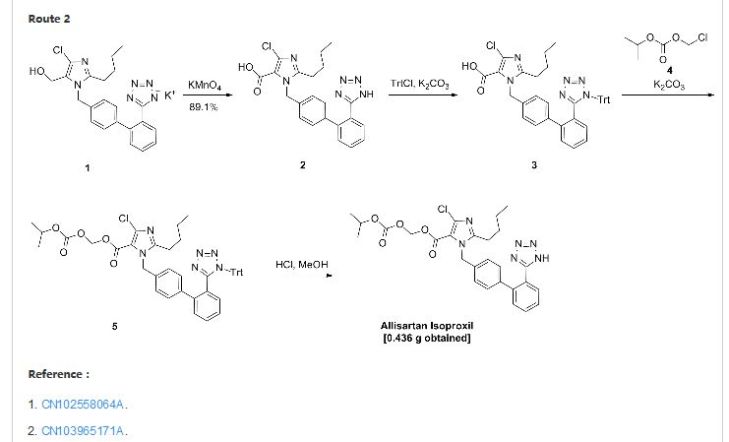
cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it

1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
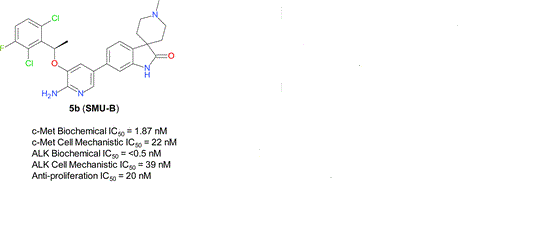
A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
ACS Med. Chem. Lett., 2013, 4 (8), pp 806–810
DOI: 10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE
CN 101851237
南方医科大学
1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set
obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie
The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one
Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one
As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one
In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N
Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P
Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P
The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P
Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one
H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,


P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

COCK WILL TEACH YOU NMR
 COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com
SWEDEN
Alfred Nobel had the unpleasant surprise of reading his own obituary, titled The merchant of death is dead, in a French newspaper.

Nyköping (Sweden)-houses.

Fjallbacka, a colorful fishing Village along the west coast of Sweden

Knights Island, Stockholm, Sweden

Stockholm, Sweden

Sweden Stockholm

Europe Örby Änger – Sweden

Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





























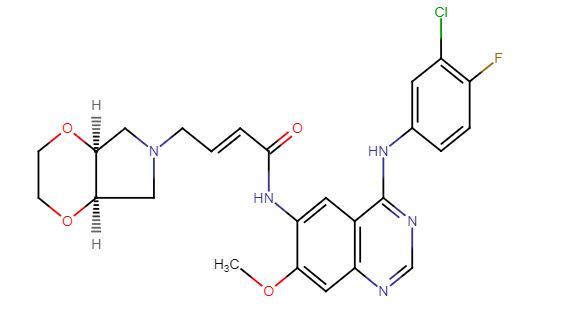
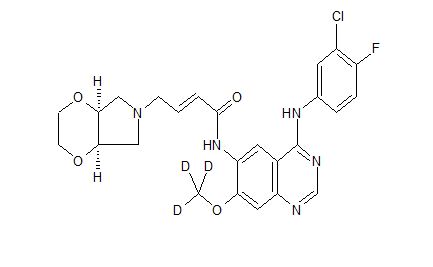
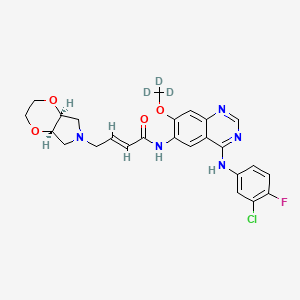















 Chidamide (Epidaza) is an
Chidamide (Epidaza) is an 



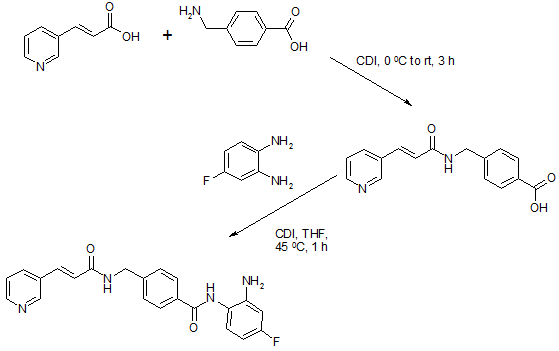
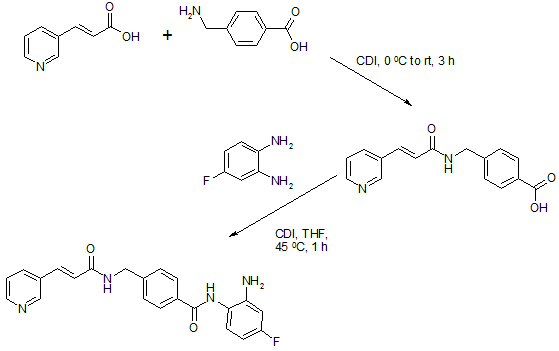




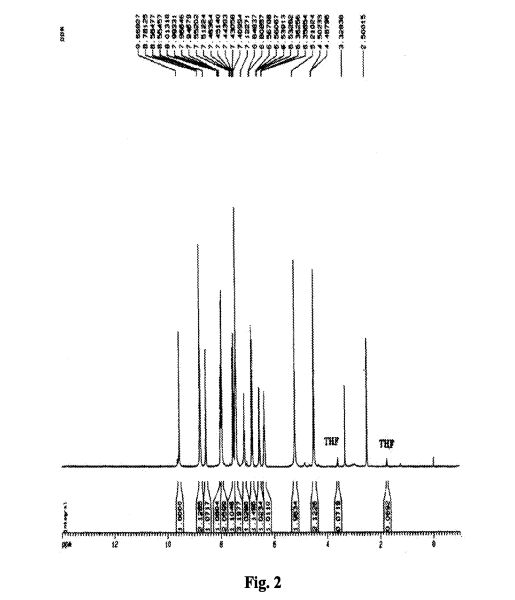
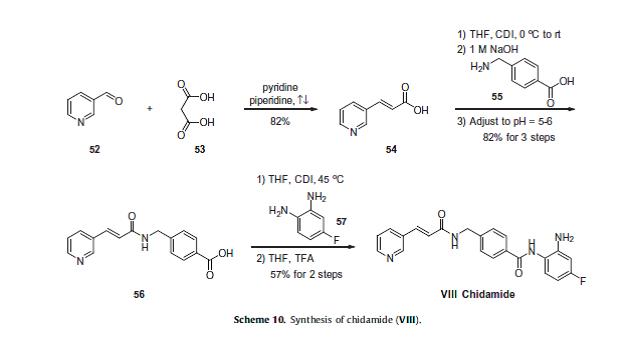
 To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.
To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.


















 CHINA FLAG
CHINA FLAG


 .
.


 AMITIZA (lubiprostone)
AMITIZA (lubiprostone)










 CHINA
CHINA