
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Febuxostat
| Febuxostat; 144060-53-7; Uloric; Adenuric; Tei 6720; 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid; | |
| Molecular Formula: | C16H16N2O3S |
|---|---|
| Molecular Weight: | 316.37484 g/mol |
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylic acid
Febuxostat is a thiazole derivative and inhibitor of XANTHINE OXIDASE that is used for the treatment of HYPERURICEMIA in patients with chronic GOUT.
CAS 144060-53-7
- 2-[3-Cyano-4-(2-methylpropoxy)phenyl]-4-methyl-5-thiazolecarboxylic acid
- 2-(3-Cyano-4-isobutyloxyphenyl)-4-methyl-5-thiazolecarboxylic acid
- FBX
- Febugood
- Feburic
- Febutaz
- TMX 67
- Zurig
Febuxostat (INN; trade names Adenuric in Europe and New Zealand, Uloric in the US, Goturic in Latin America, Feburic in Japan) is a drug that inhibits xanthine oxidase, thus reducing production of uric acid in the body. It is used in the treatment of chronicgout and hyperuricemia.
Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998. Teijin partnered the drug with TAP Pharmaceuticals in the US and Ipsen in Europe. Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008, Takeda obtained FDA approval in February 2009, and Teijin obtained approval from the Japanese “Pharmaceuticals and Medical Devices Agency” in 2011.
Medical uses
Febuxostat is used to treat chronic gout and hyperuricemia.[2] National Institute for Health and Clinical Excellence concluded that febuxostat is more effective than standard doses of allopurinol, but not more effective than higher doses of allopurinol.[2]

Uloric 40 mg tablet
Febuxostat is in the US pregnancy category C; there are no adequate and well-controlled studies in pregnant women.[3]
Side effects
The adverse effects associated with febuxostat therapy include nausea, diarrhea, arthralgia, headache, increased hepatic serum enzyme levels and rash.[3][4]
Drug interactions
Febuxostat is contraindicated with concomitant use of theophylline and chemotherapeutic agents, namely azathioprine and 6-mercaptopurine, because it could increase blood plasma concentrations of these drugs, and therefore their toxicity.[3][5]
Mechanism of action
Febuxostat is a non-purine-selective inhibitor of xanthine oxidase.[3] It works by non-competitively blocking the molybdenum pterincenter which is the active site on xanthine oxidase. Xanthine oxidase is needed to successively oxidize both hypoxanthine andxanthine to uric acid. Hence, febuxostat inhibits xanthine oxidase, therefore reducing production of uric acid. Febuxostat inhibits both oxidized as well as reduced form of xanthine oxidase because of which febuxostat cannot be easily displaced from the molybdenum pterin site.[4]

History
Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998.[6] Teijin partnered the drug withTAP Pharmaceuticals in the US and Ipsen in Europe.[7][8][9]
Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008,[10] Takeda obtained FDA approval in February 2009,[11][12] and Teijin obtained approval from the Japanese authorities in 2011.[13] Ipsen exclusively licensed its European rights to Menarini in 2009.[14] Teijin partnered with Astellas for distribution in China and southeast Asia.[15][16]
Society and culture
Cost
In the UK, NICE has found that febuxostat has a higher cost/benefit ratio than allopurinol and on that basis recommended febuxostat as a second-line drug for people who cannot use allopurinol.[2]
Trade names
Febuxostat is marketed as Adenuric in Europe and New Zealand, Uloric in the US, Goturic and Goutex in Latin America, Feburic in Japan, and is generic in several countries and is available by many names in those countries.[1]
Febuxostat (Formula I) is an inhibitor of xanthine oxidase, which was discovered by the Japanese company Teijin Pharma Ltd and it is indicated for use in the treatment of hyperuricemia and chronic gout. Its chemical name is 2-(3-cyano-4-isobutoxyphenyl)-4-methyl- l,3-thiazole-5-carboxylic acid. It is marketed under the brand names Adenuric in Europe, Feburic in Japan and Uloric in USA and Canada.

In EP0513379B1 Febuxostat is prepared from 4-hydroxy-3-nitrobenzaldehyde, according to the following scheme.

This particular process suffers from major drawbacks. Not only it is very long, including seven steps from the starting material to the final product, but, most importantly, it employs the use of cyanides, which are extremely toxic reagents. Cyanide salts are likely to generate hydrocyanide, which sets a high amount of risk in an industrial scale process.
In Japanese patent JP06345724A(JP2706037B) the intermediate ethyl ester of Febuxostat is prepared from p-cyano-nitrobenzene, in three steps. Febuxostat may, then, be prepared by alkaline hydrolysis, according to prior art.
MeCSNH,

The use of extremely toxic potassium cyanide makes this process unsuitable for manufacturing purposes.
Route A

In Japanese patent JP3202607B Febuxostat ethyl ester is prepared, according to the above scheme, through two similar routes. Route A uses flash column chromatography for the purification of the hydroxylamine reaction product, while Route B suffers from low yield and the use of chlorinated solvents for recrystallization. In addition, the reaction solvent is, in both cases, formic acid which causes severe skin burns and eye damage to humans. Formic acid is also corrosive towards metal-based materials of construction (MOC), like stainless steel and nickel alloys, limiting the options, essentially, to glass reactors or vessels. The drawbacks of using this solvent are also related to the high volumes of formic acid required per batch, which hinder the waste treatment.
In CN101723915B focus is made to the improvement of the hydroxylamine reaction. Formic acid is replaced with dimethylformamide (DMF) and other solvents. However, according to widely used organic chemistry textbooks, such as March’s Advanced Organic Chemistry, pi 287, 6th edition, M. B. Smith and J. March, ISBN 0-471-72091-7, the mechanism of the reaction involves the formation of an oxime, upon the action of hydroxylamine, which further dehydrates to form a nitrile, with the aid of a suitable reagent, for example formic acid, or acetic anhydride. In the absence of such a reagent, it is expected that the reaction will, at least, not lead to completion, thereby leading to low yields and undesired impurity levels, namely the intermediate oxime. Such impurities, arising from the reactions of the process and which exhibit similar structure of the desired product, are often difficult to remove with common industrial techniques, e.g. crystallization.
In WO2010142653A1 the intermediate Febuxostat ethyl ester is prepared from 4-cyanophenol, through a five-step process. Febuxostat can be prepared from its respective ethyl ester via alkaline hydrolysis, as in the previous case.
OH

1: patents US5614520 febuxostat synthetic process:

2: Patent JP1994329647 febuxostat synthesis


PATENT
https://www.google.com/patents/CN102936230A?cl=en
Gout occurs because the body produces too much uric acid and renal clearance capacity decreased, uric acid accumulation in the body, leading to urate crystals deposited in the joints and organs. Therefore, it means the treatment of gout usually taken to be: to promote uric acid excretion and suppression of uric acid, and the use of appropriate measures to improve symptoms. Uric acid formation and purine metabolism, the final step in the purine metabolism, hypoxanthine generation xanthine xanthine oxidoreductase (XOR) effect, further generate uric acid, inhibit the activity of the enzyme can effectively reduce uric acid production. Febuxostat is currently the world’s newly developed XOR inhibitors, which act by highly selective to the oxidase, reduce uric acid synthesis, reduce uric acid levels, so as to effectively treat the disease ventilation.
Compared with the traditional treatment of gout drug allopurinol, febuxostat has obvious advantages: (1) allopurinol reduced the XOR only inhibit rather than febuxostat of oxidized and reduced form are XOR significant inhibition, thus reducing the role of uric acid, which is more powerful and lasting; (2) Since allopurinol is a purine analogue, the inevitable result of the purine and other activity related to the impact of pyridine metabolism. So allopurinol treatment should be repeated large doses of the drug to maintain a high level. Which also brought serious or even fatal adverse reactions due to drug accumulation due.Instead of febuxostat non-purine XOR inhibitors, so it has better security.
Document TMX-67. Drugs Fut2001, 26, I, 32, and EP0513379, US5614520, W09209279, public
The detailed preparation febuxostat. Using 3-nitro-4-hydroxybenzaldehyde as the starting material is first reacted with hydroxylamine hydrochloride, to give 3-nitro-4-hydroxybenzonitrile. In effect then HCl, reaction with thioacetamide to give 3-nitro-4-hydroxy-thiobenzamide. Closed loop then reacted with 2-chloro ethyl acetoacetate to give 2- (3_ nitro-4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Followed by potassium carbonate effect, isobutane is reacted with bromo, to give 2- (3_ nitro-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester. Under the catalytic action of palladium on carbon, hydrogen reduction to give 2- (3-amino-4-isobutyloxyphenyl) -4-methyl-5-thiazole carboxylic acid ethyl ester. Followed by diazotization with sodium nitrite occur, was added cuprous cyanide and potassium cyanide, to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-thiazolecarboxylic acid ethyl ester. Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.The process route is as follows:

This route in the preparation of febuxostat, there are many disadvantages: raw 3-nitro-4-hydroxybenzaldehyde in the country is difficult to buy; requires the use of palladium-carbon catalytic hydrogenation reaction under the factory equipment higher requirements, there is a certain danger; the cyano preparation, the need to use sodium nitrite diazotization, could easily lead to corrosion of equipment; the cyano preparation, the need to use toxic cyanide copper, potassium cyanide, pollution, higher risk.
Document JP1994329647, JP1998045733, US3518279 reported another synthesis of febuxostat
Methods. From 4-hydroxy-thiobenzamide as a starting material, and the cyclization reaction to give ethyl 2-bromo-acetyl occurred
2- (4_ hydroxyphenyl) -4_ methyl-5-carboxylic acid ethyl ester in polyphosphoric acid effect, HMTA (hexamethylene tetramine) reacts with 2- (3_ aldehyde – 4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Then two cases: the first case, the effect of potassium carbonate, is reacted with isobutane to give bromo-2- (4-isobutyloxyphenyl 3_ aldehyde) -4_-methyl-5- thiazole carboxylic acid ethyl ester, and then reacted with hydroxylamine hydrochloride to give 2- (3_-cyano-4-isobutyloxyphenyl) -4_-methyl-5-thiazole carboxylic acid ethyl ester; second case is the first with hydroxylamine hydrochloride to give 2- (3_ cyano-4-hydroxyphenyl) methyl-5-thiazolecarboxylic -4_ carboxylic acid ethyl ester, and then under the effect of potassium carbonate, and reacted with isobutane to give bromo-2- (3 _-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester.
Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3_-cyano-4-isobutyloxyphenyl) -4_ methyl – thiazole-5-carboxylic acid, i.e., to obtain febuxostat . The process route is as follows:

This synthesis route febuxostat process, since the introduction of aldehyde HMTA in PPA (polyphosphoric acid) effect. So there are a lot of phosphorus wastewater, serious environmental pollution, but also because PPA has great viscosity, and therefore difficult to stir the production, operation is extremely inconvenient.
Document Heterocyclesl998, 47,2,857 JP1994345724 also reported the synthesis method of febuxostat, using p-nitrophenyl-carbonitrile as a starting material in the reaction with potassium cyanide in DMSO solvent, and then the carbonate lower potassium catalyzed reaction of isobutane and brominated 1,3-cyano-4-diisobutoxybenzene ether. By reaction with thioacetamide to afford
3-cyano-4-isobutyloxyphenyl thiobenzamide. Under heating, and 2-chloro ethyl acetoacetate, ring closure reaction occurs to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester, and finally hydrolysis under the effect of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.
The present invention febuxostat new technology system, comprising the steps of:
(1) 2-hydroxy-5-cyano – NaSH reacted with benzaldehyde to give 4-hydroxy-3- aldehyde thiobenzamide;

(2) the step (I) to give 4-hydroxy-3-aldehyde thiobenzamide reaction with ethyl 2-halo-acetyl, closed
Ring to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

X is a halogen, preferably Cl or Br;
(3) the step (2) to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole with hydroxylamine in formic acid in the reaction solution to give 2- (3- cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

(4) The step (3) to give 2- (3-cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole isobutane with halo effect in potassium carbonate, to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;
(5) in step (4) to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl-thiazol-off hydrolyzable ester group, to obtain a non-Tendon Disposition Tanzania.
[0011] Scheme of the method is as follows:

X is halogen, may be Cl, Br;
Preparation 5 febuxostat Example
To a 500ml reaction flask was added 200ml of absolute ethanol, the product of Step 4 was added with stirring (60g, O. 174mol),
5% sodium hydroxide was added 100ml. Stirring heated to 40 degrees, until it is completely dissolved. 40 degrees heat, reaction 4h. The reaction by TLC tracking. After completion of the reaction, the reaction solution was added 10% hydrochloric acid to adjust the pH to 3, the precipitated solid was filtered. And dried to give a pale yellow solid. Dried over anhydrous recrystallized from methanol to give 31. 2g of white crystals, yield 56.7%.
TLC monitoring of the reaction. Eluent: petroleum ether / ethyl acetate = 3: 1 Melting point:. 201 · 7 ~202 30C (literature value 201 ~202 ° C)
1H-NMR δ:. 1 01 (m, 6H), 2.06 (m, lH), 2.57 (m, 3 H), 3.96 (d, 2H), 7.30 (d, lH), 8.13 (m, 1H), 8. 19 (d, 1H);
MS (m / z):. 316 O (M +)
Infrared detection: 3550-3400cm_1; 2961, 2933,2874; 2227cm_1; 1680U604U511cm_1; 1425cm_1; 1296U283CHT1;
Elemental analysis for C, Η, N, S purified product actual measurement of the content of C, H, N, S content: C:. 60 57%, H:. 5 32%, N:. 8 86%, S: 10. 16%; theoretical value: In C16H16N203S calculated C: 60 74%, H: 510%, N: 885%, S: 1014%..
CLIP
Facile OnePot Transformation of Arenes into Aromatic Nitriles …

Patent




Scheme-Ill:

Clip
synthesis describes synthesis of febuxostat (I) from 4-hydroxybenzonitrile (II) in six stages. The synthesis shown is a short, concise route and does not require use of poisonous reagents such as KCN (14). Compound II was converted to 4-hydroxybenzothioamide (III) with 85% yield using NaHS in the presence of hydrated magnesium chloride as Lewis acid. Intermediate III, on cyclization with ethyl-2-chloroacetoacetate, gave thiazole ester (IV) with quantitative yield. In these two stages, the source of potential impurities was identified as an ortho isomer (i.e., 2-hydroxybenzonitrile), which can lead to Impurity VIII and subsequently to Impurity IX . Impurities VIII and IX can be controlled in starting material II with appropriate specification.
|

The ortho formylation of hydroxyl compound IV by using Duff condition (hexamine/TFA) gave aldehyde V (15). The major impurity identified in this reaction was dialdehyde X. Although we have used only 1.0 equivalence of hexamine with respect to Compound IV, the dialdehyde X impurity was formed to a 5-10% ratio in only 2.5 h. It is, therefore, impossible to get rid of this impurity during the reaction, and only effective recrystallization will eliminate it. Impurity X was minimized (≤ 2%) by recrystallization using IPA/H2O (3:5) to get aldehyde V with 50% yield and & #8805; 97% HPLC purity.
Aldehyde V, on alkylation with isobutyl bromide in the presence of potassium carbonate base, gave compound VI with 90% yield. In this stage, Impurities XI and XII were alkylations of carryover Compound IV and dialdehyde, respectively. Two more isomeric impurities n-butyl-aldehyde XIII and 1-methyl propyl-aldehyde XIV were also identified in this stage. Both isomeric impurities can be controlled with appropriate specification for isobutyl bromide. The reaction of Compound VI with hydroxylamine hydrochloride and sodium formate in formic acid at reflux temperature gave Compound VII with 85% yield. Impurities XIII and XIV will also carry forward to impurities n-butyl-nitrile XV and 1-methyl propyl-nitrile XVI, respectively.
In the final step, Compound VII was hydrolyzed using sodium hydroxide in a MeOH:THF:H2O (1:1:1) solvent combination to yield febuxostat (85%). During saponification, methyl ester Impurity XVII was identified via trans-esterification. Its hydrolysis was comparatively slower than its ethyl isomer VII. One way to avoid Impurity XVII is to replace methanol with ethanol. Carryover impurities XI, XV, and XVI were also hydrolyzed to their respective acid derivatives impurities XVIII, XIX, and XX. However, the acid derivatives of impurities X and XII were unexpectedly absent as impurities. It is believed that, because they were present in low concentrations during workup, they were eliminated in the mother liquor. Two additional impurities, amide XXI and diacid XXII, formed by the side reaction of the febuxostat nitrile group with sodium hydroxide, were identified during saponification. The amide XXI and diacid XXII impurities can be controlled by using appropriate equivalence of sodium hydroxide and controlled reaction time. Febuxostat, on acetone recrystallization and seed Crystal A at 45°C, gave pure febuxostat with 75% yield.

http://www.pharmtech.com/investigation-various-impurities-febuxostat
References
- Drugs.com Drugs.com international names for febuxostat Page accessed June 25, 2015
- Febuxostat for the management of hyperuricaemia in people with gout (TA164) Chapter 4. Consideration of the evidence
- Uloric label Updated February, 2009.
- Love BL, Barrons R, Veverka A, Snider KM (2010). “Urate-lowering therapy for gout: focus on febuxostat”. Pharmacotherapy 30 (6): 594–608. doi:10.1592/phco.30.6.594.PMID 20500048.
- Ashraf Mozayani; Lionel Raymon (2011). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science+Business Media.
- Teijin Febuxostat Story Page accessed June 25, 2015
- Tomlinson B. Febuxostat (Teijin/Ipsen/TAP). Curr Opin Investig Drugs. 2005 Nov;6(11):1168-78. PMID 16312139
- Bruce Japsen for the Chicago Tribune. August 17, 2006. FDA puts gout treatment on hold
- Note: TAP Pharmaceuticals was a joint venture between Abbott Laboratories and Takedathat was dissolved in 2008 per this press release: Takeda, Abbott Announce Plans to Conclude TAP Joint Venture
- “Adenuric (febuxostat) receives marketing authorisation in the European Union” (PDF). Retrieved 2008-05-28.
- “Uloric Approved for Gout”. U.S. News and World Report. Retrieved 2009-02-16.
- Teijin and Takeda. February 14, 2009 Press release: ULORIC® (TMX-67, febuxostat) Receives FDA Approval for the Chronic Management of Hyperuricemia in Patients with Gout
- Teijin. January 21, 2011 Press release: TMX-67 (febuxostat) Approved in Japan
- Genetic Engineering News. October 2009. Menarini to Market Takeda/Ipsen Gout Therapy in 41 European Countries
- First Word Pharma. April 1st, 2010 Teijin Pharma and Astellas Pharma enter into agreement for marketing rights of TMX-67 in China and Hong Kong
- Research Views. Aug 11 2011 Teijin Pharma Enters Into Distribution Agreement With Astellas Pharma For Febuxostat
Febuxostat is an inhibitor of xanthine oxidase, and was developed by Teijin pharma. This compound is known as a new drug that is effective against gout and hyperuricemia, and it has been 40 years since the last time a drug of this kind of drug was developed.
Febuxostat has therefore gained a lot of popularity and it has already been accepted as a drug in Europe, USA, Korea and Japan. The synthesis of this molecule have been reported in patents by Teijin pharma as shown below.[1,2]

Recently, Itami group was reported the rapoid synthesis of febxostat by using Ni-catalyzed direct coupling of azoles and arylhalides[3]
References
Sorbera, L.A.; Castaner, J.; Rabasseda, X.; Revel, L.; TMX-67. Drugs Fut 2001, 26, 1, 32
[1] Hasegawa, M.; A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile. Heterocycles 1998, 47, 2, 857. [2] Hasegawa, M.; Hasegawa, M.; Komoriya, K. (Teijin Ltd.); Cyano cpds. and their preparation method. JP 1994345724 . [3] “Nickel-Catalyzed Biaryl Coupling of Heteroarenes and Aryl Halides/Triflates”
Canivet, J.; Yamaguchi, J.; Ban, I.; Itami, K. Org. Lett. 2009, 11, 1733-1736. DOI: 10.1021/ol9001587

Ni-based catalytic systems for the arylation of heteroarenes with aryl halides and triflates have been established. Ni(OAc)2/bipy is a general catalyst for aryl bromides/iodides, and Ni(OAc)2/dppf is effective for aryl chlorides/triflates. Thiazole, benzothiazole, oxazole, benzoxazole, and benzimidazole are applicable as heteroarene coupling partners. A rapid synthesis of febuxostat, a drug for gout and hyperuricemia, is also demonstrated.
A CLIP
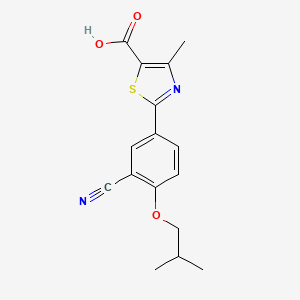
A final example of a thiazole containing drug is given in the novel xanthine oxidase inhibitor febuxostat (359, Uloric) which was approved by the FDA in 2009. This inhibitor works by blocking xanthine oxidase in a non-competitive fashion. Consequently, the amount of the oxidation product uric acid is reduced. Thus it is an efficient treatment for hyperuricemia in gout. In order to prepare febuxostat first a synthesis of the noncommercial 4-isobutoxy-1,3-dicyanobenzene building block (363), has to be conducted. An elegant way of achieving this was shown through the reaction of 4-nitrocyanobenzene (360) with potassium cyanide in dry DMSO followed by quenching with isobutyl bromide under basic conditions (Scheme 70). It is suggested that a Meisenheimer-complex intermediate 361 is initially formed, which after rearomatisation, undergoes nucleophilic aromatic substitution of the nitro group by the DMSO solvent [107]. Upon hydrolysis and O-alkylation the desired 4-isobutoxy-1,3-dicyanobenzene (363) is obtained in good overall yield. Subsequently, the less hindered nitrile is converted to the corresponding thioamide 365 in an intriguing reaction using thioacetamide (364). The thiazole ring is then formed by condensation with chloroacetoacetate 366 followed by ester hydrolysis (Scheme 70).
107 Hasegawa, M. Heterocycles 1998, 47, 857–864. doi:10.3987/COM-97-S(N)89
Paper | Special issue | Vol 47, No. 2, 1998, pp.857-864
■ A Facile One-Pot Synthesis of 4-Alkoxy-1,3-benzenedicarbonitrile
Masaichi Hasegawa
*Teijin Institute, Bio-Medical Research, Asahigaoka 4-3-2, Hino, Tokyo 191, Japan
Abstract
2-(3-Cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxlic acid (TEI-6720) was prepared. The introduction of cyano group to 4-nitrobenzonitrile with KCN in dry DMSO followed by quenching with alkyl halide afforded the key intermediates, 4-alkoky-1,3-benzenedicarbonitriles, in good yield. The reaction was completed in dry DMSO, while no reaction occurred in dry DMF. This observation can be suggested by the participation of DMSO in the reaction.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S70
A CLIP
Synthesis and characterization of process-related impurities of an anti-hyperuricemia drug-Febuxostat
Venkateswara Rao Vallu,$ Krunal Girishbhai Desai, Sandip Dhaya Patil, Rajendra Agarwal, Pratap Reddy Padi and Mahesh Reddy Ghanta
*Process Research Laboratory-I, Research & Development Centre, Macleods Pharmaceuticals Ltd, G-2, Mahakali Caves Road, Shantinagar, Andheri (East), Mumbai, Maharastra, India
$Department of Chemistry, Pacific University, Pacific Hills, Airport Road, Pratap Nagar Extension, Debari, Udaipur, Rajasthan, India _____________________________________________________________________
Der Pharma Chemica, 2014, 6(3):300-311 (http://derpharmachemica.com/archive.html)
http://derpharmachemica.com/vol6-iss3/DPC-2014-6-3-300-311.pdf
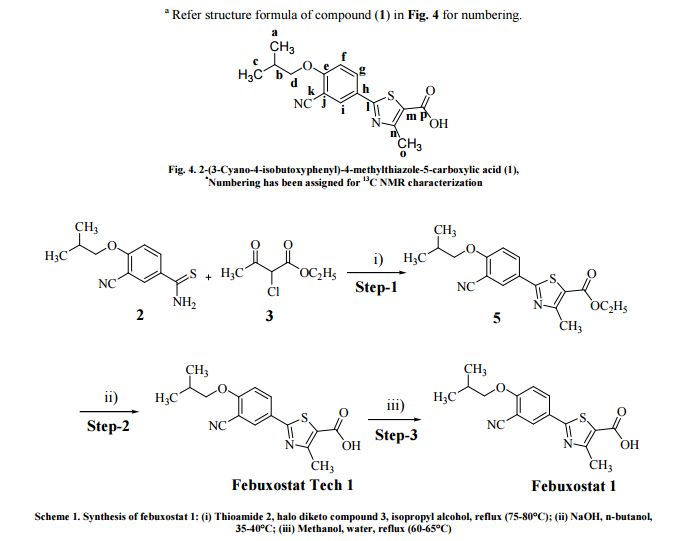
Synthesis of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1) [10] A solution of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1 tech grade, 5.0 g, 0.015 mol.) in methanol (50.0 mL) was heated the reaction mass at 60-65°C till clear solution was obtained. Water (50.0 mL) was added drop wise into reaction mass with in 30.0 min. at 60-65°C. Resultant white crystalline solid was filtrated, Mahesh Reddy Ghanta et al Der Pharma Chemica, 2014, 6 (3):300-311 _____________________________________________________________________________ 302 http://www.scholarsresearchlibrary.com washed with water (10.0 mL) and dried in vacuum tray drier at 50-55°C under vacuum to give
2-(3-cyano-4- isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1). Yield: 95.0 % (4.75 g)
mp 239°C. Purity by HPLC: 99.74 % (10.2 min. retention time),
Anal. Calcd for C16H16N2O3S: C, 60.74; H, 5.10; N, 8.85. Found: C, 60.70; H, 5.11; N, 8.87 %;
IR (KBr) υmax (in cm−1): 3834.61, 3742.03, 3680.30, 3556.85, 3456.55, 2962.76, 2877.89, 2661.85, 2546.12, 2353.23, 2229.79, 2168.06, 2029.18, 1921.16, 1790.00, 1674.27, 1604.83, 1512.24, 1427.37, 1381.08, 1280.78, 1172.76, 1118.75, 1010.73, 918.15, 833.28, 771.55, 725.26, 648.10, 524.66, 462.93; 1H NMR (300 MHz, CDCl3 or DMSO-d6) δH (in ppm): 1.00-1.02 (d, 6H, (CH3)2-CH-), 2.49-2.50 (m, 1H, (CH3)2-CH-), 3.97-3.99 (d, 2H, -CH-CH2−), 7.33–8.25 (d, dd, 3H, Ar-H), 2.64 (s, 3H, -CH3), 13.39 (s, 1H, -COOH);
13C NMR (300 MHz, DMSO–d6) δC (in ppm) (Positiona ): 166.3 (l), 162.9 (p), 162.2 (n), 159.6 (e), 133.1 (g), 131.6 (i), 125.5 (m), 123.0 (h), 115.5 (k), 114.0 (f), 101.7 (j), 75.2 (d), 27.7 (b), 18.8 (a, c), 17.1 (o);
MS m/z (%) (70 eV): m/z =317.0 (100.0 %) [M+1], 318.0 (16.0 %) [M+2], 403.0 (63.0 %), 512.0 (47.0 %), 482.0 (46.0 %), 405.0 (27.0 %), 468.0 (25.0 %), 570.0 (24.0 %).
PATENT
| WO 2012066561 |
As per the present invention, hydroxylamine hydrochloride is added to compound of Formula-Ill in presence of a polar aprotic solvent like DMSO, DMA, ACN or DMF. To this reaction mixture acetyl halides or sulfonyl chlorides are added and temperature raised to 70 -80 °C. Acetyl halides are selected from acetyl bromide or acetyl chloride. Sulfonyl chlorides are selected from methane sulfonyl chloride or para toluene sulfonyl chloride. To this reaction mixture a base selected from alkali metal carbonates like potassium carbonate or sodium carbonate, preferably potassium carbonate and alkyl halide selected from isobutyl bromide is successively added. The reaction mass is washed with water and compound of Formula-II is isolated. In one embodiment the present invention provides, process for the preparation of Febuxostat comprising the steps of:
a) reacting the compound of Formula-III(a) with hydroxylamine hydrochloride in presence of organic solvent;

Formula-III(a)
b) adding acyl halides or sulfonyl chlorides to the reaction mixture;
c) optionally isolating compound of Formula- IV (a)

Formula-IV(a)
d) reacting with isobutyl bromide in presence of base;
e) isolating the compound of Formula-II(a); and

FormuIa-II(a)
f) hydrolyzing the compound of Formula-II(a) to get Febuxostat.
The following examples are provided to illustrate the process of the present invention. They, are however, not intended to limiting the scope of the present invention in any way and several variants of these examples would be evident to person ordinarily skilled in the art. Experimental procedure:
Example – 1: Preparation of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyI thiozole -5-carboxylate
A mixture of 10. Og of Ethyl -2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 40 g of Dimethyl sulfoxide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 70 -80°C for 2-3 hours. Reaction mass was cooled to room temperature and to this 19 g of potassium carbonate and 19 g of isobutyl bromide was added successively. The reaction mass was stirred for 5 hours at 70-80°C. Reaction mass was diluted with 200 ml of purified water. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 84.0%)
Example – 2: Preparation of Ethyl-2-(3-cyano-4-hydroxyphenyl)-4-methyl thiozole – 5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 30 g of Dimethylformamide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 90°C for 2-3 hours. Reaction mass was cooled to room temperature and diluted with 100 ml of water and stir for 2 hours. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4- methyl thiozole -5-carboxyltae (yield 99.0%).
Example – 3: Preparation of Ethyl 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate, 30 g of NMP, 9.6 g of potassium carbonate and 7.2 g of isobutyl bromide were stirred for 3 hours at 90°C. Reaction mass was diluted with 100 ml of purified water. The reaction mass was filtered and washed with purified water and ethanol to give 10.5 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 88.0%). Example – 4: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxylic acid
A mixture of 10. Og of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxyltae, 2.0g of sodium hydroxide was heated at 45-60°C in 75 ml of aqueous methanol for 1 hour. Reaction mass was cooled to ambient temperature and pH adjusted to 2.0 to 2.5 with dilute hydrochloric acid and precipitated crystal was collected by filtration to give 8.8g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 95.8%).
Example – 5-13: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole – 5-carboxylic acid
The above compound was prepared by following the procedure as disclosed in Example- 4, using the below listed solvents instead of aqueous methanol.

Example – 14: Preparation of pure 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid
10.0 g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid was dissolved in 100 ml of ethanol at reflux temperature. After dissolution reaction mass was cooled and precipitated crystal was collected by filtration to give 9.6 g of pure 2-(3- cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 96%).
PATENT
KR 201603732
PATENT
WO 2015018507
https://www.google.com/patents/WO2015018507A2?cl=en



EXPERIMENTAL
of compound of formula lib

Dissolve 14.14g of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula III) in 55 ml dimethylformamide, at ambient temperature. Add 40g of potassium carbonate, along with 15.9 ml isobutyl bromide. Heat the reaction to 75-80 °C and stir for 4 hours. Cool to 25-30 °C, while 165 ml process water is added. Further cool to 0-5 °C and stir for 30 minutes at this temperature. Filter off the precipitated solid and wash the filter cake with 55 ml process water. The wet cake is dried under vacuum at 40 °C for 7 hours, to furnish 16.43 g of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula lib).
of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 1.0 g (2.88 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 3.0 mL dimethylformamide. Add 34 mg (0.19 mmol) copper acetate under stirring at 25-30 °C. Flush with oxygen (02) and add 0.66 ml (34.92 mmol) 25% aqueous ammonia. Flush again with 02. Heat the reaction mixture to 80-82 °C overnight. Check the progress of the reaction by TLC (cyclohexanerethyl acetate 3:1). Cool reaction mass to 25-30 °C. Add 25mL ethyl acetate and 25mL brine at the reaction mass, separate organic layer and extract aqueous layer twice with 25mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. The residue is purified with column chromatography (cyclohexane:ethyl acetate 9:1). afforded 0.754g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) Yield: 75.4%.
EXAMPLE 3: Preparation of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 0.17 g (0.49 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 2.5 mL tetrahydrofuran. Add 2.9 mL (153.43 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 137 mg (0.54 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Starting material is consumed. Add 2.5 mL 5% w/v aqueous sodium thiosulfate Na2S203 and 15mL ethyl acetate at the reaction mass, separate organic layer and extract twice aqueous layer with 15mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. 0.158g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) are collected.
EXAMPLE 4: Preparation of Febuxostat

In a 100 ml 2-neck round-bottomed flask charge 2.407g of ethyl-2-(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate in 20ml tetrahydrofuran under stirring, at 25-35 °C, 0.748g of sodium hydroxide and heat reaction mass to 60-65 °C for approximately 8 hrs. Check the progress of the reaction by TLC (cyclohexane:ethyl acetate 3:1). Cool reaction mass to 0-5 °C and add 50 ml process water keeping temperature within 0-5 °C. Adjust pH to 1-2 with 4.5 ml 6 N hydrochloric acid, keeping temperature within 0-5 °C. Warm up reaction mass to 25-30 °C and stir reaction mass at the above temperature for 15 min. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 2 ml process water and suck dry for 20-30 min. Transfer the crude solid in a 50 ml round-bottomed flask, charge 12 ml process water and 12 ml acetone at 25-30°C. Heat the reaction mass to 50-60 °C for 60 min. Cool down reaction mass to 0-5 °C and stir for 60 min at the above temperature. Filter off the precipitated solid though Buchner funnel under reduced pressure, spray wash with 2 ml of a 1 : 1 mixture of acetone and process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.821g of (compound I) Febuxostat are collected, Purity: 82.6%, Yield: 0.62w/w.
on of compound of formula Ilia

In a 50 mL round-bottomed flask charge under stirring 0.5g (1.72 mmol) of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate in 8.6 mL THF, at 25-30 °C. Add 10.3 mL (544.94 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 480 mg (1.89 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 1 :1). Starting material is consumed. Add 8.6 mL 5% w/v aqueous thiosulfate and 40 mL ethyl acetate at the reaction mass, separate organic layer and extract aqueous layer twice with 40 mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate to dryness. Purification of the residue with column chromatography (cyclohexane: ethyl acetate 3: 1) afforded 0.213 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula Ilia). Yield : 42.6%.
EXAMPLE 6: Preparation of compound Illb
Dissolve 2.2 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula VI) in 7 ml dimethylformamide and to this mixture add 6.6 g of potassium carbonate and 3.14 g of isobutyl bromide. Stir the reaction at 75 °C for 15 hours and then cool to 40 °C. Add 15 ml process water and cool to 0-5 °C. Filter the precipitated solid off and wash with 15 ml process water, which, after drying, affords 2.28 g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb).
EXAMPLE 7: Preparation of compound I (Febuxostat)
In a 100 ml 2-neck round-bottomed flask charge 2.131 g of ethyl-2(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate, 64 ml methanol and 2.5 ml process water are added under stirring at 25-35 °C. Add 1.718 g potassium carbonate and heat reaction mass to reflux for approximately 2-3 hrs. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Cool reaction mass to 20-25 °C. Concentrate solvent at below 40 °C. To the residue add 43 ml process water, 21 ml ethyl acetate and stir for 30 min at 25-35 °C. Separate layers and transfer aqueous layer in a 100 ml round-bottomed flask. Adjust pH to 2.3-2.7 with 25 ml 1 N hydrochloric acid, at 25-35 °C. Warm up reaction mass to 40 °C and stir reaction mass at this temperature for 60-90 min. Cool down reaction mass to 25-35 °C. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 5 ml process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.708g of (compound I) Febuxostat are collected, Purity: 86.7%, Yield: 0.69w/w.
EXAMPLE 8: Preparation of Febuxostat crystalline form III
In a 250 mL round-bottomedflask charge under stirring at 25-30 °C 10 g of crude 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (Febuxostat) in 200 mL ethyl acetate. Heat reaction mass to reflux and stir for 30 min. Cool reaction mass to 25-30°C. Warm again reaction mass and partially distill off solvent from the reaction mass at temperature below 40 °C under reduced pressure. Cool reaction mass to 25-30°C. Filter off the precipitated solid through Buchner funnel under reduced pressure and spray wash with 10 mL ethyl acetate. Dry under vacuum at 60°C. 8.5 g of Febuxostat are collected. Yield: 85 % w/w. XRPD of crystalline compound is in accordance with the one reported in Chinese patent CN101412700B.
PATENT
CN 104418823
https://www.google.com/patents/CN104418823A?cl=zh

PATENT
CN 103588723
https://www.google.com/patents/CN103588723A?cl=zh
Chinese patent CN1275126 described by the Japanese company Teijin invention relates febuxostat Form A, B, C, D, G, and six kinds of amorphous and crystalline preparation method, reported in the literature Form A relatively stable . The method used is a solvent of methanol and water, patent phase diagram (Figure 7 Zone I) can be obtained in anhydrous crystalline Form A (hereinafter referred to as “Form A”), the mixing process by a temperature and the formation of methanol and water to determine the composition of the solvent, and the need to add a certain amount of Form a as a seed crystal to induce precipitation of crystals to control crystallization conditions are very harsh, operable range is very small, easy to form methanol solvate, hydrate or stable crystalline type C, to obtain reproducible single crystal type a low, it is difficult to achieve industrial production, and no mention of the preparation of Form a yield and purity in this patent.
[0011]
[0012] Chinese patent CN102267957A invention discloses a method for preparing febuxostat Form A, the solvent is preferably acetone, dissolved into 25 ~ 40 ° C was allowed to stand, when there began to crystallize when stirred for 20 to 40 minutes, then placed in -15 ~ 0 ° C to continue the crystallization of 8 to 10 hours. The crystallization process need to well below zero, when industrial mass production, resulting in high production costs, is not conducive to industrial production, the process yield up to 95.4%.
[0013] Chinese patent CN101139325 of Example 7 discloses the preparation of Form A with acetone method, although the process is simple, but the yield is low, only 50%.
[0014] Although the Chinese patent CN101684108A isopropyl alcohol as a solvent is disclosed a method for preparing crystalline form, the crystalline form of preparation is used to cool and heat a phased manner was allowed to stand, the crystallization temperature, long crystallization time, about 30 hours, the yield is low, and its products are not crystalline Form A.
[0015] In addition, Chinese patent CN101525319A, CN101805310, CN101926795A, CN101926794, W02012020272A2 are disclosed ethanol as a solvent or aqueous ethanol as a solvent preparation methods, and its products are crystalline ethanol solvate.
[0016] World Patent W02011139886A2 discloses the use of a mixed solvent of alcohol, and its products are not obtained polymorph A0
PAPER
Letters in Organic Chemistry (2015), 12(3), 217-221
Synthesis of the Major Metabolites of Febuxostat
Author(s): Xiao Long Li, Rui Qiu, Wei Li Wan, Xu Cheng, Li Hai and Yong Wu
Affiliation: Key Laboratory of Drug Targeting of Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China.
Graphical Abstract:

Abstract:
Total synthesis of three Febuxostat metabolites, named 67M-1, 67M-2, and 67M-4,is described in this article. Through condensation of the key intermediate compound A with different side chains, and then oxidation and hydrolysis, we obtained three target compounds with an overall yield of 19.5%-28.0%.
VOLUME: 12
ISSUE: 3
Page: [217 – 221]
Pages: 5
DOI: 10.2174/1570178612666150108000805, http://www.eurekaselect.com/127479/article

ULORIC (febuxostat) is a xanthine oxidase inhibitor. The active ingredient in ULORIC is 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid, with a molecular weight of 316.38. The empirical formula is C16H16N2O3S.
The chemical structure is:
 |
Febuxostat is a non-hygroscopic, white crystalline powder that is freely soluble in dimethylformamide; soluble in dimethylsulfoxide; sparingly soluble in ethanol; slightly soluble in methanol and acetonitrile; and practically insoluble in water. The melting range is 205°C to 208°C.
LORIC tablets for oral use contain the active ingredient, febuxostat, and are available in two dosage strengths, 40 mg and 80 mg. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide and magnesium stearate. ULORIC tablets are coated with Opadry II, green.
| CN1642546A * | Mar 28, 2003 | Jul 20, 2005 | Teijin Ltd. | Containing a single crystalline solid preparation |
| CN102471295A * | Jul 14, 2010 | May 23, 2012 | Teijin Pharma Ltd. | The method of manufacturing the poor solvent additive method of 2- (3-cyano-4-isobutyl-phenyl) -4-methyl-5-carboxylic acid crystalline polymorph of |
| EP2502920A1 * | Mar 25, 2011 | Sep 26, 2012 | Sandoz Ag | Crystallization process of Febuxostat from A |
| JP2011020950A* | Title not available |
| WO2015018507A3 * | Jul 30, 2014 | Oct 22, 2015 | Pharmathen S.A. | A novel process for the preparation of febuxostat |
| CN103304512A * | Jun 4, 2013 | Sep 18, 2013 | 华南理工大学 | Preparation method for febuxostat |
| WO2011031409A1 * | Aug 12, 2010 | Mar 17, 2011 | Teva Pharmaceutical Industries Ltd. | Processes for preparing febuxostat |
| JP2834971B2 | Title not available | |||
| JP3202607B2 | Title not available | |||
| JPH1045733A * | Title not available | |||
| US5614520 | Jan 30, 1995 | Mar 25, 1997 | Teijin Limited | 2-arylthiazole derivatives and pharmaceutical composition thereof |
| CN102229581A * | Nov 15, 2010 | Nov 2, 2011 | 邹巧根 | Preparation method for febuxostat intermediate |
| JPH1045733A * | Title not available |
| CN101497589A * | Feb 26, 2009 | Aug 5, 2009 | 沈阳药科大学 | Method for synthesizing 2-(3-cyano-4- isobutoxy phenyl)-4-methyl-carboxylate |
| CN101863854A * | Jun 29, 2010 | Oct 20, 2010 | 沈阳药科大学 | Synthesis method of 2-(3-cyan-4-isobutoxy) phenyl-4-methyl-5-thiazole formic acid |
| JP2706037B2 * | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | HASEGAWA, M. ET AL.: ‘A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile‘ HETEROCYCLES vol. 47, no. 2, 1998, pages 857 – 864 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2012131590A1 * | Mar 28, 2012 | Oct 4, 2012 | Sandoz Ag | An improved process for preparation of febuxostat and its polymorphic crystalline form c thereof |
| WO2014009817A1 * | Mar 19, 2013 | Jan 16, 2014 | Alembic Pharmaceuticals Limited | Pharmaceutical composition of febuxostat |
| WO2014057461A1 | Oct 10, 2013 | Apr 17, 2014 | Ranbaxy Laboratories Limited | Process for the preparation of crystalline form g of febuxostat |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(3-cyano-4-isobutoxyphenyl)-4-methyl-
1,3-thiazole-5-carboxylic acid |
|
| Clinical data | |
| Trade names | Uloric, Adenuric, Atenurix, Feburic, Goturic, Goutex. Generic in several countries.[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609020 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | ~49% absorbed |
| Protein binding | ~99% to albumin |
| Metabolism | via CYP1A2, 2C8, 2C9,UGT1A1, 1A3, 1A9, 2B7 |
| Biological half-life | ~5-8 hours |
| Excretion | Urine (~49% mostly as metabolites, 3% as unchanged drug); feces (~45% mostly as metabolites, 12% as unchanged drug) |
| Identifiers | |
| CAS Number | 144060-53-7 |
| ATC code | M04AA03 (WHO) |
| PubChem | CID 134018 |
| IUPHAR/BPS | 6817 |
| DrugBank | DB04854 |
| ChemSpider | 118173 |
| UNII | 101V0R1N2E |
| KEGG | D01206 |
| ChEMBL | CHEMBL1164729 |
| Chemical data | |
| Formula | C16H16N2O3S |
| Molar mass | 316.374 g/mol |
/////////Febuxostat, 144060-53-7, Uloric, Adenuric, Tei 6720, thiazole derivative, inhibitor of XANTHINE OXIDASE, treatment of HYPERURICEMIA, chronic GOUT, FBX, Febugood, Feburic, Febutaz, TMX 67, Zurig
CC1=C(SC(=N1)C2=CC(=C(C=C2)OCC(C)C)C#N)C(=O)O

