited use by the military.[42] In February 2021, China approved the vaccine for general use.[11]
In February, Malaysia‘s Solution Biologics agreed to supply 3.5 million doses to the government.[43] The doses would be delivered starting in April with 500,000 complete doses, with the rest in bulk to be finished by Solution Biologics.[13]
In October, Indonesia reached an agreement with CanSino to deliver 100,000 doses in November 2020, with the expectation that an additional 15 to 20 million doses would be delivered in 2021.[44]
In February, Pakistan approved the vaccine for emergency use.[45] The country purchased 20 million doses of the vaccine[12] of which the first 3 million doses are to arrive in May.[12]
Europe
In March, Hungary granted emergency use approval for the vaccine.[14]
In March, Moldova authorized use of the vaccine.[46]
North America
In December 2020, Mexico‘s Foreign Minister Marcelo Ebrard signed an agreement for 35 million doses.[47] In February, Mexico approved the vaccine for emergency use.[48] Mexico received active ingredients for 2 million doses with a total of 6 million doses expected to arrive in February.[16]
South America
In June, Argentina approved emergency use of the vaccine and ordered 5.4 million doses.[17]
In June, Brazil announced plans to purchase 60 million doses.[49] In May, Brazil began reviewing the vaccine for emergency use.[50]
In March, Chile signed a deal for 1.8 million doses for delivery between May and June,[51] for which emergency use approval was granted in April.[18]
In June, Ecuador approved emergency use and ordered 6 million doses for delivery between June and August 2021.[52]
References
- ^ Jump up to:a b c “It’s not just Johnson & Johnson: China has a single-dose COVID-19 vaccine that has 65% efficacy”. Fortune. Retrieved 2021-02-11.
- ^ Jump up to:a b “Comenzará en la Argentina un nuevo estudio de vacuna recombinante contra el SARS-CoV-2”. infobae (in Spanish). 14 December 2020. Retrieved 2020-12-15.
- ^ Jump up to:a b “Gob.cl – Article: Science Minister: “We Work With Maximum Rigor So That Science And Technology Benefit People’S Health””. Government of Chile. Retrieved 2020-11-21.
- ^ Jump up to:a b “Chinese Covid vaccine trials to be expanded to five more states”. Mexico News Daily. 2020-11-10. Retrieved 2020-11-11.
- ^ Jump up to:a b “Phase III Trial of A COVID-19 Vaccine of Adenovirus Vector in Adults 18 Years Old and Above – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2020-10-21.
- ^ Jump up to:a b Reuters Staff (2020-12-07). “Russia approves clinical trials for Chinese COVID-19 vaccine Ad5-Ncov: Ifax”. Reuters. Retrieved 2020-12-07.
- ^ Jump up to:a b Eltahir N (9 August 2020). “CanSino to start Phase III trial of COVID-19 vaccine in Saudi”. Reuters. Retrieved 9 August 2020.
- ^ Jump up to:a b “CanSinoBIO’s COVID-19 vaccine 65.7% effective in global trials, Pakistan official says”. Reuters. 8 February 2021. Retrieved 2021-02-08.
- ^ Jump up to:a b c d “China’s CanSino Covid Vaccine Shows 65.7% Efficacy”. Bloomberg.com. 2021-02-08. Retrieved 2021-02-10.
- ^ Jump up to:a b Zimmer C, Corum J, Wee SL (2020-06-10). “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 2020-12-12.
- ^ Jump up to:a b Liu R (2021-02-25). “China approves two more domestic COVID-19 vaccines for public use”. Reuters. Retrieved 2021-02-26.
- ^ Jump up to:a b c “Pakistan purchases over 30 million COVID doses from China: sources”. ARY NEWS. 2021-04-25. Retrieved 2021-04-26.
- ^ Jump up to:a b c d “Malaysia to receive CanSino vaccine this month | The Malaysian Insight”. http://www.themalaysianinsight.com. Retrieved 2021-04-03.
- ^ Jump up to:a b Ashok R (2021-03-22). “UPDATE 2-China’s CanSino Biologics COVID-19 vaccine receives emergency use approval in Hungary”. Reuters. Retrieved 2021-03-22.
- ^ “Membrii NITAG au venit cu recomandări privind utilizarea vaccinurilor împotriva COVID-19 în Republica Moldova”. Ministerul Sănătății, Muncii și Protecţiei Sociale. 2021-03-03. Retrieved 2021-05-21.
- ^ Jump up to:a b “‘Our gratitude always’: From China’s CanSino, Mexico welcomes biggest vaccine shipment yet”. Reuters. 2021-02-11. Retrieved 2021-02-11.
- ^ Jump up to:a b “Argentina issues emergency approval to China’s single-dose Cansino COVID-19 vaccine”. Reuters. 2021-06-11. Retrieved 2021-06-11.
- ^ Jump up to:a b “ISP Approves Emergency Use And Importation Of Cansino Vaccine To Fight COVID-19”. Institute of Public Health of Chile. Retrieved 2021-04-08.
- ^ Jump up to:a b “China can hit 500-mln-dose annual capacity of CanSinoBIO COVID-19 vaccine this year”. finance.yahoo.com. Retrieved 2021-02-28.
- ^ Jump up to:a b Solomon DB (2021-02-28). “China’s CanSino says first vaccines packaged in Mexico will be ready in March”. Reuters. Retrieved 2021-03-12.
- ^ “Pakistan develops homemade anti-Covid vaccine ‘PakVac'”. The Express Tribune. 2021-05-24. Retrieved 2021-05-25.
- ^ Jump up to:a b Constable P, Hussain S. “Defying fears and skepticism, thousands in Pakistan volunteer for Chinese vaccine trials”. The Washington Post. ISSN 0190-8286. Retrieved 2021-01-01.
- ^ Cui J (23 March 2020). “Human vaccine trial gets underway”. China Daily. Retrieved 18 April 2020.
- ^ Xie J (15 April 2020). “China Announces Phase 2 of Clinical Trials of COVID-19 Vaccine”. Voice of America. Retrieved 18 April2020.
- ^ Zhu FC, Guan XH, Li YH, Huang JY, Jiang T, Hou LH, et al. (August 2020). “Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial”. Lancet. 396 (10249): 479–488. doi:10.1016/S0140-6736(20)31605-6. PMC 7836858. PMID 32702299.
- ^ O’Brien E (2020-09-25). “Covid Antibodies Endure Over Six Months in China Trial Subjects”. http://www.bloomberg.com. Retrieved 2020-09-29.
- ^ “Phase IIb Clinical Trial of A COVID-19 Vaccine Named Recombinant Novel Coronavirus Vaccine (Adenovirus Type 5 Vector) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2020-10-21.
- ^ Yu S (17 August 2020). “China grants country’s first COVID-19 vaccine patent to CanSino: state media”. Reuters. Retrieved 17 August 2020.
- ^ Bogart N (16 May 2020). “Health Canada approves first clinical trial for potential COVID-19 vaccine”. CTV News. Retrieved 7 September 2020.
- ^ Jump up to:a b Ryan H (May 16, 2020). “Canada’s first COVID-19 vaccine trials approved for Halifax university”. CBC News. Retrieved January 4, 2021.
- ^ Cooke A (26 August 2020). “Canadian COVID-19 clinical trial scrapped after China wouldn’t ship potential vaccine”. CBC News. Retrieved 7 September 2020.
- ^ “A Clinical Trial of a Recombinant Adenovirus 5 Vectored COVID-19 Vaccine (Ad5-nCoV) With Two Doses in Healthy Adults – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 25 September 2020.
- ^ Cao X, Liu Y (2021-06-04). “陈薇院士:雾化吸入式新冠疫苗正在申请紧急使用”. Sci Tech Daily. Chinanews.com. Retrieved 2021-06-04.
- ^ “México recibe el primer lote de la vacuna candidata de CanSino Biologics; alistan pruebas”. EL CEO (in Spanish). 2020-11-03. Retrieved 2020-11-03.
- ^ “Clinical Trial of Recombinant Novel Coronavirus Vaccine (Adenovirus Type 5 Vector) Against COVID-19 – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2020-10-21.
- ^ Bloomberg News (2020-11-25). “Russia’s Richest Man Seeks Global Market for Local Covid-19 Drug – BNN Bloomberg”. BNN. Retrieved 2020-11-28.
- ^ Yáñez PL (2020-11-15). “Así funcionan las cuatro vacunas que se probarán en Chile”. La Tercera. Retrieved 2020-11-17.
- ^ “China trials mixing of CanSinoBIO’s and Zhifei Longcom’s COVID-19 vaccines -data”. Reuters. 2021-04-19. Retrieved 2021-06-16.
- ^ “Covid vaccine: Pakistan starts production of CanSino, China’s single-dose jab”. Khaleej Times. Retrieved 2021-05-28.
- ^ “Russian Recruits Show ‘No Side Effects’ in Chinese Coronavirus Vaccine Trials”. The Moscow Times. 2020-09-21. Retrieved 2020-09-22.
- ^ “Status of COVID-19 Vaccines within WHO EUL/PQ evaluation process”. World Health Organization (WHO).
- ^ Reuters Staff (2020-06-29). “CanSino’s COVID-19 vaccine candidate approved for military use in China”. Reuters. Retrieved 2020-12-13.
- ^ Reuters Staff (2021-02-04). “Malaysia’s Solution Group to supply 3.5 million doses of CanSino vaccine to government”. Reuters. Retrieved 2021-02-04.
- ^ Taufiqurrahman M. “Indonesia can be manufacutring hub for COVID-19 vaccine, says Chinese foreign minister”. Jakarta Post. Retrieved 13 October 2020.
- ^ Shahzad A (2021-02-12). “Pakistan approves Chinese CanSinoBIO COVID vaccine for emergency use”. Reuters. Retrieved 2021-02-12.
- ^ “Membrii NITAG au venit cu recomandări privind utilizarea vaccinurilor împotriva COVID-19 în Republica Moldova”. Ministerul Sănătății, Muncii și Protecţiei Sociale. 2021-03-03. Retrieved 2021-05-21.
- ^ Reuters Staff (2020-12-10). “Mexico agrees to buy 35 million doses of CanSino COVID vaccine”. Reuters. Retrieved 2020-12-10.
- ^ “Mexico approves China’s CanSino and Sinovac COVID-19 vaccines”. Reuters. 10 February 2021.
- ^ “Brazil to buy single-shot Chinese COVID-19 vaccine”. Reuters. 2021-06-15. Retrieved 2021-06-16.
- ^ “Brazil in vaccine talks with Moderna, reviewing CanSino shot”. Reuters. 2021-05-19. Retrieved 2021-05-21.
- ^ Sherwood D (2021-03-30). “Chile inks deal for 1.8 million doses of CanSino COVID-19 vaccine as inoculation drive plows ahead”. Reuters. Retrieved 2021-03-30.
- ^ Valencia A. “Ecuador authorizes use of China’s CanSino vaccine against COVID-19”. Reuters. Retrieved 2021-06-16.
Further reading
- Zhu FC, Li YH, Guan XH, Hou LH, Wang WJ, Li JX, et al. (June 2020). “Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial”. Lancet. 395 (10240): 1845–1854. doi:10.1016/S0140-6736(20)31208-3. PMC 7255193. PMID 32450106.
External links
| Scholia has a profile for Ad5-nCoV (Q96695265). |
/////////Convidicea, Ad5-nCoV, Recombinant vaccine, adenovirus type 5 vector, CanSino Biologics, china, SARS-CoV-2, corona virus, vaccine, covid 19
Convidecia
Convidecia is a viral vector vaccine[478] produced by the Chinese company CanSino Biologics and the Beijing Institute of Biotechnology of the Academy of Military Medical Sciences.Full (1)
- China[479]
Emergency (8)
- Argentina[480]
- Chile[481]
- Ecuador[482]
- Hungary[483][272]
- Malaysia[484]
- Mexico[436]
- Moldova[229]
- Pakistan[485]
NEW DRUG APPROVALS
one time
$10.00
Sinovac COVID-19 vaccine, CoronaVac,
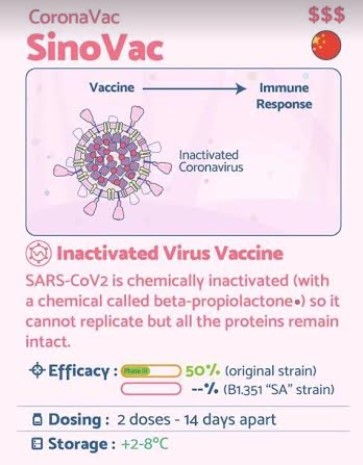

Sinovac COVID-19 vaccine, CoronaVac,
- PiCoVacc
CoronaVac, also known as the Sinovac COVID-19 vaccine,[1] is an inactivated virus COVID-19 vaccine developed by the Chinese company Sinovac Biotech.[2] It has been in Phase III clinical trials in Brazil,[3] Chile,[4] Indonesia,[5] the Philippines,[6] and Turkey.[7]
It relies on traditional technology similar to BBIBP-CorV and BBV152, other inactivated-virus COVID-19 vaccines in Phase III trials.[8] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[9]
Brazil announced results on 13 January 2021 showing 50.4% effective at preventing symptomatic infections, 78% effective in preventing mild cases needing treatment, and 100% effective in preventing severe cases.[10] Final Phase III results from Turkey announced on 3 March 2021 showed an efficacy of 83.5%.[11] Interim results in Indonesia were announced on 11 January 2021 with an efficacy of 65.3%.[12] A detailed report containing confidence intervals, efficacy by age and side effects has not yet been released.
CoronaVac is being used in vaccination campaigns by certain countries in Asia,[13][14][15] South America,[16][17][18] North America,[19][20] and Europe.[21] In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June 2021.[22] As of March 21, 70 million doses of CoronaVac had been administered worldwide.[23
Technology
CoronaVac is an inactivated vaccine. It uses a similar, more traditional technology as in BBIBP-CorV and BBV152, other inactivated-virus vaccines for COVID-19 in Phase III trials.[24][25] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[26] CoronaVac could remain stable for up to three years in storage, which might offer some advantage in vaccine distribution to regions where cold chains are not developed.[27]
NEW DRUG APPROVALS
one time
$10.00
Efficacy

Empty bottle of CoronaVac
On 7 January 2021, results from Phase III trials in Brazil among 13,000 volunteers revealed the vaccine was 78% effective in preventing symptomatic cases of COVID-19 requiring medical assistance (grade 3 on the WHO Clinical Progression Scale[28]) and 100% effective against moderate and severe infections.[29] After mounting pressure from scientists, Butantan said on 12 January that these rates only included volunteers who had mild to severe cases of COVID-19.[30] The overall efficacy, including asymptomatic cases and symptomatic cases not requiring medical assistance (WHO grade 2), was 50.38%.[31] Of the 220 participants infected, 160 cases were in the placebo group and 60 cases in the group that received CoronaVac.[32]
On 3 March 2021, final Phase III results from Turkey showed an efficacy of 83.5%. The final efficacy rate was based on 41 infections, 32 of which had received a placebo, said Murat Akova, head of the Phase III trials in Turkey. He added the vaccine prevented hospitalization and severe illness in 100% of cases, saying six people who were hospitalized were all in the placebo group. The final results were based on a 10,216 participants, 6,648 of whom received the vaccine as part of the Phase III study that began mid-September. Turkey had announced an interim result with 29 infections in December, which placed the efficacy at 91.25%.[33][34]
On 11 January, Indonesia released Phase III results from an interim analysis of 25 cases which showed an efficacy rate of 65.3% based on data of 1,600 participants in the trial.[35] The trial was conducted in the city of Bandung, and it was not clear how Indonesian scientists made their calculations.[30]
Variability in results
Officials said the lowered figure of 50.4% included “very light” cases of COVID-19 among participants omitted in the earlier analysis. Ricardo Palácios, Medical Director of Instituto Butantan said Sinovac’s relatively low efficacy rate of 50% was due to more rigorous standards for what counts as an infection among trial participants. The Institute included six types of cases in its results: asymptomatic, very mild, mild, two levels of moderate, and severe, while western vaccine makers generally included only mild, moderate, and severe categories. Brazil’s trial was also largely made up of frontline health care workers. “They are more exposed to the virus and may explain the relatively low efficacy rate,” said Yanzhong Huang, a senior fellow for global health at the Council on Foreign Relations.[36]
The release of more definitive data on CoronaVac’s efficacy was delayed because Sinovac needed to reconcile results from different trials using varying protocols.[32] According to Instituto Butantan director Dimas Covas, the Brazilian group was considered more vulnerable to infection and exposure to higher viral loads. In Turkish and Indonesian Phase III trials, the composition of volunteers was similar to that of the general population.[37]
COVID-19 variants
On March 10, Instituto Butantan Director Dimas Covas said CoronaVac was efficient against three variants of COVID-19 in the country; British B.1.1.7, South African 501.V2, and Brazil’s P.1, of which are derived variants P.1 from Manaus state, and P.2 from Rio de Janeiro.[38]
CoronaVac and other inactivated virus vaccines have all parts of the virus. Butantan said this may generate a more comprehensive immune response compared to other vaccines using only a part of the spike protein used by COVID-19 to infect cells. Tests run by Butantan used the serum of vaccinated people, which are placed in a cell culture and subsequently infected with the variants. The neutralization consists of determining whether antibodies generated from the vaccine will neutralize the virus in the culture.[38]
Clinical trials
For broader coverage of this topic, see COVID-19 vaccine.
Phase I–II
In a Phase II clinical trial completed in July 2020 and published in The Lancet, CoronaVac showed seroconversion of neutralising antibodies for 109 (92%) of 118 participants in the 3 μg group, 117 (98%) of 119 in the 6 μg group, after the days 0 and 14 schedule; whereas at day 28 after the days 0 and 28 schedule, seroconversion was seen in 114 (97%) of 117 in the 3 μg group, 118 (100%) of 118 in the 6 μg group.[39]
In May, CoronaVac began Phase I–II trials in China on adults over the age 60, and in September CoronaVac began Phase I–II trials in China on children ages 3–17.[40] Phase II results for older adults published in The Lancet showed CoronaVac was safe and well tolerated in older adults, with neutralising antibody induced by a 3 μg dose were similar to those of a 6 μg dose.[41]
Phase III
Latin America
In late July 2020, Sinovac began conducting a Phase III vaccine trial to evaluate efficacy and safety on 9,000 volunteer healthcare professionals in Brazil, collaborating with Butantan Institute.[42][43] On 19 October, São Paulo Governor João Doria said the first results of the clinical study conducted in Brazil proved that among the vaccines being tested in the country, CoronaVac is the safest, the one with the best and most promising immunization rates.[44] On 23 October, São Paulo announced the creation of six new centers for trials of CoronaVac, increasing the number of volunteers in the trials to 13,000.[45]
Brazil briefly paused Phase III trials on 10 November after the suicide of a volunteer before resuming on 11 November. Instituto Butantan said the suicide had no relation to the vaccine trial.[46][47]
In August, a Phase III trial was started in Chile, headed by Pontifical Catholic University of Chile, which was expected to include 3,000 volunteers between the ages of 18 and 65.[48]
Europe
In September, Turkey began Phase III trials with 13,000 volunteers on a two-dose 14-day interval.[49] The monitoring process for CoronaVac is underway at 25 centers in 12 cities across the country.[50]

The Governor of West Java Ridwan Kamil participating in phase 3 trial of the Sinovac COVID-19 vaccine in Indonesia.
Asia
In August, Sinovac began Phase III trials in Indonesia with Bio Farma in Bandung involving 1,620 volunteers.[51] In November, Padjadjaran University Medical School provided an update that the trials were running smoothly and that “at most, they found a slight body fever which disappeared within two days”.[52]
In October, Saudi Arabia signed an agreement with Sinovac to distribute CoronaVac to 7,000 healthcare workers, after conducting Phase III trials with the Saudi Arabian National Guard.[53]
Manufacturing
_2021_A.jpg)
Brazilian version of CoronaVac, manufactured by Butantan
In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June. The figure is double the capacity of 1 billion doses in bulk ingredients the firm said it could reach by February.[22]
After Indonesia’s Phase III trials, Bio Farma plans to ramp up production to 250 million doses a year.[54]
On 9 November, São Paulo began building a facility to produce 100 million doses a year.[55] On 10 December, João Doria said Butantan aimed to fill and finish 1 million doses per day on its production line for a vaccination campaign starting 25 January. Doria said 11 Brazilian states have contacted Butantan seeking doses of CoronaVac.[56]
In Malaysia, Pharmaniaga will manufacture, fill, and finish CoronaVac. Pharmaniaga signed a deal to obtain bulk supply of the vaccine as well as technology transfer from Sinovac.[57]
In Egypt, the government was in “advanced stage” discussions with Sinovac to manufacture CoronaVac for local use and export to African countries.[58]
Market and deployment
As of March 21, 70 million doses of CoronaVac had been administered worldwide.[23]
| show Full authorizationshow Emergency authorization Eligible COVAX recipient (assessment in progress)[80] |
South America
.jpg)
São Paulo State Secretary of Health Jean Gorinchteyn (left) and Instituto Butantan chairman Dimas Covas (right) holding single-dose prefilled syringes of CoronaVac, part of the fourth shipment of Sinovac-manufactured vaccine to arrive in Brazil
In Brazil, São Paulo governor João Doria signed a $90 million contract with Sinovac in September to receive the initial 46 million doses of CoronaVac.[81] The price for CoronaVac was announced to be US$10.3 (about R$59).[82] In January, Brazil announced it would obtain 100 million total doses.[83] On 17 January, ANVISA approved emergency use of CoronaVac, with a 54-year-old nurse in São Paulo being the first to receive a vaccine outside of clinical trials in the country.[16] In early February, Brazil said it intends to buy an additional 30 million doses to be produced locally on top of the existing 100 million doses.[84]
In January, Bolivia authorized use of CoronaVac. Butantan Institute had opened negotiations with South American countries to sell the vaccine, which would be produced in São Paulo.[85]
In October, Chile signed an agreement to purchase 20 million doses of CoronaVac[86] which was approved for emergency use on 20 January.[87] By early March, the country had received 10 million doses of CoronaVac and had vaccinated 4.1 million people.[88]
In February, Colombia had purchased 5 million doses of CoronaVac and was in talks for an additional 5 million doses,[89] which had been approved for emergency use on February 5.[90]
In February, Ecuador signed a deal for 2 million doses of CoronaVac which had been approved for emergency use.[91] Chile donated 20,000 doses of CoronaVac to Ecuador on March 6.[92]
In March, Paraguay received a donation of 20,000 doses of CoronaVac from Chile.[92] Paraguay began vaccinations with CoronaVac on March 10.[93]
In January, Uruguay announced the purchased of 1.75 million doses of CoronaVac.[94] The first 192,000 doses arrived on 25 February and vaccinations started on 1 March.[18]
Europe
In March, Albania received 192,000 doses of a first batch of 1 million doses purchased through Turkey.[95]
In November, Turkey signed a contract to buy 50 million doses of CoronaVac.[96] Turkey approved emergency use on 13 January[97] and President Recep Tayyip Erdoğan received his first dose at Ankara City Hospital.[98] In February, Turkey signed a deal for another 50 million doses for a total of 100 million doses.[21] By March 10.7 million doses had been administered, and 852 of the 1.3 million people who had received both doses were later diagnosed with the disease. 53 were hospitalized, but none of those hospitalized were intubated or died.[99]
In December, Ukraine signed a contract to purchase 1.8 million doses of CoronaVac. One dose of CoronaVac would cost 504 hryvnias (around $18).[100] On March 9, Ukraine granted approval for use of CoronaVac.[101]
Asia
On 19 January, Azerbaijan launched its vaccination campaign with CoronaVac. Azerbaijan plans to receive 4 million doses of the vaccine and aims to vaccinate 40% of the population.[102]
In February, Cambodia approved Coronavac[103] for emergency use and later ordered 1.5 million doses to arrive on March 26.[104]
In late August, China approved CoronaVac for emergency use to vaccinate high-risk groups such as medical staff.[105] In early February, China approved CoronaVac for general use.[15]
In December, Hong Kong ordered 7.5 million doses of CoronaVac.[106] The vaccination campaign with CoronaVac began on 26 February.[107]
In August, Indonesia’s Foreign Minister Retno Marsudi said an agreement was signed with Sinovac for 50 million doses,[108] which later increased to 140 million doses.[109] Indonesia approved emergency use authorization on 11 January and[35] President Joko Widodo received the first shot of the vaccine, which would be free for all Indonesian citizens.[13] By March, Indonesia had received 53.5 million doses of CoronaVac.[110]
On 26 January, Malaysia ordered 12 million doses.[57] CoronaVac was approved for emergency use on 2 March.[111] Malaysian Science, Technology and Innovation Minister Khairy Jamaluddin received the first dose with CoronaVac on 18 March as part of the vaccination campaign.[112]
In January, the Philippine’s announced the country had secured 25 million doses.[113] The vaccine was approved on 22 February but not for all health workers as it had lower efficacy when used with health workers compared to healthy individuals aged 18-59. The first 600,000 doses of CoronaVac arrived on 28 February.[114]
Singapore has signed advance purchase agreements for CoronaVac.[115] In February, the first doses arrived in the country.[116]
In early January, Thailand’s Ministry of Public Health announced an order for 2 million doses of CoronaVac,[117] which was approved for emergency use on 22 February.[118] Thailand started its vaccination program on 27 February.[14] In March, Thailand was in talks to purchase an additional 5 million doses.[119]
North America
By March 8, Dominican Republic had vaccinated 400,000 people and had reserved delivery for 10 million additional doses of CoronaVac.[19]
In February, Mexico approved emergency use of CoronaVac.[120] The country has ordered 20 million doses,[121] of which the first 200,000 doses arrived on 20 February.[122] It is currently used as part of the national vaccination campaign.[20]
Africa
In March, Benin received 203,000 doses of CoronaVac with vaccinations to start with health workers and the medically vulnerable.[123]
In March, South Africa’s drug regulator began assessing CoronaVac for use in the country.[124] South African firm Numolux said it could supply 5 million doses once it secured regulatory clearances.[125]
In March, Tunisia’s Ministry of Health approved marketing authorization of CoronaVac in the country.[126]
In March, Zimbabwe approved CoronaVac for emergency use.[127]
Oceania
In March, Fiji said it would be receiving a donation of CoronaVac.[128]
Controversies
Politicization
CoronaVac has been championed by the governor of São Paulo, João Doria, who many believe will challenge Jair Bolsonaro for the presidency in 2022.[129] A political showdown began in October 2020, when Bolsonaro vetoed a deal between the Brazilian health ministry and the São Paulo government for the purchase of 46 million doses of the vaccine.[130] After Instituto Butantan announced CoronaVac’s efficacy rate, Bolsonaro mocked the vaccine’s effectiveness against COVID-19.[131] Critics against the politicization of vaccines have warned that failure to follow international testing and safety protocols risks undermining public trust and can increase people’s hesitancy to inoculation.[129] Doctors in São Paulo said they were struggling to convince patients that CoronaVac would be safe.[132]
In March 2021, the Paraná Pesquisas opinion polling institute found that the vaccines preferred by Brazilians are CoronaVac and the Oxford–AstraZeneca vaccine, chosen by 23.6% and 21.2% of Brazilians interviewed, respectively, against 11.3% of those who would prefer the Pfizer–BioNTech vaccine.[133]
Delays in releasing results
On 23 December 2020, researchers in Brazil said the vaccine was more than 50% effective, but withheld full results at Sinovac’s request, raising questions again about transparency as it was the third delay in releasing results from the trials.[134] São Paulo Health Secretary Jean Gorinchteyn later said the vaccine didn’t reach 90% efficacy. Turkey said its trial showed an estimated efficacy rate of 91.25%, though that was based on only 29 infected cases.[32] When São Paulo state officials announced the protection rate, they declined to provide a more detailed breakdown of the trial, such as information about age groups and side effects of the vaccine.[32] Scientists said the lack of transparency about the data ran the risk of damaging CoronaVac’s credibility, with Brazilians and others world-wide already reluctant to take it.[30] Nikolai Petrovsky, a professor at the College of Medicine and Public Health at Flinders University said, “There is enormous financial and prestige pressure for these trials to massively overstate their results.”[135]
References
- ^ Corum, Jonathan; Zimmer, Carl. “How the Sinovac Vaccine Works”. The New York Times. ISSN 0362-4331. Retrieved 1 March 2021.
- ^ Nidhi Parekh (22 July 2020). “CoronaVac: A COVID-19 Vaccine Made From Inactivated SARS-CoV-2 Virus”. Retrieved 25 July2020.
- ^ “New coronavirus vaccine trials start in Brazil”. AP News. 21 July 2020. Retrieved 7 October 2020.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Chile Reports. 4 August 2020. Retrieved 7 October 2020.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. 30 August 2020. Retrieved 7 October2020.
- ^ “DOH eyes 5 hospitals for Sinovac vaccine Phase 3 clinical trial”. PTV News. 16 September 2020. Retrieved 7 October 2020.
- ^ “Turkey begins phase three trials of Chinese Covid-19 vaccine”. TRT World News. 1 September 2020. Retrieved 7 October 2020.
- ^ Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 12 February 2021.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 1 November 2020. Retrieved 12 February 2021.
- ^ “Sinovac: Brazil results show Chinese vaccine 50.4% effective”. BBC News. 13 January 2021. Retrieved 12 February 2021.
- ^ AGENCIES, DAILY SABAH WITH (25 December 2020). “Turkey set to receive ‘effective’ COVID-19 vaccine amid calls for inoculation”. Daily Sabah. Retrieved 12 February 2021.
- ^ hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 12 February 2021.
- ^ Jump up to:a b TARIGAN, EDNA; MILKO, VICTORIA (13 January 2021). “Indonesia starts mass COVID vaccinations over vast territory”. Associated Press. Retrieved 15 January 2021.
- ^ Jump up to:a b “Thailand Kicks Off Covid-19 Vaccine Program With Sinovac Shots”. Bloomberg.com. Retrieved 28 February 2021.
- ^ Jump up to:a b “China approves Sinovac vaccines for general public use”. South China Morning Post. 6 February 2021. Retrieved 6 February2021.
- ^ Jump up to:a b Fonseca, Jamie McGeever, Pedro (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Miranda, Natalia A. Ramos (28 January 2021). “Chile receives two million-dose first delivery of Sinovac COVID-19 vaccine”. Reuters. Retrieved 30 January 2021.
- ^ Jump up to:a b “BNamericas – Uruguay prepares to launch COVID-19 vaccinat…” BNamericas.com. Retrieved 1 March 2021.
- ^ Jump up to:a b “Anticovid vaccines run out as Dominican Republic awaits arrival of more doses”. Dominican Today. Retrieved 10 March2021.
- ^ Jump up to:a b “Venustiano Carranza next up for Covid vaccination in Mexico City”. Mexico News Daily. 15 March 2021. Retrieved 16 March2021.
- ^ Jump up to:a b “Turkey aims to vaccinate 60 percent of population: Minister – Turkey News”. Hürriyet Daily News. Retrieved 12 February 2021.
- ^ Jump up to:a b Liu, Roxanne (3 March 2021). “Sinovac eyes two billion doses in annual capacity of virus vaccine by June”. Reuters. Retrieved 3 March 2021.
- ^ Jump up to:a b Liu, Roxanne (21 March 2021). “China steps up COVID-19 vaccination, considers differentiated visa policies”. Reuters. Retrieved 21 March 2021.
- ^ Tan Y (16 December 2020). “Covid: What do we know about China’s coronavirus vaccines?”. BBC News. Retrieved 18 December 2020.
- ^ Zimmer C, Corum J, Wee SL (10 June 2020). “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 27 December 2020.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 1 November 2020. Archivedfrom the original on 16 December 2020. Retrieved 1 November2020.
- ^ Staff (7 September 2020). “China’s Sinovac coronavirus vaccine candidate appears safe, slightly weaker in elderly”. Reuters. Archived from the original on 7 October 2020. Retrieved 6 October 2020.
- ^ WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection (2020). “A minimal common outcome measure set for COVID-19 clinical research”. The Lancet Infectious Diseases. 20 (8): e192–e197. doi:10.1016/S1473-3099(20)30483-7. PMC 7292605. PMID 32539990.
- ^ Mariz, Fabiana; Caires, Luiza (7 January 2021). “Eficaz em prevenir doença grave e morte por covid, Coronavac deve ter impacto em frear pandemia”. Jornal da USP (in Portuguese). Retrieved 7 January 2021.
- ^ Jump up to:a b c Pearson, Samantha; Magalhaes, Luciana (12 January 2021). “Chinese Covid-19 Vaccine Is Far Less Effective Than Initially Touted in Brazil”. The Wall Street Journal. Retrieved 12 January2021.
- ^ Gielow, Igor; Lopes Batista, Everton; Bottallo, Ana (12 January 2021). “Coronavac tem eficácia geral de 50,38% no estudo feito pelo Butantan”. Folha de S. Paulo (in Portuguese). Retrieved 12 January 2021.
- ^ Jump up to:a b c d “Sinovac’s Covid Shot Proves 78% Effective in Brazil Trial”. Bloomberg L.P. 7 January 2021. Retrieved 7 January 2021.
- ^ Kucukgocmen, Ali (3 March 2021). “Turkish study revises down Sinovac COVID-19 vaccine efficacy to 83.5%”. Reuters. Retrieved 7 March 2021.
- ^ “China’s Sinovac vaccine efficacy 83.5 percent: Turkish researchers – Turkey News”. Hürriyet Daily News. Retrieved 7 March 2021.
- ^ Jump up to:a b hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 11 January 2021.
- ^ “Why did the efficacy of China’s top vaccine drop from 78% to 50%?”. Fortune. Retrieved 14 January 2021.
- ^ “Coronavac tem eficácia de 78% contra a Covid-19 em estudo no Brasil”. Folha de S.Paulo (in Portuguese). 7 January 2021. Retrieved 7 January 2021.
- ^ Jump up to:a b “Estudos mostram eficácia da CoronaVac contra três variantes do vírus”. Agência Brasil (in Portuguese). 10 March 2021. Retrieved 18 March 2021.
- ^ Zhang Y, Zeng G, Pan H, Li C, Hu Y, Chu K, et al. (November 2020). “Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial”. The Lancet. Infectious Diseases. 21 (2): 181–192. doi:10.1016/S1473-3099(20)30843-4. PMC 7832443. PMID 33217362. S2CID 227099817. Archived from the original on 16 December 2020. Retrieved 18 November 2020.
- ^ Clinical trial number NCT04551547 for “A Randomized, Double-Blinded, Placebo-Controlled, Phase I/II Clinical Trial, to Evaluate the Safety and Immunogenicity of the SARS-CoV-2 Inactivated Vaccine (Vero Cell) in Healthy Population Aged 3–17 Years” at ClinicalTrials.gov
- ^ Wu, Zhiwei; Hu, Yaling; Xu, Miao; Chen, Zhen; Yang, Wanqi; Jiang, Zhiwei; Li, Minjie; Jin, Hui; Cui, Guoliang; Chen, Panpan; Wang, Lei (3 February 2021). “Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine (CoronaVac) in healthy adults aged 60 years and older: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial”. The Lancet Infectious Diseases. 0. doi:10.1016/S1473-3099(20)30987-7. ISSN 1473-3099. PMC 7906628. PMID 33548194.
- ^ Savarese M (21 July 2020). “New coronavirus vaccine trials start in Brazil”. Associated Press. Archived from the original on 13 August 2020. Retrieved 15 August 2020.
- ^ Palacios R, Patiño EG, de Oliveira Piorelli R, Conde MT, Batista AP, Zeng G, et al. (October 2020). “Double-Blind, Randomized, Placebo-Controlled Phase III Clinical Trial to Evaluate the Efficacy and Safety of treating Healthcare Professionals with the Adsorbed COVID-19 (Inactivated) Vaccine Manufactured by Sinovac – PROFISCOV: A structured summary of a study protocol for a randomised controlled trial”. Trials. 21 (1): 853. doi:10.1186/s13063-020-04775-4. PMC 7558252. PMID 33059771. Archived from the original on 16 December 2020. Retrieved 28 October 2020.
- ^ “World’s vaccine testing ground deems Chinese COVID candidate ‘the safest, most promising'”. Fortune. Archived from the original on 31 October 2020. Retrieved 9 November 2020.
- ^ “Doria says it guarantees purchase of 100 million doses of CoronaVac…” AlKhaleej Today (in Arabic). 29 October 2020. Archived from the original on 1 November 2020. Retrieved 30 October 2020.
- ^ “Brazil Clears Sinovac Trial to Resume Two Days After Halting It”. Bloomberg L.P. 11 November 2020. Archived from the original on 11 November 2020. Retrieved 11 November 2020.
- ^ “Brazil’s health regulator says China’s Sinovac can resume Covid-19 vaccine trial after suspension”. CNBC. 12 November 2020. Archived from the original on 13 November 2020. Retrieved 17 November 2020.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Government of Chile. 4 August 2020. Archived from the original on 11 October 2020. Retrieved 28 August 2020.
- ^ Health Institutes of Turkey (8 October 2020). “Randomized, Double-Blind, Placebo-Controlled Phase III Clinical Trial For Evaluation of Efficacy and Safety of SARS-CoV-2 Vaccine (Vero Cell), Inactivated”. ClinicalTrials. Archived from the original on 20 October 2020. Retrieved 21 October 2020.
- ^ “Chinese COVID-19 vaccine to be free, 1st doses to be delivered soon: Turkey’s health minister”. Daily Sabah. 23 November 2020. Archived from the original on 23 November 2020. Retrieved 23 November 2020.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. Archived from the original on 30 September 2020. Retrieved 22 September 2020.
- ^ antaranews.com. “Phase 3 Sinovac clinical trial running smoothly: research team”. Antara News. Archived from the original on 3 November 2020. Retrieved 3 November 2020.
- ^ “Virus vaccine waiting on Saudi ‘green light'”. Arab News. 31 October 2020. Archived from the original on 16 December 2020. Retrieved 1 November 2020.
- ^ hermesauto (12 October 2020). “Indonesia aims to start administering coronavirus vaccines in early November”. The Straits Times. Archived from the original on 13 October 2020. Retrieved 12 October 2020.
- ^ “Sao Paulo starts building production plant for China’s Sinovac vaccine – governor”. Financial Post. Archived from the original on 29 November 2020. Retrieved 9 November 2020.
- ^ Mano A, Simões (10 December 2020). “Chinese vaccine draws demand across Latin America, say Brazilian officials”. Reuters. Archived from the original on 10 December 2020. Retrieved 10 December 2020.
- ^ Jump up to:a b Choong, Jerry (26 January 2021). “Health Ministry: Malaysia secures 18.4 million doses of Russian, Chinese Covid-19 vaccines”. The Malay Mail. Retrieved 26 January 2021.
- ^ Mourad, Mahmoud (22 March 2021). “Egypt aims for deal to produce Sinovac COVID-19 vaccines”. Reuters. Retrieved 22 March 2021.
- ^ Jump up to:a b c Liu R (6 February 2021). “China approves Sinovac Biotech COVID-19 vaccine for general public use”. Reuters. Retrieved 7 February 2021.
- ^ Sipalan, Joseph; Donovan, Kirsten (3 March 2021). “Malaysia approves Sinovac, AstraZeneca COVID-19 vaccines for use”. Reuters. Retrieved 7 March 2021.
- ^ Aliyev J. “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency. Retrieved 7 February 2021.
- ^ “Bolívia autoriza uso de vacinas Sputnik V e CoronaVac contra covid-19”. noticias.uol.com.br (in Portuguese). Retrieved 6 January 2021.
- ^ McGeever J, Fonseca P (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Chanritheara, Torn. “Cambodia Approves AstraZeneca and Sinovac Vaccines for COVID-19 Emergency Use”. Cambodianess. Retrieved 12 February 2021.
- ^ “Chile aprueba el uso de emergencia de la vacuna china de Sinovac contra covid-19”. France 24. 20 January 2021. Retrieved 30 January 2021.
- ^ Aliyev J. “Colombia approves emergency use of CoronaVac vaccine”. Anadolu Agency. Retrieved 7 February 2021.
- ^ “Anticovid vaccines run out as Dominican Republic awaits arrival of more doses”. DominicanToday. Retrieved 10 March 2021.
- ^ “Ecuador signs agreement with Sinovac for 2 million COVID-19 vaccine: minister”. National Post. Retrieved 26 February 2021.
- ^ “Use of Sinovac vaccine authorised”. Government of Hong Kong. 18 February 2021. Retrieved 19 February 2021.
- ^ Soeriaatmadja W (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 11 January 2021.
- ^ “BPOM Grants Emergency Use Authorization for Sinovac Vaccine”. Tempo. 11 January 2021. Retrieved 11 January 2021.
- ^ Barrera, Adriana (11 February 2021). “Mexico approves China’s CanSino and Sinovac COVID-19 vaccines”. Reuters. Retrieved 11 February 2021.
- ^ “CoronaVac, vacuna de alta eficacia”. Ministerio de Salud Publica Y Bienestar Social.
- ^ “Philippines approves Sinovac’s COVID-19 vaccine for emergency use”. Reuters. 22 February 2021.
- ^ Thepgumpanat, Panarat (22 February 2021). “Thailand allows emergency use of Sinovac’s COVID-19 vaccine”. Reuters. Retrieved 23 February 2021.
- ^ “Tunisia approva vaccino cinese Sinovac” (in Italian). Agenzia Nazionale Stampa Associata (in Italian). 5 March 2021. Retrieved 7 March 2021.
- ^ “Turkey to begin COVID-19 vaccine jabs by this weekend”. Anadolu. 11 January 2021. Retrieved 11 January 2021.
- ^ Zinets, Natalia (9 March 2021). “Ukraine approves China’s Sinovac COVID-19 vaccine”. Reuters. Retrieved 10 March 2021.
- ^ “Covid-19: Zimbabwe authorises Sputnik V, Sinovac vaccines for emergency use”. news24.com. 9 March 2021.
- ^ “Regulation and Prequalification”. World Health Organization. Retrieved 12 March 2021.
- ^ Simoes E (30 September 2020). “Brazil’s Sao Paulo signs agreement with Sinovac for COVID vaccine doses”. Reuters. Archived from the original on 1 October 2020. Retrieved 1 October 2020.
- ^ Fonseca I (30 October 2020). “CoronaVac May Be Four Times More Costly Than Flu Vaccine”. The Rio Times. Archived from the original on 3 November 2020. Retrieved 30 October 2020.
- ^ “Em meio a críticas por atrasos, Pazuello diz que Brasil está preparado para iniciar vacinação em janeiro”. Folha de S.Paulo(in Portuguese). 6 January 2021. Retrieved 7 January 2021.
- ^ Rochabrun, Marcelo. “Brazil health ministry says plans to order 30 million more Coronavac doses | The Chronicle Herald”. http://www.thechronicleherald.ca. Retrieved 26 February 2021.
- ^ “Bolívia autoriza uso de vacinas Sputnik V e CoronaVac contra covid-19”. noticias.uol.com.br (in Portuguese). Retrieved 7 January 2021.
- ^ “Government meets with Sinovac for first COVID-19 vaccine clinical trial in Chile”. Government of Chile. 13 October 2020. Archived from the original on 17 October 2020. Retrieved 8 November 2020.
- ^ Presse, AFP-Agence France. “Chile Approves Chinese Coronavirus Vaccine”. barrons.com. Retrieved 21 January 2021.
- ^ “Fifth shipment with over two million Sinovac vaccines arrives to Chile”. Chile Reports. Retrieved 12 March 2021.
- ^ “Colombia extends health state of emergency, seeks more Sinovac vaccines”. Reuters. Retrieved 26 February 2021.
- ^ MENAFN. “Colombia declares emergency use of Sinovac vaccines”. menafn.com. Retrieved 4 February 2021.
- ^ “Ecuador signs agreement with Sinovac for 2 million COVID-19 vaccine: minister”. nationalpost. Retrieved 26 February 2021.
- ^ Jump up to:a b Valencia, Alexandra (7 March 2021). “Chile donates 40,000 doses of Sinovac vaccine to Ecuador and Paraguay”. Reuters. Retrieved 7 March 2021.
- ^ “CoronaVac, vacuna de alta eficacia”. Ministerio de Salud Publica Y Bienestar Social.
- ^ “Uruguay will receive first batches of Pfizer and Sinovac vaccines late February or early March: US$ 120 million investment”. MercoPress. Retrieved 24 January 2021.
- ^ “Albania gets 192,000 doses of Chinese Sinovac vaccine”. CNA. Retrieved 25 March 2021.
- ^ “Turkey signs 50 million dose COVID-19 vaccine deal, health minister says”. Reuters. 25 November 2020. Archived from the original on 1 December 2020. Retrieved 27 November 2020.
- ^ “Turkey grants emergency authorization to Sinovac’s CoronaVac: Anadolu”. Reuters. 13 January 2021. Retrieved 15 January 2021.
- ^ “Turkish president gets COVID-19 vaccine”. Anadolu Agency. 14 January 2021. Retrieved 20 January 2021.
- ^ SABAH, DAILY (12 March 2021). “Few virus infections reported among vaccinated people in Turkey”. Daily Sabah. Retrieved 12 March 2021.
- ^ “Ukraine signs up for China’s Sinovac vaccine, with doses expected soon”. Reuters. 30 December 2020. Retrieved 30 December 2020.
- ^ Zinets, Natalia (9 March 2021). “Ukraine approves China’s Sinovac COVID-19 vaccine”. Reuters. Retrieved 9 March 2021.
- ^ Aliyev, Jeyhun (19 January 2021). “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency.
- ^ “Cambodian PM okays two more Covid-19 vaccines – Sinovac and AstraZeneca – for emergency use | The Star”. http://www.thestar.com.my. Retrieved 19 March 2021.
- ^ “Have no fear about shortage of vaccines, 1.5 million doses of Sinovac arriving on March 26”. Khmer Times. 19 March 2021. Retrieved 19 March 2021.
- ^ “Sinovac’s coronavirus vaccine candidate approved for emergency use in China – source”. Reuters. 29 August 2020. Archived from the original on 31 August 2020. Retrieved 30 August 2020.
- ^ “Government announces latest development of COVID-19 vaccine procurement” Archived 11 December 2020 at the Wayback Machine (Hong Kong Government Press Releases, 12 December 2020)
- ^ “Hong Kong kicks off COVID-19 vaccinations with Sinovac jab”. AP NEWS. 26 February 2021. Retrieved 7 March 2021.
- ^ “Indonesia books 50 million coronavirus vaccine doses from Sinovac”. Reuters. 21 August 2020. Archived from the original on 29 August 2020. Retrieved 21 August 2020.
- ^ “Sinovac vaccine has no critical side effects, BPOM says”. The Jakarta Post. Retrieved 21 December 2020.
- ^ Arkyasa, Mahinda (25 March 2021). “16 Million Sinovac Vaccines Material Arrives in Indonesia”. Tempo. Retrieved 25 March 2021.
- ^ “Malaysia’s NPRA Approves AstraZeneca, Sinovac Covid-19 Vaccines”. CodeBlue. 2 March 2021. Retrieved 2 March 2021.
- ^ Babulal, Veena (18 March 2021). “KJ gets first dose of Sinovac vaccine [NSTTV] | New Straits Times”. NST Online. Retrieved 19 March 2021.
- ^ “Duque says deal sealed for 25M doses of Sinovac COVID-19 vaccine”. GMA News Online. Retrieved 10 January 2021.
- ^ “Philippines receives COVID-19 vaccine after delays”. AP NEWS. 28 February 2021. Retrieved 28 February 2021.
- ^ Chen F (24 December 2020). “Brazil joins ranks of Chinese vaccine backers”. Asia Times Online. Retrieved 30 December2020.
- ^ “Singapore receives China’s Sinovac vaccine ahead of approval”. The Star. 25 February 2021. Retrieved 26 February2021.
- ^ “Thailand to get 2 million shots of China’s Sinovac”. Bangkok Post. Bangkok Post Public Company. Retrieved 4 January 2021.
- ^ “Thailand gives emergency use authorisation for Sinovac’s COVID-19 vaccine – official”. Reuters. 22 February 2021. Retrieved 23 February 2021.
- ^ Limited, Bangkok Post Public Company. “Thailand in talks to buy another 5m Sinovac shots”. Bangkok Post. Retrieved 20 March2021.
- ^ “Mexico approves China’s CanSino and Sinovac COVID-19 vaccines”. Reuters. 11 February 2021. Retrieved 11 February2021.
- ^ Jorgic, Drazen (10 March 2021). “Mexico leans on China after Biden rules out vaccines sharing in short term”. Reuters. Retrieved 10 March 2021.
- ^ Exteriores, Secretaría de Relaciones. “The Mexican Government receives 200,000 Sinovac COVID-19 vaccines”. gob.mx (in Spanish). Retrieved 7 March 2021.
- ^ “Lutte contre la Covid-19 : 203.000 doses de vaccins s dont 100.000 offertes par la Chine au Bénin”. Concentrées d’informations sur le Bénin et le monde à votre service depuis 2009(in French). 23 March 2021. Retrieved 25 March 2021.
- ^ Winning, Alexander. “South Africa’s drugs regulator to start assessing Sinovac COVID-19 vaccine”. U.S. Retrieved 12 March2021.
- ^ Nijini, Felix (18 March 2021). “Sinovac May Supply South Africa With 5 Million Vaccines: Report – BNN Bloomberg”. BNN. Retrieved 19 March 2021.
- ^ “Covid: Tunisia approva vaccino cinese Sinovac”. Agenzia Nazionale Stampa Associata (in Italian). 5 March 2021. Retrieved 7 March 2021.
- ^ Dzirutwe, MacDonald (10 March 2021). “Zimbabwe authorises Sputnik V, Sinovac coronavirus vaccines for emergency use”. Reuters. Retrieved 13 March 2021.
- ^ “China to donate Sinovac Vaccine to Fiji”. Fiji Broadcasting Corporation. Retrieved 17 March 2021.
- ^ Jump up to:a b Phillips, Tom (10 November 2020). “Jair Bolsonaro claims ‘victory’ after suspension of Chinese vaccine trial”. The Guardian. Retrieved 18 January 2021.
- ^ Baptista, Eduardo (11 December 2020). “China-made coronavirus vaccine at heart of political showdown in Brazil”. South China Morning Post. Retrieved 18 January 2021.
- ^ Carvalho, Daniel (14 January 2021). “‘Is 50% Good?’, Asks Bolsonaro, Mocking Coronavac’s Effectiveness”. Folha de S.Paulo. Retrieved 18 January 2021.
- ^ Pearson, Samantha; Magalhaes, Luciana (10 November 2020). “Brazil’s Medical Experts Worry Politics Is Hampering Covid-19 Vaccine Progress”. The Wall Street Journal. Retrieved 18 January 2021.
- ^ “Covid: 70% dos brasileiros não fazem questão de escolher vacina” [Covid: 70% of Brazilians do not make a point of choosing vaccine]. R7.com (in Portuguese). 3 March 2021. Retrieved 9 March2021.
- ^ Fonseca P. “Brazil institute says CoronaVac efficacy above 50%, but delays full results”. Reuters. Retrieved 25 December 2020.
- ^ Hong, Jinshan (12 January 2021). “How Effective Is China’s Sinovac Vaccine? Data Confuse Experts”. Bloomberg News. Retrieved 12 January 2021.
External links
- Clinical Research Protocol for CoronaVac Phase III Trials in Brazil
- Clinical Research Protocol for CoronaVac Phase III Trials in Chile
- “How the Sinovac Covid-19 Vaccine Works”. The New York Times.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Routes of administration | Intramuscular injection |
| ATC code | None |
| Legal status | |
| Legal status | Emergency authorization for use in China, Indonesia, Brazil and Turkey |
| Identifiers | |
| DrugBank | DB15806 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
Sinovac Biotech Ltd. (Chinese: 北京科兴生物制品有限公司, Nasdaq: SVA) is a Chinese biopharmaceutical company that focuses on the research, development, manufacture and commercialization of vaccines that protect against human infectious diseases. The company is based in Haidian District, Beijing.[2] The company is listed on the NASDAQ but the exchange halted Sinovac’s trading in February 2019 due to a proxy fight.[3][4]
Vaccines
Sinovac’s commercialized vaccines include Healive (hepatitis A), Bilive (combined hepatitis A and B), Anflu (influenza), Panflu (H5N1) and PANFLU.1 (H1N1). Sinovac is currently developing a Universal Pandemic Influenza vaccine and a Japanese encephalitis vaccine.[5][better source needed]
Sinovac is also developing vaccines for enterovirus 71 and human rabies. Its wholly owned subsidiary, Tangshan Yian, is conducting field trials for independently developed inactivated animal rabies vaccines.[citation needed]
COVID-19 vaccine development
Main article: CoronaVac
CoronaVac is an inactivated virus COVID-19 vaccine developed by Sinovac.[6] It has been in Phase III clinical trials in Brazil,[7] Chile,[8] Indonesia,[9] Malaysia,[10] Philippines,[11] and Turkey.[12]
It relies on traditional technology similar to BBIBP-CorV and BBV152, other inactivated-virus COVID-19 vaccines in Phase III trials.[13] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[14]
Brazil announced results on January 13, 2021 showing 50.4% effective at preventing symptomatic infections, 78% effective in preventing mild cases needing treatment, and 100% effective in preventing severe cases.[15] Final Phase III results from Turkey announced on 3 March 2021 showed an efficacy of 83.5%.[16] Interim results in Indonesia were announced on 11 January 2021 with an efficacy of 65.3%.[17]
CoronaVac is being used in vaccination campaigns by certain countries in Asia,[18][19][20] South America,[21][22] and Europe.[23] In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June 2021.[24] As of 27 February 36 million doses had been administered in total.[25]
See also
References
- ^ “China’s Vaccine Front-Runner Aims to Beat Covid the Old-Fashioned Way”. Bloomberg. 24 August 2020.
- ^ “Home (English)”. Sinovac. Retrieved 2021-03-06.
Add: No. 39 Shangdi Xi Road, Haidian District, Beijing, P.R.C. 100085
– Chinese address: “地址:中国· 北京 海淀区上地西路39号北大生物城(100085)” - ^ Dou, Eva (December 4, 2020). “As China nears a coronavirus vaccine, bribery cloud hangs over drugmaker Sinovac”. The Washington Post. ISSN 0190-8286. Archived from the original on December 4, 2020. Retrieved 2020-12-06.
- ^ Levine, Matt (May 22, 2020). “A Vaccine With a Poison Pill”. Bloomberg News. Archived from the original on June 21, 2020. Retrieved December 6, 2020.
- ^ Google Finance, url=https://www.google.com/finance?q=Sinovac
- ^ Nidhi Parekh (22 July 2020). “CoronaVac: A COVID-19 Vaccine Made From Inactivated SARS-CoV-2 Virus”. Retrieved 25 July2020.
- ^ “New coronavirus vaccine trials start in Brazil”. AP News. 21 July 2020. Retrieved 2020-10-07.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Chile Reports. 4 August 2020. Retrieved 2020-10-07.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. 30 August 2020. Retrieved 2020-10-07.
- ^ “Malaysia Receives China’s Sinovac Vaccine For Regulatory Testing”. Bloomberg.com. 2021-02-27. Retrieved 2021-03-02.
- ^ “DOH eyes 5 hospitals for Sinovac vaccine Phase 3 clinical trial”. PTV News. 16 September 2020. Retrieved 2020-10-07.
- ^ “Turkey begins phase three trials of Chinese Covid-19 vaccine”. TRT World News. 1 September 2020. Retrieved 2020-10-07.
- ^ Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 2021-02-12.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 2020-11-01. Retrieved 2021-02-12.
- ^ “Sinovac: Brazil results show Chinese vaccine 50.4% effective”. BBC News. 2021-01-13. Retrieved 2021-02-12.
- ^ AGENCIES, DAILY SABAH WITH (25 December 2020). “Turkey set to receive ‘effective’ COVID-19 vaccine amid calls for inoculation”. Daily Sabah. Retrieved 12 February 2021.
- ^ hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 12 February 2021.
- ^ TARIGAN, EDNA; MILKO, VICTORIA (13 January 2021). “Indonesia starts mass COVID vaccinations over vast territory”. Associated Press. Retrieved 15 January 2021.
- ^ Aliyev, Jeyhun (19 January 2021). “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency.
- ^ “China approves Sinovac vaccines for general public use”. South China Morning Post. 6 February 2021. Retrieved 6 February2021.
- ^ Fonseca, Jamie McGeever, Pedro (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Miranda, Natalia A. Ramos (28 January 2021). “Chile receives two million-dose first delivery of Sinovac COVID-19 vaccine”. Reuters. Retrieved 30 January 2021.
- ^ “Turkey aims to vaccinate 60 percent of population: Minister – Turkey News”. Hürriyet Daily News. Retrieved 12 February 2021.
- ^ Liu, Roxanne (2021-03-03). “Sinovac eyes two billion doses in annual capacity of virus vaccine by June”. Reuters. Retrieved 2021-03-03.
- ^ “Malaysia receives first batch of Sinovac Covid-19 vaccine today”. Bernama. 27 February 2021. Retrieved 27 February 2021– via The Malay Mail.
External links
- Official website
- Business data for Sinovac Biotech:
| Type | Public |
|---|---|
| Traded as | Nasdaq: SVA (American Depository Receipts) |
| Founded | 1999; 22 years ago |
| Founder | Yin Weidong[1] |
| Headquarters | Beijing,China |
| Website | http://www.sinovac.com/ |
| Sinovac Biotech | |
|---|---|
| Simplified Chinese | 北京科兴生物制品有限公司 |
| Traditional Chinese | 北京科興生物製品有限公司 |
| hideTranscriptionsStandard MandarinHanyu PinyinBěijīng Kē Xìng Shēngwù Zhìpǐn Yǒuxiàn Gōngsī |
/////////Sinovac COVID-19 vaccine, CoronaVac, corona virus, covid 19, vaccine, china, Sinovac Biotech, PiCoVacc
#Sinovac COVID-19 vaccine, #CoronaVac, #corona virus, #covid 19, #vaccine, #china, #Sinovac Biotech, #PiCoVacc
BBIBP-CorV, Sinopharm COVID-19 vaccine


BBIBP-CorV, Sinopharm COVID-19 vaccine
- Inactivated novel coronavirus (2019-CoV) vaccine (Vero cells)
- Purified inactivated SARS-CoV-2 Vaccine
ref Lancet Infectious Diseases (2021), 21(1), 39-51.
BBIBP-CorV, also known as the Sinopharm COVID-19 vaccine,[1] is one of two inactivated virus COVID-19 vaccines developed by Sinopharm. In late December 2020, it was in Phase III trials in Argentina, Bahrain, Egypt, Morocco, Pakistan, Peru, and the United Arab Emirates (UAE) with over 60,000 participants.[2]
On December 9, the UAE announced interim results from Phase III trials showing BBIBP-CorV had a 86% efficacy against COVID-19 infection.[3] In late December, Sinopharm announced that its internal analysis indicated a 79% efficacy.[4] While mRNA vaccines like the Pfizer–BioNTech COVID-19 vaccine and mRNA-1273 showed higher efficacy of +90%, those present distribution challenges for some nations as they require deep-freeze facilities and trucks. BIBP-CorV could be transported and stored at normal refrigerated temperatures.[5]
BBIBP-CorV shares similar technology with CoronaVac and BBV152, other inactivated virus vaccines for COVID-19 being developed in Phase III trials.[6][7]
BBIBP-CorV is being used in vaccination campaigns by certain countries in Asia,[8][9][10] Africa,[11][12][13] South America,[14][15] and Europe.[16][17][18] Sinopharm expects to produce one billion doses of BBIBP-CorV in 2021.[19] By February 21, Sinopharm said more than 43 million doses of the vaccine had been administered in total.[20]
BBIBP-CorV vaccine contains a SARS-CoV-2 strain inactivated inside Vero Cells. Investigation shows this vaccine induces neutralizing antibodies in several mammalian species while also showing protective efficacy with SARS-CoV-2 challenge in rhesus macaques2. As of August 2020, this vaccine is being tested for prophylaxis against COVID-19 in human clinical trials.

A vaccination certificate of BBIBP-CorV (Beijing Institute of Biological Products, Sinopharm).
Clinical research
Main article: COVID-19 vaccine
Phases I and II
In April 2020, China approved clinical trials for a candidate COVID-19 vaccine developed by Sinopharm‘s Beijing Institute of Biological Products[21] and the Wuhan Institute of Biological Products.[22] Both vaccines are chemically-inactivated whole virus vaccines for COVID-19.
On October 15, the Beijing Institute of Biological Products published results of its Phase I (192 adults) and Phase II (448 adults) clinical studies for the BBIBP-CorV vaccine, showing BBIBP-CorV to be safe and well-tolerated at all tested doses in two age groups. Antibodies were elicited against SARS-CoV-2 in all vaccine recipients on day 42. These trials included individuals older than 60.[21]
On August 13, the Wuhan Institute of Biological Products published interim results of its Phase I (96 adults) and Phase II (224 adults) clinical studies. The report noted the inactivated COVID-19 vaccine had a low rate of adverse reactions and demonstrated immunogenicity, but longer-term assessment of safety and efficacy would require Phase III trials.[22]
BIBP-CorV may have characteristics favorable for vaccinating people in the developing world. While mRNA vaccines, such as the Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine showed higher efficacy of +90%, mRNA vaccines present distribution challenges for some nations, as some may require deep-freeze facilities and trucks. By contrast, BIBP-CorV can be transported and stored at normal refrigeration temperatures.[23] While Pfizer and Moderna are among developers relying on novel mRNA technology, manufacturers have decades of experience with the inactivated virus technology Sinopharm is using.[23]
Phase III
Africa and Asia
On July 16, Sinopharm began conducting a Phase III vaccine trial of 31,000 volunteers in the UAE in collaboration with G42 Healthcare, an Abu Dhabi-based company.[24] By August, all volunteers had received their first dose and were to receive the second dose within the next few weeks.[25] On December 9, UAE’s Ministry of Health and Prevention announced the official registration of BBICP-CorV, after an interim analysis of the Phase III trial showed BBIBP-CorV to have a 86% efficacy against COVID-19 infection.[26] The vaccine had a 99% sero-conversion rate of neutralizing antibodies and 100% effectiveness in preventing moderate and severe cases of the disease.[27]
On September 2, Sinopharm began a Phase III trial in Casablanca and Rabat on 600 people.[28][29] In September, Egypt opened registration for a Phase III trial to last one year and enroll 6,000 people.[30]
In August 2020, Sinopharm began a Phase III clinical trial in Bahrain on 6,000 citizens and resident volunteers.[31][32] In a November update, 7,700 people had volunteered in the trials.[33] Also in late August, Sinopharm began a Phase III clinical trial in Jordan on 500 volunteers at Prince Hamzah Hospital.[34][35]
In Pakistan, Sinopharm began working with the University of Karachi on a trial with 3,000 volunteers.[36]
South America
On September 10, Sinopharm began a Phase III trial in Peru with the long-term goal of vaccinating a total of 6,000 people between the ages of 18 and 75.[37] In October, the trials were expanded to include an additional 6,000 volunteers.[38] On January 26, a volunteer in the placebo group of the vaccine trials had died.[39]
On September 16, Argentina began a Phase III trial with 3,000 volunteers.[40]
Manufacturing
Sinopharm’s Chariman Yang Xioyun has said the company could produce one billion doses in 2021.[19]
In October, Dubai’s G42 Healthcare reached manufacturing agreements to provide UAE and other regional states with BBIBP-CorV, with the UAE producing 75 to 100 million doses in 2021.[41]
In December, Egypt announced an agreement between Sinopharm and Egyptian Holding Company for Biological Products & Vaccines (VACSERA) for the vaccine to be manufactured locally,[42] which would also be exported to other African countries.[43]
In December, AP reported Morocco plans to produce BBIBP-CorV locally.[44]
In March, Serbia announced plans to produce 24 million doses of BBIBP-CorV annually starting in October. The production volume would be sufficient to meet the needs of Serbia and all of its neighbors, deputy prime minister Branislav Nedimović noted.[45]
In March, Belarus was looking to produce BBIBP-CorV locally.[18]
Marketing and Distribution
| show Full authorizationshow Emergency authorizationshow Received donated doses Eligible COVAX recipient (assessment in progress)[86] |
On February 21, 2021 Sinopharm said more than 43 million doses of BBIBP-CorV had been administered so far, including more than 34 million administered in China and the rest internationally.[20]
Asia
In February, Afghanistan was pledged 400,000 doses of BBIBP-CorV by China.[82]
In November 3, 2020 Bahrain granted emergency use authorization of BBIBP-CorV for frontline workers.[33] In December, Bahrain approved Sinopharm’s vaccine, citing data from Phase III clinical trials that showed an 86% efficacy rate.[87]
In February, Brunei received the first batch of Sinopharm vaccines donated by China.[84]
In January, Cambodia said China would provide a million doses.[88] Cambodia granted emergency use authorization on February 4[89] and started the vaccination campaign on February 10 with the first 600,000 doses.[90]
In China, Sinopharm obtained an EUA in July.[91] In October, it began offering the vaccine for free to students going abroad for higher studies.[92] On December 30, China‘s National Medical Products Administration approved BBIBP-CorV for general use.[93][8] In February, Macau received the first 100,000 doses of 400,000 doses.[94]
In October, Indonesia reached an agreement with Sinopharm to deliver 15 million dual-dose vaccines in 2020.[95]
In February, Iran approved emergency use of BBIBP-CorV,[96] and received the first batch of 250,000 doses on February 28.[97]
In January, Iraq approved BBIBP-CorV for emergency use[98] and has signed agreements for 2 million doses. The first doses arrived on March 2.[99]
In January, Jordan approved BBIBP-CorV for emergency use[100] and started its vaccination campaign on January 13.[101]
In March, Kyrgyzstan received a donation of 150,000 doses of the vaccine.[102]
In January, Laos began vaccinating medical workers at hospitals in Vientiane [103] and received another 300,000 doses in early February.[104]
In March, Lebanon received a donation of 50,000 doses at its request,[105] for which it granted emergency use authorization on March 2.[106]
In March, Maldives granted emergency approval for use. At the time of approval, the country had received 18,000 doses and was awaiting 200,000 additional doses.[107]
In February, Mongolia received a donation of 300,000 doses.[108] On March 10, Governor of Ulaanbaatar D. Sumiyabazar and Deputy Prime Minister S. Amarsaikhan received the first doses of BBIBP-CorV.[109]
In February, Nepal approved the vaccine for emergency use, allowing a donation of 500,000 doses to enter the country.[110]
In December, Pakistan‘s purchased 1.2 million doses,[111] which was approved for emergency use on January 18,[112] and began a vaccination campaign on February 2.[10]
In March, Palestine said it would receive 100,000 doses donated by China.[113]
In March 19, Sri Lanka approved the vaccine for emergency use, allowing a donation of 600,000 doses by China to enter the country.[114]
On 14 September 2020, the United Arab Emirates approved the vaccine for front-line workers following successful interim Phase III trials.[24] In December, the country registered BBIBP-CorV after it reviewed the results of the interim analysis.[26] In March, a small number of people who have reduced immunity against diseases, have chronic illnesses, or belong to high-risk groups have been given a 3rd booster shot.[115]
Africa
In February, Algeria received a donation of 200,000 doses.[83]
Egypt plans to buy 40 million doses of Sinpharm’s vaccine[116] which was approved for regulatory use on January 3.[116] President Abdel Fattah el-Sisi announced a vaccination campaign starting 24 January.[11]
In February, Equatorial Guinea received a donation of 100,000 doses which arrived on February 10. The country began vaccinations on February 15.[56]
In March, Gabon received a donation of 100,000 doses which was the second vaccine approved for use in the country.[117]
Morocco placed orders for 41 million vaccine doses from Sinopharm and 25 million from AstraZeneca, for a total of 66 million doses.[118] Morocco granted emergency use approval on January 23,[119] and the first 500,000 doses arrived on January 27.[12]
In February, Mozambique received a donation of 200,000 doses[120] and planned to start vaccinations on March 8.[121]
In March, Namibia received a donation of 100,000 doses and announced the start of vaccinations in the Khomas and Erongo regions.[122]
In March, Niger received a donation of 400,000 doses with vaccinations to begin on March 27.[123]
In February, Senegal received 200,000 doses in Dakar[124] and began vaccinating health workers on February 22.[125]
In February, Sierra Leone received a donation of 200,000 doses.[126] It was approved for emergency use and vaccinations began on March 15.[127]
In January, Seychelles said it would begin administering vaccinations on January 10 with 50,000 doses it had received as a gift from the UAE.[128]
In March, Republic of the Congo received 100,000 doses with vaccinations prioritizing the medically vulnerable and those over 50.[129]
In February, Zimbabwe purchased 600,000 doses on top of 200,000 doses donated by China,[130] and started vaccinations on February 18.[13] Zimbabwe later purchased an additional 1.2 million doses.[131]
North America
In February, the Dominican Republic ordered 768,000 doses of BBIBP-CorV.[132]
In March, Dominica received 20,000 doses of BBIBP-CorV which it began using in its vaccination campaign on March 4.[133]
In March, Mexico announced it would order 12 million doses of BBIBP-CorV pending approval by its health regulator.[134]
South America
In February, Argentina authorized emergency use of BBIBP-CorV[135] ahead of the arrival of 904,000 doses on February 26.[136]
In February, Bolivia purchased 400,000 doses on top of 100,000 doses donated by China,[137] and started its vaccination campaign on February 26.[15]
In March, Guyana received a donation of 20,000 doses of BBIBP-CorV.[138] Vaccinations were to start on March 7.[139]
In January, Peru purchased 38 million doses of BBIBP-CorV.[140] Peru granted emergency approval for BBIBP-CorV on January 27[141] and started vaccinations on February 9 with the first 300,000 doses.[14]
In March, Venezuela granted approval for BBIBP-CorV to be used in the country.[142] The first 500,000 doses arrived on March 2.[143]
Europe
In February, Belarus received a donation of 100,000 doses[144] and began using the vaccine on March 15.[18]
In January, Hungary became first EU member to approve BBIBP-CorV, signing a deal for 5 million doses.[145] The first 550,000 doses arrived in Budapest on February 16[146] and vaccinations started on February 24.[17] Prime Minister Viktor Orbán was vaccinated with BBIBP-CorV on February 28.[147]
In March, Moldova received 2,000 doses donated by the UAE[148] which will be used to vaccinate doctors at the State University of Mediecne and Pharmacy starting on March 22.[149]
In March 3, Montenegro received a donation of 30,000 doses of BBIBP-CorV.[85]
In February, North Macedonia signed an agreement for 200,000 doses of BBIBP-CorV, with which they hoped to launch their vaccination program later that month.[150]
In January, Serbia received one million doses, making it the first country in Europe to receive BBIBP-CorV.[151] On January 19, Serbia approved the vaccine and Health Minister Zlatibor Lončar became the first person to receive a shot.[16]
Controversies
Lack of public data
Unlike Moderna‘s MRNA-1273, Oxford–AstraZeneca‘s AZD1222, and Johnson & Johnson‘s Ad26.COV2.S, there is little public information about the Chinese vaccine’s safety or efficacy.[152] The UAE said it had reviewed Sinopharm’s interim data analysis which showed the vaccine was 100% effective to prevent moderate and severe instances of COVID-19, but did not say whether it had independently analyzed the case data in its review. It was unclear how Sinopharm drew conclusions, since the UAE announcement of the approval for BBIBP-CorV noticeably lacked details such as the number of COVID-19 cases in the placebo or active group or the volunteers ages.[153]
As of December 30, 2020, no detailed efficacy data of the vaccine has been released to the public. A Sinopharm executive said detailed data would be released later and published in scientific journals in China and internationally.[8]
Sinopharm president Wu Yonglin said the trial results exceeded the WHO’s requirements, but a director at a large pharmaceutical company in Shanghai expressed skepticism over the trials and the expectation that drug regulators in Bahrain and the UAE would not hold the same standard as the U.S. Food and Drug Administration.[154]
Unauthorized use in Asia
On December 30, Philippine Defense Secretary Delfin Lorenzana said in an interview that at least one minister and president Rodrigo Duterte‘s bodyguards were provided BBIBP-CorV which were “smuggled” but that he felt what happened was “justified”. Brigadier General Jesus Durante, head of the Presidential Security Guard (PSG), said he felt compelled and “took the risk” to have some of his men vaccinated because they provide close-in security to Duterte, who at 75 is highly vulnerable to COVID-19.[155] Ingming Aberia, an author at The Manila Times commented that FDA director-general Enrique Domingo had reason to believe Sinopharm may cause harm to the consuming public given that no COVID-19 vaccine license was issued, but out of “self-preservation”, he would not initiate charges against PSG.[156]
On January 1, Mainichi Shimbun reported that 18 wealthy people, including several owners of leading Japanese companies, have been vaccinated with Sinopharm vaccines since November 2020. The vaccines were brought in by a Chinese consultant close to a senior member of the Chinese Communist Party.[157] The Chinese embassy in Japan later expressed its dissatisfaction at the unverified claims by Japanese news media.[158]
References
- ^ https://www.nytimes.com/interactive/2020/health/sinopharm-covid-19-vaccine.html
- ^ Reuters Staff (2020-11-19). “China Sinopharm’s coronavirus vaccine taken by about a million people in emergency use”. Reuters. Retrieved 2020-12-09.
- ^ “UAE: Ministry of Health announces 86 per cent vaccine efficacy”. gulfnews.com. Retrieved 2020-12-13.
- ^ Wee, Sui-Lee; Qin, Amy (2020-12-30). “China Approves Covid-19 Vaccine as It Moves to Inoculate Millions”. The New York Times. ISSN 0362-4331. Retrieved 2021-02-12.
- ^ “China State-Backed Covid Vaccine Has 86% Efficacy, UAE Says”. Bloomberg.com. 2020-12-09. Retrieved 2020-12-09.
- ^ Cohen J (December 2020). “China’s vaccine gambit”. Science. 370 (6522): 1263–1267. Bibcode:2020Sci…370.1263C. doi:10.1126/science.370.6522.1263. PMID 33303601.
- ^ Tan Y (16 December 2020). “Covid: What do we know about China’s coronavirus vaccines?”. BBC News. Retrieved 18 December 2020.
- ^ Jump up to:a b c Liu R (2020-12-31). “China gives its first COVID-19 vaccine approval to Sinopharm”. Reuters. Retrieved 2020-12-31.
- ^ Turak, Natasha (2021-01-18). “The UAE is on track to have half its population vaccinated by the end of March”. CNBC. Retrieved 2021-01-21.
- ^ Jump up to:a b Dawn.com (2021-02-02). “PM Imran kicks off Pakistan’s Covid-19 vaccination drive”. DAWN.COM. Retrieved 2021-02-03.
- ^ Jump up to:a b Reuters Staff (2021-01-24). “Sisi says Egypt to begin COVID-19 vaccinations on Sunday”. Reuters. Retrieved 2021-01-24.
- ^ Jump up to:a b Dumpis, Toms (2021-01-27). “Morocco Receives Half a Million Doses of Chinese Sinopharm Vaccine”. Morocco World News. Retrieved 2021-01-28.
- ^ Jump up to:a b “Zimbabwe starts administering China’s Sinopharm vaccines”. thestar.com. 2021-02-18. Retrieved 2021-02-20.
- ^ Jump up to:a b Aquino, Marco (2021-02-10). “‘The best shield’: Peru launches inoculation drive with Sinopharm vaccine”. Reuters. Retrieved 2021-02-10.
- ^ Jump up to:a b “Bolivia begins inoculation with Sinopharm jabs | The Star”. http://www.thestar.com.my. Retrieved 2021-02-28.
- ^ Jump up to:a b “Serbia Becomes First European Nation To Use China’s Sinopharm Vaccine”. RadioFreeEurope/RadioLiberty. Retrieved 2021-01-21.
- ^ Jump up to:a b “Hungary first EU nation to use China’s Sinopharm vaccine against COVID”. euronews. 2021-02-24. Retrieved 2021-02-26.
- ^ Jump up to:a b c d “Belarus begins COVID-19 vaccinations with Chinese shots”. eng.belta.by. 2021-03-15. Retrieved 2021-03-16.
- ^ Jump up to:a b “Which companies will likely produce the most COVID-19 vaccine in 2021?”. Pharmaceutical Processing World. 2021-02-05. Retrieved 2021-02-28.
- ^ Jump up to:a b hermesauto (2021-02-22). “More than 43 million doses of China’s Sinopharm Covid-19 vaccines used globally”. The Straits Times. Retrieved 2021-02-22.
- ^ Jump up to:a b Xia S, Zhang Y, Wang Y, Wang H, Yang Y, Gao GF, et al. (October 2020). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial”. The Lancet. Infectious Diseases. 21 (1): 39–51. doi:10.1016/s1473-3099(20)30831-8. PMC 7561304. PMID 33069281.
- ^ Jump up to:a b Xia S, Duan K, Zhang Y, Zhao D, Zhang H, Xie Z, et al. (September 2020). “Effect of an Inactivated Vaccine Against SARS-CoV-2 on Safety and Immunogenicity Outcomes: Interim Analysis of 2 Randomized Clinical Trials”. JAMA. 324 (10): 951–960. doi:10.1001/jama.2020.15543. PMC 7426884. PMID 32789505.
- ^ Jump up to:a b “China State-Backed Covid Vaccine Has 86% Efficacy, UAE Says”. Bloomberg.com. 2020-12-09. Retrieved 2020-12-09.
- ^ Jump up to:a b Maxwell C. “Coronavirus: UAE authorises emergency use of vaccine for frontline workers”. The National. Retrieved 14 September 2020.
- ^ “Coronavirus: 15,000 register as volunteers for Covid-19 vaccine trial in UAE”. The National. 13 August 2020. Retrieved 15 August2020.
- ^ Jump up to:a b Reuters Staff (2020-12-09). “UAE says Sinopharm vaccine has 86% efficacy against COVID-19”. Reuters. Retrieved 2020-12-09.
- ^ “UAE: Ministry of Health announces 86 per cent vaccine efficacy”. gulfnews.com. Retrieved 2020-12-09.
- ^ “Morocco orders R-Pharm Covid-19 vaccine | The North Africa Post”. northafricapost.com. Retrieved 2020-10-07.
- ^ “Chinese Clinical Trial Register (ChiCTR) – The world health organization international clinical trials registered organization registered platform”. http://www.chictr.org.cn. Retrieved 2020-10-21.
- ^ “Egypt to start receiving volunteers for COVID-19 vaccine trials”. Egypt Independent. 2020-09-12. Retrieved 2020-09-21.
- ^ “Bahrain starts Phase III trial of Sinopharm’s Covid-19 vaccine”. Clinical Trials Arena. 24 August 2020.
- ^ Manama TD. “Vaccine trial continues | THE DAILY TRIBUNE | KINGDOM OF BAHRAIN”. DT News. Retrieved 2020-10-22.
- ^ Jump up to:a b Barrington L (3 November 2020). “Bahrain allows Sinopharm COVID-19 vaccine candidate use in frontline workers”. Reuters. Retrieved 3 November 2020.
- ^ Liu R (5 September 2020). “China’s CNBG, Sinovac find more countries to test coronavirus vaccines”. Reuters. Retrieved 6 September 2020.
- ^ “Jordan starts phase 3 trial of China’s COVID-19 vaccine”. Jordan Times. 2020-08-30. Retrieved 2020-10-22.
- ^ “Coronavirus vaccine should be available in Pakistan ‘within 6-8 weeks'”. http://www.geo.tv. Retrieved 2020-11-14.
- ^ “Third Phase of Human Trials for Coronavirus Vaccine Underway in Peru | Voice of America – English”. http://www.voanews.com. Retrieved 2020-09-11.
- ^ “6,000 additional volunteers required for trials of Sinopharm’s COVID-19 vaccine” (in Spanish). Andina. Retrieved 17 October2020.
- ^ Aquino, Marco (2021-01-27). “Peru volunteer in Sinopharm vaccine trial dies of COVID-19 pneumonia, university says”. Reuters. Retrieved 2021-01-27.
- ^ “Clinical Trial to Evaluate the Efficacy, Immunogenicity and Safety of the Inactivated SARS-CoV-2 Vaccine (COVID-19) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2020-09-28.
- ^ Greaves J (2020-10-08). “UAE company nears end of Chinese Covid-19 vaccine trial”. Reuters. Retrieved 2020-10-10.
- ^ “Chinese COVID-19 vaccine effective: Egypt’s MoH”. EgyptToday. 2020-12-13. Retrieved 2020-12-15.
- ^ “Health Min: a new production line to produce Sinopharm’s Chinese vaccine in Egypt and will be exported to African countries”. EgyptToday. 2020-12-12. Retrieved 2020-12-31.
- ^ “Morocco acquires 65 million vaccine doses from China, UK”. ABC News. Retrieved 2020-12-26.
- ^ “Serbia to produce 24 mln doses of China’s Sinopharm vaccine annually – deputy PM”. seenews.com. Retrieved 2021-03-17.
- ^ “Bahrain approves China’s Sinopharm coronavirus vaccine”. Arabian Business Industries. 13 December 2020.
- ^ Kuo L. “China approves Sinopharm coronavirus vaccine, the country’s first for general use”. The Washington Post.
- ^ “President Ramkalawan and First Lady receives second dose SinoPharm Vaccine”. statehouse.gov.sc. Retrieved 5 February2021.
- ^ Wee S (9 December 2020). “Chinese Covid-19 Vaccine Gets Key Push, but Doubts Swirl”. The New York Times. Retrieved 12 December 2020.
- ^ “Coronavirus: UAE authorises emergency use of vaccine for frontline workers”. The National. Retrieved 24 November 2020.
- ^ Biannchi, Walter (21 February 2021). “Argentina approves Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 23 February 2021.
- ^ “Bolivia begins inoculation with Sinopharm jabs | The Star”. The Star. Malaysia. Retrieved 28 February 2021.
- ^ Rinith T (4 February 2021). “Health Ministry grants Emergency Use Authorization to China’s Sinopharm vaccine”. Khmer Times. Retrieved 4 February 2021.
- ^ “Dominica: Melissa Skerrit receives the Sinopharm COVID-19 vaccine”. WIC News. 4 March 2021. Retrieved 7 March 2021.
- ^ “Egypt licenses China’s Sinopharm COVID-19 vaccine for emergency use: health minister – Xinhua | English.news.cn”. Xinhua News Agency.
- ^ Jump up to:a b “Equatorial Guinea President receives 1st dose of Chinese COVID-19 vaccine”. dailynewsegypt.com. Retrieved 2021-03-10.
- ^ “Gabon receives 100,000 doses of Sinopharm’s vaccine from China”. Gabon 24. 2021-03-12. Retrieved 2021-03-13.
- ^ “Sinopharm vaccine rollout starts this weekend”. Stabroek News. 6 March 2021. Retrieved 7 March 2021.
- ^ “Hungary signs deal for Chinese Sinopharm’s COVID-19 vaccine, first in EU”. National Post.
- ^ “Iran Launches Phase Two of Mass Inoculation Campaign”. Financial Tribune. 22 February 2021. Retrieved 23 February 2021.
- ^ “Iraq approves Sinopharm, AstraZeneca vaccines”. Big News Network.com. Retrieved 30 January 2021.
- ^ “First batch of Chinese Sinopharm vaccine arrives in Jordan”. Roya News. Retrieved 9 January 2021.
- ^ “Laos declares Covid-19 vaccinations safe, more to be inoculated next week | The Star”. The Star. Malaysia. Retrieved 19 February2021.
- ^ Crean, Rosabel. “China donates 50,000 doses of Sinopharm vaccine to Lebanon | News , Lebanon News | THE DAILY STAR”. Daily Star. Retrieved 2 March 2021.
- ^ “Macau receives first batch of COVID-19 vaccines”. IAG. 7 February 2021. Retrieved 24 February 2021.
- ^ “MFDA approves Pfizer, Sinopharm Covid-19 vaccines for emergency use”. raajje.mv. Retrieved 2021-03-15.
- ^ Cristina (2021-03-19). “O mie de studenți și medici-rezidenți din cadrul USMF vor fi imunizați anti-COVID cu vaccinul BBIBP-CorV, produs de către Sinopharm Beijing Institute of Biological Products”. Ziarul de Gardă (in Romanian). Retrieved 2021-03-19.
- ^ “Deputy PM and City Governor get the first dose of Sinopharm vaccine”. MONTSAME News Agency. Retrieved 2021-03-15.
- ^ “Covid-19: Morocco authorizes use of the Sinopharm vaccine”. en.yabiladi.com.
- ^ Reuters Staff (6 March 2021). “Mozambique expects to vaccinate 16 million against coronavirus by 2022”. Reuters. Retrieved 7 March 2021.
- ^ Namibian, The. “Khomas, Erongo first to get vaccinated”. The Namibian. Retrieved 2021-03-17.
- ^ Poudel, Arjun. “China’s Shinopharm vaccine gets emergency use authorisation in Nepal”. Kathmandu Post. Retrieved 19 February2021.
- ^ “Covid-19 : Le Niger réceptionne 400.000 doses de vaccin SINOPHARM, un don de la Chine | Agence Nigérienne de Presse”. http://www.anp.ne. Retrieved 2021-03-21.
- ^ Shahzad A (19 January 2021). “Pakistan approves Chinese Sinopharm COVID-19 vaccine for emergency use”. Reuters.
- ^ “Peru grants ‘exceptional’ approval for Sinopharm COVID-19 vaccine – government sources”. Reuters. 27 January 2021.
- ^ Asala, Kizzi. “Senegal Kicks Off COVID-19 Vaccination Campaign with China’s Sinopharm”. Africanews. Retrieved 23 February2021.
- ^ “Serbia Becomes First European Nation To Use China’s Sinopharm Vaccine”. RadioFreeEurope/RadioLiberty.
- ^ Thomas, Abdul Rashid (2021-03-15). “Sierra Leone’s President Bio leads the way in taking COVID-19 Vaccine”. SIERRA LEONE TELEGRAPH. Retrieved 2021-03-15.
- ^ “NMRA approves sinopharm vaccine for emergency use”. Colombo Gazette. 2021-03-19. Retrieved 2021-03-20.
- ^ Sequera, Vivian (1 March 2021). “Venezuela approves use of China’s Sinopharm coronavirus vaccine”. Reuters. Retrieved 2 March 2021.
- ^ Mutsaka, Farai (18 February 2021). “Zimbabwe starts administering China’s Sinopharm vaccines”. The Star. Retrieved 20 February 2021.
- ^ Jump up to:a b “China ‘to provide 400,000 COVID vaccine doses’ to Afghanistan”. http://www.aljazeera.com. Retrieved 2021-03-01.
- ^ Jump up to:a b Presse, AFP-Agence France. “Algeria Receives 200,000 Coronavirus Jabs From China”. http://www.barrons.com. Retrieved 2021-02-26.
- ^ Jump up to:a b “Vaccine donation from China arrives | The Star”. http://www.thestar.com.my. Retrieved 2021-02-12.
- ^ Jump up to:a b Radomir, Ralev. “Montenegro receives 30,000 doses of China’s COVID-19 vaccine”.
- ^ “Regulation and Prequalification”. World Health Organization. Retrieved 18 March 2021.
- ^ “Bahrain approves Chinese COVID-19 vaccine for use”. ABC News. Retrieved 2020-12-13.
- ^ Reuters Staff (2021-01-15). “Cambodia says China donates 1 million doses of COVID-19 vaccines”. Reuters. Retrieved 2021-01-16.
- ^ “Health Ministry grants Emergency Use Authorization to China’s Sinopharm vaccine”. Khmer Times. 2021-02-04. Retrieved 2021-02-04.
- ^ “Lt Gen Manet first to be inoculated today with the Sinopharm vaccine”. Khmer Times. 2021-02-09. Retrieved 2021-02-10.
- ^ “Sinovac’s coronavirus vaccine candidate approved for emergency use in China – source”. Reuters. 2020-08-29. Retrieved 2020-08-30.
- ^ Vivek V (15 October 2020). “China’s Sinopharm offers experimental COVID-19 vaccines to students: WSJ”. Reuters. Retrieved 15 October 2020.
- ^ “China gives conditional approval to coronavirus vaccine made by Sinopharm”. Global News. Retrieved 2020-12-31.
- ^ “First Sinopharm Covid-19 vaccines to arrive today”. Macau Business. 2021-02-06. Retrieved 2021-02-07.
- ^ Taufiqurrahman M. “Indonesia can be manufacturing hub for COVID-19 vaccine, says Chinese foreign minister”. Jakarta Post. Retrieved 13 October 2020.
- ^ “Iran Launches Phase Two of Mass Inoculation Campaign”. Financial Tribune. 2021-02-22. Retrieved 2021-02-23.
- ^ “Iran Gets Chinese Vaccine for Coronavirus – Society/Culture news”. Tasnim News Agency. Retrieved 2021-02-28.
- ^ Jangiz, Khazan. “Iraq approves the emergency use of two more COVID-19 vaccines”. http://www.rudaw.net. Retrieved 2021-01-21.
- ^ “Iraq receives first Covid vaccines, gift from China”. France 24. 3 March 2021.
- ^ “Jordan approves China’s Sinopharm Covid vaccine”. France 24. 2021-01-09. Retrieved 2021-03-07.
- ^ Omari, Raed. “Jordan begins COVID-19 vaccination drive as physician, 87, gets first jab”. Arab News.
- ^ KHARIZOV, Ruslan (2021-03-19). “150,000 doses of Sinopharm coronavirus vaccine delivered to Kyrgyzstan”. 24.kg. Retrieved 2021-03-20.
- ^ Thanabouasy, Phayboune (2021-01-27). “Laos Begins Vaccinations for Over 600 Medical Workers”. Laotian Times. Retrieved 2021-01-27.
- ^ Limited, Bangkok Post Public Company. “Laos receives 300,000 vaccine doses from China”. Bangkok Post. Retrieved 2021-02-10.
- ^ “China donates 50,000 doses of Sinopharm vaccine to Lebanon | News , Lebanon News | THE DAILY STAR”. http://www.dailystar.com.lb. Retrieved 2021-03-02.
- ^ March 2021, Naharnet Newsdesk 01; 20:39. “Lebanon Authorizes Use of Chinese Vaccine Sinopharm”. Naharnet. Retrieved 2021-03-07.
- ^ “MFDA approves Pfizer, Sinopharm Covid-19 vaccines for emergency use”. raajje.mv. Retrieved 2021-03-15.
- ^ “Mongolia receives COVID-19 vaccine donation from China – The Manila Times”. http://www.manilatimes.net. Retrieved 2021-02-28.
- ^ “Deputy PM and City Governor get the first dose of Sinopharm vaccine”. MONTSAME News Agency. Retrieved 2021-03-15.
- ^ “China’s Shinopharm vaccine gets emergency use authorisation in Nepal”. kathmandupost.com. Retrieved 2021-02-19.
- ^ Peshimam GN (2020-12-31). “Pakistan to purchase 1.2 million COVID-19 vaccine doses from China’s Sinopharm”. Reuters. Retrieved 2020-12-31.
- ^ Shahzad, Asif (2021-01-19). “Pakistan approves Chinese Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 2021-01-21.
- ^ “Palestine to receive 100,000 doses of Sinopharm Covid-19 vaccine”. WAFA Agency. Retrieved 2021-03-12.
- ^ “NMRA approves sinopharm vaccine for emergency use”. Colombo Gazette. 2021-03-19. Retrieved 2021-03-20.
- ^ Sircar, Nandini. “UAE Covid vaccine: Third dose to help those with weak immunity”. Khaleej Times. Retrieved 2021-03-19.
- ^ Jump up to:a b “Egypt approves Chinese COVID vaccine, roll-out likely this month”. http://www.aljazeera.com. Retrieved 2021-01-03.
- ^ “Gabon receives 100,000 doses of Sinopharm’s vaccine from China”. Gabon 24. 2021-03-12. Retrieved 2021-03-13.
- ^ Eljechtimi, Ahmed (2021-01-26). “Morocco prepares to launch COVID-19 vaccination programme”. Reuters. Retrieved 2021-01-27.
- ^ “Moroccan health ministry grants emergency approval to Sinopharm’s Covid-19 vaccine”. wam. Retrieved 2021-01-27.
- ^ “China, Africa and the Vaccine Donations”. Modern Ghana. Retrieved 2021-03-05.
- ^ Mucari, Manuel (2021-03-06). “Mozambique expects to vaccinate 16 million against coronavirus by 2022”. Reuters. Retrieved 2021-03-07.
- ^ Namibian, The. “Khomas, Erongo first to get vaccinated”. The Namibian. Retrieved 2021-03-17.
- ^ “Covid-19 : Le Niger réceptionne 400.000 doses de vaccin SINOPHARM, un don de la Chine | Agence Nigérienne de Presse”. http://www.anp.ne. Retrieved 2021-03-21.
- ^ Staff, Reuters (2021-02-18). “Senegal takes delivery of China’s Sinopharm vaccine”. Reuters. Retrieved 2021-02-19.
- ^ AfricaNews (2021-02-23). “Senegal begins covid-19 vaccination with doses from China’s Sinopharm”. Africanews. Retrieved 2021-02-23.
- ^ AFP. “Sierra Leone to receive 200,000 virus vaccine doses”. ewn.co.za. Retrieved 2021-02-26.
- ^ Thomas, Abdul Rashid (2021-03-15). “Sierra Leone’s President Bio leads the way in taking COVID-19 Vaccine”. SIERRA LEONE TELEGRAPH. Retrieved 2021-03-15.
- ^ “Seychelles to start vaccinations with Chinese-made Sinopharm”. AP NEWS. 2021-01-08. Retrieved 2021-01-08.
- ^ “Covid-19: le Congo-Brazzaville reçoit des milliers de doses du vaccin chinois Sinopharm”. RFI (in French). 2021-03-10. Retrieved 2021-03-12.
- ^ Banya, Nelson (2021-02-11). “Zimbabwe purchases 600,000 Sinopharm COVID-19 vaccinations -information minister”. Reuters. Retrieved 2021-02-11.
- ^ Staff, Reuters (2021-02-24). “Zimbabwe to buy 1.2 million more COVID-19 vaccine doses from China”. Reuters. Retrieved 2021-02-26.
- ^ Lopez, Ezequiel Abiu (2021-02-16). “Dominican Republic launches COVID-19 vaccination campaign”. Reuters. Retrieved 2021-02-28.
- ^ “Dominica: Melissa Skerrit receives the Sinopharm COVID-19 vaccine”. WIC News. 2021-03-04. Retrieved 2021-03-05.
- ^ Jorgic, Drazen (2021-03-10). “Mexico leans on China after Biden rules out vaccines sharing in short term”. Reuters. Retrieved 2021-03-10.
- ^ Biannchi, Walter (2021-02-21). “Argentina approves Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 2021-02-22.
- ^ “Buenos Aires Times | Shipment of 900,000 Sinopharm vaccine doses arrives in Argentina”. http://www.batimes.com.ar. Retrieved 2021-02-26.
- ^ Ramos, Danny (2021-02-11). “Bolivia signs deal with China´s Sinopharm for coronavirus vaccine”. Reuters. Retrieved 2021-02-11.
- ^ “China-donated Sinopharm vaccine received”. Guyana Chronicle. 2021-03-03. Retrieved 2021-03-03.
- ^ “Sinopharm vaccine rollout starts this weekend”. Stabroek News. 2021-03-06. Retrieved 2021-03-07.
- ^ Reuters Staff (2021-01-06). “Peru inks deals with Sinopharm, AstraZeneca for coronavirus vaccines -president”. Reuters. Retrieved 2021-01-07.
- ^ Aquino, Marco (2021-01-27). “Peru grants ‘exceptional’ approval for Sinopharm COVID-19 vaccine – government sources”. Reuters. Retrieved 2021-01-28.
- ^ Sequera, Vivian (2021-03-01). “Venezuela approves use of China’s Sinopharm coronavirus vaccine”. Reuters. Retrieved 2021-03-02.
- ^ Sequera, Vivian (2021-03-02). “Venezuela receives donated coronavirus vaccine from China”. Reuters. Retrieved 2021-03-02.
- ^ “China sends 100,000 coronavirus vaccines to Belarus”. eng.belta.by. 2021-02-19. Retrieved 2021-02-19.
- ^ “Hungary signs deal for Chinese Sinopharm’s COVID-19 vaccine, first in EU”. nationalpost. Retrieved 2021-01-29.
- ^ Staff, Reuters (2021-02-16). “First 550,000 doses of Chinese Sinopharm’s vaccine arrive in Hungary”. Reuters. Retrieved 2021-02-18.
- ^ “Hungary’s PM Viktor Orbán vaccinated against COVID with Chinese Sinopharm vaccine”. euronews. 2021-02-28.
- ^ “Emiratele Arabe Unite au donat Republicii Moldova un lot de vaccin împotriva COVID-19”. TV8 (in Romanian). 13 March 2021.
- ^ Cristina (2021-03-19). “O mie de studenți și medici-rezidenți din cadrul USMF vor fi imunizați anti-COVID cu vaccinul BBIBP-CorV, produs de către Sinopharm Beijing Institute of Biological Products”. Ziarul de Gardă (in Romanian). Retrieved 2021-03-19.
- ^ “Vaccine delay in North Macedonia stirs political tension”. ABC News. Retrieved 2021-02-12.
- ^ Reuters Staff (2021-01-16). “Serbia receives million doses of China’s Sinopharm COVID-19 vaccine”. Reuters. Retrieved 2021-01-16.
- ^ “Abu Dhabi starts COVID-19 vaccinations”. Arab News. 2020-12-14. Retrieved 2020-12-17.
- ^ Wee SL (9 December 2020). “Chinese Covid-19 Vaccine Gets Key Push, but Doubts Swirl”. The New York Times. Retrieved 21 December 2020.
- ^ Yu, Sun (December 31, 2020). “China approves first domestic Covid-19 vaccine for general use”. Financial Times. Retrieved January 12, 2021.
- ^ Dancel R. “Philippine officials under fire from critics, health authorities for unsanctioned Covid-19 vaccinations”. The Straits Times.
- ^ Aberia, Ingming (6 January 2021). “Did Sinopharm forget that Duque exists?”. The Manila Times. Retrieved 9 January 2021.
- ^ “水面下で出回る中国ワクチン 富裕層から永田町へ? 狙われる日本市場”. Mainichi Daily News (in Japanese). 2020-12-31. Retrieved 2021-01-02.
- ^ Elmer, Keegan (January 3, 2021). “Beijing responds to claims Japanese were given unapproved Sinopharm jabs”. South China Morning Post. Retrieved January 9, 2021.
External links
- “How the Sinopharm Covid-19 Vaccine Works”. The New York Times.
| A vial of the BBIBP-CorV COVID‑19 vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | Authorization for use in Bahrain, China, Egypt, Iraq, Pakistan, Serbia, United Arab Emirates, Iran (emergency use) |
| Identifiers | |
| CAS Number | 2503126-65-4 |
| DrugBank | DB15807 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
How the Sinopharm Vaccine Works
By Jonathan Corum and Carl ZimmerUpdated March 22, 2021Leer en español

In early 2020, the Beijing Institute of Biological Products created an inactivated coronavirus vaccine called BBIBP-CorV. Clinical trials run by the state-owned company Sinopharm showed that it had an efficacy rate of 79 percent. China approved the vaccine and soon began exporting it to other countries.
A Vaccine Made From Coronaviruses
BBIBP-CorV works by teaching the immune system to make antibodies against the SARS-CoV-2 coronavirus. The antibodies attach to viral proteins, such as the so-called spike proteins that stud its surface.

Spikes
Spike
protein
gene
CORONAVIRUS
To create BBIBP-CorV, the Beijing Institute researchers obtained three variants of the coronavirus from patients in Chinese hospitals. They picked one of the variants because it was able to multiply quickly in monkey kidney cells grown in bioreactor tanks.
Killing the Virus
Once the researchers produced large stocks of the coronaviruses, they doused them with a chemical called beta-propiolactone. The compound disabled the coronaviruses by bonding to their genes. The inactivated coronaviruses could no longer replicate. But their proteins, including spike, remained intact.

Beta-
propiolactone
INACTIVATED
CORONAVIRUS
Inactivated
genes
The researchers then drew off the inactivated viruses and mixed them with a tiny amount of an aluminum-based compound called an adjuvant. Adjuvants stimulate the immune system to boost its response to a vaccine.
Inactivated viruses have been used for over a century. Jonas Salk used them to create his polio vaccine in the 1950s, and they’re the bases for vaccines against other diseases including rabies and hepatitis A.
Prompting an Immune Response
Because the coronaviruses in BBIBP-CorV are dead, they can be injected into the arm without causing Covid-19. Once inside the body, some of the inactivated viruses are swallowed up by a type of immune cell called an antigen-presenting cell.

INACTIVATED
CORONAVIRUS
Engulfing
the virus
ANTIGEN-
PRESENTING
CELL
Digesting
virus proteins
Presenting
virus protein
fragments
HELPER
T CELL
The antigen-presenting cell tears the coronavirus apart and displays some of its fragments on its surface. A so-called helper T cell may detect the fragment. If the fragment fits into one of its surface proteins, the T cell becomes activated and can help recruit other immune cells to respond to the vaccine.
Making Antibodies
Another type of immune cell, called a B cell, may also encounter the inactivated coronavirus. B cells have surface proteins in a huge variety of shapes, and a few might have the right shape to latch onto the coronavirus. When a B cell locks on, it can pull part or all of the virus inside and present coronavirus fragments on its surface.
A helper T cell activated against the coronavirus can latch onto the same fragment. When that happens, the B cell gets activated, too. It proliferates and pours out antibodies that have the same shape as their surface proteins.

ACTIVATED
HELPER
T CELL
INACTIVATED
CORONAVIRUS
Activating
the B cell
Matching
surface proteins
B CELL
SECRETED
ANTIBODIES
Stopping the Virus
Once vaccinated with BBIBP-CorV, the immune system can respond to an infection of live coronaviruses. B cells produce antibodies that stick to the invaders. Antibodies that target the spike protein can prevent the virus from entering cells. Other kinds of antibodies may block the virus by other means.

ANTIBODIES
LIVE
VIRUS
Remembering the Virus
Sinopharm’s clinical trials have demonstrated that BBIBP-CorV can protect people against Covid-19. But no one can yet say how long that protection lasts. It’s possible that the level of antibodies drops over the course of months. But the immune system also contains special cells called memory B cells that might retain information about the coronavirus for years or even decades.
Vaccine Timeline
January, 2020 Sinopharm begins developing an inactivated vaccine against the coronavirus.
June Researchers report the vaccine produces promising results in monkeys. A Phase 1/2 trial shows that the vaccine doesn’t cause any serious side effects and enables people to make antibodies against the coronavirus.

A Sinopharm production plant in Beijing.Zhang Yuwei/Xinhua, via Associated Press
July A Phase 3 trial begins in the United Arab Emirates.
August Phase 3 trials begin in Morocco and Peru.

Preparing a Sinopharm dose in Lima, Peru.Ernesto Benavides/Agence France-Presse
Sept. 14 The U.A.E. gives emergency approval for Sinopharm’s vaccine to use on health care workers. Government officials and others begin to receive it.
November The chairman of Sinopharm says almost a million people in China have received Sinopharm vaccines.
Nov. 3 The ruler of Dubai, Sheikh Mohammed bin Rashid al-Maktoum, announces he received the vaccine.

Sheikh Mohammed before receiving the vaccine.Agence France-Presse
Dec. 9 The U.A.E. gives full approval to BBIBP-CorV, announcing it has an efficacy rate of 86 percent. But the government did not release any details with their announcement, leaving it unclear how they had come to their conclusions.
Dec. 13 Bahrain also approves the vaccine.

Vials of the Sinopharm vaccine at a packaging plant.Zhang Yuwei/Xinhua, via Associated Press
Dec. 30 Sinopharm announces that the vaccine has an efficacy of 79.34 percent, leading the Chinese government to approve it. The company has yet to publish detailed results of their Phase 3 trial.
Jan. 3, 2021 Egypt authorizes the vaccine for emergency use.
Sources: National Center for Biotechnology Information; Science; The Lancet; Lynda Coughlan, University of Maryland School of Medicine; Jenna Guthmiller, University of Chicago.
Data
/////////////BBIBP-CorV, Sinopharm, COVID-19 vaccine, china, covid 19, corona virus, vaccine
#BBIBP-CorV, #Sinopharm, #COVID-19 vaccine, #china, #covid 19, #corona virus, #vaccine
SULCARDINE SULPHATE
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
Coblopasvir

Coblopasvir
CAS: 1312608-46-0
Chemical Formula: C41H50N8O8
Molecular Weight: 782.89
methyl {(2S)-1-[(2S)-2-(4-{4-[7-(2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl)-2H-1,3-benzodioxol-4-yl]phenyl}-1Himidazol-2-yl)pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
hepatitis C virus infection
KW-136
Coblopasvir is an antiviral drug candidate.
Coblopasvir dihydrochloride

CAS 1966138-53-3
| C41 H50 N8 O8 . 2 Cl H | |
| Molecular Weight | 855.806 |
| PHASE 3 | Beijing Kawin Technology Share-Holding |
Beijing Kawin Technology Share-Holding, in collaboration with Beijing Fu Rui Tiancheng Biotechnology and Ginkgo Pharma , is developing coblopasvir as an oral capsule formulation of dihydrochloride salt (KW-136), for treating hepatitis C virus infection. In June 2018, an NDA was filed in China by Beijing Kawin Technology and Sichuan Qingmu Pharmaceutical . In August 2018, the application was granted Priority Review in China . Also, Beijing Kawin is investigating a tablet formulation of coblopasvir dihydrochloride.
PATENT
WO2011075607 , claiming substituted heterocyclic derivatives as HCV replication inhibitors useful for treating HCV infection and liver fibrosis, assigned to Beijing Kawin Technology Share-Holding Co Ltd and InterMune Inc ,
PATENT
CN 108675998
PATENT
WO-2020001089
Novel crystalline and amorphous forms of methyl carbamate compound, particularly coblopasvir dihydrochloride , (designated as Forms H) processes for their preparation, compositions and combinations comprising them are claimed. Also claim is an article or kit comprising a container and a package insert, wherein the container contains coblopasvir dihydrochloride.

///////////////Coblopasvir , KW-136, hepatitis C virus infection, CHINA, Beijing Kawin Technology, NDA, Phase III
O=C(OC)N[C@@H](C(C)C)C(N1[C@H](C2=NC(C3=CC=C(C4=C5OCOC5=C(C6=CNC([C@H]7N(C([C@@H](NC(OC)=O)C(C)C)=O)CCC7)=N6)C=C4)C=C3)=CN2)CCC1)=O
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F
SY-008


![Acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91758795&t=l)
SY-008
CAS 1878218-66-6
FREE FORM 1480443-32-0
SGLT1 inhibitor (type 2 diabetes),
β-D-Glucopyranoside, 4-[[4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)-1-buten-1-yl]-2-methylphenyl]methyl]-5-(1-methylethyl)-1H-pyrazol-3-yl, acetate (1:1)
acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol
MF H50 N4 O6 . C2 H4 O2
MW 58.8 g/mol,C35H54N4O8
Originator Eli Lilly
- Developer Eli Lilly; Yabao Pharmaceutical Group
- Class Antihyperglycaemics; Small molecules
- Mechanism of Action Sodium-glucose transporter 1 inhibitors
- Phase I Diabetes mellitus
- 28 Aug 2018 No recent reports of development identified for phase-I development in Diabetes-mellitus in Singapore (PO)
- 24 Jun 2018 Biomarkers information updated
- 12 Mar 2018 Phase-I clinical trials in Diabetes mellitus (In volunteers) in China (PO) (NCT03462589)
-
Eli Lilly is developing SY 008, a sodium glucose transporter 1 (SGLT1) inhibitor, for the treatment of diabetes mellitus. The approach of inhibiting SGLT1 could be promising because it acts independently of the beta cell and could be effective in both early and advanced stages of diabetes. Reducing both glucose and insulin may improve the metabolic state and potentially the health of beta cells, without causing weight gain or hypoglycaemia. Clinical development is underway in Singapore and China.
As at August 2018, no recent reports of development had been identified for phase-I development in Diabetes-mellitus in Singapore (PO).
Suzhou Yabao , under license from Eli Lilly , is developing SY-008 , an SGLT1 inhibitor, for the potential oral capsule treatment of type 2 diabetes in China. By April 2019, a phase Ia trial was completed
PATENT
WO 2013169546
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 201 1 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLTl is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLTl may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLTl inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 201 1/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides certain novel inhibitors of SGLTl which may be suitable for the treatment of diabetes.
Accordingly, the present invention provides a compound of Formula II:
Preparation 1
Synthesis of (4-bromo-2-methyl-phenyl)methanol.
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)methanol.
Borane-dimethyl sulfide complex (2M in THF; 1 16 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.1 13 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
Synthesis of 4-bromo- l-2-methyl-benzene.
Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4-bromo-2-methyl-phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and
-Cl-
dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. XH NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo- 1 -chloromethyl-2-methyl-benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). XH NMR (300.1 1 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
Synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol.
Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo- l-chloromethyl-2-methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction.
Extract with ethyl acetate (200 mL), wash extract with water (200 rnL) and brine (200 mL), dry over a2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l).
Alternative synthesis of 4-r(4-bromo-2-methyl-phenyl)methyl1-5-isopropyl- !H-pyrazol- 3-oL
A solution of 4-bromo- 1 -chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (1 1.94 g, 71.94 mmol) and methyl 4-methyl-3-oxo valerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2O5 at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/31 1 (M+l).
Preparation 4
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (ml 2): 889.2 (M+l), 887.2 (M-l).
Preparation 5
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
Synthesis of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2-dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+1).
Preparation 8
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D- glucopyranoside dihydrochloride.
Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3is)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Example 1
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40 °C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 um C18XBridge ODB column, solvent A – 1¾0 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Preparation 9
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-acetyl-beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammomum chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate
(32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l).
Preparation 10
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol). MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0- acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2-dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
Synthesis oftert-butyl 2-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,8- diazaspiro[4.5]decane-8-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 852.8, 853.6 (M+l), 850.8, 851.6 (M-l).
Preparation 14
Synthesis oftert-butyl 9-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-3,9- diazaspiro[5.5]undecane-3-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 866.8, 867.6 (M+l), 864.8, 865.6 (M-l).
Preparation 15
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D- glucopyranoside dihydrochloride.
Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Preparation 16
Synthesis of 4-{4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5- (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
dihydrochloride.
The title compound is prepared essentially by the method of Preparation 15. MS (m/z): 752.8, 753.8 (M+1), 750.8 (M-1).
First alternative synthesis of Example 1
First alternative synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en- 2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 urn C18XBridge ODB column, solvent A – H20 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+1), 596.8 (M-1). 1H MR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=1.3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.1 1 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Example 2
Synthesis of 4- {4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbi
(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
O H
The title compound is prepared essentially by the method of the first alternative synthesis of Example 1. MS (m/z): 584.7 (M+l), 582.8 (M-l).
Example 3
Synthesis of 4- {4-[( 1 E)-4-(3 ,9-diazaspiro[5.5]undec-3 -yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
The title compound is prepared essentially by first treating the compound of Prearation 14 with HC1 as discussed in Preparation 15 then treating the resulting hydrochloride salt with triethyl amine as discussed in the first alternative synthesis of Example 1. MS (m/z): 598.8, 599.8 (M+l), 596.8, 597.8 (M-l).
Example 1 Preparation 17
Synthesis of tert-butyl 4-but-3- nyl-4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgSC^, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). iH MR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H),
2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 18
Synthesis of tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]- 4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5, 5-tetramethyl-1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield), !H NMR (300.1 1 MHz, CDC13): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H),
3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 19
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2-dicyclohexylphosphino-2′,4′,6′-tri-i-propyl-l, r-biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).
Second alternative Synthesis of Example 1
Second alternative synthesis of 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200ml) and concentrated in vacuo. This is repeated several times give the title compound (12.22 g, yield 96%). MS (m/z): 599 (M+l). [a]D20 = -12 ° (C=0.2, MeOH).
PATENT
WO 2015069541
https://patents.google.com/patent/WO2015069541A1
4-{4-[(1 E)-4-(2,9-DIAZASPIRO[5.5]UNDEC-2-YL)BUT-1 -EN-1
-YL]-2-METHYLBENZYL}-5-(PROPAN-2-YL)-1 H-PYRAZOL-3-YL
BETA-D- GLUCOPYRANOSIDE ACETATE
The present invention relates to a novel SGLT1 inhibitor which is an acetate salt of a pyrazole compound, to pharmaceutical compositions comprising the compound, to methods of using the compound to treat physiological disorders, and to intermediates and processes useful in the synthesis of the compound.
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 2011 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLT1 is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLT1 inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 2011/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides an acetate salt of a pyrazole compound, which is an SGLT1 inhibitor, and as such, may be suitable for the treatment of certain disorders, such as diabetes. Accordingly, the present invention provides a compound of Formula I:

or hydrate thereof.
Preparation 1
(4-bromo-2-methyl-phenyl)methanol

Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). !H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)mefhanol.
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
4-bromo- 1 -chloromethyl -2 -methyl -benzene

Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4- bromo-2 -methyl -phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. !H NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo-l-chloromethyl-2-methyl -benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). !H NMR (300.11 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
4- [(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-3 -ol

Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo-l-chloromethyl-2- methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction. Extract with ethyl acetate (200 mL), wash extract with water (200 mL) and brine (200 mL), dry over Na2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l). Alternative synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-
3-ol.
A solution of 4-bromo-l-chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (11.94 g, 71.94 mmol) and methyl 4-methyl-3-oxovalerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2Os at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/311 (M+l).
Preparation 4
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl- beta-D-glucopyranoside
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (m/z): 889.2 (M+l), 887.2 (M-l).
Preparation 5
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- lH-pyrazol-3 -yl 2,3 ,4,6-tetra-O-benzoyl-beta-D- glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
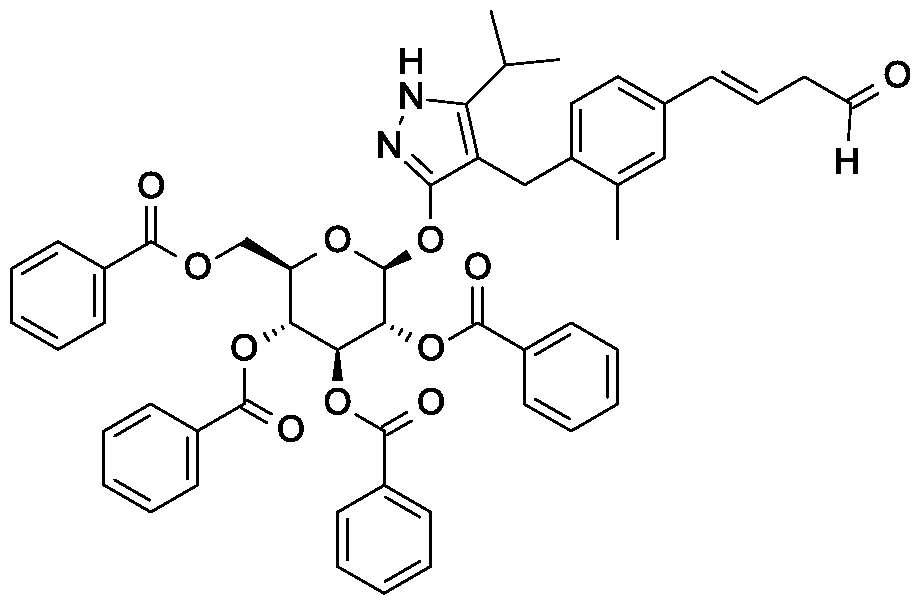
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate

Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2- dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+l).
Preparation 8
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride

Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
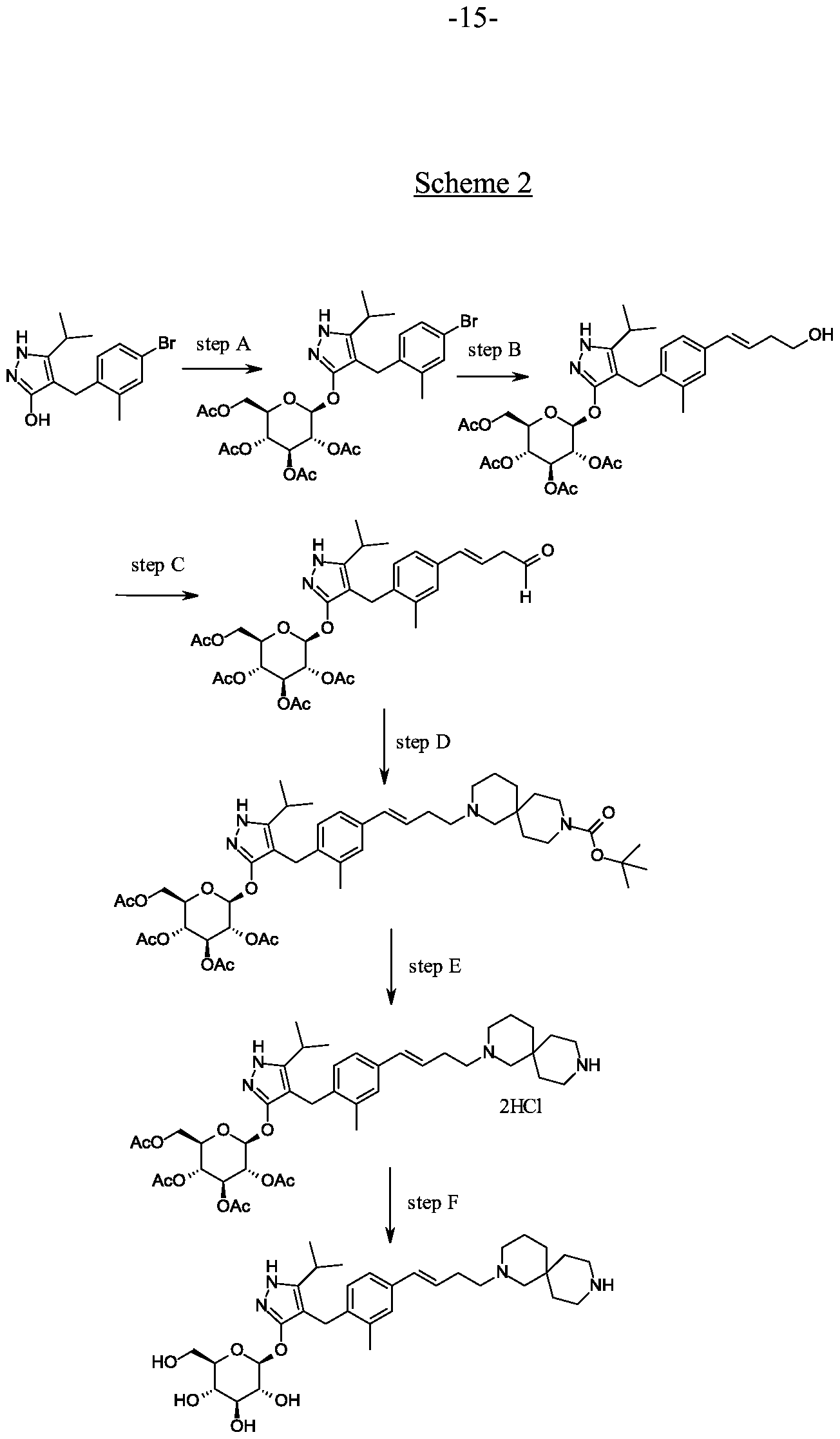
Preparation 9
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl- beta-D-glucopyranoside.
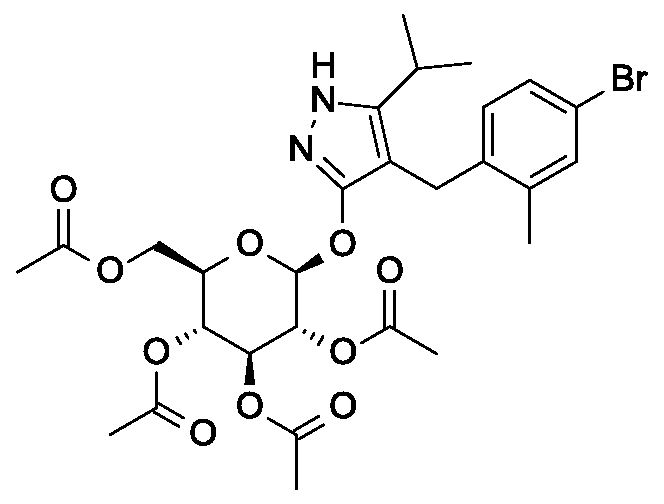
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)mefhyl]-5- isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D- glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate (32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l). Preparation 10
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
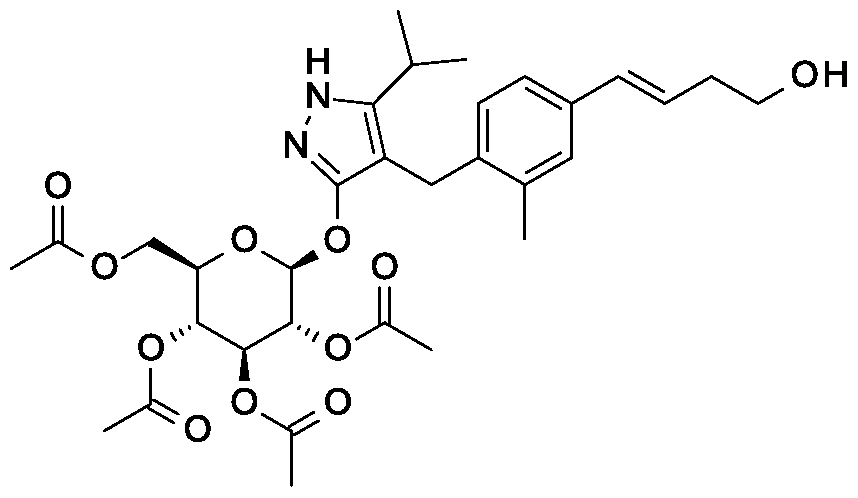
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- 1 H-pyrazol-3 -yl 2,3 ,4,6-tetra-O-acetyl-beta-D- glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol) MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside

Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12a
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy] -lH-pyrazol-4-yl}methyl)phenyl]but-3-en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate
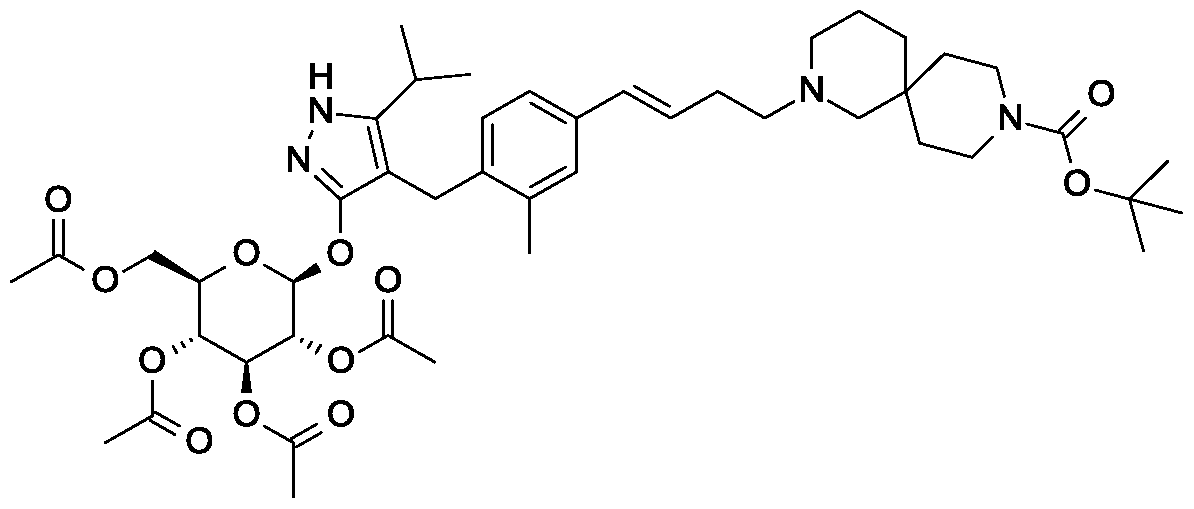
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2- dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride

Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6- tetra-0-acetyl-beta-D-glucopyranosyl)oxy] – lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 – yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Scheme 3
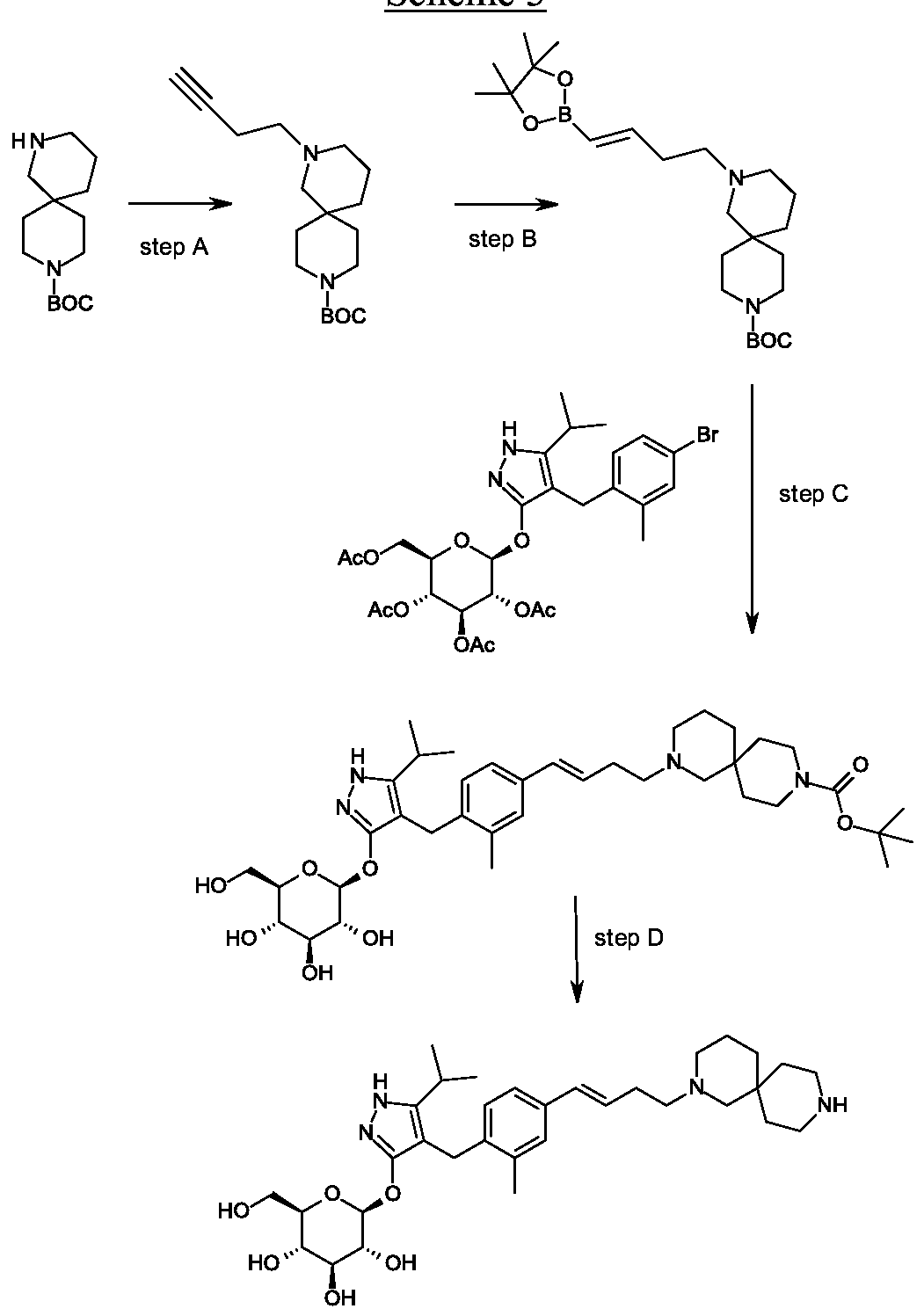
Preparation 14
tert-butyl 4-but-3-ynyl-4,9-diazas iro[5.5]undecane-9-carboxylate

Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgS04, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). lH NMR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H), 2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 15
tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9- diazaspiro[5.5]undecane-9-carboxylate
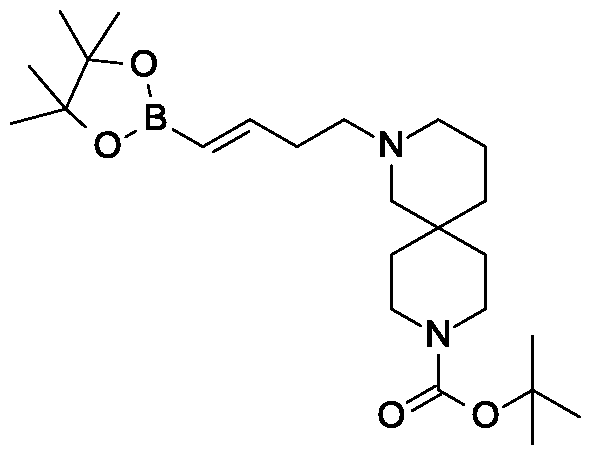
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield). 1H NMR (300.11 MHz, CDCI3): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H), 3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 16
tert-butyl 2-{(3£’)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH- pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9-diazaspiro [5.5]undecane-9-carboxylate

Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert- butyl 4-[(£)-4-(4,4,5 ,5 -tetramethyl- 1 ,3,2-dioxaborolan-2-yl)but-3 -enyl] -4,9- diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2- dicyclohexylphosphino-2′,4′,6′-tri-i -propyl- Ι, -biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).

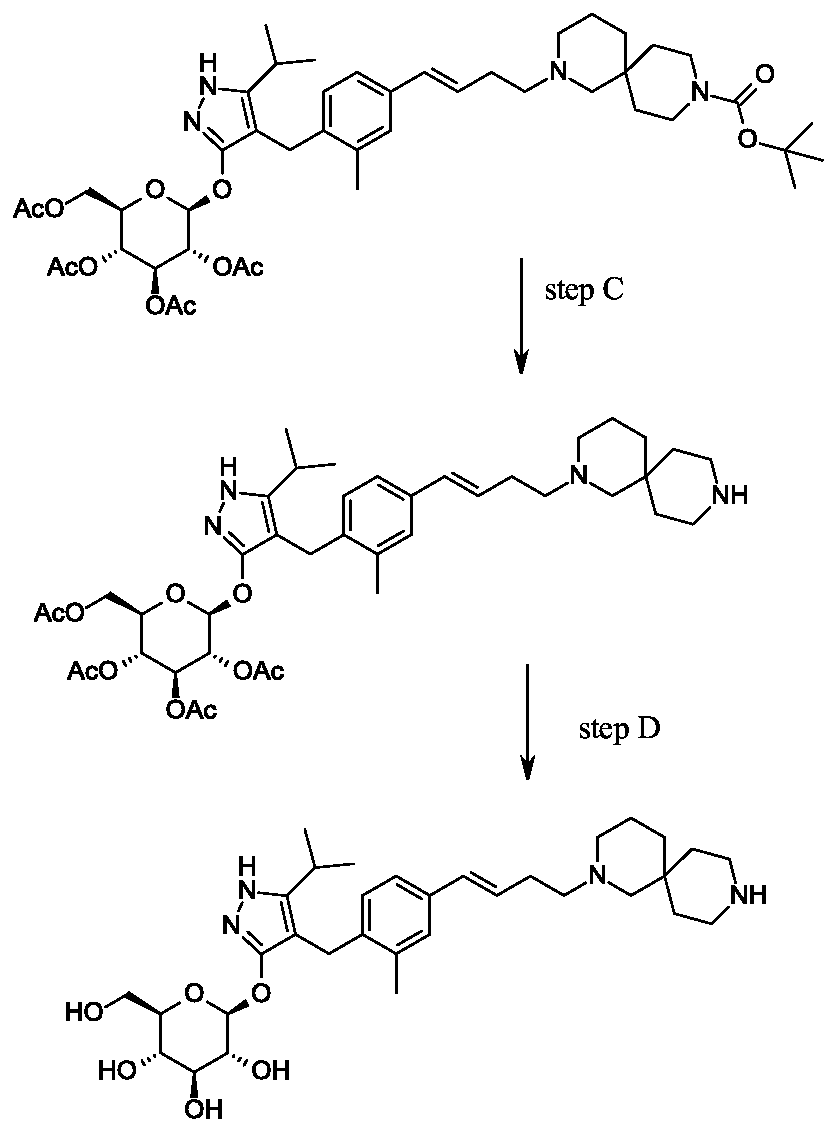
Preparation 17
tert-butyl 4- [(E)-4- [4- [(3 -hydroxy-5-isopropyl- 1 H-pyrazol-4-yl)methyl] -3 -methyl- phenyl]but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate

Scheme 4, step A: Add tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (35.8 kg, 82.4 mol) in methanol (130 L) to a solution of (4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (23.9 kg, 77.3 mol) in methanol (440 L) at room temperature. Add water (590 L) and tripotassium phosphate (100 kg, 471.7 mol) and place the reaction under nitrogen atmosphere. To the stirring solution, add a suspension of
tris(dibenzylideneacetone) dipalladium (1.42 kg, 1.55 mol) and di-tert- butylmethylphosphonium tetrafluoroborate (775 g, 3.12 mol) in methanol (15 L). The resulting mixture is heated at 75 °C for 2 hours. Cool the mixture and filter over diatomaceous earth. Rinse the the filter cake with methanol (60 L), and concentrate the filtrate under reduced pressure. Add ethyl acetate (300 L), separate the layers, and wash the organic layer with 15% brine (3 x 120 L). Concentrate the organic layer under reduced pressure, add ethyl acetate (300 L), and stir the mixture for 18 to 20 hours. Add heptane (300 L), cool the mixture to 10 °C, and stir the mixture for an additional 18 to 20 hours. Collect the resulting solids by filtration, rinse the cake with ethyl acetate/heptane (2:3, 2 x 90 L), and dry under vacuum at 40°C to give the title compound (29.3 kg, 70.6% yield) as a white solid. lH NMR (400 MHz, CD3OD): δ 7.14 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 7.6 Hz, 1H), 6.39 (d, J= 16.0 Hz, 1H), 6.25-6.12 (m, 1H), 3.63 (s, 2H), 3.45-3.38 (bs, 3H), 3.34 (s, 3 H), 3.33 (s, 3H), 2.85-2.75 (m, 1H), 2.49-2.40 (m, 5 H), 2.33 (s, 3H), 1.68-1.62 (m, 2H), 1.60-1.36 (m, 15H), 1.11 (s, 3H), 1.10 (s, 3H).
Preparation 12b
Alterternative preparation of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but- 3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate.

Scheme 4, step B: Combine tert-butyl 4-[(E)-4-[4-[(3-hydroxy-5-isopropyl-lH- pyrazol-4-yl)methyl] -3-methyl-phenyl]but-3 -enyl] -4,9-diazaspiro [5.5]undecane-9- carboxylate (17.83 kg, 33.2 moles), acetonitrile (180 L), and benzyltributylammonium chloride (1.52 kg, 4.87 moles) at room temperature. Slowly add potassium carbonate (27.6 kg, 199.7 moles) and stir the mixture for 2 hours. Add 2,3,4,6-tetra-O-acetyl-alpha- D-glucopyranosyl bromide (24.9 kg, 60.55 mol), warm the reaction mixture to 30°C and stir for 18 hours. Concentrate the mixture under reduced pressure and add ethyl acetate (180 L), followed by water (90 L). Separate the layers, wash the organic phase with 15% brine (3 x 90 L), concentrate the mixture, and purify using column chromatography over silica gel (63 kg, ethyl acetate/heptanes as eluent (1 :2→1 :0)) to provide the title compound (19.8 kg, 94% purity, 68.8% yield) as a yellow foam, !H NMR (400 MHz, CDC13): δ 7.13 (s, 1H), 7.03 (d, J= 8.0 Hz, 1H), 6.78 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 16.0,
1H), 6.25-6.13 (m, 1H), 5.64 (d, J= 8.0 Hz, 1H), 5.45-5.25 (m, 2H), 5.13-4.95 (m, 2H), 4.84-4.76 (m, 1H), 4.25-4.13 (m, 2H), 4.10-4.00 (m, 2H), 3.90-3.86 (m, 1H), 3.58-3.50 (m, 2H), 3.40-3.22 (m, 4H), 2.89-2.79 (m, 1H), 2.10-1.90 (m, 18 H), 1.82 (s, 3H), 1.62- 0.82 (m, 22H).
Preparation 18
2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane

Scheme 4, step C: Combine tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)- 3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (19.6 kg, 22.6 moles) with dichloromethane (120 L) and cool to 0°C. Slowly add trifluoroacetic acid (34.6 L, 51.6 kg, 452 moles) and stir for 9 hours. Quench the reaction with ice water (80 L), and add ammonium hydroxide (85-90 L) to adjust the reaction mixture to pH (8- 9). Add dichloromethane (120 L), warm the reaction mixture to room temperature, and separate the layers. Wash the organic layer with water (75 L), brine, and concentrate under reduced pressure to provide the title compound (16.2 kg, 95.0% purity, 93% yield) as a yellow solid. lH NMR (400 MHz, CDC13): δ 7.08 (s, IH), 6.99 (d, J= 8.0 Hz, IH),
6.76 (d, J= 7.6 Hz, IH), 6.38 (d, J=15.6 Hz, IH), 6.00-5.83 (m, IH), 5.31 (d, J= 7.6 Hz, IH), 5.25-5.13 (m, 4H), 4.32 (dd, J= 12.8, 9.2 Hz, IH), 4.14 (d, J= 11.2 Hz, IH), 3.90 (d, J= 10.0 Hz, IH), 3.75-3.50 (m, 3H), 3.30-3.00 (m, 5 H), 2.85-2.75 (m, IH), 2.70-2.48 (m, 3H), 2.25 (s, IH), 2.13-1.63 (m, 19H), 1.32-1.21 (m, IH), 1.14 (s, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Example 1
Hydrated crystalline 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside acetate
First alternative preparation of 4-{4-[(l£’)-4-(2.9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl| -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (free base).
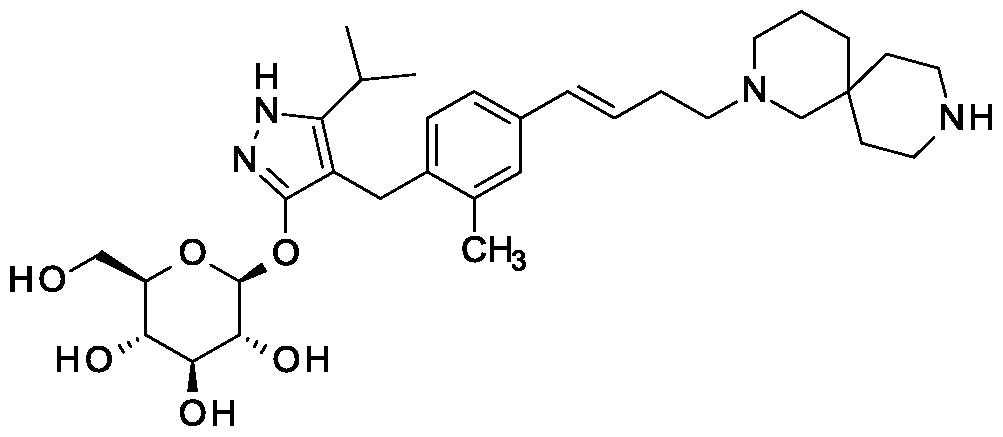
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4- {4-[( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} – 5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40°C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H.0 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Second alternative preparation of 4-{4-r(l-£’)-4-(2.9-diazaspiror5.51undec-2-yl)but-l-en- 1 -yl] -2-methylbenzyl I -5 -(propan-2-yl)- lH-pyrazol-3 -yl beta-D-glucopyranoside (free base“).

Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4- {4-[( lJE)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H20 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+l), 596.8 (M-l). 1H NMR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=l .3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.11 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Third alternative preparation of 4-{4-[(l£,)-4-(2,9-diazaspiro[5.51undec-2-yl)but-l-en-l- yll-2-methylbenzyl|-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200mL) and concentrated in vacuo. This is repeated several times to give the title compound (free base) (12.22 g, yield 96%). MS (m/z): 599 (M+l); [a]D 20 = -12 ° (C=0.2, MeOH).
Preparation of final title compound, hydrated crystalline 4-{4-|YlE)-4-(2.9- diazaspiro [5.5“|undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-vD- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.

4- {4- [(1 E)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (902 mg) is placed in a round bottom flask (100 mL) and treated with wet ethyl acetate (18 mL). [Note – wet ethyl acetate is prepared by mixing ethyl acetate (100 mL) and dionized water (100 mL). After mixing, the layers are allowed to separate, and the top wet ethyl acetate layer is removed for use. Acetic acid is a hydrolysis product of ethyl acetate and is present in wet ethyl acetate.] The compound dissolves, although not completely as wet ethyl acetate is added. After several minutes, a white precipitate forms. An additional amount of wet ethyl acetate (2 mL) is added to dissolve remaining compound. The solution is allowed to stir uncovered overnight at room temperature during which time the solvent partially evaporates. The remaining solvent from the product slurry is removed under vacuum, and the resulting solid is dried under a stream of nitrogen to provide the final title compound as a crystalline solid. A small amount of amorphous material is identified in the product by solid-state NMR. This crystalline final title compound may be used as seed crystals to prepare additional crystalline final title compound.
Alternative preparation of final title compound, hvdrated crystalline 4-{4-[(lE)-4-(2.,9- diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-yl)- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.
Under a nitrogen atmosphere combine of 4-{4-[(lE)-4-(2,9-diazaspiro[5.5]undec- 2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan-2-yl)- 1 H-pyrazol-3-yl 2,3,4,6-tetra-O- acetyl-beta-D-glucopyranoside (2.1 kg, 2.74 mol), methanol (4.4 L), tetrahydrofuran (4.2 L), and water (210 mL). Add potassium carbonate (460 g, 3.33 moles) and stir for four to six hours, then filter the reaction mixture to remove the solids. Concentrate the filtrate under reduced pressure, then add ethanol (9.0 L) followed by acetic acid (237 mL, 4.13 mol) and stir at room temperature for one hour. To the stirring solution add wet ethyl acetate (10 L, containing approx. 3 w/w% water) slowly over five hours, followed by water (500 mL). Stir the suspension for twelve hours and add wet ethyl acetate (4.95 L, containing approx. 3 w/w% water) over a period of eight hours. Stir the suspension for twelve hours and add additional wet ethyl acetate (11.5 L, containing approx. 3 w/w% water) slowly over sixteen hours. Stir the suspension for twelve hours, collect the solids by filtration and rinse the solids with wet ethyl acetate (3.3 L, containing approx. 3 w/w% water). Dry in an oven under reduced pressure below 30°C to give the title compound as an off-white crystalline solid (1.55 kg, 2.35 mol, 96.7% purity, 72.4 w/w% potency, 68.0% yield based on potency). HRMS (m/z): 599.3798 (M+l).
PATENT
CN105705509
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN175101669&tab=PCTDESCRIPTION
The present invention is in the field of treatment of diabetes and other diseases and conditions associated with hyperglycemia. Diabetes is a group of diseases characterized by high blood sugar levels. It affects approximately 25 million people in the United States, and according to the 2011 National Diabetes Bulletin, it is also the seventh leading cause of death in the United States (US Department of Health and Human Resources Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 can result in a decrease in glucose absorption in the small intestine, thus providing a useful method of treating diabetes.
Alternative medicines and treatments for diabetes are needed. The present invention provides an acetate salt of a pyrazole compound which is an SGLT1 inhibitor, and thus it is suitable for treating certain conditions such as diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives having human SGLT1 inhibitory activity, which are also disclosed for use in the prevention or treatment of diseases associated with hyperglycemia, such as diabetes. Moreover, WO 2011/039338 discloses certain pyrazole derivatives having SGLT1/SGLT2 inhibitor activity, which are also disclosed for use in the treatment of bone diseases such as osteoporosis.
PATENT
WO-2019141209
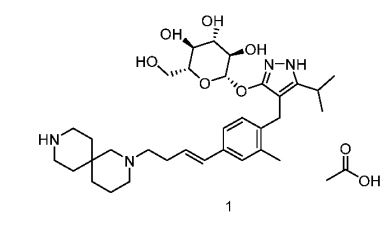
Process for preparing pyranoglucose-substituted pyrazole compound, used as a pharmaceutical intermediate in SGLT inhibitor for treating diabetes.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9573970 | 4–5-(PROPAN-2-YL)-1H-PYRAZOL-3-YL BETA-D GLUCOPYRANOSIDE ACETATE | 2014-10-30 | 2016-07-28 |
/////////////SY-008 , SY 008 , SY008, ELI LILY, PHASE 1, GLT1 inhibitor, type 2 diabetes, Yabao Pharmaceutical, CHINA, DIABETES
CC(=O)O.Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4O
Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4
O
Fluazolepali, 氟唑帕利 , Fluzoparib

Fluazolepali
CAS 2170504-09-1
Fluzoparib; SHR-3162, (HS10160)
- HS 10160
- SHR 3162
An orally available inhibitor of poly(ADP-ribose) polymerase 1 and 2 (PARP-1/2) for treatment of solid tumors (Jiangsu Hengrui Medicine Co. Ltd., Lianyungang, China)
Fluazolepali, developed by Hengrui and Howson, is intended for the treatment of recurrent ovarian cancer, triple-negative breast cancer, advanced gastric cancer and other advanced solid tumors. Currently, the drug has been introduced into China for recurrent ovarian cancer. Clinical stage.
In February 2019, a randomized, double-blind, controlled, multicenter, phase III clinical study (CTR20190294) of flazopril capsule versus placebo for maintenance of recurrent ovarian cancer was initiated in China and was sponsored by Hengrui Medicine.
Jiangsu Hansoh Pharmaceutical , in collaboration with Jiangsu Hengrui Medicine , is developing an oral capsule formulation of fluazolepali (fluzoparib; SHR-3162), a small molecule inhibitor to PARP-1 and PARP-2, for the treatment of solid tumors including epithelial ovarian, fallopian tube or primary peritoneal, breast and gastric cancer.
- Originator Jiangsu Hengrui Medicine Co.
- Class Antineoplastics
- Mechanism of Action Poly(ADP-ribose) polymerase 1 inhibitors; Poly(ADP-ribose) polymerase 2 inhibitors
- Phase II Ovarian cancer
- Phase I Breast cancer; Fallopian tube cancer; Gastric cancer; Peritoneal cancer; Solid tumours
- 09 Jul 2019 Jiangsu HengRui Medicine initiates a phase I trial in Solid tumors in China (NCT04013048) [14C]-Fluzoparib
- 01 Jul 2019 Jiangsu HengRui Medicine plans a phase I drug-drug interaction trial (In volunteers) in China (PO) (NCT04011124)
- 12 Jun 2019 Jiangsu HengRui Medicine completes a phase I trial in Gastric cancer (Combination therapy, Recurrent, Metastatic disease, Second-line therapy or greater, Late-stage disease) in China (PO) (NCT03026881)
Fluzoparib (SHR 3162) is a selective poly [ADP-ribose] polymerase 1 (PARP1) and poly [ADP-ribose] polymerase 2 inhibitor (PARP2), being developed by Jiangsu HengRui Medicine, for the treatment of cancer. PARP enzymes play a vital role in repair of DNA damage and maintaining genomic stability. Fluzoparib inhibits PARP enzymes and induces DNA-double strands breaks, G2/M arrest and apoptosis in homologous recombination repair (HR)-deficient cells. Clinical development for ovarian cancer, breast cancer, fallopian tube cancer, peritoneal cancer, gastric cancer and solid tumours is underway in China and Australia.
An orally available inhibitor of poly (ADP-ribose) polymerase (PARP) types 1 and 2, with potential antineoplastic activity. Upon oral administration, fluzoparib inhibits PARP 1 and 2 activity, which inhibits PARP-mediated repair of damaged DNA via the base excision repair (BER) pathway, enhances the accumulation of DNA strand breaks, promotes genomic instability, and leads to an induction of apoptosis. The PARP family of proteins catalyze post-translational ADP-ribosylation of nuclear proteins, which then transduce signals to recruit other proteins to repair damaged DNA. PARP inhibition may enhance the cytotoxicity of DNA-damaging agents and may reverse tumor cell chemoresistance and radioresistance. Check for active clinical trials using this agent. (NCI Thesaurus)
PATENT
WO-2019137358
Process for preparing heterocyclic compounds (presumed to be fluazolepali ) and its intermediates as PARP inhibitors useful for treating cancer.


PATENT
WO2019109938
claiming synergistic combination comprising PARP inhibitor fluazolepali and apatinib mesylate .
PATENT
WO 2018005818
WO 2018129553
WO 2018129559
WO 2018208968
WO 2018213732
WO 2018191277
WO 2018201096
WO 2018085469
WO 2018085468
WO 2019090227
WO 2019133697
WO 2019067978
WO 2019071123
WO 2019090141
///////////Fluazolepali, Jiangsu Hansoh Pharmaceutical, Jiangsu Hengrui Medicine, fluzoparib, SHR-3162, 氟唑帕利 , Phase II, Ovarian cancer, HS10160, CHINA, HS 10160
HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CS 3001
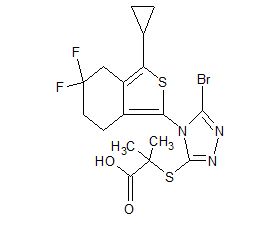
CS-3001
BB 7, VX 033
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in



WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
_K_(cropped).jpeg){kind=link}