
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
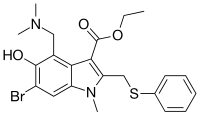
Umifenovir
- Molecular FormulaC22H25BrN2O3S
- Average mass477.414 Da
Umifenovir[2] (trade names Arbidol Russian: Арбидол, Chinese: 阿比朵尔) is an antiviral treatment for influenza infection used in Russia[3] and China. The drug is manufactured by Pharmstandard (Russian: Фармстандарт). Although some Russian studies have shown it to be effective, it is not approved for use in other countries. It is not approved by FDA for the treatment or prevention of influenza.[4] Chemically, umifenovir features an indole core, functionalized at all but one positions with different substituents. The drug is claimed to inhibit viral entry into target cells and stimulate the immune response. Interest in the drug has been renewed as a result of the SARS-CoV-2 outbreak.
Umifenovir is manufactured and made available as tablets, capsules and syrup.
.jpg)
Status
Testing of umifenovir’s efficacy has mainly occurred in China and Russia,[5][6] and it is well known in these two countries.[7] Some of the Russian tests showed the drug to be effective[5] and a direct comparison with Tamiflu showed similar efficiency in vitro and in a clinical setting.[8] In 2007, Arbidol (umifenovir) had the highest sales in Russia among all over-the-counter drugs.
Mode of action
Biochemistry
Umifenovir inhibits membrane fusion.[3] Umifenovir prevents contact between the virus and target host cells. Fusion between the viral envelope (surrounding the viral capsid) and the cell membrane of the target cell is inhibited. This prevents viral entry to the target cell, and therefore protects it from infection.[9]
Some evidence suggests that the drug’s actions are more effective at preventing infections from RNA viruses than infections from DNA viruses.[10]
As well as specific antiviral action against both influenza A and influenza B viruses, umifenovir exhibits modulatory effects on the immune system. The drug stimulates a humoral immune response, induces interferon-production, and stimulates the phagocytic function of macrophages.[11]
Clinical application
Umifenovir is used primarily as an antiviral treatments for influenza. The drug has also been investigated as a candidate drug for treatment of hepatitis C.[12]
More recent studies indicate that umifenovir also has in vitro effectiveness at preventing entry of Ebolavirus Zaïre Kikwit, Tacaribe arenavirus and human herpes virus 8 in mammalian cell cultures, while confirming umifenovir’s suppressive effect in vitro on Hepatitis B and poliovirus infection of mammalian cells when introduced either in advance of viral infection or during infection.[13][14]
Research
In February 2020, Li Lanjuan, an expert of the National Health Commission of China, proposed using Arbidol (umifenovir) together with darunavir as a potential treatment during the 2019–20 coronavirus pandemic.[15] Chinese experts claim that preliminary tests had shown that arbidol and darunavir could inhibit replication of the virus.[16][17] So far without additional effect if added on top of recombinant human interferon α2b spray.[18]
Side effects
Side effects in children include sensitization to the drug. No known overdose cases have been reported and allergic reactions are limited to people with hypersensitivity. The LD50 is more than 4 g/kg.[19]
Criticism
In 2007, the Russian Academy of Medical Sciences stated that the effects of Arbidol (umifenovir) are not scientifically proven.[20]
Russian media criticized lobbying attempts by Tatyana Golikova (then-Minister of Healthcare) to promote umifenovir,[21] and the unproven claim that Arbidol can speed up recovery from flu or cold by 1.3-2.3 days.[22] They also debunked claims that the efficacy of umifenovir is supported by peer-reviewed studies.[23][24]
PAPER
Bioorganic & medicinal chemistry (2017), 25(1), 327-337.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84983071&_cid=P12-KZB0QP-98324-1
| Arbidol hydrochloride, chemical name: 6-bromo-4-(dimethylaminomethyl)-5-hydroxy-1-methyl-2-(phenylthiomethyl)-1H- Indole-3-carboxylic acid ethyl ester hydrochloride, the structural formula is as follows: |
| |
| Arbidol hydrochloride is an antiviral drug developed by the Soviet Medicinal Chemistry Research Center. It was first listed in Russia in 1993. It is used as a monohydrate for medicinal purposes. This product not only has immunomodulatory and interferon-inducing effects, but also has good anti-influenza virus activity, and is clinically used for the prevention and treatment of influenza and other acute viral respiratory tract infections. |
| The preparation of Arbidol hydrochloride has multiple synthetic routes, Chinese patent CN1687033A and Wang Dun, Wu Xiujing, Gong Ping’s “Synthesis of Arbidol Hydrochloride” bibliographical report in Chinese Pharmaceutical Industry Magazine 2004,35(8) are Taking p-benzoquinone and 3-aminocrotonic acid ethyl ester as starting materials, through Neitzescu reaction, O-acylation, N-alkylation, bromination, thiophenol reaction, Mannich amine methylation reaction, hydrochloric acid acidification to obtain hydrochloric acid Arbidol, the total reaction yield was 22.9%. |
| The synthetic route is as follows: |
| |
| The Nenitzescu reaction used in the synthesis of indole rings in this method, the reaction yield of this step is about 60%, resulting in a total yield of 22.9%. |
| U.S. Patent US5198552 and World Patent WO9008135 reported that 5-hydroxy-1,2-dimethylindole-3-ethyl carboxylate was used as raw material, and arbidol hydrochloride was prepared through bromination, condensation, Mannich reaction and salt-forming reaction you. |
| |
| Although the synthesis steps of this method are short, the raw material 5-hydroxy-1,2-dimethylindole-3-carboxylic acid ethyl ester is not easy to obtain, and the large-scale application is difficult. |
| Song Yanling, Zhao Yanfang, Gong Pingren reported in the 3rd National Symposium on Pharmaceutical Engineering Technology and Education “Synthesis Research on Arbidol Hydrochloride” in the literature report using thiophenol as the starting material, and chloroacetoacetic acid. After the substitution reaction of the ethyl ester, the thiophenyl fragment in the molecule is introduced, which is then condensed with methylamine, followed by the Neitzescu reaction with p-benzoquinone, and the dimethylamine methyl group is introduced through the Mannich reaction. Reaction, then carry out deprotection reaction, and finally obtain the final product Arbidol hydrochloride through salification reaction |
| Its synthetic route is as follows: |
| |
| Since the Nenitzescu reaction yield in this method is only 33.7%, the total yield is only 11.2%. |
| There are also bibliographical reports (Wen Yanzhen, Gao Zhiwei, Wei Wenlong, Zhi Cuimei, Wang Qi etc. in China Pharmaceutical Industry Journal 2006, “The Synthetic Route Diagram of Arbidol Hydrochloride” reported in 2006,37(12)) is based on ethyl acetoacetate. Ester and methylamine are used as starting materials, and Arbidol hydrochloride is obtained by Neitzescu reaction, acylation to protect hydroxyl group, bromination, thiophenol reaction, Mannich reaction, and acidification. |
| The synthetic route is as follows: |
| |
| The method has relatively mild reaction conditions and relatively easy-to-obtain raw materials, but the total yield is still low, about 20%. |
| The above synthesis methods of Arbidol hydrochloride all use the Nenitzescu indole ring synthesis method to synthesize the indole ring of Arbidol hydrochloride, resulting in a low total reaction yield of about 10% to 20%. |
| SUMMARY OF THE INVENTION |
| In view of the above-mentioned problems, the object of the present invention is to provide a preparation method of Arbidol hydrochloride, the raw materials are easy to obtain, the reaction technical conditions are relatively simple, the reaction conditions are mild, and the total reaction yield is relatively high, reaching more than 30%. The cost is low, and it is suitable for industrial production. The method of the invention is based on the starting material of 3-iodo-4-nitrophenol, which is protected by a hydroxyl group, synthesized by indole ring, N-methylated, brominated, thiophenolated, and Mannich amine. Methylation reaction, acidification with hydrochloric acid, and purification to obtain Arbidol hydrochloride. |
| The reaction formula of the inventive method is as follows: |
| |
| |
| Fe stands for iron powder |
| CH 3 COOH stands for acetic acid |
| H 2 O is for water |
| (CH 3 ) 2 SO 4 Represents dimethyl sulfate |
| K 2 CO 3 stands for potassium carbonate |
| Example 1: |
| A preparation method of Arbidol hydrochloride, its steps are (preparation of compound 1): |
| A. Preparation of compound 1: 53 g of 3-iodo-4-nitrophenol was added to 160 g of acetone (drying over anhydrous potassium carbonate), 30.3 g of triethylamine was added, and 37.7 g of triethylamine was added dropwise at room temperature (20-25° C., the same below). g acetyl chloride, dripped in 1 hour, the reaction solution was automatically raised to reflux temperature T=56°C, reacted for 0.5h, cooled to room temperature T=25°C naturally, the reaction solution was poured into 1000g ice water, stirred, filtered, and the filter cake was washed with water , and vacuum-dried to obtain 57.4 g of compound 1 crude product with a yield of 93.6%. The next reaction was carried out directly without further purification. |
| B. Preparation of compound 2: 48.6 g of ethyl acetoacetate and 180 ml of freshly distilled tetrahydrofuran were added to a dry flask. Over 2 hours, 41.9 g of potassium tert-butoxide was added in portions with stirring. The temperature was raised to T=70°C (reflux), and the solution of 57.4 g of compound 1 obtained in the step and 75 mL of freshly distilled tetrahydrofuran was added dropwise, and the drop was completed in 2 hours. TLC plates monitor the reaction endpoint. After the reaction mixture was cooled to room temperature T=25°C, 93.5 ml of a 4 mol/L hydrochloric acid solution was added dropwise. The precipitated potassium chloride was removed by filtration, the solvent was evaporated under reduced pressure, and the obtained solid was washed with 45 mL of water and 60 mL of petroleum ether in turn to obtain 56.6 g of a crude product of compound 2 with a yield of 98%. The crude product can be recrystallized from the mixed solution of petroleum ether and ethyl acetate to obtain pure product. |
| C. Preparation of compound 3: add 56.6 g of compound 2, 160 mL of acetic acid and 160 mL of water to the flask, stir under nitrogen protection, add 30.8 g of iron powder in batches, stir vigorously, and heat the reaction mixture to T=80 °C for 4 h. End (TLC plate detection). Iron and its oxides were removed by filtration, water and acetic acid were distilled off under reduced pressure, neutralized with saturated sodium carbonate solution to weakly alkaline, extracted with ethyl acetate, dried over anhydrous magnesium sulfate, and concentrated to obtain 44.1 g of compound 3 crude product in a yield of 44.1 g. 92.3%. |
| D. Preparation of compound 3: add 10.0 g of compound 2, 28 mL of acetic acid and 28 mL of water to the flask, stir under nitrogen protection, add 7.2 g of iron powder in batches, stir vigorously, and simultaneously heat the reaction mixture to T=80 ° C, 4h reaction End (TLC plate detection). Iron and its oxides were removed by filtration, water and acetic acid were evaporated under reduced pressure, neutralized with saturated sodium carbonate solution to weakly alkaline, extracted with ethyl acetate, dried over anhydrous magnesium sulfate, and concentrated to obtain 7.5 g of compound 3 crude product, yield 89.4%. |
| E. Preparation of compound 4: 44.1 g of compound 3 prepared in step C was added to 230 ml of DMF, and after adding 35.0 g of anhydrous potassium carbonate, 31.9 g of dimethyl sulfate was slowly added dropwise at 100° C. under stirring, and the same temperature T= The reaction was carried out at 100 °C for 4 h. The reaction solution was cooled to room temperature of T=25°C, 280 ml of water was added under stirring, left to stand for crystallization, suction filtered, the filter cake was washed with water and dried to obtain 44.6 g of a crude compound 4, which was recrystallized with methanol to obtain 36.8 g of a refined compound of compound 4, Yield 79.3% |
| F. Preparation of compound 5: 36.8g of compound 4 was added to 200ml of carbon tetrachloride, 0.1g of benzoyl peroxide was added, heated to T=76°C and refluxed, 45.0g of bromine was added dropwise, and the reaction was completed within 2h. For 4 h, the reaction solution was cooled in an ice-water bath, filtered, and the filter cake was washed with a small amount of carbon tetrachloride, and dried to obtain 47.5 g of compound 5 crude product, with a yield of 82%. |
| G. Preparation of compound 6: dissolve 15.4 g of potassium hydroxide in 360 ml of methanol, stir, cool to 0-10° C. in an ice-water bath, add 12.7 g of thiophenol, react for 10 min, add 47.5 g of compound 5, and warm to room temperature, The reaction was carried out for 3 to 3.5 h, the reaction solution was poured into 1500 ml of ice water, adjusted to pH 2 with hydrochloric acid under stirring, filtered, the filter cake was washed with water, and dried in vacuo to obtain 42.9 g of crude compound 6 with a yield of 93.1%. The crude product was recrystallized with ethyl acetate, 10 g of activated carbon was decolorized, and 36.0 g of the dried compound 6 was purified, with a purification yield of 84%. The mother liquor of recrystallization is concentrated and recovered. Or to prepare compound 6, dissolve 3.3 g of potassium hydroxide in 75 ml of methanol, stir, cool to 0-10° C. in an ice-water bath, add 2.7 g of thiophenol, react for 10 min, add 10.0 g of compound 5, warm to room temperature, and react For 3-3.5 h, the reaction solution was poured into 300 ml of ice water, adjusted to pH 2 with hydrochloric acid under stirring, filtered, the filter cake was washed with water, and dried in vacuo to obtain 9.0 g of crude compound 6 with a yield of 93.1%. The crude product was recrystallized with isopropanol, 2 g of activated carbon was decolorized, and 6.3 g of the dried refined product of compound 6 was obtained, with a purification yield of 70%. The mother liquor of recrystallization is concentrated and recovered. |
| H. Preparation of compound 7: |
| In 320ml of ethanol, add, (33%) dimethylamine aqueous solution 29.2g, (37-40%) formaldehyde aqueous solution 23.8g, stir for 10min, add 36.0g compound 6, react at 60°C for 5h, the reaction is completed, 5.0g activated carbon decolorization, Filtration while hot, tetrahydrofuran was distilled off from the filtrate under reduced pressure, and dried to obtain 40.4 g of compound 7 crude product, with a yield of 99.0%. Or to prepare compound 7, under stirring and cooling conditions, 8.1 g of (33%) dimethylamine aqueous solution, 6.6 g of (37-40%) formaldehyde solution and 10 g of compound 6 were sequentially added to 100 ml of glacial acetic acid, and placed in 70 The reaction was carried out at °C for 6 hours. After the completion of the reaction, the reaction solution was concentrated under reduced pressure, 100 ml of water was added, and the pH was adjusted to 12 with trimethylamine solution. The aqueous phase was extracted three times with dichloromethane (20 ml×3), and the organic phase was dried over anhydrous sodium sulfate. Concentrate under reduced pressure and dry to obtain 9.6 g of crude compound 7, with a yield of 85.0%. |
| J. Preparation of compound 8: |
| 40.4 g of the crude product of compound 7 obtained in the above step H was heated and dissolved in 150 ml of acetone, adjusted to pH=2 with hydrochloric acid while hot, a solid was precipitated, cooled to about 0°C in an ice-water bath, filtered, and the filter cake was washed with frozen acetone and dried in vacuo to obtain compound 8 Crude product 40.5g, yield 89.8%. |
| The above crude compound J was recrystallized from acetone-ethanol-water (3:1:1). 36.5 g of product were obtained, and the yield was 90.0%. |
| 1 H- NMR of the product (400MHz, DMSO-d 6 ) : 1.25 (t , 3H , -OCH 2 CH 3 ),2.74[s,6H,-N(CH 3 ) 2 ],3.70(s,3H, =NCH 3 ) , 4.19 (q , 2H , -OCH 2 CH 3 ),4.74(s,2H,-CH 2 S-),4.91(s,2H,-CH2N=),7.31~ 7.36(m,5H,-PhH),8.03(s,1H,indole-7-H),9.11(br s,1H,HCl),9.43(s,1H,-0H)。 |
| Mass spectrum MS (m/z) of the product: 477 (M+1, Br=79), 479 (M+1, Br=81). |
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN304759079&_cid=P12-KZB0LB-96613-1
| Research and develop a kind of method for efficient green synthesis of arbidol hydrochloride intermediate, the structural formula of arbidol hydrochloride is as follows: |
| |
| Example 1: Ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate |
| |
| After the device was installed, 150 mL of acetic anhydride solvent was added to the three-necked flask, and then solid ethyl 5-hydroxy-1,2-dimethylindole-3-carboxylate (23.3 g, 0.1 mol) was added while stirring. After all dissolved, heated to reflux for 4 h, after the reaction was completed, the reaction solution was cooled, and the solid was obtained by suction filtration. Wash the solid with water for 4 times (150 mL-200 mL of water each time), and slowly add 0.15 mol/L ammonia water to the solution in the third time to control the pH of the mixed system by adding water to the solid to be 8 to 9. Finally, suction filtration A solid was obtained, which was dried in an oven at 70° C. for 5 h to obtain a crude product. Recrystallization from methanol gave 18.8 g of ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate as brown crystals. Yield 65%. |
| Example 2: Ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate |
| |
| After installing the device, in a three-necked flask, ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate (17.9g, 0.065mol), catalyst (p-cymene)- Ruthenium dichloride dimer (4.0g, 0.0065moL), N-bromosuccinimide NBS (46.28g, 0.26moL) and 200mL dimethylacetamide DMA, slowly warmed to 90°C in oil bath under nitrogen protection , maintain the reaction temperature for 24h, after the reaction is over; cool the reaction solution to room temperature, add an appropriate amount of water to the reaction solution, extract 5 times with ethyl acetate, combine the organic phases, dry, spin dry the solvent to obtain a solid, use acetone After recrystallization, a white powdery solid was precipitated, which was dried in vacuo to obtain 23.3 g of ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate. Yield 80%. |
| Example 3: Ethyl 6-bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate |
| |
| Install the device, add 150 mL of methanol solvent to the three-necked flask, slowly add 8.6 g of solid potassium hydroxide under stirring, cool to room temperature after all dissolved, then add thiophenol (6.2 g, 0.05 mL) under stirring, After about 15 min, ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate (23.3 g, 0.05 moL) was finally added, and the reaction was stirred at room temperature for 4 h. After the reaction is completed. 10% acetic acid was added dropwise to the reaction solution until the pH of the reaction solution was 3-4. After a large amount of yellow solid was precipitated, the solid was obtained by suction filtration, washed once with water, filtered with suction, and dried at 70 °C for 5 h in a drying box. get crude products. Recrystallization from ethyl acetate gave 12.6 g of ethyl 6-bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate as yellow-white crystals. Yield 60%. |
| Example 4: Arbidol |
| |
| After installing the device, add 100 mL of glacial acetic acid solution to the three-necked flask, cool it to 0 °C, slowly add 40 mL of 40% methylamine aqueous solution, and then add 10 mL of 37% formaldehyde aqueous solution, and after the reaction is stirred for 15 min, add 6- Ethyl bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate (12.6g, 0.03moL), stirred uniformly for 10 min, then began to heat up to 80°C, maintaining the reaction temperature , and react for 4 h after complete dissolution. After the reaction is over, pour the reaction solution into water, add an appropriate amount of 20% potassium hydroxide solution to neutralize it with stirring, adjust the pH of the solution to 7.0, precipitate solids, filter with suction, and wash with water once. The solid was obtained by suction filtration, and dried in an oven at 70 °C for 5 h to obtain a crude product. Recrystallize with acetonitrile, after complete dissolution, add 1 g of activated carbon to reflux for 30 min, filter hot, cool, and precipitate 8.5 g of brown solid Arbidol. Yield 60%. |
| Example 5: Arbidol hydrochloride |
| |
| Install the device, add an appropriate amount of acetone solvent to the three-necked flask, add Arbidol (8.5g, 0.018moL) under stirring, heat to reflux, add 10mL of concentrated hydrochloric acid dropwise, reflux for 30 min, and after the reaction is over, cool the reaction The liquid was brought to room temperature, and filtered with suction to obtain crude Arbidol hydrochloride, which was dried in an oven at 50 °C for 3 h. Recrystallize with acetone:ethanol (3:2) solvent, cool at room temperature for 10 h, freeze in refrigerator for 10 h, suction filtration, wash the solid with a small amount of acetone, and obtain 7.0 g of refined Arbidol hydrochloride in a yield of 75%. MS (EI): m/z: 513.8754 ([M]+). |
Clip
https://www.sciencedirect.com/science/article/pii/S0960894X1730687X

CLIP
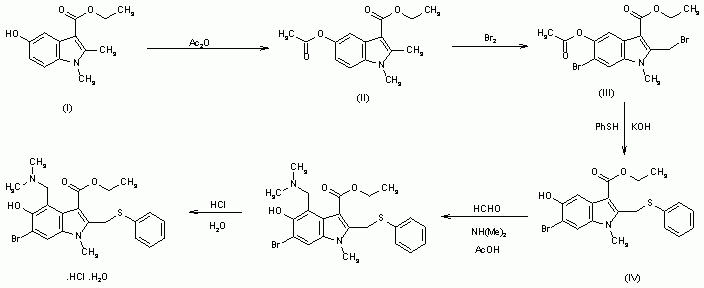
1,2-Dimethyl-5-hydroxyindole-3-acetic acid ethyl ester (I) is acetylated with acetic anhydride affording the O-acyl derivative (II) , which is brominated to the corresponding dibromide compound (III) . The reaction of (III) with thiophenol in KOH yields (IV) , which is then submitted to a conventional Mannich condensation with formaldehyde and dimethylamine in acetic acid, giving the free base of arbidol (V), which is treated with aqueous hydrochloric acid .
CLIP
Umifenovir
https://www.sciencedirect.com/science/article/pii/S0045206820317867
Umifenovir (Arbidol®) is an indole derivative first marketed in 1993 for the prophylactic treatment of infections caused by influenza A and B viruses [74]. Produced by Pharmstandard, it is still currently used in Russia and China to treat influenza infections [75]. Umifenovir is marketed in 50 and 100 mg capsules, being administered orally. The pharmacokinetics is limited, presenting rapid absorption and reaching the maximum concentration in 1.6–1.8 h. It is a slow elimination drug, with a half-life of 16 to 21 h, and may be administered twice a day [76].
The drug’s anti-influenza mechanism of action is related to arbidol’s ability to bind to the haemagglutinin (HA) protein [77]. The haemagglutinin (HA) protein is a homotrimeric glycoprotein found on the surface of the influenza virus, and it is essential for its infectivity. This protein is responsible for allowing the influenza virus binding to the sialic acid present on the surface of the target cells (respiratory tract cells or erythrocytes). As a result of this interaction, the virus is internalized in the host cell. Once umifenovir binds to the HA protein, this glycoprotein is prevented from binding to sialic acid, so the virus is no longer able to penetrate the host cell [78].
The structural similarity between the SARS-CoV-2 peak and the influenza virus (H3N2) HA glycoproteins justifies the fact that drugs that are capable of binding to HA can also do so to the SARS-CoV-2 spike protein. This fact was evidenced by molecular modeling studies, wherein was demonstrated that umifenovir is able to bind to the protein peak, preventing its trimerization, which would be a determining factor for the mechanism of cell adhesion (Fig. 8) [78].

Fig. 8. Umifenovir (in orange) binding region in SARS-CoV-2 spike glycoprotein. Reprinted from International Journal of Antimicrobial Agents, 56, N. Vankadari, “Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein”, Page 2, with permission of Elsevier. Copyright 2020. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Recently in 2020, in vitro studies performed with Vero cells confirmed that arbidol efficiently inhibits SARS-CoV-2 infection with an EC50 of 4.11 μM. The author also determined that arbidol was able to efficiently block both viral entry and post-entry stages, and also concluded that the drug prevented the viral attachment and release of SARS-CoV-2 from the intracellular vesicles. Importantly, the EC50 of arbidol against SARS-CoV-2 led the authors to suggest that the dose of arbidol currently recommended by the Chinese Guidelines (200 mg, 3 times/day) should be elevated in order to achieve ideal therapeutic efficacy to inhibit the SARS-CoV-2 infection [79].
A clinical trial was conducted at Wuhan Jinyintan Hospital, in 2020, from February 2 to March 20 conducted to evaluate the effectiveness and safety of umifenovir in the treatment of COVID-19 patients. In this study, 81 patients were evaluated: 45 received 200 mg of umifenovir three times a day, and 36 were in the control group. The authors concluded that baseline clinical and laboratory characteristics were similar in the two groups, and patients in the umifenovir group had a longer hospital stay than those in the control [80]. Although such results may seem discouraging, further clinical trials with higher doses of umifenovir may be required in order to verify its clinical efficiency against the SARS-CoV-2 infection.
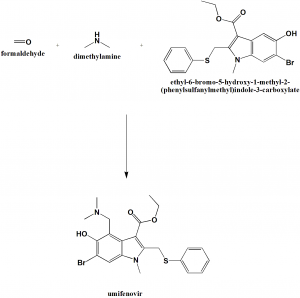
The synthesis of umifenovir was described in 2006 starting from the reaction between ethyl acetoacetate 63 and methylamine, giving enaminone 64, which next undergoes a Nentizescu condensation reaction with 1,4-benzoquinone to produce indole derivative 65 (Scheme 9). Then, an acetylation reaction is carried out to protect the hydroxyl group in 65, producing 66, which is converted to 67 after a bromination step. The reaction of intermediate 67 with thiophenol in basic medium leads to the formation of 68, which finally affords umifenovir after a Mannich reaction [81].

Scheme 9. Synthetic route towards umifenovir.
Q. Guo, M. Xu, S. Guo, F. Zhu, Y. Xie, J. Shen
Y.S. Boriskin, I.A. Leneva, E.I. Pecheur, S.J. Polyak
I.A. Leneva, R.J. Russell, Y.S. Boriskin, A.J. Hay
P. Deng, D. Zhong, K. Yu, Y. Zhang, T. Wang, X. Chena
N. Vankadari
T.O. Edinger, M.O. Pohl, S. Stertz
X. Wang, R. Cao, H. Zhang, J. Liu, M. Xu, H. Hu, Y. Li, L. Zhao, W. Li, X. Sun, X. Yang, Z. Shi, F. Deng, Z. Hu, W. Zhong, M. Wang
N. Lian, H. Xie, S. Lin, J. Huang, J. Zhao, Q. Lin

Drug-repurposing studies are testing a range of compounds to treat COVID-19, but manufacturers may struggle to meet demand if any of these candidates prove effective against SARS-CoV-2. The pandemic has already strained global supply chains and limited the availability of a number of products, including hand sanitizer and diagnostic test reagents. The raw materials needed to make a new antiviral drug would most likely face similar pressures. But a team led by Tim Cernak of the University of Michigan has used an AI-based retrosynthesis program called Synthia to devise alternative routes to 12 leading drug candidates under investigation. The work appears on a preprint server and has not been peer reviewed (ChemRxiv 2020, DOI: 10.26434/chemrxiv.12765410.v1). “If the world runs out of one of the drugs currently in the clinic, we are providing a backup recipe,” Cernak says. Using alternative starting materials that are readily available, the researchers aimed to find routes of similar length and cost to those of existing syntheses. For each compound, the researchers whittled down a long list of options offered by Synthia to identify the most suitable synthetic strategies. Then the team tested some of these syntheses in the lab, including four new routes to the antiviral umifenovir, currently being investigated in eight clinical trials against COVID-19. Cernak says this approach could be used more generally to rapidly identify alternative synthetic routes whenever crises cause supply chain disruptions in drug manufacturing.
Artificial intelligence finds alternative routes to COVID-19 drug candidates
If drug-repurposing studies hit pay dirt, backup recipes could help antiviral manufacturers avoid supply chain problems
by Mark Peplow, special to C&EN

PATENT
https://patents.google.com/patent/WO2018112128A1/en


Arbidol Core Synthesis
Ethyl 5-acetoxy-2-methyl-1H-indole-3-carboxylate 1a: Acetic anhydride (25.9 ml, 274 mmol 20 eq.) was added to a stirred solution of ethyl 5-hydroxy-2-methyl-1H-indole- 3-carboxylate 1 (3.00 g, 13.6 mmol, 1.0 eq.) in pyridine (3.32 mL, 41.1 mmol, 3.0 eq.) and the reaction heated to reflux. After 1 h, the reaction was allowed to cool back to rt before pouring the mixture into a solution of aqueous saturated sodium bicarbonate (40 mL). The product was extracted with ethyl acetate (3 x 40 ml) and the combined organic layers were washed with water (40 mL), dried (Na 2 SO 4 ) and concentrated In vacua to yield the product as a white solid which was used without further purification (3.4g, 96%). NMR: δH ( 400 MHz, CDCl 3) 8.34 (1H, s; NH), 7.75 (1H, s, H 4 ), 7.21 (1H, d, J 8.5, H 6 ), 6.89 (1H, d, J 8.5, H 7 ), 4.38 (2H , q, J 7.1 , CO 2 CH 2 CH 3 ), 2.71 (3H, s, C 1 CH 3 ), 2.34 (3H, s, CO 2 CH 3 ), 1.43 (3H, t, J 7.1, CO 2 CH 2 CH 3 ). δ c (100 MHz, CDCl 3 ) 170.8<a name=”
(CO 2 CH 3 ), 165.8 (CO 2 Et), 1.45.9 (C 5 ), 145.3 (C 2 ), 132.4 (C 8 ). 127.9 (C 3 ), 116.3 (C 6 ), 113.8 (C 4 ), 111.2 (C 7 ), 99.8 (C 1 ), 59.7 (CO 2 ΡΗ 2 ΡΗ 3 ), 21.4 (C 1 CH 3 ). 14.8 (CO 2 PH 2 CH 3 ), 14.4 (CO 2 CH 3 ). Rf : _
0.31 (40% ethyl acetate in hexane), HRMS. (ESI-TOF): C 14 H 15 O 4 N ([M+H] + ) requires 262.1074, found 262.1074.
Ethyl 5-acetoxy-1,2-dimethyl-1H-indole-3-carboxylate 1b: Protected indole 1b (1.35 g, 5.17 mmol, 1 eq.) was dissolved in DMF (15 mL). To this solution, methyl iodide (0.965 ml, 15.5 mmol, 3.0 eq.) was added and the resulting mixture was cooled on ice. Sodium hydride (0.186 g, 7.75 mmol, 1.5 eq.) was added and the reaction was left to stir on ice for 1.5 h. After this time, a small amount of water (5.0 mL) was added to the reaction and the solvents removed in vacuo. The resultant brown oil was then purified directly by column chromatography (30% ethyl acetate in petrol) to yield the title compound as a pale yellow solid (1.50 g, 95%). NMR: δ Η (500 MHz, CDCl 3 ) 7.79 (s, 1H, H 4 ), 7.26 (m, 1H, H 6 ), 6.96 (ddd, J = 8.8, 2.4, 0.8 Hz, 1H, H7 ), 4.38 (f, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.69 (s, 3H, NCH 3 ), 2.77 (d, J = 1.3 Hz, 3H, ΑrCΗ 3 ), 2.33 (s , 3H, CO 2 CH 3 ), 1.43 (t, J= 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ C (150 MHz, CDCl 3 ) 170.5 (CO 2 CH 3 ), 166.0 (CO 2 Et), 146.5 (C 5 ), 146.0 (C 2 ), 134.5 (C 8 ), 127.2 (C 3 ), 116.2 ( C6 ), 114.0 (C4 ) , 109.6 (C7 ), 104.4 (C 1 ), 59.6 (CO 2 CH 2 CH 3 ), 29.9 (NCH 3 ), 21.3 (C 1 CH 3 ), 14.8 (CO 2 CH 2 CH 3 ), 12.1 (CO 2 CH 3 ) . R f : 0.4 (30% ethyl acetate in hexane). HRMS (ESI-TOF): C 15 H 17 O 4 N ,([M+H] + ) requires 276.1230, found 276.1229.
Ethyl 6-bromo-2-(bromomethyl)-5-hydroxy-1-methyl-1H-indole-3-carboxylate 2: Bromine (558 μL, 10.9 mmol, 3.0 eq.) was added to a stirred solution of protected indole ( 1b, 1.00 g, 3.63 mmol, 1.0 eq.) in carbon tetrachloride (100 mL). After refluxing for 16 h, the reaction was cooled and aqueous sodium thlosulphate (10%. w/v, 100 mL) was added and left to stir for 20 min until the orange color disappeared. After this time, the organic layer was separated, washed with water (2 x 100 mL), dried (Na 2 SO 4 and concentrated in vacuo to yield a pale yellow solid, which was used without further purification (1.40 g, 99%) NMR: δ Η (400 MHz, PDCl 3 ) 7.86 (1H, s, H 4 ), 7.54 (1H, s, H 7), 5.05 (2H, s, CH 2 Br), 4.41 (2H, q, J 7.1, CO 2 CH 2 CH 3 ), 3.69 (3H, s, NCH 3 ), 2.39 (3H, s, CO 2 CH 3 ), 1.45 (3H, t, J 7.1, CO 2 CH 2 CH 3 ), δ C (100 MHz, CDCl 3 )
169.4 (CO 2 CH 3 ), 164.3 (CO 2 Et), 143.4 (C 5 ), 142.6 (C 2 ), 135.5 (C 3 ), 125.7 (C 8 ), 116.3 (C 4 ), 113.9 (C 7 ) , 112.0 (C 6 ), 105.6 (C 1 ), 60.3 (CO 2 CΒ 2 CΗ 3 ), 30.1 (NCH 3 ), 20.9 (CO 2 CH 3 ), 20.6 (C 1 CH 2 Br),
1.4.5 (CO 2 CH 2 CH 3 ), R f : 0.75 (CH 2 Cl 2 ). HRMS (ESI-TOF): C 15 Η 15 O 3 ΝΒr ([M+H] + ) requires 431.9441, found 431.9441.<a name=”
Ethyl 6-bromo-5-hydroxy-1-methyl-2-((phenylthio)methyl)-1H-indole-3-carboxylate 3: Thiophenol (99.8 μL, 0.972 mmol, 1.0 eq.) was added to a solution of potassium hydroxide (164 mg, 2.92 mmol, 3.0 eq.) in methanol (2 ml) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (880 mg, 0.972 mmol, 10 eq.) in CH 2 Cl 2 (5 mL) was added. The reaction was left to stir for 3 h before neutralization with acetic acid. The solvent was removed in vacuo and columned directly (20% EtOAc in petrol) to yield the title product as a pale yellow solid (362 mg, 86%). NMR: δ Η (600 MHz, CDCl 3 ) 7.74 (s, 1 H, Hr), 7.43 (s, 1H, H 4 ), 7.36 (dq, J = 5.2, 3.4, 2.4 Hz, 2H, H10 ), 7.25 (dd, J = 5.2, 1.9 Hz, 3H, H 11 and 5.33 (s, 2H, SCH 2 ), 4.29 (q, J = 7.3 Hz, 2H, CO 2 CH 2 CH 3 ), 3.60 (d, J = 18.1 Hz, 3H, NCH 3 ), 1.38 (t, J = 7.3 Hz, 3H, CO 2 CH 2 CH 3 ), δ c (150 MHz, CDCl 3 ) 165.1, 147.7, 144.2, 134.1 R f : 0.35 (20% EtOAc in petrol) HRMS (ESI-TOF)-: C 19 H 18 BrNO 3 S ([M+H] + ) requires 420.0263, found 420.0260.
Arbidol [Ethyl 6-bromo-4((dimethylamino)methyl)-5-hydroxy-1-methyl-2-((phenylthio)methyl)-1H-indole-3-carboxylate] 4: 1 Indole 3 (200 mg, 0.476 mmol, 1.0 eq.) and N, N, N’, N’-tetramethylaminomethane (1-95 μL, 1.43 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2 mL). The reaction was. heated to reflux for 3.5 h before removing the solvent in vacuo. The reaction was then re-dissolved in ethyl acetate and 1 M HCl was added to the solution causing the title product to crash out as a pale yellow solid (117 mg, 51%). NMR-: δ B (500 MHz, MeOD) 7.87 (s, 1 H, H 7 ), 7.39 (dd, J = 7.4, 2.2 Hz, 2H, H 10 ), 7.35 – 7.31 (m, 3H, H 11 and H12 ) . 4.87 (s, 2H, SCH 2), 4.71 (s, 2H, CH 2 NMe 2 ), 4.33 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CB 3 ), 3.63 (s, 3H, NCH 3 ), 2.97 (s, 6H, N (CH 3 ) 2 ), 1.39 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 169.7 (CO 2 Et), 152.7 (C s ), 149.0 (C), 137.7 (C 10 ), 136.4 (C 3 ), 136.0 (C 8 ), 132.3 (C 11 ), 131.3 (CH), 129.3 (C12 ) , 119.8 (C7 ) , 113.1 (C2), 111.3 ( C6), 108.3 (C4 ) , 64.2. (CO 2 CH 2 CH 3 ), 57.4 (CH 2 NMe 2 ), 45.4 (CH 2 N(CR 3 ) 2 ); 33.7 (CH 2 SPh), 32.9 (NCH 3 ), 16.6 (CO 2 CH 2 CH 3 ). R f : 0.25 (EtOAc). HRMS (ESI-TOF): G 22 H 25 BrN 2 O 3 S ([M+H] + ) requires 477.0842, found 477.0844.
Synthesis of Arbidol Analogues
Ethyl 5-acetoxy-6-bromo-2-(((3-hydroxyhpenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 8a: 3-hydroxythiophenol. (117 μL, 1.15 mmol, 1.0 eq.) was added to a solution of sodium carbonate (367 mg, 3.46 mmol, 3.0: eq.) and bromo indole 2 (500 mg, 1.15 mmol, 1.0 eq.) in dry ethyl acetate (10mL). The reaction was heated to 100°C and stirred for 5 h before<a name=”cooling, filtering and removing the solvent in vacuo. The compound was purified by column chromatography (40% EtOAc in Hexanes) to produce the title product as a pale yellow solid (240 mg, 44%). NMR: δ H (500 MHz,. CDCl 3 ) 7.85 (s, 1H, H 7 ), 7.56 (s, 1 H, H 4 ), 7.12 (t, J = 7.9 Ηz, 1Η, H 13 ), 6.95 – 6.90.(m, 1H, Η 14 ), 6.78 (s, 1H, H 10 ), 6.75-6.71 (m, 1H,.H 12 ), 4.69 (s, 2H, SCH 2 ), 4.30 (q, J = 7.4 Hz, 3H, CO 2 CH 2 CH 3 ), -3.66 (s, 3H, NGH 3 ), 2.4Q (s, 3H, COCH 3), 1.38 (t, J = 7.4 Hz, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, CDCL 3 ) 169.8, 165.0, 156.1, 144.6, 143.3, 135.6, 135.1, 130.1, 126.1, 124.8, 119.3, 113.9, 111.1, 105.8, 60.1, 30.4, 29.9, 21.0, 14. . R f : OAS (30% EtOAc in Hexane). HRMS (ESS-TOF): C 21 H 20 SrNO 5 S ([M+H] + ) requires 473.0318, found 478.0317.
Ethyl 6-bromo-5-hydroxy-24(((3-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 8: Sodium carbonate (106 mg. 1.00 mmol, 2.0 eq.) was added to a stirred solution of meta-hydroxy indole 8a (240 mg, 0.502 mmol, 1.0 eq.) in methanol (40 ml) and left to stir for 2h, The solution was then filtered and the solvent removed in vacuo, The product was re -dissolved in ethyl acetate (10 mL) and washed once with water (40 mL) before drying (Na 2 SO 4 and concentrating in vacuo to give the title product as a white solid, which could be used without further purification (160 mg, 67%), NMR-: δ H (600 MHz, MeOD) 7.60 (s, 1H, H 7 ), 7.58 (s, 1 H, H 4 ), 7.07 (dd, J = 8.2, 7.7 Hz, 1H, H 13), 6.83 – 6.81 (m, 1Η, Η 14 ), 6.79 (ddd, J = 7.7, 1.8, 0.9, 1H, H 10 ), 6.7.0 (dd, J = 8.2, 1.8, 0.9 Hz, 1H, H 12 ), 4.70 (s, 2H, SCH 2 ), 4.26 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.64 (s, 3H, NCH 3 ), 1.39 (t, J = 7.1 Hz , 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 166.9, 158.9, 150.6, 145.5, 136.4, 133.6, 131.0,. 130.7, 128.0, 1.24.9, 120.5, 116.0, 114.8, 107.9, 104.8, 60.8, 30.5, 30.4, 14.8. R f : 0.45 (1% MeOH in CΗ 2 Cl 2 ). HRMS (ESI-TOF): C 7 PM18 BrNO 4 S ([M+H] + ) requires 436.0213, found 436.0215.
Ethyl 2-(((3-aminophenyl)thio)methyl)-6-bromo-5-hydroxy-1-methyl-1H-indole-3-carboxyiate 9: 3-aminothiophenol (54.3 μL, 0.511 mmol, 1.0 eq.) was added to a solution of potassium hydroxide (86 mg, 1.53 mmol, 3.0 eq.) in methanol (2 ml) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (200 mg, 0.511 mmol, 1.0 eq.) in CH 2 Cl 2 (5 ml) was added. The reaction was left to stir for 3 h before neutralization with acetic-acid. The solvent was removed in vacuo and purified directly by preparative TLC (1% MeOH in CH 2 Cl 2 ) to yield the title product as a pale yellow solid (138 mg, 62%), NMR: δ H (500 MHz, CDCl 3) 7.74 (d, J = 1.8 Hz, 1H, H 7 ), .7.42 (d, J = 1.8 Hz., 1H, H 4 ), 7.03 (t, J = 8.1 Hz, 1 H, H 13 ), 6.75 (d, J= 7.7 Hz, 1H, H 14 ), 6.68 (s, 1H, H 10 ), 6.55 (d, J = 6.1 Hz, 1H, H 12 ), 4.68 (d, J = 1.9 Hz, 2H, SCH 2 ), 4.35 ™ 4.30 (m, 2H, COCH 2 GH 3 ),. 3.60 (d, J = 1.9 Hz, 3H, NCH 3 ), 1.40 (td, J = 7.1, 1.8 Hz, 3H, COCH 2 CH 3 ). δ c (150 MHz, CDCl 3 ) 1-66.9, 150.6, 149.6,<a name=”
146.0, 136.0, 133.5, 1 . 30.5, 128.1, 123.1, 120.2, 117.6, 115.8, 114.8, 107.9, 104.6, 68.1, 60.8, 30.4, 14.8. R f : 0.85 (1% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 19 H 19 BrN 2 O 3 S ([M+H] + ) requires 435.0372, found 435.0370.
Ethyl 2-(((3-aminophenyl)thio)methyl)-6-bromo-5-hydroxy-1-methyl-1H-indole-3-carboxylate 10: 2-napthalenethiol (82.0 mg, 0.511 mmot, 1.0 eq.) was added to a solution of potassium hydroxide (86 mg, 1.53 mmol, 3.0 eq.) in methanol (2 mL) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (200 mg, 0.511 mmol, 1.0 eq.) in CH 2 Cl 2 (5 mL) was added. The reaction was left to stir for 3 h before neutralization with acetic acid. The solvent was removed in vacuo and purified directly by preparative TLC (1% MeOH in CH 2 Cl 2 ) to yield the title product as a pale yellow solid (118 mg, 50%). NMR: δR(600 MHz, DMSO) 9.77 (s, 1H, OH), 7.83 (d, J = 1.8 Hz, 1H, Ar), 7.81 -7.79 (m, 1H, Ar), 7.75 (d, J = 8.6 Hz, 1H , Ar), 7.72 – 7.70 (m, 1H, Ar), 7.66 (s, 1H, Hz), 7.46 (s, 1H, H 4 ), 7.45 – 7.40 (m, 2H, Ar), 7.34 (dd, J = 8.5, 1.9 Hz, 1H, Ar), 4.82 (s, 2H, CH 2 SPh), 4.04 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.63 (s, 3H, NC. %), 1.14 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, DMSO) 164.3, 149.3, 143.4, 133.2, 132.0, 131.7, 131.6, 128.9, 128.4, 128.4, 127.7, 127.2, 126.8, 126.3, 1:26.0, 116.3, 116.3, 116.3 06.3, 103.3, 59.2, 30.3, 28.1, 14.3. R f : 0.75 (1% MeOH in CH2 Cl 2 ). HRMS (ESI-TOF): C 23 H 20 BrNO 3 S (fM+Hf) requires 470.0420, found 470.0420.
Ethyl 6-bromo-4-((dimethylammino)methyl)-5-hydroxy-2-(((3-hydroxyohenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 11; Meta-hydroxy indole 8 (30.0 mg, 0.069 mmol, 1.0 eq,) and N, N,N’,N’-tetramethyldiaminomethane (47.0 μL, 0.344 mmol, 5.0 eq.) were dissolved in CH 2 Cl 2 (30 mL) . The reaction was heated to reflux for 3.5 h before removing the solvent in vacuo to, yield the title product as a pale yellow solid (34 mg. 99%). NMR; δH (500 MHz, CDCl 3 ) 7.47 (s, 1H, H 7 ), 7.12 (t, J = 7.9 Hz, 1H, H 13 ), 6.90 (d, J = 7.9 Hz, 1H, H 14 ), 6.90 ( d, J = 7.9 Hz, 1H, H 12 ), 6.66 (s, 1H, H 10 ), 4.41 (s, 2H, CH 2 NMe2 ), 4.34 (s, 2H, CH 2 SPh), 4.15 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.60 (s, 3H, NCH 3 ), 2.55 (s, 6H, CH 2N (CH 3 ) 2 ), . 1.33 – 1.21 (m, 3H, CO 2 CH 2 CH 3 ). δ c ( . 150 MHz, CDCl 3 ) 165.9, 156.7, 150.9, 142.6, 135.1, 132.2, 131.0, 130.0, 128.9, 124.6, 124.3, 119.3, 115.5, 113.4, 106.8, 106.8, 106.8, 106.8, 165.5 58.7, 44.0, 30.4, 29.9, 14.3, R f : 0.15 (10% MeOH in CH 2 Cl 2 ). HRMS (ESS-TOF): C22 H 25 BrN 2 O 4 S ([M+H] + ) requires 493.0791, found 493.0792,
Ethyl 22-(((3-aminophenyl)thio)methyl)-6-bromo-4-((dimethylamino)methyl)-5-hydroxy-1-methyl-1H-indole-3-carboxylate 12: Meta-amino Indole 14 (104 mg, 0.238 mmol, 1.0 eq.) and<a name=”
N,N,N’,N’-tetramethyldiaminomethane (1.95 μL, 1.43 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (1.5 ml). The reaction was heated to reflux for 3.5 h before removing the solvent in vacuo. The product was purified using preparative TIC to yield a pale yellow solid (13 mg, 11%). NMR: δ Β (500 MHz, MeOD.) 7.61 (s, 1H, H 7 ), 7.00 (t, J = 7.8 Hz, 1 H, H13), 8.69 (t, J = 2.0 Hz, 1. H , H 10 ), 6.61 – 6.66 (m, 2H, H 12 and H 14 ), 4.57 (s, 2H, CH 2 SPh), 4.40 (s, 2H, CH 2 NMe 2 ), 4.24 (q, J = 7.2 Hz, 2H, CO 2 CH 2 CH 3 ), 3.61 (s, 3H, NCH 3), 2.55 (s, 6H, CH 2 N(CH 3 ) 2 X 1.36 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 167.6, 149.7, 144.5, 136.0, 132.6, 130.6 , 129.8, 126.1, 123.0, 120.2, 115.9, 114.8, 112.5, 111.4, 110.7, 61.5, 59.8, 43.5, 31.2, 30.5, 14.6. in CH 2 Cl 2 ).HRMS (ESi-TOF): C 22 H 26 BrN 3 O 3 S ([M+H] + ) requires 492.0951, found 492.0951.
Ethyl 6-bromo-4-((dimethylaminom)ethyl)-5-hydroxy-1-methyl-2-((naphthalen-2-ylthio)methyl)-1H-indole-3-carboxylate 13: Naphthatenyl indole 10 (112 mg , 0.238 mmol, 1.0 eq.) and N,N,N’,N’-tetramethyldiaminomethane (1.95 μL, 1.43 mmol, 3.0 eq.) were dissolved in 1,4-dioxane ( 1.5 mL). The reaction was heated, to reflux for 3.5 h .before removing the solvent in vacuo. The product was purified using preparative TIC to yield a pale yellow solid (26 mg, 20%). NMR: δH (600 MHz, MeOD) 7.83 – 7.60 (m, 1H, Ar), 7.74 (d, J = 7.8 Hz, 2H, Ar), 7.68 -7.65 (m, 1H, Ar), 7.60 (s, 1 H, Hr), 7.49 -7.43 (m, 2H, Ar), 7.34 (dd. J = 8.6, 1.7 Hz, 1 H. Ar), 4.69 (s, 2H, CH 2 SPh), 4.27 (s, 2H, CH 2 NMe 2 ), 4.04 – 3.98 (m, 2H, CO 2 CH 2 CH3 ), 3.65 (s, 3H, NCW 3 ), 2.45 (s, 6H, CH 2 N(CH 3 ) 2 ), 1.21 – 1.15 (m, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, MEOD) 167.3, 154.9, 143.8, 133.2, 132.6, 132.6, 131.3, 129.5, 128.6, 128.6, 125.9, 114, 106.6, 61.4, 114, 127.6, 127. 60.0, 43.5, 31.0, 30.8, 30.6, 14.5. R f : 0.65 (10% MeOH in CH 2 Cl 2 ). HRMS (ESI-TOF): C 26 H 27 BrN 2 O 3 S ([M+H] + ) requires 527.0998, found 527.0996.
Boc piparazinyl arbidol [Ethyl 5-acetoxy-6-bromo-4-((4-tert-butoxycarbonyl)piperazin-1-yl)methyl)-1-methyl-2-((phenylthio)moethyl)-1H-indole-3 -carboxylate] 14; Arbidol 4 (20mg,
0.0419 mmol, 1.0 eq.) and 1-boc-plperazilne (24 mg, 0.126 mmol, 3.0 eq. ) were dissolved in 1,4-dioxane. (2 mL) and refluxed overnight. The solvent was then removed in vacuo and the reaction columned directly to yield the title product as a yellow solid (20 mg, 77%). MMR: δH (400 MHz, MeOD) 7.55 (s, 1H, H 7 ), 7.30 – 7.20 (m, 5H, SPh), 4.54 (s, 2H, CH 2 SPh), 4.25 – 4.07 (m, 4H, CH 2 NR 2 and CO 2 CH 2 CH 3 ), 3.58 (d, J= 8.2 Hz, 3H, NCH 3 ), 3.47 (br s, 4H, BocN(CH 2 ) 2 ), 2.53 (brs, 4H , BoeN(CH 2 CH 2)2). 1.44 (s, 9H, Boc), 1.30 – 1.26 (m, 3H, CO 2 CH 2 CH 3 ). δC (150 MHz, MeOD ) 167.2, 156.2, 151.3, 144.0, 1.35.4, 134.3, 134.1, 133.6,. 130.1, 129.9,. 129.0, 128.9, 125.6, 114.8, 114.3, 113.8, 108.7, 107.9, 167.1, 81.5, 61.5, 60.7, 58.6, 53.0, 30.9, 30.6, 28.6,<a name=”14.8, 14.6. R f : 0.5 (30% EtOAc in petrol). HRMS (ESI-TOF): C 29 H 36 BrN 3 O 5 S ([M + H]+) requires 618.1632, found 618.1631.
Alkoxy piperazinyl Arbidol [Ethyl 5-acetoxy-6-bromo-4-((4-(2-hydroxyethyl)piperazin-1-yl)methyl)-1-methyl-2-((phenylthio)methyl)-1H-indole- 3-carboxylate] 15: Arfaidol 4 (20mg,
0.0419 mmol, 1.0 eq.) and 1-(2-((trimethylsilyl)oxy)ethyl)piperazine (25 mg, 0.126 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2 mL) and refluxed overnight. The solvent was then removed in vacuo and the reaction columned directly to yield the title product as a yellow solid (12 mg, 51%). NMR: δ Η (400 MHz, MeOD) 7.56 (s, 1.H, H 7 ), 7.22 – 7.30 (m, 5H, SPh), 4.57 (s, 2H, CH 2 SPh), 4.14 – 4.19 (m, 4H, CH 2 NR 2 and CΟ 2 CH 2 CΗ 3 ), 3.68 (t, J = 5.9 Hz, 2H, . CH 2 OH), 3.60 (s, 3H, NCH 3 ), 2.53 – 2.70 (m, 10H, piperazine ring and CH 2 CH2 CH 2 OH), . 134 – 1.30 (rn, 3H, CO 2 CH 2 CH 3 ), δ c (150 MHz, MeOD) 167.2, 151.7, 143.9, 135.4, 134.2, 133.4, 130.1, 129.0, 125.5, 114.1, 113.6, 107.0, 108.9, 108.9, 108.9 61.5, 61.1, 59.8, 59.1, 54.3, 52.9, 30.9, 30.5, 14.7. R f : 0.15 (5% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 26 H 32 BrN 3 O 4 S ([M+H] + ) requires 562.1370, found 562.1368,
Ethyl 6-bromo-5-hydroxy-2-(((2-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 16: 2-hydroxythiophenol (26.0 μL, 0.256 mmol, 1.0 eq.) was added to a solution of sodium carbonate (81.0 mg, 0.767 mmol, 3.0 eq.) and promo indole 2 (100 mg, 0.256 mmol, 1.0 eq.) in ethyl acetate (2 mL). The reaction was heated to 50°C and stirred for 2 h before cooling and removing the solvent in vacuo. The product was then re-dissolved in methanol (2 mL) and potassium hydroxide (21.5 mg, 0.384 mmol, 1.5 eq.) was added. The reaction was stirred at room temperature for 3 h before direct purification by preparative TLC (2% MeOH in CH 2 Cl 2 ) to yield the title product as. a white solid (20.5 mg, 1.8%), NMR: δ H (500 MHz, MeOD) 7.59 (s, 1H, H 7), 7.54 (s, 1H, Η 4 ), 7.17 (t, J = 7.7 Hz, 1H, H 12 ), 7.08 (d, J = 7.7 Hz, 1H, H 14 ), 6.85 (d, J =7.7 Hz , 1H, H 13 ), 6.66 (t, J = 7.7 Hz, 1H, H 15 ), 4.58 (s, 2H, SCH 2 ), 4.24 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ) , 3.59 (s, 3H, NCH 3 ), 1.40 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 161.3, 146.4, 143.3, 134.8, 132.0, 130.4, 129.4, 123.7, 123.0, 121.8, 120.5, 114.4, 113.6, 110.0, 102.7, 60.04, 390.2, 390.2, 390.04 R f : 0.6 (2% MeOH in CH 2 Cl2 ). HRMS (ESI-TOF): C 19 H 18 BrNO 4 S ([M+H] + ) requires 436.0213, found 436.0212.
Ethyl 6-bromo-5-hydroxy-2-(((4-hydroxyphenyl)thmetihoyl)-)1-methyl-1H-indole-3-carboxylate 17: 4-hydroxythiophenol (26.0 μL, 0.256 mmol, 1.0 eq.) was added to a solution of sodium carbonate (81.0 mg, 0.767 mmol, 3.0 eq.) and bromo indole 2 (100 mg, 0.256 mmol, 1.0 eq.) in ethyl acetate (2 mL). The reaction was heated to 50°C. and stirred for 2 hours before cooling<a name=”and removing the solvent in vacuo. The product was then re-dissolved in methanol (2 mL) and potassium hydroxide (21.5 mg, 0.384 mmol, 1.5 eq.) was added. The reaction was stirred at room temperature for 3 h before direct purification by preparative TLC (2% MeOH in CH 2 Cl 2 ) to yield the title product as a white solid (2.5 mg, 2%). NMR: δ Η (600 MHz, DMSO) 7.65 (s, 1H, H 7 ), 7.49 (s, 1 H, H 4 ), 7.03 (d, J = 8.7 Hz, 2H , H 12 ), 6.58 (d, J = 8.7 Hz, 2. HH 13 ) , 4.52 (s, 2H, SCH 2 ), 4.08 (q, J = 7.2 Hz, 2H, CO 2 CH 2 CH 3 ), 3.51 (s, 3H, NCH3 ), 1.23 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, DMSO) 164.2, 157.9, 149.2, 144.3, 135.7, 131.4, 126.1, 1214, 115.9, 114.1, 106.3, 102.9, 79.2, 59.0, 55.4, 30.0, R 14.3, f ; 0.5 (2% MeOH in CH 2 Cl 2 ). HRMS (ESI-TOF): C 19 H 18 BrNO 4 S ([Μ+H] + ) requires 436.0213, found 436.0213.
Ethyl 5-acetoxy-6-bromo-2-(((3-methoxyphenyl) thio)methyl)-1-methyl-1H-indole-3-carboxylate 18a: 3-methpxythiophenol (14.6 μL, 0.118 mmol, 1.0 eq.) was added to a solution of sodium carbonate (37.4 mg, 0.353 mmol, 3.0 eq.) and bromo indoles 2 (46.0 mg, 0.118 mmol, 1.0 eq.) in dry ethyl acetate (20 ml). The reaction was heated to 50°C and stirred for 2 h before addition of water. The organic layer was separated, dried (Na 2 SO 4 ) and concentrated in vacuo. The compound was purified by column chromatography (20% EtOAc in Hexanes) to produce the title product as a white solid (34 mg, 59%). UMR; δ Η (600 MHz, DMSO) 7.92 (s, 1 H, H 7 ), 7.6.6 (s, 1 H, H 4 ), 7.13 (t, J = 7.9 Hz, 1H, H 13), 6.8.7 – 6.84 (m, 1 H, H 14 , 6.79 – 6.74 (m, 2H, H 10 and H 1 2 ), 4.77 (s, 2H, SCH 2 ), 4.13 (q, J = 7.1 Hz , 2H, CO 2 CH 2 CH 3 ), 3.70 (s, 3H, NCH 3 ), 3.58 (s, 3H, SPhOCH 3 ), 2.27 (s, 3H, COCH 3 ), 1.20 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CW 3 ).δ c (150 MHz, DMSO) 169.1, 163.9, 159.4, 144.9, 142.6, 135.1, 135.0, 129.9, 125.2, 123.3, 116.2, 115.1, 114.73, 110.73, 110.2 104.3, 59.5, 55.1, 30.6, 28.1, 20.7, 14.3 R f : 0.4 (20% EtOAc in Hexane) HRMS (ESI-TOF): C 22H 22 BrNO s S ([M+H] + ) requires 492.0475, found 492.0472.
Ethyl 6-bromo-5-hydroxy-2-((3(-methoxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 18: Sodium carbonate (41.3 mg, 0.390 mmol, 2.0 eq.) was added to a stirred solution of meta-methoxy indole 18a (96.0 mg, 0.195 mmot, 1.0 eq.) in methanol (10 mL) and left to stir for 2h, the solution was then filtered and the solvent removed in vacuo. The product-was re-dissolved in ethyl acetate (10 mL) and washed once with water (10 mL) before drying (Na 2 SO 4 ) and concentrating in vacuo to give the title product as a white solid, which could he used without further purification (80 mg, 91%), NMR: δ Η (600 MHz, CDCl 3 ) 7.73 (s, 1H, Η 7 ), 7.41 (s, 1H, H 4 ), 7.17 – 7.13 (m, H 13), 7.07 (m, 1H, H 14 ), 6.96 (dt, J = 7.7, 1.3, 1H, H 10 ), 6.85 (m, 1H, H 12 ),4.71 (s, 2H, SCH 2 ), 4.30 ( q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.63 (s, 3H, NCH 3 ), 1.41 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, CDCL 3 ) 165.2, 159.8, 147.7, 135.4, 132.6, 129.8, 12.7.2, 124.6, 119.7, 117.3, 114.1, 112.5, 107.5, 107.9, 55.4, 29.9, 29.6, 14.7 Rf :. _<a name=”0.55 (1% MeOH in CH 2 Cl 2 ). HRMS (ESl-TOF): C 20 H 20 BrNO 4 S ([M+H] + ) requires 450.0369, found 450.0367.
Ethyl 6-bromo-4-((dimethylamino)methy-5-hydroxy-2-(((2-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylatete 19: Ortho-hydroxy indole: 16 (13.5 mg, 0.0309 mmol, 1.0 eq.) and N, N, N’,N’-tetramethyldiaminomethane (12.7 μL, 0.0928 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2.0 mL). reaction was heated io reflux for 3.5 h before removing the solvent in vacuo to yield the title product as a white solid (13 mg, 85 %).HMR: δ H (500 MHz, MeOD) 7.53 (s, 1H, H 7 ) , 7.19 – 7.11 (m, 1H, H 4 ), 7.03 (dd, J = 7.6, 1.7 Hz, 1 H, H 12 ), 6.86 – 6.78 (m, TH, H 14 ), 6.63 (dt, J = 13.7 , 7.6 Hz, 1H, H 15 ), 4.48 (s, 2H, CH 2 Sph), 4.34 (s, 2H, CH 2NMe 2 ), 4.22 (dq, J = 10.8, 7.1, 6.3 Hz, 2H, CO 2 CH 2 CH 3 ), 3.56 (s, 3H, NCH 3 ), 2.49 (d, J = 11.4 Hz, 6H, CH 2 N(CH 3 ) 2 ), 1.42 – 1.37 (m, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, MEOD) 167.5, 160.4, 137.0, 136.6, 132.5, 131.6, 130.6, 126.1, 114.5, 112.6, 110.6, 106.0, 68.1, 61.4, 60.1, 43.5, 29.9, 29.9 14.6. R f : 0.4 (5% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 22 H 25 BrN 2 O4 S ([M+H] + ) requires 493.0791, found 493.0793.
Ethyl 6-bromo-4-((dimethylamino)methyl-5-hydroxy-2-(((3-methoxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 21: Sodium carbonate (17.5 mg, 0165 mmol, 3.0 eq.) was added to a stirred solution of meta-methoxy indole 18 (27.0 mg, 0.055 mmol, 1 .0 eq.) in ethyl acetate (8 mL) and methanol (1 mL). to stir for 3h before filtering and removing the solvent in vacuo.The compound was then re-dissolved in 1,4-dioxane (5 mL) and N, N, N’,N’-tetramethyldiaminomethane (5.5 μL, 0.04 mmol, 3.0 eq.) as added. The reaction was heated to reflux overnight before removing the solvent in vacuo. Purification by preparative TLC (5% MeOH In CH 2 Cl 2 ) yielded the title product as a pale yellow solid (7 mg, 24%) .NMR: δ Μ (600 MHz, CDCl 3) 7.44 (s, 1H, H 7 ), 7.19 (t, J = 7.9 Hz, 1 H, H 15 ), 6.96 (rn, 1H, H 14 ), 6.82 (m, 1H, W 10 ), 6.77 (m , 1H, H 12 ), 4.52 (s, 2H, CH 2 SPh), 4.21 (qd, J = 7.2, 0.8 Hz, 2H, CO 2 CH 2 CH 3 ), 4.17 (s, 2H, CH 2 NMe 2 ), 3.66 (s, 3H, NCH 3 ), 3.58 (s, 3H, OCH 3 ), 2.38 (s, 6H, CH 2 H(CH 3 ) 2 ), 1.34 (m, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, CDCl 3) 165.6, 159.9, 151.7, 141.7, 135.5, 131.9, 129.9, 124.7, 117.5, 114.2, 113.1, 112.6, 108.6. 106.2, 60.5, 59.9, 55.3, 44.2, 30.5, 29.9, 14.5. R f : 0.35 (5% MeOH in CH 2 Cl 2 ). HUMS (ESI-TOF): C 23 H 27 BrN 2 O 5 S ((M+H] + ) requires 523.0897, found
References
- ^ “Full Prescribing Information: Arbidol® (umifenovir) film-coated tablets 50 and 100 mg: Corrections and Additions”. State Register of Medicines (in Russian). Open joint-stock company “Pharmstandard-Tomskchempharm”. Retrieved 3 June 2015.
- ^ Recommended INN: List 65., WHO Drug Information, Vol. 25, No. 1, 2011, page 91
- ^ Jump up to:a b Leneva IA, Russell RJ, Boriskin YS, Hay AJ (February 2009). “Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol”. Antiviral Research. 81 (2): 132–40. doi:10.1016/j.antiviral.2008.10.009. PMID 19028526.
- ^ “FDA Approved Drugs for Influenza”. U.S. Food and Drug Administration.
- ^ Jump up to:a b Leneva IA, Fediakina IT, Gus’kova TA, Glushkov RG (2005). “[Sensitivity of various influenza virus strains to arbidol. Influence of arbidol combination with different antiviral drugs on reproduction of influenza virus A]”. Terapevticheskii Arkhiv (Russian translation). ИЗДАТЕЛЬСТВО “МЕДИЦИНА”. 77 (8): 84–8. PMID 16206613.
- ^ Wang MZ, Cai BQ, Li LY, Lin JT, Su N, Yu HX, Gao H, Zhao JZ, Liu L (June 2004). “[Efficacy and safety of arbidol in treatment of naturally acquired influenza]”. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. Acta Academiae Medicinae Sinicae. 26 (3): 289–93. PMID 15266832.
- ^ Boriskin YS, Leneva IA, Pécheur EI, Polyak SJ (2008). “Arbidol: a broad-spectrum antiviral compound that blocks viral fusion”. Current Medicinal Chemistry. 15 (10): 997–1005. doi:10.2174/092986708784049658. PMID 18393857.
- ^ Leneva IA, Burtseva EI, Yatsyshina SB, Fedyakina IT, Kirillova ES, Selkova EP, Osipova E, Maleev VV (February 2016). “Virus susceptibility and clinical effectiveness of anti-influenza drugs during the 2010-2011 influenza season in Russia”. International Journal of Infectious Diseases. 43: 77–84. doi:10.1016/j.ijid.2016.01.001. PMID 26775570.
- ^ Boriskin YS, Pécheur EI, Polyak SJ (July 2006). “Arbidol: a broad-spectrum antiviral that inhibits acute and chronic HCV infection”. Virology Journal. 3: 56. doi:10.1186/1743-422X-3-56. PMC 1559594. PMID 16854226.
- ^ Shi L, Xiong H, He J, Deng H, Li Q, Zhong Q, Hou W, Cheng L, Xiao H, Yang Z (2007). “Antiviral activity of arbidol against influenza A virus, respiratory syncytial virus, rhinovirus, coxsackie virus and adenovirus in vitro and in vivo”. Archives of Virology. 152 (8): 1447–55. doi:10.1007/s00705-007-0974-5. PMID 17497238.
- ^ Glushkov RG, Gus’kova TA, Krylova LIu, Nikolaeva IS (1999). “[Mechanisms of arbidole’s immunomodulating action]”. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk (in Russian) (3): 36–40. PMID 10222830.
- ^ Pécheur EI, Lavillette D, Alcaras F, Molle J, Boriskin YS, Roberts M, Cosset FL, Polyak SJ (May 2007). “Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol”. Biochemistry. 46 (20): 6050–9. doi:10.1021/bi700181j. PMC 2532706. PMID 17455911.
- ^ Pécheur EI, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M, Mire CE, Kawaoka Y, Geisbert TW, Polyak SJ (January 2016). “The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses”. Journal of Virology. 90 (6): 3086–92. doi:10.1128/JVI.02077-15. PMC 4810626. PMID 26739045.
- ^ Hulseberg CE, Fénéant L, Szymańska-de Wijs KM, Kessler NP, Nelson EA, Shoemaker CJ, Schmaljohn CS, Polyak SJ, White JM. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J Virol. 2019 Apr 3;93(8). pii: e02185-18. doi:10.1128/JVI.02185-18 PMID 30700611
- ^ Ng E (4 February 2020). “Coronavirus: are cocktail therapies for flu and HIV the magic cure?”. South China Morning Post.
Bangkok and Hangzhou hospitals put combination remedies to the test.
- ^ Zheng W, Lau M (4 February 2020). “China’s health officials say priority is to stop mild coronavirus cases from getting worse”. South China Morning Post.
- ^ Lu H (January 2020). “Drug treatment options for the 2019-new coronavirus (2019-nCoV)”. Bioscience Trends. doi:10.5582/bst.2020.01020. PMID 31996494.
- ^ “Efficacies of lopinavir/ritonavir and abidol in the treatment of novel coronavirus pneumonia”. 4 February 2020. Retrieved 24 February 2020.
- ^ “АРБИДОЛ® (ARBIDOL)”. Vidal. Archived from the originalon 4 February 2009.
- ^ “Resolution”. Meetings of the Presidium of the Formulary Committee. Russian Academy of Medical Sciences. 16 March 2007.
- ^ “How do we plant federal ministers”. MKRU. 21 April 2011.
- ^ Golunov I (23 December 2013). “13 most popular flu cures: do they work?”. Professional Journalism Platform.
- ^ Reuters S. “Stick in the wheel”. Esquire.
- ^ “Repetition – the mother of learning”. Esquire.
- “How do we plant federal ministers”. MKRU (in Russian). 21 April 2011.
- ^ Golunov I (23 December 2013). “13 most popular flu cures: do they work?”. Professional Journalism Platform (in Russian).
- ^ Reuters S. “Stick in the wheel”. Esquire (in Russian).
- ^ “Repetition – the mother of learning”. Esquire (in Russian).
- ^ Recommended INN: List 65., WHO Drug Information, Vol. 25, No. 1, 2011, page 91
External links
- “Мастерлек” Pharmaceuticals, Moscow, Russia. Patent number No. 2033157, Registry number No. 003610/01.
- (in Russian) Arbidol
- English published clinical studies and translations for Arbidol 1973–2016
- “Umifenovir”. Drug Information Portal. U.S. National Library of Medicine.
- “Compendium”. Drug Information Portal. Drug Information Portal.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Arbidol |
| Pregnancy category |
|
| Routes of administration |
Oral (hard capsules, tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 40% |
| Metabolism | Hepatic |
| Elimination half-life | 17–21 hours |
| Excretion | 40% excrete as unchanged umifenovir in feces (38.9%) and urine (0.12%)[1] |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.247.800 |
| Chemical and physical data | |
| Formula | C22H25BrN2O3S |
| Molar mass | 477.41 g/mol g·mol−1 |
| 3D model (JSmol) | |
Umifenovir is an indole-based, hydrophobic, dual-acting direct antiviral/host-targeting agent used for the treatment and prophylaxis of influenza and other respiratory infections.13 It has been in use in Russia for approximately 25 years and in China since 2006. Its invention is credited to a collaboration between Russian scientists from several research institutes 40-50 years ago, and reports of its chemical synthesis date back to 1993.13 Umifenovir’s ability to exert antiviral effects through multiple pathways has resulted in considerable investigation into its use for a variety of enveloped and non-enveloped RNA and DNA viruses, including Flavivirus,2 Zika virus,3 foot-and-mouth disease,4 Lassa virus,6 Ebola virus,6 herpes simplex,8, hepatitis B and C viruses, chikungunya virus, reovirus, Hantaan virus, and coxsackie virus B5.13,9 This dual activity may also confer additional protection against viral resistance, as the development of resistance to umifenovir does not appear to be significant.13
Umifenovir is currently being investigated as a potential treatment and prophylactic agent for COVID-19 caused by SARS-CoV2 infections in combination with both currently available and investigational HIV therapies.1,16,17
Indication
Umifenovir is currently licensed in China and Russia for the prophylaxis and treatment of influenza and other respiratory viral infections.13 It has demonstrated activity against a number of viruses and has been investigated in the treatment of Flavivirus,2 Zika virus,3 foot-and-mouth disease,4 Lassa virus,6 Ebola virus,6 and herpes simplex.8 In addition, it has shown in vitro activity against hepatitis B and C viruses, chikungunya virus, reovirus, Hantaan virus, and coxsackie virus B5.13,9
Umifenovir is currently being investigated as a potential treatment and prophylactic agent for the prevention of COVID-19 caused by SARS-CoV-2 infections.1,16
Pharmacodynamics
Umifenovir exerts its antiviral effects via both direct-acting virucidal activity and by inhibiting one (or several) stage(s) of the viral life cycle.13 Its broad-spectrum of activity covers both enveloped and non-enveloped RNA and DNA viruses. It is relatively well-tolerated and possesses a large therapeutic window – weight-based doses up to 100-fold greater than those used in humans failed to produce any pathological changes in test animals.13
Umifenovir does not appear to result in significant viral resistance. Instances of umifenovir-resistant influenza virus demonstrated a single mutation in the HA2 subunit of influenza hemagglutinin, suggesting resistance is conferred by prevention of umifenovir’s activity related to membrane fusion. The mechanism through which other viruses may become resistant to umifenovir requires further study.13
Mechanism of action
Umifenovir is considered both a direct-acting antiviral (DAA) due to direct virucidal effects and a host-targeting agent (HTA) due to effects on one or multiple stages of viral life cycle (e.g. attachment, internalization), and its broad-spectrum antiviral activity is thought to be due to this dual activity.13 It is a hydrophobic molecule capable of forming aromatic stacking interactions with certain amino acid residues (e.g. tyrosine, tryptophan), which contributes to its ability to directly act against viruses. Antiviral activity may also be due to interactions with aromatic residues within the viral glycoproteins involved in fusion and cellular recognition,5,7 with the plasma membrane to interfere with clathrin-mediated exocytosis and intracellular trafficking,10 or directly with the viral lipid envelope itself (in enveloped viruses).13,12 Interactions at the plasma membrane may also serve to stabilize it and prevent viral entry (e.g. stabilizing influenza hemagglutinin inhibits the fusion step necessary for viral entry).13
Due to umifenovir’s ability to interact with both viral proteins and lipids, it may also interfere with later stages of the viral life cycle. Some virus families, such as Flaviviridae, replicate in a subcellular compartment called the membranous web – this web requires lipid-protein interactions that may be hindered by umifenovir. Similarly, viral assembly of hepatitis C viruses is contingent upon the assembly of lipoproteins, presenting another potential target.13
Absorption
Umifenovir is rapidly absorbed following oral administration, with an estimated Tmax between 0.65-1.8 hours.14,15,13 The Cmax has been estimated as 415 – 467 ng/mL and appears to increase linearly with dose,14,15 and the AUC0-inf following oral administration has been estimated to be approximately 2200 ng/mL/h.14,15
Volume of distribution
Data regarding the volume of distribution of umifenovir are currently unavailable.
Protein binding
Data regarding protein-binding of umifenovir are currently unavailable.
Metabolism
Umifenovir is highly metabolized in the body, primarily in hepatic and intestinal microsomess, with approximately 33 metabolites having been observed in human plasma, urine, and feces.14 The principal phase I metabolic pathways include sulfoxidation, N-demethylation, and hydroxylation, followed by phase II sulfate and glucuronide conjugation. In the urine, the major metabolites were sulfate and glucuronide conjugates, while the major species in the feces was unchanged parent drug (~40%) and the M10 metabolite (~3.0%). In the plasma, the principal metabolites are M6-1, M5, and M8 – of these, M6-1 appears of most importance given its high plasma exposure and long elimination half-life (~25h), making it a potentially important player in the safety and efficacy of umifenovir.14
Enzymes involved in the metabolism of umifenovir include members of the cytochrome P450 family (primarily CYP3A4), flavin-containing monooxygenase (FMO) family, and UDP-glucuronosyltransferase (UGT) family (specifically UGT1A9 and UGT2B7).14,11
- Lu H: Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci Trends. 2020 Jan 28. doi: 10.5582/bst.2020.01020. [PubMed:31996494]
- Haviernik J, Stefanik M, Fojtikova M, Kali S, Tordo N, Rudolf I, Hubalek Z, Eyer L, Ruzek D: Arbidol (Umifenovir): A Broad-Spectrum Antiviral Drug That Inhibits Medically Important Arthropod-Borne Flaviviruses. Viruses. 2018 Apr 10;10(4). pii: v10040184. doi: 10.3390/v10040184. [PubMed:29642580]
- Fink SL, Vojtech L, Wagoner J, Slivinski NSJ, Jackson KJ, Wang R, Khadka S, Luthra P, Basler CF, Polyak SJ: The Antiviral Drug Arbidol Inhibits Zika Virus. Sci Rep. 2018 Jun 12;8(1):8989. doi: 10.1038/s41598-018-27224-4. [PubMed:29895962]
- Herod MR, Adeyemi OO, Ward J, Bentley K, Harris M, Stonehouse NJ, Polyak SJ: The broad-spectrum antiviral drug arbidol inhibits foot-and-mouth disease virus genome replication. J Gen Virol. 2019 Sep;100(9):1293-1302. doi: 10.1099/jgv.0.001283. Epub 2019 Jun 4. [PubMed:31162013]
- Kadam RU, Wilson IA: Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci U S A. 2017 Jan 10;114(2):206-214. doi: 10.1073/pnas.1617020114. Epub 2016 Dec 21. [PubMed:28003465]
- Hulseberg CE, Feneant L, Szymanska-de Wijs KM, Kessler NP, Nelson EA, Shoemaker CJ, Schmaljohn CS, Polyak SJ, White JM: Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J Virol. 2019 Apr 3;93(8). pii: JVI.02185-18. doi: 10.1128/JVI.02185-18. Print 2019 Apr 15. [PubMed:30700611]
- Zeng LY, Yang J, Liu S: Investigational hemagglutinin-targeted influenza virus inhibitors. Expert Opin Investig Drugs. 2017 Jan;26(1):63-73. doi: 10.1080/13543784.2017.1269170. Epub 2016 Dec 14. [PubMed:27918208]
- Li MK, Liu YY, Wei F, Shen MX, Zhong Y, Li S, Chen LJ, Ma N, Liu BY, Mao YD, Li N, Hou W, Xiong HR, Yang ZQ: Antiviral activity of arbidol hydrochloride against herpes simplex virus I in vitro and in vivo. Int J Antimicrob Agents. 2018 Jan;51(1):98-106. doi: 10.1016/j.ijantimicag.2017.09.001. Epub 2017 Sep 7. [PubMed:28890393]
- Pecheur EI, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M, Mire CE, Kawaoka Y, Geisbert TW, Polyak SJ: The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses. J Virol. 2016 Jan 6;90(6):3086-92. doi: 10.1128/JVI.02077-15. [PubMed:26739045]
- Blaising J, Levy PL, Polyak SJ, Stanifer M, Boulant S, Pecheur EI: Arbidol inhibits viral entry by interfering with clathrin-dependent trafficking. Antiviral Res. 2013 Oct;100(1):215-9. doi: 10.1016/j.antiviral.2013.08.008. Epub 2013 Aug 25. [PubMed:23981392]
- Song JH, Fang ZZ, Zhu LL, Cao YF, Hu CM, Ge GB, Zhao DW: Glucuronidation of the broad-spectrum antiviral drug arbidol by UGT isoforms. J Pharm Pharmacol. 2013 Apr;65(4):521-7. doi: 10.1111/jphp.12014. Epub 2012 Dec 24. [PubMed:23488780]
- Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Bockmann A, Meier BH, Penin F, Pecheur EI: Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS One. 2011 Jan 25;6(1):e15874. doi: 10.1371/journal.pone.0015874. [PubMed:21283579]
- Blaising J, Polyak SJ, Pecheur EI: Arbidol as a broad-spectrum antiviral: an update. Antiviral Res. 2014 Jul;107:84-94. doi: 10.1016/j.antiviral.2014.04.006. Epub 2014 Apr 24. [PubMed:24769245]
- Deng P, Zhong D, Yu K, Zhang Y, Wang T, Chen X: Pharmacokinetics, metabolism, and excretion of the antiviral drug arbidol in humans. Antimicrob Agents Chemother. 2013 Apr;57(4):1743-55. doi: 10.1128/AAC.02282-12. Epub 2013 Jan 28. [PubMed:23357765]
- Liu MY, Wang S, Yao WF, Wu HZ, Meng SN, Wei MJ: Pharmacokinetic properties and bioequivalence of two formulations of arbidol: an open-label, single-dose, randomized-sequence, two-period crossover study in healthy Chinese male volunteers. Clin Ther. 2009 Apr;31(4):784-92. doi: 10.1016/j.clinthera.2009.04.016. [PubMed:19446151]
- Wang Z, Chen X, Lu Y, Chen F, Zhang W: Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci Trends. 2020 Feb 9. doi: 10.5582/bst.2020.01030. [PubMed:32037389]
- Nature Biotechnology: Coronavirus puts drug repurposing on the fast track [Link]
/////////////////Arbidol, umifenovir, covid 19, corona virus, Арбидол , 阿比朵尔 ,
CCOC(=O)C1=C(CSC2=CC=CC=C2)N(C)C2=CC(Br)=C(O)C(CN(C)C)=C12

https://eurekalert.org/pub_releases/2020-02/nuos-edm022620.php

NEW DRUG APPROVALS
ONE TIME HELP TO SUSTAINPAYMENTS FOR THIS BLOG AS SUBSCRIPTION
$10.00

