
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















-Facebook.png)









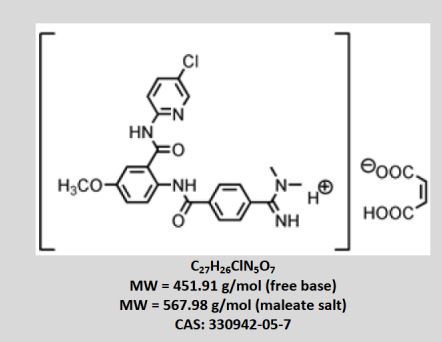
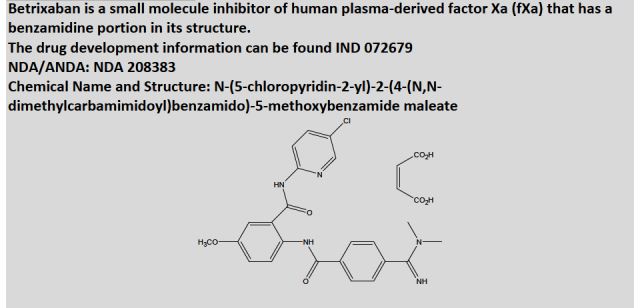



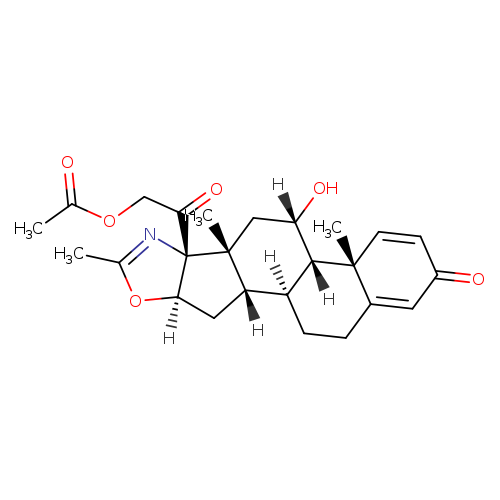
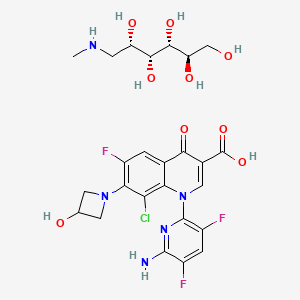
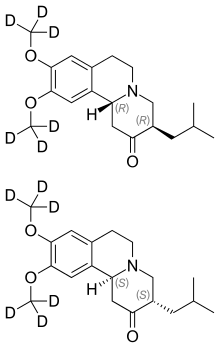































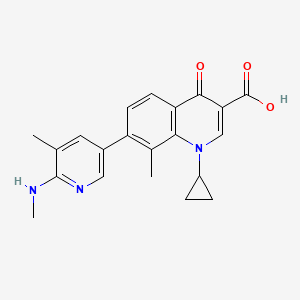





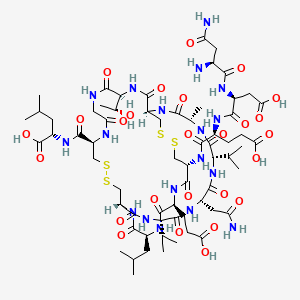



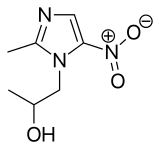


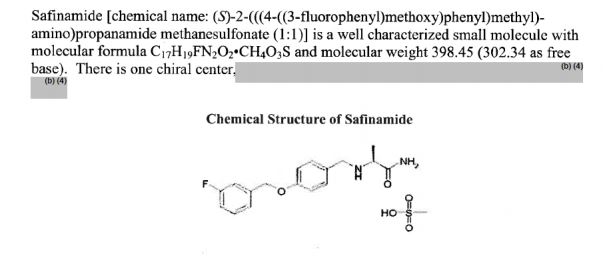






































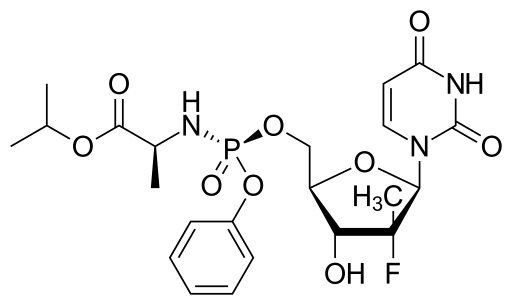











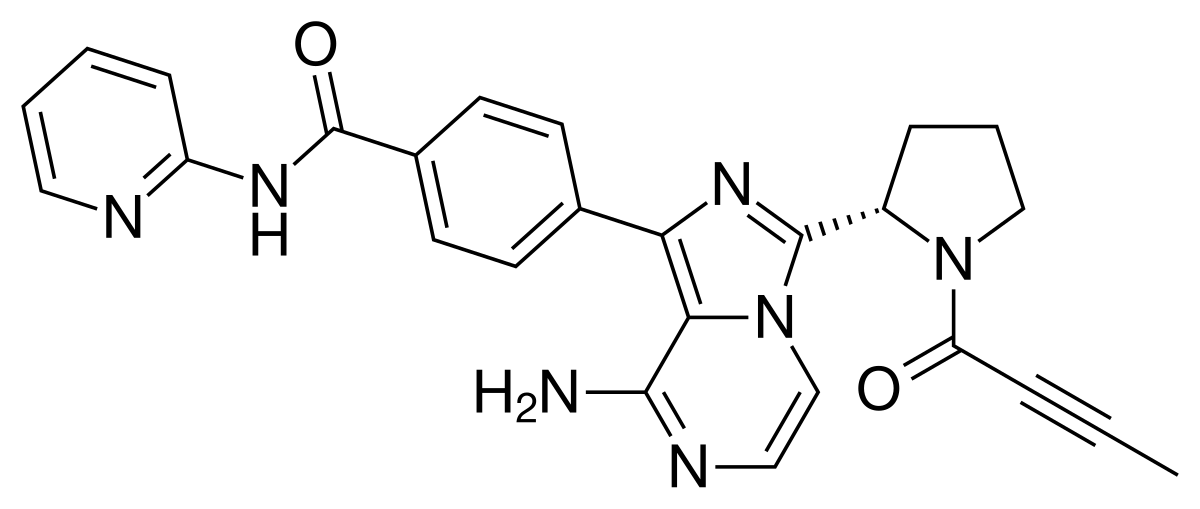
 approved by the FDA")












(WO2015139602) 2′-SUBSTITUTED-2,2′-DEHYDRATED URIDINE OR 2′-SUBSTITUTED-2,2′-DEHYDRATED CYTIDINE COMPOUND AND PREPARATION METHOD AND USE THEREOF
ZHANG, Rongxia
A further object of the present invention to provide a method for preparing a compound of formula I.
The present invention provides a process for preparing a compound I 2′-deoxy-2′-fluoro-2′-substituted uridine or 2′-deoxy-2′-fluoro-cytidine using the following formula or 2′-deoxy-2′-substituted 2′-2′-substituted nitrile or uridine 2′-deoxy-2′-substituted-2′-carbonitrile The method of cytidine compound,
2′-deoxy-2′-fluoro-2′-methyl-uridine (IIIa) is the preparation of anti-hepatitis C drugs Sofosbuvir key intermediate.
Sofosbuvir developed by Gilead Science Company, FDA on December 6, 2013 Sofosbuvir formally approved for the treatment of chronic hepatitis C virus (HCV) infection. Sofosbuvir is first used to treat certain types of HCV infection without the use of interferon effective and safe drugs. Clinical trials have shown, sofosbuvir can achieve very high proportion of sustained virologic response (clinical cure). More revolutionary breakthrough that, sofosbuvir without joint peginterferon α situation is still very significant effect, such as sofosbuvir ribavirin genotype 2 and genotype 3 patients with previously untreated chronic hepatitis C continued virological response rate of 100%. Sofosbuvir is a prodrug is metabolized in vivo to 2′-deoxy-2′-fluoro-2′-methyl-uridine-5′-monophosphate.
Currently reported 2′-deoxy-2′-fluoro-2′-methyl uridine synthetic methods are as follows:
In the literature (Journal of Medicinal Chemistry, 2005,48,5504) in order cytidine as a raw material, first selectively protected 3 ‘, 5′-hydroxyl group, and then oxidizing the 2′-hydroxyl to a carbonyl group, and the reaction of methyllithium get the 2’-hydroxyl compound, and then removing the protective group, use benzoyl protected 3 ‘, 5’-hydroxyl group, and then reacted with DAST fluorinated compound, followed by hydrolysis and aminolysis reaction products, such as the following Reaction Scheme. The method of route length, the need to use expensive silicon ether protecting group molecule relatively poor economy; conducting methylation time will generate a non-methyl enantiomer beta bits.

In Patent (WO2005003147, WO2006031725A2, US20040158059) using 2′-fluoro-2′-methyl – ribose derivative with N- benzoyl cytosine for docking the reaction, then after the hydrolysis, aminolysis reaction to obtain the final product, As shown in the following reaction scheme. Raw material of the process is not readily available, synthetic steps cumbersome, expensive; the reaction product obtained contained docking base for the alpha position isomers, need purification removed to form waste.
SUMMARY OF THE INVENTION
The present inventors have designed and synthesized a compound of formula I as shown, the compound may be a fluorinated or nitrile reaction of 2′-deoxy-2′-fluoro-2′-get-substituted uridine or 2 under appropriate conditions’ – 2′-deoxy-2′-fluoro-2′-deoxy-2′-substituted cytidine or nitrile uridine or 2′-substituted-2′-deoxy-2′-substituted-2′-cytidine nitrile compound; or a compound of formula I or a nitrile group by fluoro reaction, followed by deprotection reaction to give 2′-deoxy-2′-fluoro-2′-substituted uridine or 2′-deoxy-2′-fluoro–2 ‘- cytidine or 2′-substituted-2′-deoxy-2′-nitrile-substituted uridine or 2′-deoxy-2′-substituted-2′-cytidine compound nitrile group; or a compound of formula I through the opening cyclization reaction, and then through the group of fluoro or nitrile, and finally deprotection reaction to give 2′-deoxy-2′-fluoro-2′-substituted uridine or 2′-deoxy-2′-fluoro-2’-substituted Cellular glycoside or 2 ‘substituted-2′-deoxy-2′-carbonitrile 2′-deoxy-uridine or 2′-substituted-2’-cytidine compound nitrile group; or a compound of formula I through a ring-opening reaction, and then 2 ‘- hydroxyl forming a leaving group, and then after a nitrile group or a fluorinated reaction, the final deprotection reaction of 2′-deoxy-2′-fluoro-2′-substituted uridine or 2′-deoxy-2′- cytidine or 2′-fluoro-2′-substituted-2′-deoxy-2′-nitrile-substituted uridine or 2′-deoxy-2′-substituted-2’-cytidine nitrile compound.
It is therefore an object of the present invention is to provide a compound of the general formula I prepared 2′-deoxy-2′-fluoro-2′-substituted uridine or 2′-deoxy-2′-fluoro-2′-substituted cytidine or 2′-substituted-2′-deoxy-2′-carbonitrile uridine or 2′-deoxy-2′-substituted-2′-carbonitrile The method of cytidine compound.
Example 1:
The 2′-C- methyl uridine (18.4g, 0.07mol), N, N’- carbonyldiimidazole (216.2g, 0.10mol), sodium bicarbonate (8.4g, 0.10mol) was suspended N, N- two dimethylformamide (50ml), the temperature was raised to 130 ℃, reaction for 4 hours, cooled and filtered to remove inorganic salts, the filtrate was added ethyl acetate (200ml), analyze the material at room temperature, suction filtered, washed with ethyl acetate cooled to, drying to give a yellow solid (19.9g, yield: 83%).
Ia: 1 H NMR (300 MHz, CD 3 OD): [delta] 7.80 (d, 1H, J = 7.5 Hz), 6.05 (d, 1H, J = 7.5 Hz), 5.91 (s, 1H), 4.34 (d, 1H, J = 4.8Hz), 4.07 (m, 1H), 3.56 (m, 2H), 1.63 (s, 3H); ESI-MS m / z (M + 1) 241.
Example 2:
The compound of Example 1 Ia (0.24g, 1mmol)) was dissolved in 70% HF in pyridine was heated to 140 ~ 150 ℃, stirred for 3 hours, cooled and the solvent was removed under reduced pressure, the residue was added acetone, beating, and filtered to give solid (0.18g, yield: 70%).
IIIa: 1 H NMR (300 MHz, DMSO-d 6 ): [delta] 11.48 (s, 1H), 7.82 (d, 1H, J = 6.0 Hz), 6.00 (d, 1H, J = 15.6 Hz), 5.67 (m , 2H), 5.30 (s, 1H), 3.85 (m, 3H), 3.62 (s, 1H), 1.25 (d, 3H, J = 16.8Hz), ESI-MS m / z (M-1) 259.
Example 3:
Compound Ib (0.45g, 1mmol) was dissolved in a mixture of dichloromethane and pyridine, was added DAST (0.32g), stirred for 24 hours, added dichloromethane (20ml) was diluted with water (30ml × 2), dried over anhydrous dried over sodium sulfate, filtered and the solvent removed under reduced pressure to give the residue was subjected to column chromatography to give the product (0.36g, yield: 78%).
IIa: 1 H NMR (400 MHz, CDCl 3 and DMSO-d 6 ): [delta] 7.99 (d, J = 7.6 Hz, 2H), 7.90 (d, J = 7.6 Hz, 2H), 7.34 ~ 7.61 (m, 7H ), 6.10 (brs, 1H), 5.64 (brs, 1H), 5.42 (d, J = 8.0Hz, 1H), 4.53-4.68 (m, 3H), 1.40 (d, J = 22.8Hz, 3H); ESI -MS m / z (M + 1) 469.
Example 4:
The compound of Example 3 IIa (0.47g, 1mmol) dissolved in 10% methanol solution of ammonia and stirred overnight, the solvent was removed under reduced pressure, and the residue was slurried in ethyl acetate, filtered to give a white solid (0.2g, yield : 77%).
IIIa: 1 H NMR (300 MHz, DMSO-d 6 ): [delta] 11.48 (s, 1H), 7.82 (d, 1H, J = 6.0 Hz), 6.00 (d, 1H, J = 15.6 Hz), 5.67 (m , 2H), 5.30 (s, 1H), 3.85 (m, 3H), 3.62 (s, 1H), 1.25 (d, 3H, J = 16.8Hz), ESI-MS m / z (M-1) 259.
Example 5:
Compound IVa (0.57g, 1mmol) was dissolved in dichloroethane (20ml) was added trifluoromethanesulfonic acid trimethylsilyl ester (1ml), was heated for 12 hours, cooled, and the reaction solution was concentrated dryness, added two dichloromethane (100ml) was dissolved, washed successively with water (50ml) and saturated brine (50ml), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated to dryness to give an oil which was purified by column chromatography to give a white solid (0.3g, yield : 67%).
Ib: 1 H NMR (300 MHz, CDCl 3 ): δ7.96-8.10 (m, 6H), 7.41-7.65 (m, 9H), 7.32 (d, 1H, J = 5.4 Hz), 6.09 (d, 1H, J = 5.4Hz), 5.79 (m, 2H), 4.67 (m, 1H), 4.48 (m, 2H), 1.81 (s, 3H); ESI-MS m / z (M-1) 447.
Example 6:
N The compound of Example 1 Ia (1.3g, 5.4mmol) dissolved in dry, N- dimethylformamide (10ml) was added p-toluenesulfonic acid monohydrate (1.12g, 5.9mmol) and 3,4- dihydropyran (1.28ml, 14.04mmol), The reaction was stirred for 5 hours at room temperature, water was added and the methylene chloride solution was separated, the organic layer was concentrated and purified by silica gel chromatography to give the product 1.3g.
Ic: 1 H NMR (300 MHz, CDCl 3 ): [delta] 7.29 (m, 1H), 6.08 (m, 1H), 5.61 (m, 1H), 4.33-4.72 (m, 4H), 3.37-3.90 (m, 6H), 1.43-1.82 (m, 12H), 1.25 (s, 3H); ESI-MS m / z (M + 1) 427.
Example 7:
The solvent was removed, the residue was purified compound of Example 6 Ic (0.43g, 1mmol) was dissolved in 70% HF in pyridine was heated to 100 ~ 120 ℃, stirred for 5 hours, cooled, reduced pressure was purified through silica gel column to give a solid ( 0.18g, yield: 72%).
IIIa: 1 H NMR (300 MHz, DMSO-d 6 ): [delta] 11.48 (s, 1H), 7.82 (d, 1H, J = 6.0 Hz), 6.00 (d, 1H, J = 15.6 Hz), 5.67 (m , 2H), 5.30 (s, 1H), 3.85 (m, 3H), 3.62 (s, 1H), 1.25 (d, 3H, J = 16.8Hz), ESI-MS m / z (M-1) 259.
Example 8:
The compound of Example 6 Ic (50mg, 0.122mmol) was dissolved in methanol (1ml) was added 1N sodium hydroxide solution (0.2ml), stirred at room temperature overnight, water was added and the methylene chloride solution was separated, the organic layer was concentrated after purified by column chromatography to give the product (45mg, yield: 87%).
VA: 1 H NMR (300 MHz, CDCl 3 ): [delta] 7.89 (d, 1H, J = 4.5Hz), 6.01 (s, 1H), 5.95 (d, 1H, J = 4.5Hz), 5.65 (m, 2H ), 4.73 (m, 3H), 4.59 (m, 1H), 3.52-4.30 (m, 4H), 1.56-1.80 (m, 12H), 1.32 (s, 3H); ESI-MS m / z (M + 35) 461.
Example 9:
The mixture of Example 8 Compound Va (0.43g, 1mmol) was dissolved in dichloromethane and pyridine, was added DAST (0.32g), stirred for 24 hours, added dichloromethane (20ml) was diluted with water (30ml × 2) and washed , dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain compound IIb. Compound IIb is dissolved in methanol (10ml) was added p-toluenesulfonic acid (200mg), stirred for 6 hours at room temperature, the methanol was removed under reduced pressure, silica gel column chromatography to give the product IIIa (180mg, yield: 75%).
IIIa: 1 H NMR (300 MHz, DMSO-d 6 ): [delta] 11.48 (s, 1H), 7.82 (d, 1H, J = 6.0 Hz), 6.00 (d, 1H, J = 15.6 Hz), 5.67 (m , 2H), 5.30 (s, 1H), 3.85 (m, 3H), 3.62 (s, 1H), 1.25 (d, 3H, J = 16.8Hz), ESI-MS m / z (M-1) 259.
Example 10:
The 2′-C- methyl uridine (0.2g, 0.8mmol) was dissolved in N, N- dimethylformamide (4ml) was added N, N’- carbonyldiimidazole (0.194g, 1.2mmol) and sodium bicarbonate (55mg, 0.66mmol), was heated to 130 ℃, stirred for 4 hours, cooled and the solvent was removed under reduced pressure, and the residue was dissolved in 70% HF in pyridine was heated to 140 ~ 150 ℃, stirred for 3 hours, cooled, The solvent was removed under reduced pressure, the residue was added to acetone and filtered to obtain a solid IIIa (0.12g, yield: 60%).
Example 11:
The 2′-C- methyl uridine (0.2g, 0.8mmol) was dissolved in N, N- dimethylformamide (4ml) was added diphenyl carbonate (0.256g, 1.2mmol) and sodium bicarbonate ( 55mg, 0.66mmol), was heated to 150 ℃, stirred for 6 hours, cooled and the solvent was removed under reduced pressure, and the residue was dissolved in 70% HF in pyridine was heated to 140 ~ 150 ℃, stirred for 3 hours, cooled and the solvent was removed under reduced pressure The residue was added to acetone and filtered to obtain a solid IIIa (0.13g, yield: 65%).
Example 12:
Under nitrogen, the compound of Example 9 Example Va (4.26g, 10mmol) was dissolved in dry tetrahydrofuran (100ml) was added triethylamine (6g, 60mmol), cooled to -78 ℃, was added trifluoromethanesulfonic anhydride (4.23g , 15mmol), stirred for 1 hour, the reaction system was added saturated ammonium chloride solution, extracted three times with methylene chloride, organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and the residue was subjected to silica gel column chromatography to give the product Vb ( 4g, yield: 72%). ESI-MS m / z (M-1) 557.
Compound Vb (4g) was dissolved in dry tetrahydrofuran, was added tetrabutylammonium fluoride (1.87g, 7.1mmol), warmed to reflux, cooled to room temperature after heating for 1 hour, water was added to the reaction system, and extracted with methylene chloride three times, the combined organic phase was dried over anhydrous sodium sulfate, concentrated, and the residue was subjected to silica gel column chromatography to give the product IIb (2.7g, yield: 88%). ESI-MS m / z (M-1) 427.
Compound IIb (2.7g) was dissolved in methanol (20ml) was added 3M hydrochloric acid (10ml), 50 ℃ stirred for 8 hours, and concentrated to give a solid, was added acetonitrile, beating, and filtered to give the product IIIa (1g, yield: 61%).
IIIa: 1 H NMR (300 MHz, DMSO-d 6 ): [delta] 11.48 (s, 1H), 7.82 (d, 1H, J = 6.0 Hz), 6.00 (d, 1H, J = 15.6 Hz), 5.67 (m , 2H), 5.30 (s, 1H), 3.85 (m, 3H), 3.62 (s, 1H), 1.25 (d, 3H, J = 16.8Hz), ESI-MS m / z (M-1) 259.
UPDATE DEC2015………….








