
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


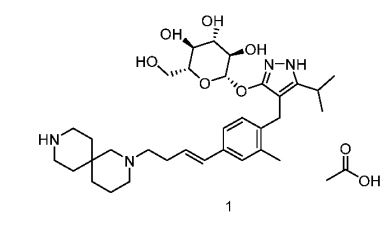
![Acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91758795&t=l)
SY-008
CAS 1878218-66-6
FREE FORM 1480443-32-0
SGLT1 inhibitor (type 2 diabetes),
β-D-Glucopyranoside, 4-[[4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)-1-buten-1-yl]-2-methylphenyl]methyl]-5-(1-methylethyl)-1H-pyrazol-3-yl, acetate (1:1)
acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol
MF H50 N4 O6 . C2 H4 O2
MW 58.8 g/mol,C35H54N4O8
Originator Eli Lilly
- Developer Eli Lilly; Yabao Pharmaceutical Group
- Class Antihyperglycaemics; Small molecules
- Mechanism of Action Sodium-glucose transporter 1 inhibitors
- Phase I Diabetes mellitus
- 28 Aug 2018 No recent reports of development identified for phase-I development in Diabetes-mellitus in Singapore (PO)
- 24 Jun 2018 Biomarkers information updated
- 12 Mar 2018 Phase-I clinical trials in Diabetes mellitus (In volunteers) in China (PO) (NCT03462589)
-
Eli Lilly is developing SY 008, a sodium glucose transporter 1 (SGLT1) inhibitor, for the treatment of diabetes mellitus. The approach of inhibiting SGLT1 could be promising because it acts independently of the beta cell and could be effective in both early and advanced stages of diabetes. Reducing both glucose and insulin may improve the metabolic state and potentially the health of beta cells, without causing weight gain or hypoglycaemia. Clinical development is underway in Singapore and China.
As at August 2018, no recent reports of development had been identified for phase-I development in Diabetes-mellitus in Singapore (PO).
Suzhou Yabao , under license from Eli Lilly , is developing SY-008 , an SGLT1 inhibitor, for the potential oral capsule treatment of type 2 diabetes in China. By April 2019, a phase Ia trial was completed
PATENT
WO 2013169546
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 201 1 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLTl is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLTl may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLTl inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 201 1/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides certain novel inhibitors of SGLTl which may be suitable for the treatment of diabetes.
Accordingly, the present invention provides a compound of Formula II:
Preparation 1
Synthesis of (4-bromo-2-methyl-phenyl)methanol.
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)methanol.
Borane-dimethyl sulfide complex (2M in THF; 1 16 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.1 13 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
Synthesis of 4-bromo- l-2-methyl-benzene.
Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4-bromo-2-methyl-phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and
-Cl-
dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. XH NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo- 1 -chloromethyl-2-methyl-benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). XH NMR (300.1 1 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
Synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol.
Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo- l-chloromethyl-2-methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction.
Extract with ethyl acetate (200 mL), wash extract with water (200 rnL) and brine (200 mL), dry over a2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l).
Alternative synthesis of 4-r(4-bromo-2-methyl-phenyl)methyl1-5-isopropyl- !H-pyrazol- 3-oL
A solution of 4-bromo- 1 -chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (1 1.94 g, 71.94 mmol) and methyl 4-methyl-3-oxo valerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2O5 at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/31 1 (M+l).
Preparation 4
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (ml 2): 889.2 (M+l), 887.2 (M-l).
Preparation 5
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
Synthesis of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2-dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+1).
Preparation 8
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D- glucopyranoside dihydrochloride.
Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3is)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Example 1
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40 °C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 um C18XBridge ODB column, solvent A – 1¾0 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Preparation 9
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-acetyl-beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammomum chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate
(32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l).
Preparation 10
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol). MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12
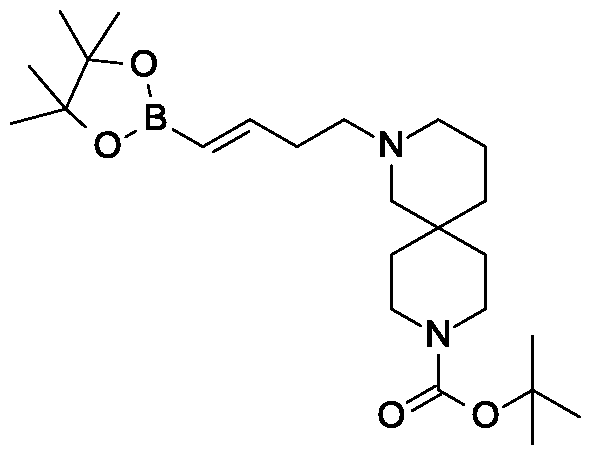
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0- acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2-dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
Synthesis oftert-butyl 2-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,8- diazaspiro[4.5]decane-8-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 852.8, 853.6 (M+l), 850.8, 851.6 (M-l).
Preparation 14
Synthesis oftert-butyl 9-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-3,9- diazaspiro[5.5]undecane-3-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 866.8, 867.6 (M+l), 864.8, 865.6 (M-l).
Preparation 15
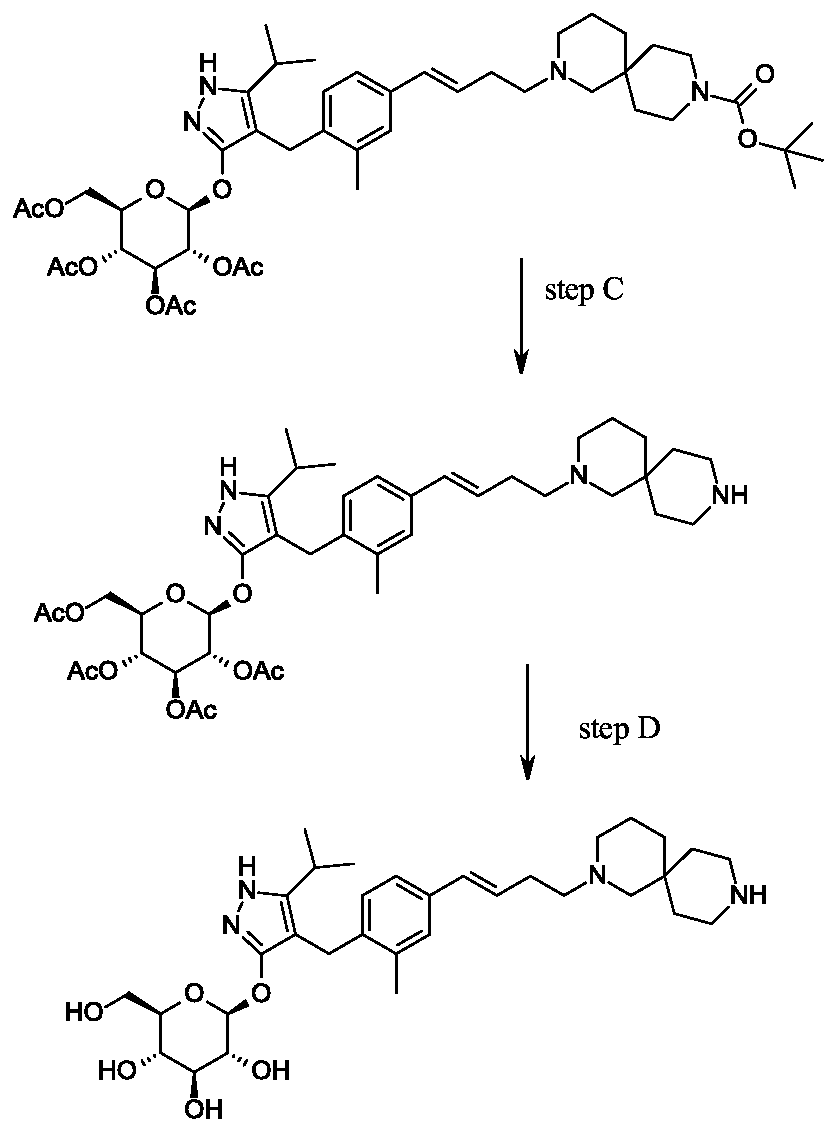
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D- glucopyranoside dihydrochloride.
Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Preparation 16
Synthesis of 4-{4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5- (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
dihydrochloride.
The title compound is prepared essentially by the method of Preparation 15. MS (m/z): 752.8, 753.8 (M+1), 750.8 (M-1).
First alternative synthesis of Example 1
First alternative synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en- 2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 urn C18XBridge ODB column, solvent A – H20 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+1), 596.8 (M-1). 1H MR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=1.3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.1 1 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Example 2
Synthesis of 4- {4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbi
(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
O H
The title compound is prepared essentially by the method of the first alternative synthesis of Example 1. MS (m/z): 584.7 (M+l), 582.8 (M-l).
Example 3
Synthesis of 4- {4-[( 1 E)-4-(3 ,9-diazaspiro[5.5]undec-3 -yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
The title compound is prepared essentially by first treating the compound of Prearation 14 with HC1 as discussed in Preparation 15 then treating the resulting hydrochloride salt with triethyl amine as discussed in the first alternative synthesis of Example 1. MS (m/z): 598.8, 599.8 (M+l), 596.8, 597.8 (M-l).
Example 1 Preparation 17
Synthesis of tert-butyl 4-but-3- nyl-4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgSC^, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). iH MR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H),
2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 18
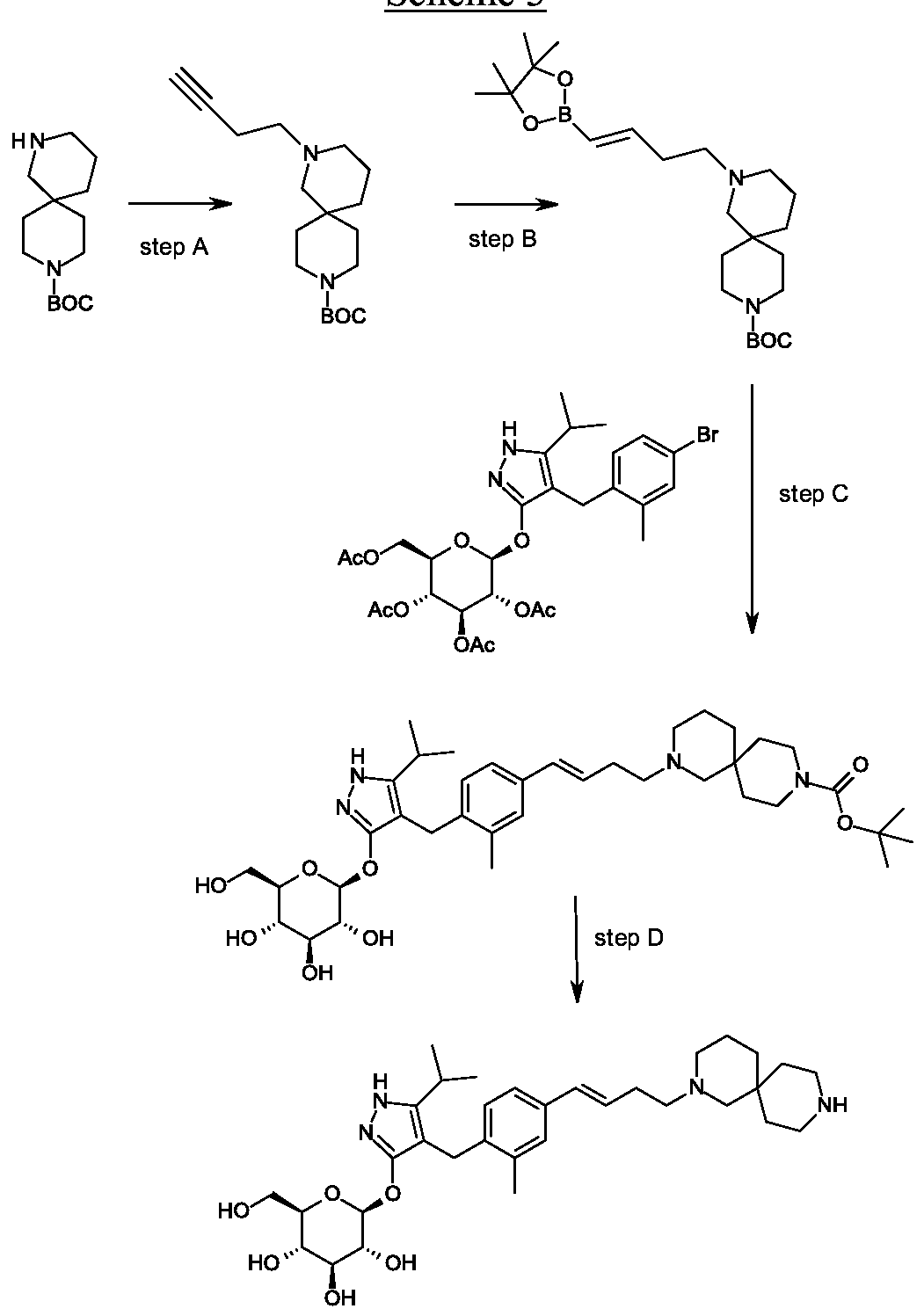
Synthesis of tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]- 4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5, 5-tetramethyl-1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield), !H NMR (300.1 1 MHz, CDC13): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H),
3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 19
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2-dicyclohexylphosphino-2′,4′,6′-tri-i-propyl-l, r-biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).
Second alternative Synthesis of Example 1
Second alternative synthesis of 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200ml) and concentrated in vacuo. This is repeated several times give the title compound (12.22 g, yield 96%). MS (m/z): 599 (M+l). [a]D20 = -12 ° (C=0.2, MeOH).
PATENT
WO 2015069541
https://patents.google.com/patent/WO2015069541A1
4-{4-[(1 E)-4-(2,9-DIAZASPIRO[5.5]UNDEC-2-YL)BUT-1 -EN-1
-YL]-2-METHYLBENZYL}-5-(PROPAN-2-YL)-1 H-PYRAZOL-3-YL
BETA-D- GLUCOPYRANOSIDE ACETATE
The present invention relates to a novel SGLT1 inhibitor which is an acetate salt of a pyrazole compound, to pharmaceutical compositions comprising the compound, to methods of using the compound to treat physiological disorders, and to intermediates and processes useful in the synthesis of the compound.
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 2011 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLT1 is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLT1 inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 2011/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides an acetate salt of a pyrazole compound, which is an SGLT1 inhibitor, and as such, may be suitable for the treatment of certain disorders, such as diabetes. Accordingly, the present invention provides a compound of Formula I:

or hydrate thereof.
Preparation 1
(4-bromo-2-methyl-phenyl)methanol

Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). !H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)mefhanol.
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
4-bromo- 1 -chloromethyl -2 -methyl -benzene

Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4- bromo-2 -methyl -phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. !H NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo-l-chloromethyl-2-methyl -benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). !H NMR (300.11 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
4- [(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-3 -ol

Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo-l-chloromethyl-2- methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction. Extract with ethyl acetate (200 mL), wash extract with water (200 mL) and brine (200 mL), dry over Na2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l). Alternative synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-
3-ol.
A solution of 4-bromo-l-chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (11.94 g, 71.94 mmol) and methyl 4-methyl-3-oxovalerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2Os at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/311 (M+l).
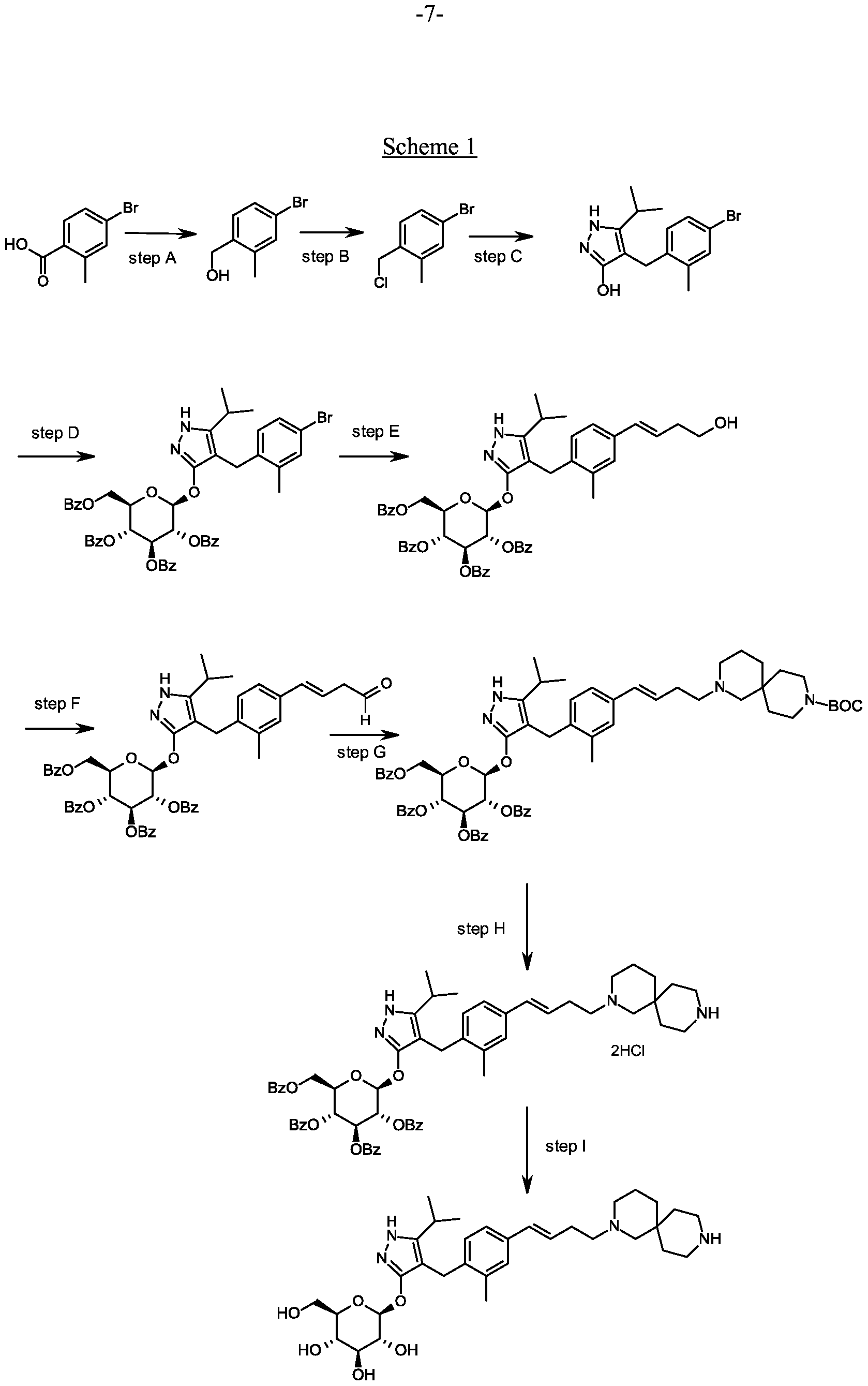
Preparation 4
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl- beta-D-glucopyranoside
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (m/z): 889.2 (M+l), 887.2 (M-l).
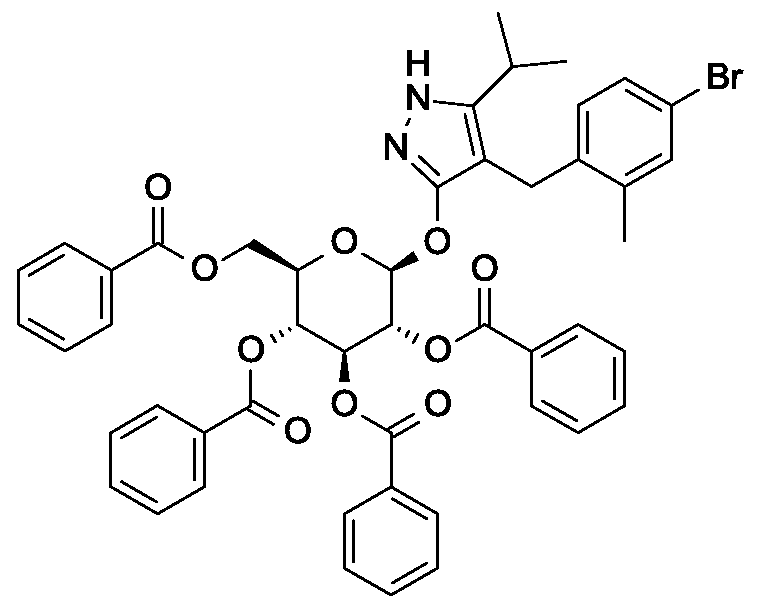
Preparation 5
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- lH-pyrazol-3 -yl 2,3 ,4,6-tetra-O-benzoyl-beta-D- glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
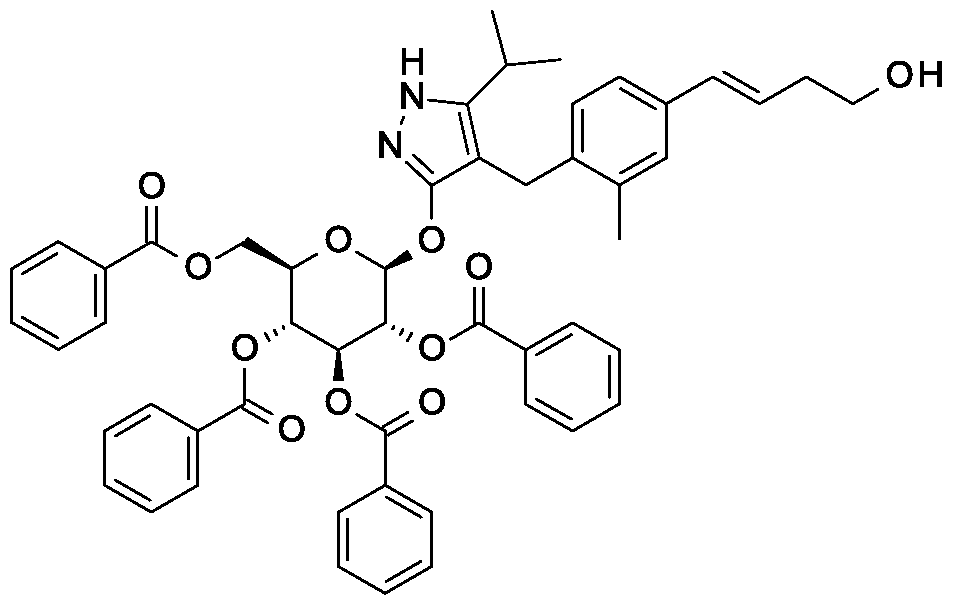
Preparation 6
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
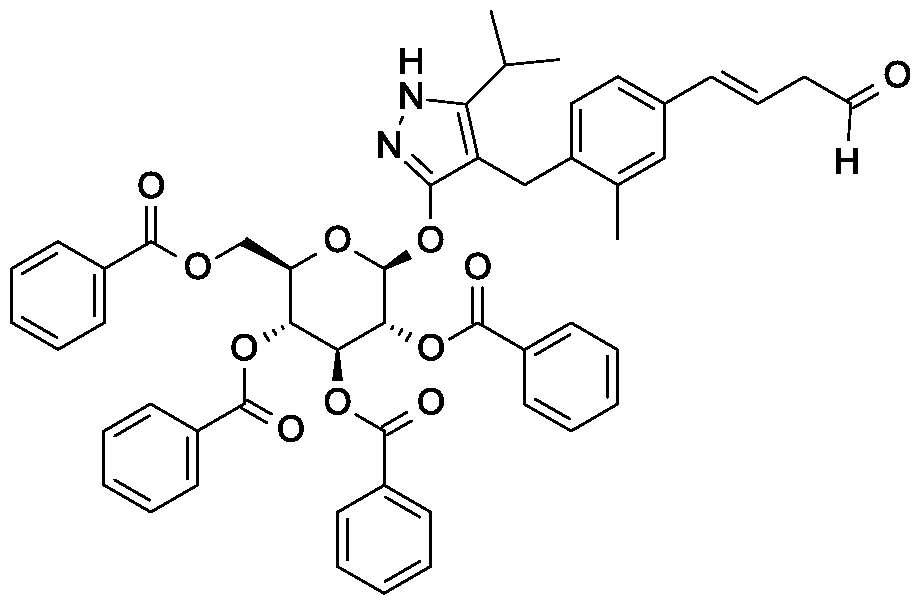
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate

Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2- dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+l).
Preparation 8
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride

Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
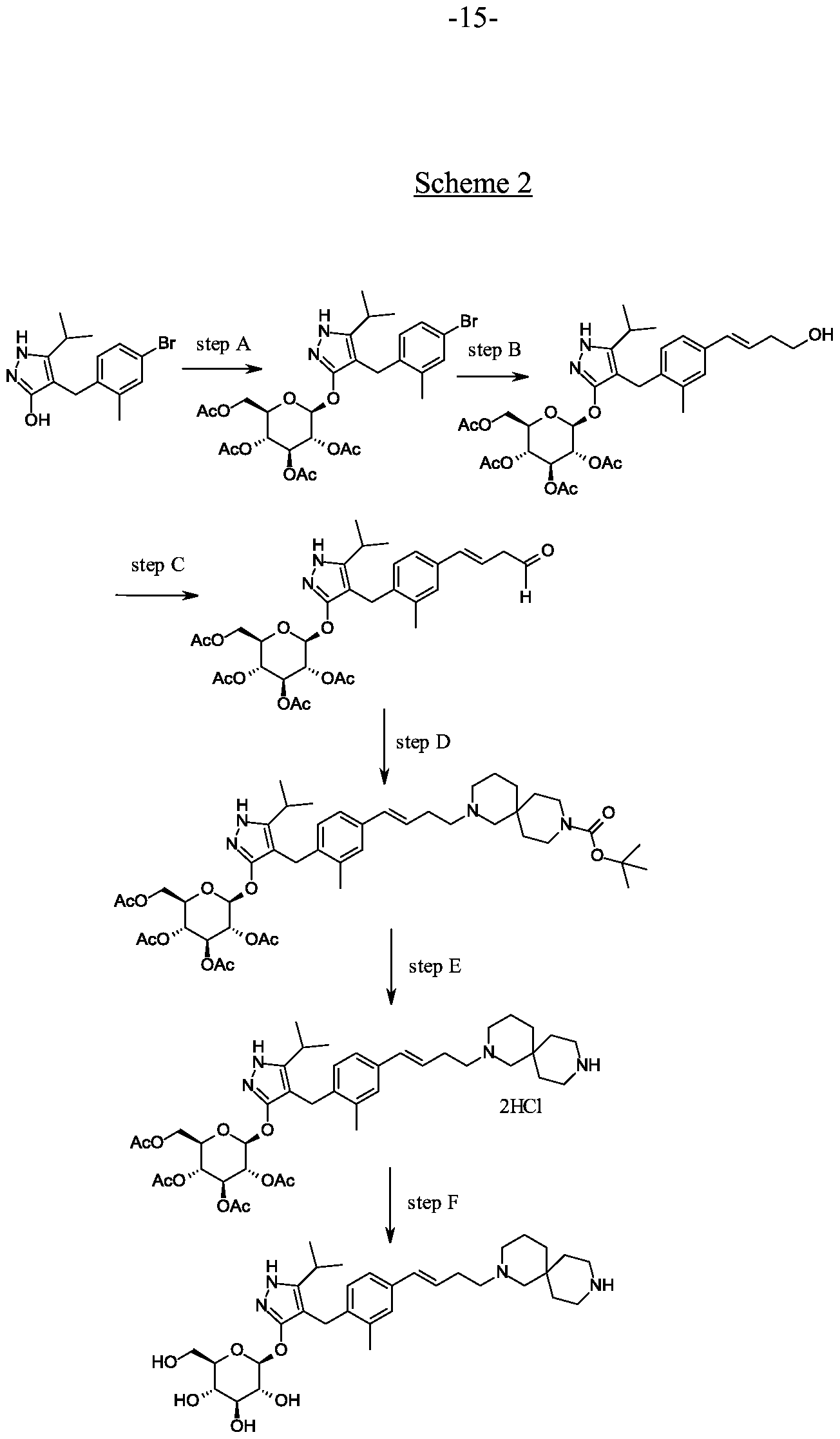
Preparation 9
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl- beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)mefhyl]-5- isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D- glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate (32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l). Preparation 10
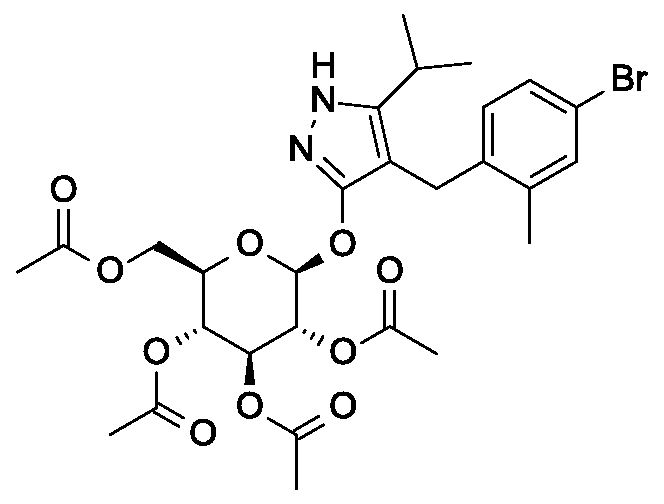
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- 1 H-pyrazol-3 -yl 2,3 ,4,6-tetra-O-acetyl-beta-D- glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol) MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside

Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
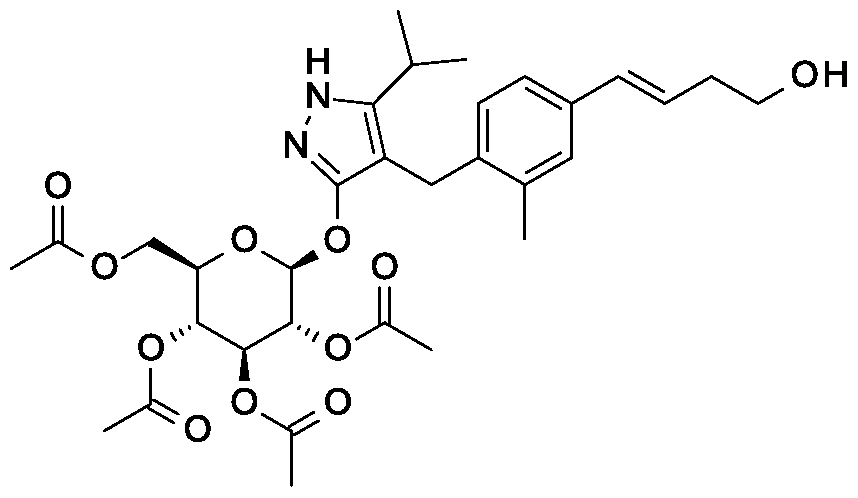
Preparation 12a
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy] -lH-pyrazol-4-yl}methyl)phenyl]but-3-en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2- dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride

Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6- tetra-0-acetyl-beta-D-glucopyranosyl)oxy] – lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 – yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Scheme 3
Preparation 14
tert-butyl 4-but-3-ynyl-4,9-diazas iro[5.5]undecane-9-carboxylate

Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgS04, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). lH NMR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H), 2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 15
tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9- diazaspiro[5.5]undecane-9-carboxylate
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield). 1H NMR (300.11 MHz, CDCI3): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H), 3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 16
tert-butyl 2-{(3£’)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH- pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9-diazaspiro [5.5]undecane-9-carboxylate

Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert- butyl 4-[(£)-4-(4,4,5 ,5 -tetramethyl- 1 ,3,2-dioxaborolan-2-yl)but-3 -enyl] -4,9- diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2- dicyclohexylphosphino-2′,4′,6′-tri-i -propyl- Ι, -biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).

Preparation 17
tert-butyl 4- [(E)-4- [4- [(3 -hydroxy-5-isopropyl- 1 H-pyrazol-4-yl)methyl] -3 -methyl- phenyl]but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate

Scheme 4, step A: Add tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (35.8 kg, 82.4 mol) in methanol (130 L) to a solution of (4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (23.9 kg, 77.3 mol) in methanol (440 L) at room temperature. Add water (590 L) and tripotassium phosphate (100 kg, 471.7 mol) and place the reaction under nitrogen atmosphere. To the stirring solution, add a suspension of
tris(dibenzylideneacetone) dipalladium (1.42 kg, 1.55 mol) and di-tert- butylmethylphosphonium tetrafluoroborate (775 g, 3.12 mol) in methanol (15 L). The resulting mixture is heated at 75 °C for 2 hours. Cool the mixture and filter over diatomaceous earth. Rinse the the filter cake with methanol (60 L), and concentrate the filtrate under reduced pressure. Add ethyl acetate (300 L), separate the layers, and wash the organic layer with 15% brine (3 x 120 L). Concentrate the organic layer under reduced pressure, add ethyl acetate (300 L), and stir the mixture for 18 to 20 hours. Add heptane (300 L), cool the mixture to 10 °C, and stir the mixture for an additional 18 to 20 hours. Collect the resulting solids by filtration, rinse the cake with ethyl acetate/heptane (2:3, 2 x 90 L), and dry under vacuum at 40°C to give the title compound (29.3 kg, 70.6% yield) as a white solid. lH NMR (400 MHz, CD3OD): δ 7.14 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 7.6 Hz, 1H), 6.39 (d, J= 16.0 Hz, 1H), 6.25-6.12 (m, 1H), 3.63 (s, 2H), 3.45-3.38 (bs, 3H), 3.34 (s, 3 H), 3.33 (s, 3H), 2.85-2.75 (m, 1H), 2.49-2.40 (m, 5 H), 2.33 (s, 3H), 1.68-1.62 (m, 2H), 1.60-1.36 (m, 15H), 1.11 (s, 3H), 1.10 (s, 3H).
Preparation 12b
Alterternative preparation of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but- 3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate.

Scheme 4, step B: Combine tert-butyl 4-[(E)-4-[4-[(3-hydroxy-5-isopropyl-lH- pyrazol-4-yl)methyl] -3-methyl-phenyl]but-3 -enyl] -4,9-diazaspiro [5.5]undecane-9- carboxylate (17.83 kg, 33.2 moles), acetonitrile (180 L), and benzyltributylammonium chloride (1.52 kg, 4.87 moles) at room temperature. Slowly add potassium carbonate (27.6 kg, 199.7 moles) and stir the mixture for 2 hours. Add 2,3,4,6-tetra-O-acetyl-alpha- D-glucopyranosyl bromide (24.9 kg, 60.55 mol), warm the reaction mixture to 30°C and stir for 18 hours. Concentrate the mixture under reduced pressure and add ethyl acetate (180 L), followed by water (90 L). Separate the layers, wash the organic phase with 15% brine (3 x 90 L), concentrate the mixture, and purify using column chromatography over silica gel (63 kg, ethyl acetate/heptanes as eluent (1 :2→1 :0)) to provide the title compound (19.8 kg, 94% purity, 68.8% yield) as a yellow foam, !H NMR (400 MHz, CDC13): δ 7.13 (s, 1H), 7.03 (d, J= 8.0 Hz, 1H), 6.78 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 16.0,
1H), 6.25-6.13 (m, 1H), 5.64 (d, J= 8.0 Hz, 1H), 5.45-5.25 (m, 2H), 5.13-4.95 (m, 2H), 4.84-4.76 (m, 1H), 4.25-4.13 (m, 2H), 4.10-4.00 (m, 2H), 3.90-3.86 (m, 1H), 3.58-3.50 (m, 2H), 3.40-3.22 (m, 4H), 2.89-2.79 (m, 1H), 2.10-1.90 (m, 18 H), 1.82 (s, 3H), 1.62- 0.82 (m, 22H).
Preparation 18
2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane

Scheme 4, step C: Combine tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)- 3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (19.6 kg, 22.6 moles) with dichloromethane (120 L) and cool to 0°C. Slowly add trifluoroacetic acid (34.6 L, 51.6 kg, 452 moles) and stir for 9 hours. Quench the reaction with ice water (80 L), and add ammonium hydroxide (85-90 L) to adjust the reaction mixture to pH (8- 9). Add dichloromethane (120 L), warm the reaction mixture to room temperature, and separate the layers. Wash the organic layer with water (75 L), brine, and concentrate under reduced pressure to provide the title compound (16.2 kg, 95.0% purity, 93% yield) as a yellow solid. lH NMR (400 MHz, CDC13): δ 7.08 (s, IH), 6.99 (d, J= 8.0 Hz, IH),
6.76 (d, J= 7.6 Hz, IH), 6.38 (d, J=15.6 Hz, IH), 6.00-5.83 (m, IH), 5.31 (d, J= 7.6 Hz, IH), 5.25-5.13 (m, 4H), 4.32 (dd, J= 12.8, 9.2 Hz, IH), 4.14 (d, J= 11.2 Hz, IH), 3.90 (d, J= 10.0 Hz, IH), 3.75-3.50 (m, 3H), 3.30-3.00 (m, 5 H), 2.85-2.75 (m, IH), 2.70-2.48 (m, 3H), 2.25 (s, IH), 2.13-1.63 (m, 19H), 1.32-1.21 (m, IH), 1.14 (s, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Example 1
Hydrated crystalline 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside acetate
First alternative preparation of 4-{4-[(l£’)-4-(2.9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl| -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (free base).
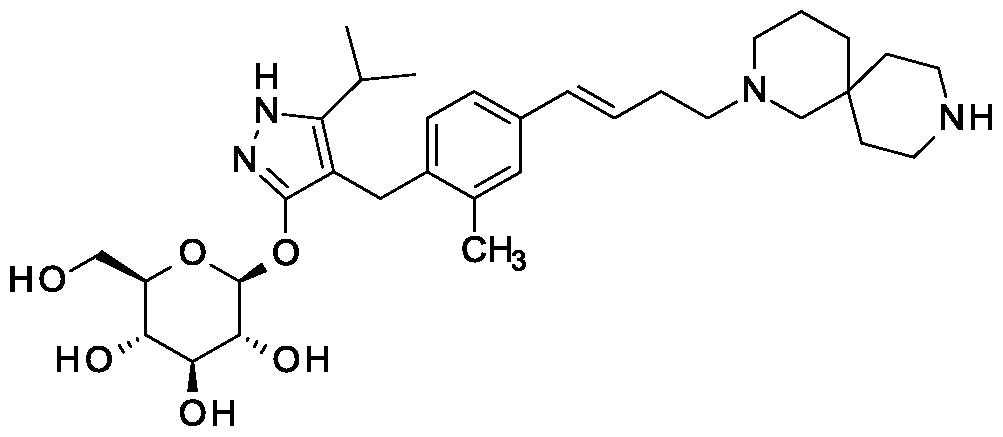
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4- {4-[( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} – 5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40°C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H.0 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Second alternative preparation of 4-{4-r(l-£’)-4-(2.9-diazaspiror5.51undec-2-yl)but-l-en- 1 -yl] -2-methylbenzyl I -5 -(propan-2-yl)- lH-pyrazol-3 -yl beta-D-glucopyranoside (free base“).

Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4- {4-[( lJE)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H20 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+l), 596.8 (M-l). 1H NMR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=l .3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.11 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Third alternative preparation of 4-{4-[(l£,)-4-(2,9-diazaspiro[5.51undec-2-yl)but-l-en-l- yll-2-methylbenzyl|-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200mL) and concentrated in vacuo. This is repeated several times to give the title compound (free base) (12.22 g, yield 96%). MS (m/z): 599 (M+l); [a]D 20 = -12 ° (C=0.2, MeOH).
Preparation of final title compound, hydrated crystalline 4-{4-|YlE)-4-(2.9- diazaspiro [5.5“|undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-vD- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.

4- {4- [(1 E)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (902 mg) is placed in a round bottom flask (100 mL) and treated with wet ethyl acetate (18 mL). [Note – wet ethyl acetate is prepared by mixing ethyl acetate (100 mL) and dionized water (100 mL). After mixing, the layers are allowed to separate, and the top wet ethyl acetate layer is removed for use. Acetic acid is a hydrolysis product of ethyl acetate and is present in wet ethyl acetate.] The compound dissolves, although not completely as wet ethyl acetate is added. After several minutes, a white precipitate forms. An additional amount of wet ethyl acetate (2 mL) is added to dissolve remaining compound. The solution is allowed to stir uncovered overnight at room temperature during which time the solvent partially evaporates. The remaining solvent from the product slurry is removed under vacuum, and the resulting solid is dried under a stream of nitrogen to provide the final title compound as a crystalline solid. A small amount of amorphous material is identified in the product by solid-state NMR. This crystalline final title compound may be used as seed crystals to prepare additional crystalline final title compound.
Alternative preparation of final title compound, hvdrated crystalline 4-{4-[(lE)-4-(2.,9- diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-yl)- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.
Under a nitrogen atmosphere combine of 4-{4-[(lE)-4-(2,9-diazaspiro[5.5]undec- 2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan-2-yl)- 1 H-pyrazol-3-yl 2,3,4,6-tetra-O- acetyl-beta-D-glucopyranoside (2.1 kg, 2.74 mol), methanol (4.4 L), tetrahydrofuran (4.2 L), and water (210 mL). Add potassium carbonate (460 g, 3.33 moles) and stir for four to six hours, then filter the reaction mixture to remove the solids. Concentrate the filtrate under reduced pressure, then add ethanol (9.0 L) followed by acetic acid (237 mL, 4.13 mol) and stir at room temperature for one hour. To the stirring solution add wet ethyl acetate (10 L, containing approx. 3 w/w% water) slowly over five hours, followed by water (500 mL). Stir the suspension for twelve hours and add wet ethyl acetate (4.95 L, containing approx. 3 w/w% water) over a period of eight hours. Stir the suspension for twelve hours and add additional wet ethyl acetate (11.5 L, containing approx. 3 w/w% water) slowly over sixteen hours. Stir the suspension for twelve hours, collect the solids by filtration and rinse the solids with wet ethyl acetate (3.3 L, containing approx. 3 w/w% water). Dry in an oven under reduced pressure below 30°C to give the title compound as an off-white crystalline solid (1.55 kg, 2.35 mol, 96.7% purity, 72.4 w/w% potency, 68.0% yield based on potency). HRMS (m/z): 599.3798 (M+l).
PATENT
CN105705509
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN175101669&tab=PCTDESCRIPTION
The present invention is in the field of treatment of diabetes and other diseases and conditions associated with hyperglycemia. Diabetes is a group of diseases characterized by high blood sugar levels. It affects approximately 25 million people in the United States, and according to the 2011 National Diabetes Bulletin, it is also the seventh leading cause of death in the United States (US Department of Health and Human Resources Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 can result in a decrease in glucose absorption in the small intestine, thus providing a useful method of treating diabetes.
Alternative medicines and treatments for diabetes are needed. The present invention provides an acetate salt of a pyrazole compound which is an SGLT1 inhibitor, and thus it is suitable for treating certain conditions such as diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives having human SGLT1 inhibitory activity, which are also disclosed for use in the prevention or treatment of diseases associated with hyperglycemia, such as diabetes. Moreover, WO 2011/039338 discloses certain pyrazole derivatives having SGLT1/SGLT2 inhibitor activity, which are also disclosed for use in the treatment of bone diseases such as osteoporosis.
PATENT
WO-2019141209
Process for preparing pyranoglucose-substituted pyrazole compound, used as a pharmaceutical intermediate in SGLT inhibitor for treating diabetes.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9573970 | 4–5-(PROPAN-2-YL)-1H-PYRAZOL-3-YL BETA-D GLUCOPYRANOSIDE ACETATE | 2014-10-30 | 2016-07-28 |
/////////////SY-008 , SY 008 , SY008, ELI LILY, PHASE 1, GLT1 inhibitor, type 2 diabetes, Yabao Pharmaceutical, CHINA, DIABETES
CC(=O)O.Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4O
Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4
O

