
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

TEZACAFTOR, VX 661
CAS : 1152311-62-0;
- Molecular FormulaC26H27F3N2O6
- Average mass520.498 Da
l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide).
(R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl]-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)cyclopropanecarboxamide
Vertex (INNOVATOR)
UNII: 8RW88Y506K
![]()
In July 2016, this combination was reported to be in phase 3 clinical development.
Update
Symdeko (tezacaftor/ivacaftor) ; Vertex; For the treatment of cystic fibrosis , Approved February 2018
Urology
Tezacaftor, also known asVX-661, is CFTR modulator. VX-661 is potentially useful for treatment of cystic fibrosis disease. Cystic fibrosis (CF) is a genetic disease caused by defects in the CF transmembrane regulator (CFTR) gene, which encodes an epithelial chloride channel. The most common mutation, Δ508CFTR, produces a protein that is misfolded and does not reach the cell membrane. VX-661 can correct trafficking of Δ508CFTR and partially restore chloride channel activity. VX-661 is currently under Phase III clinical trial.
VX-661 is an orally available deltaF508-CFTR corrector in phase III clinical trials at Vertex for the treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
Novel deuterated analogs of a cyclopropanecarboxamide ie tezacaftor (VX-661), as modulators of cystic fibrosis transmembrane conductance regulator (CFTR) proteins, useful for treating a CFTR-mediated disorder eg cystic fibrosis.
VX-661 (CAS #: 1152311-62-0; l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide). VX-661 is a cystic fibrosis transmembrane conductance regulator modulator. VX-661 is currently under investigation for the treatment of cystic fibrosis. VX-661 has also shown promise in treating sarcoglycanopathies, Brody’s disease, cathecolaminergic polymorphic ventricular tachycardia, limb girdle muscular dystrophy, asthma, smoke induced chronic obstructive pulmonary disorder, chronic bronchitis, rhinosinusitis, constipation, pancreatitis, pancreatic insufficiency, male infertility caused by congenital bilateral absence of the vas deferens (CBAVD), mild pulmonary disease, idiopathic pancreatitis, allergic bronchopulmonary aspergillosis (ABPA), liver disease, hereditary emphysema, hereditary hemochromatosis, coagulation-fibrinolysis deficiencies, such as protein C deficiency, type 1 hereditary angioedema, lipid processing deficiencies, such as familial hypercholesterolemia, type 1 chylomicronemia, abetalipoproteinemia, lysosomal storage diseases, such as I-cell disease/pseudo-Hurler, mucopolysaccharidoses, Sandhof/Tay-Sachs, Crigler-Najjar type II, polyendocrinopathy/hyperinsulinemia, diabetes mellitus, Laron dwarfism, myeloperoxidase deficiency, primary hypoparathyroidism, melanoma, glycanosis CDG type 1, congenital hyperthyroidism, osteogenesis imperfecta, hereditary hypofibrinogenemia, ACT deficiency, diabetes insipidus (DI), neurohypophyseal DI, nephrogenic DI, Charcot-Marie tooth syndrome, Pelizaeus-Merzbacher disease, neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, progressive supranuclear palsy, Pick’s disease, polyglutamine neurological disorders such as Huntington’s, spinocerebellar ataxia type I, spinal and bulbar muscular atrophy, dentatombral pallidoluysian, and myotonic dystrophy, as well as spongifiorm encephalopathies, such as hereditary Creutzfeldt- Jakob disease (due to prion protein processing defect), Fabry disease, Gerstrnarm-Straussler-Scheinker syndrome, chronic obstructive pulmonary disorder, dry-eye disease, or Sjogren’s disease, osteoporosis, osteopenia, bone healing and bone growth (including bone repair, bone regeneration, reducing bone resorption and increasing bone deposition), Gorham’s Syndrome, chloride channelopathies such as myotonia congenita (Thomson and Becker forms), Bartter’s
syndrome type III, Dent’s disease, hyperekplexia, epilepsy, lysosomal storage disease, Angelman syndrome, and primary ciliary dyskinesia (PCD), a term for inherited disorders of the structure and/or function of cilia, including PCD with situs inversus (also known as Kartagener syndrome), PCD without situs inversus, and ciliary aplasia. WO 2014086687; WO2013185112.

VX-661

VX-661 is likely subject to extensive CYP45o-mediated oxidative metabolism. These, as well as other metabolic transformations, occur in part through polymorphically-expressed enzymes, exacerbating interpatient variability. Additionally, some metabolites of VX-661 may have undesirable side effects. In order to overcome its short half-life, the drug likely must be taken several times per day, which increases the probability of patient incompliance and discontinuance.Deuterium Kinetic Isotope Effect
PATENT
Scheme I


EXAMPLE 1
(R)-l-(2,2-difluorobenzo[dl[l,31dioxol-5-vn-N-(l-q,3-dihvdroxypropyn-6-fluoro-2-(l- hvdroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cvclopropanecarboxamide
(VX-661)

![]()
Methyl 2.2-difluorobenzo[dl [1.31dioxole-5-carboxylate: To a 200 mL pressure tank reactor (10 atm. in CO), was placed 5-bromo-2,2-difluoro-2H-l,3-benzodioxole (20.0 g, 84.4 mmol, 1.00 equiv), methanol (40 mL), triethylamine (42.6 g, 5.00 equiv.), Pd2(dba)3 (1.74 g, 1.69 mmol, 0.02 equiv), Pd(dppf)Cl2 (1.4 g, 1.69 mmol, 0.02 equiv.). The resulting solution was stirred at 85 °C under an atmosphere of CO overnight and the reaction progress was monitored by GCMS. The reaction mixture was cooled. The solids were filtered out. The organic phase was concentrated under vacuum to afford 17.5 g of methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate as a crude solid, which was used directly in the next step. Step 2
![]()
2 step 2 3
(2.2-difluorobenzo[dl [ 1.31 dioxol-5 -vDmethanol : To a 500mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen were placed methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate (17.5 g, 81.01 mmol, 1.00 equiv.), tetrahydrofuran (200 mL). This was followed by the addition of L1AIH4 (6.81 mg, 162.02 mmol, 2.00 equiv.) at 0 °C. The resulting solution was stirred for 1 h at 25 °C and monitored by GCMS. The reaction mixture was cooled to 0 °C until GCMS indicated the completion of the reaction. The pH value of the solution was adjusted to 8 with sodium hydroxide (1 mol/L). The solids were filtered out. The organic layer combined and concentrated under vacuum to afford 13.25 g (87%) of (2,2-difluoro-2H-l,3-benzodioxol-5-yl)methanol as yellow oil.
Step 3
![]()
step 3
5-(chloromethyl)-2.2-difluorobenzo[diri.31dioxole: (2.2-difluoro-2H-1.3-benzodioxol-5-yl)methanol (13.25 g, 70.4 mmol, 1.00 equiv.) was dissolved in DCM (200 mL). Thionyl chloride (10.02 g, 1.20 equiv.) was added to this solution. The resulting mixture was stirred at room temperature for 4 hours and then concentrated under vacuum. The residue was then diluted with DCM (500 mL) and washed with 2 x 200 mL of sodium bicarbonate and 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate, filtered and evaporated to afford 12.36 g (85%) of 5-(chloromethyl)-2,2-difluoro-2H-l ,3-benzodioxole as yellow oil.
Step 4
![]()
step 4 5
[00160] 2-(2.2-difluorobenzordi ri .31dioxol-5-yl)acetonitrile: 5-(chloromethyl)-2,2-difluoro-2H-l,3-benzodioxole (12.36 g, 60 mmol, 1.00 equiv.) was dissolved in DMSO (120 mL). This was followed by the addition of NaCN (4.41 g, 1.50 equiv.) with the inert temperature below 40 °C. The resulting solution was stirred for 2 hours at room temperature. The reaction progress was monitored by GCMS. The reaction was then quenched by the addition of 300 mL of water/ice. The resulting solution was extracted with 3 x 100 mL of ethyl acetate. The organic layers combined and washed with 3 x 100 mL brine dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.84 g (92%) of 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile as brown oil.
Step 5

l -(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l -carbonitrile: To a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, were placed 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile (10.84 g, 55 mmol, 1.00 equiv.),
NaOH (50%) in water), 1 -bromo-2-chloroethane (11.92g, 82.5 mmol, 1.50 equiv.), BmNBr
(361 mg, 1.1 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 °C. The reaction progress was monitored by GCMS. The reaction mixture was cooled. The resulting solution was extracted with 3 x 200 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.12g of 1 -(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile as brown oil.
Step 6

[00162] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 250-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile (10.12 g, 45.38 mmol, 1.00 equiv), 6 N NaOH (61 mL) and EtOH (60 mL). The resulting solution was stirred for 3 h at 100 °C. The reaction mixture was cooled and the pH value of the solution was adjusted to 2 with hydrogen chloride (1 mol/L) until LCMS indicated the completion of the reaction. The solids were collected by filtration to afford 9.68 g (88%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid as a light yellow solid.
Step 7

[00163] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carbonyl chloride; To a solution of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid (687 mg, 2.84 mmol, 1.00 equiv.) in toluene (5 mL) was added thionyl chloride (1.67 g, 5.00 equiv.). The resulting solution was stirred for 3h at 65 °C. The reaction mixture was cooled and concentrated under vacuum to afford 738 mg (99%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride as a yellow solid.
Step 8
![]()
9 STEP 8 10
2-methyl-4-(trimethylsilyl)but-3-vn-2-ol: To a solution of ethynyltrimethylsilane (20 g, 203.63 mmol, 1.00 equiv) in THF (100 mL) was added n-BuLi (81 mL, 2.5M in THF)
dropwise with stirring at -78 °C. Then the resulting mixture was warmed to 0 °C for 1 h with stirring and then cooled to -78 °C. Propan-2-one (11.6 g, 199.73 mmol, 1.00 equiv.) was added dropwise with the inert temperature below -78 °C. The resulting solution was stirred at -78 °C for 3 h. The reaction was then quenched by the addition of 100 mL of water and extracted with 3 x 100 mL of MTBE. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 28 g (90%) of 2-methyl-4-(trimethylsilyl)but-3-yn-2-ol as an off-white solid. ¾ NMR (400 MHz, CDCh) δ: 1.50 (s, 6H), 1.16-1.14 (m, 9H).
Step 9
![]()
step 9
10
(3-chloro-3-methylbut-l-vnvntrimethylsilane: To a lOOmL round-bottom flask, was placed 2-methyl-4-(trimethylsilyl) but-3-yn-2-ol (14 g, 89.57 mmol, 1.00 equiv.), cone. HC1 (60 mL, 6.00 equiv.). The resulting solution was stirred for 16 h at 0 °C. The resulting solution was extracted with 3 x 100 mL of hexane. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 8 g (51%) of (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 1.84 (s, 6H), 1.18-1.16 (m, 9H).
Step 10
![]()
step 10
11 12
(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)trimethylsilane: Magnesium turnings (1.32 g, 1.20 equiv) were charged to a 250-mL 3-necked round-bottom flask and then suspended in THF (50 mL). The resulting mixture was cooled to 0 °C and maintained with an inert atmosphere of nitrogen. (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane (8 g, 45.78 mmol, 1.00 equiv.) was dissolved in THF (50 mL) and then added dropwise to this mixture with the inert temperature between 33-37 °C. The resulting solution was stirred at room temperature for an addition 1 h before BnOCH2Cl (6.45 g, 41.33 mmol, 0.90 equiv.) was added dropwise with the temperature below 10 °C. Then the resulting solution was stirred for 16 h at room temperature. The reaction was then quenched by the addition of 50 mL of water and extracted with 3 x 100 mL of hexane. The combined organic layers was dried over
anhydrous sodium sulfate and concentrated under vacuum to afford 10 g (84%) of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 7.37-7.35 (m, 5H), 4.62 (s, 2H), 3.34 (s, 2H), 1.24 (s, 6H), 0.17-0.14 (m, 9H).
Step 11
![]()
((2.2-dimethylbut-3-vnyloxy)methyl)benzene: To a solution of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane (10 g, 38.40 mmol, 1.00 equiv) in methanol (100 mL) was added potassium hydroxide (2.53 g, 38.33 mmol, 1.30 equiv). The resulting solution was stirred for 16 h at room temperature. The resulting solution was diluted with 200 mL of water and extracted with 3 x 100 mL of hexane. The organic layers combined and washed with 1 x 100 mL of water and then dried over anhydrous sodium sulfate and concentrated under vacuum to afford 5 g (69%) of [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene as light yellow oil. ¾ NMR (300 MHz, D20) δ: 7.41-7.28 (m, 5H) , 4.62 (s, 2H), 3.34 (s, 2H), 2.14 (s, 1H), 1.32-1.23 (m, 9H).
Step 12
![]()
14 15
methyl 2.2-difluorobenzo[d1[1.31dioxole-5-carboxylate: To a solution of 3-fluoro-4-nitroaniline (6.5 g, 41.64 mmol, 1.00 equiv) in chloroform (25 mL) and AcOH (80 mL) was added Bn (6.58 g, 41.17 mmol, 1.00 equiv.) dropwise with stirring at 0 °C in 20 min. The resulting solution was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 150 mL of water/ice. The pH value of the solution was adjusted to 9 with sodium hydroxide (10 %). The resulting solution was extracted with 3 x 50 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was re-crystallized from PE/EA (10: 1) to afford 6 g (61%) of 2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 13

(R)-l-(benzyloxy)-3-(2-bromo-5-fluoro-4-nitrophenylamino)propan-2-ol: 2-bromo-5-fluoro-4-nitroaniline (6.00 g, 25.56 mmol, 1.00 equiv.), Zn(C104)2 (1.90 g, 5.1 mmol, 0.20 equiv.), 4A Molecular Sieves (3 g), toluene (60 mL) was stirred at room temperature for 2 h and maintain with an inert atmosphere of N2 until (2R)-2-[(benzyloxy)methyl]oxirane (1.37 g, 8.34 mmol, 2.00 equiv.) was added. Then the resulting mixture was stirred for 15 h at 85 °C. The reaction progress was monitored by LCMS. The solids were filtered out and the resulting solution was diluted with 20 mL of ethyl acetate. The resulting mixture was washed with 2 x 20 mL of Sat. NH4CI and 1 x 20 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :5) to afford 7.5 g (70%) of N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 14

[00170] (R)-l-(4-amino-2-bromo-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 250-mL round-bottom flask, was placed N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline (7.5 g, 18.84 mmol, 1.00 equiv.), ethanol (80 mL), water (16 mL), NH4CI (10 g, 189 mmol, 10.00 equiv.), Zn (6.11 g, 18.84 mmol, 5.00 equiv.). The resulting solution was stirred for 4 h at 85 °C. The solids were filtered out and the resulting solution was concentrated under vacuum and diluted with 200 mL of ethyl acetate. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :3) to afford 4.16 g (60%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine as light yellow oil.
Step 15
TsO

(R)-4-(3-(benzyloxy)-2-hvdroxypropylamino)-5-bromo-2-fluorobenzenaminium 4-methylbenzenesulfonate: l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine (2 g, 5.42 mmol, 1.00 equiv.) was dissolved in dichloromethane (40 mL) followed by the addition of TsOH (1 g, 5.81 mmol, 1.10 equiv.). The resulting mixture was stirred for 16 h at room temperature and then concentrated under vacuum to afford 2.8 g (95%) of 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate as an off-white solid.
Step 16

(R)-l-(4-amino-2-(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 100-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate (2.9 g, 5.36 mmol, 1.00 equiv.), [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene (1.2 g, 6.37 mmol, 1.20 equiv.), Pd(OAc)2 (48 mg, 0.21 mmol, 0.04 equiv.), dppb (138 mg, 0.32 mmol, 0.06 equiv.), potassium carbonate (2.2 g, 15.92 mmol, 3.00 equiv.) and MeCN (50 mL). The resulting solution was stirred for 16 h at 80 °C. The solids were filtered out and the resulting mixture was concentrated under vacuum until LCMS indicated the completion of the reaction. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :4) to afford 2.2 g (86%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l ,4-diamine as a light brown solid.
Step 17

l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 40-mL vial purged and maintained with an inert atmosphere of nitrogen, was placed 1-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l,4-diamine (1 g, 2.1 mmol, 1.00 equiv.), MeCN (10 mL), Pd(MeCN)2Cl2 (82 mg, 0.32 mmol, 0.15 equiv.). The resulting solution was stirred for 12 h at 85 °C. The reaction progress was monitored by LCMS. The resulting mixture was concentrated under vacuum to afford 900 mg (crude) of (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol as a brown solid, which was used for next step without further purification.
Step 18

(R)-N-(l-(3-(benzyloxy)-2-hvdroxypropyl)-2-(l-(benzyloxy)-2-methylpropan-2-yl)-6-fluoro- lH-indol-5-yl)- 1 -(2.2-difluorobenzo[dl [ 1.31 dioxol-5-vDcvclopropanecarboxamide: To a 40 mL vial purged and maintained with an inert atmosphere of nitrogen, was placed (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol (800 mg, 1.68 mmol, 1.00 equiv.), dichloromethane (20 mL), TEA (508 mg, 5.04 mmol, 3.00 equiv.). l-(2,2-difiuoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride (524 mg, 2 mmol, 1.20 equiv.) was added to this mixture at 0 °C. The resulting solution was stirred for 2 h at 25 °C. The reaction progress was monitored by LCMS. The resulting solution was diluted with 20 mL of DCM and washed with 3 xlO mL of brine. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1:5) to afford 400 mg (30%) of N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide as a light yellow solid.
Step 19

(R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide: To a 100-mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of H2, were placed N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide (400 mg, 0.77 mmol, 1.00 equiv.) dry Pd/C (300 mg) and MeOH (5 Ml, 6M HC1). The resulting mixture was stirred at room temperature for 2 h until LCMS indicated the completion of the reaction. The solids were filtered out and the resulting mixture was concentrated under vacuum. The residue was purified by prep-HPLC with the following conditions: Column, XBridge Prep C18 OBD Column 19 x 150 mm, 5um; mobile phase and Gradient, Phase A: Waters (0.1%FA ), Phase B: ACN; Detector, UV 254 nm to afford 126.1 mg (42.4%) of (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide as a light yellow solid.
¾ NMR (400 MHz, OMSO-de) δ: 8.32 (s, 1H), 7.54 (s, 1H), 7.41-7.38 (m, 2H), 7.34-7.31 (m, 2H), 6.22 (s, 1H), 5.03-5.02 (m, 1H), 4.93-4.90 (m, 1H), 4.77-4.75 (m, 1H), 4.42-4.39 (m, 1H), 4.14-4.08 (m, 1H), 3.91 (brs, 1H) , 3.64-3.57 (m, 2H), 3.47-3.40 (m, 2H), 1.48-1.46 (m, 2H), 1.36-1.32 (m, 6H), 1.14-1.12 (m, 2H).
LCMS: m/z = 521.2[M+H]+.
PATENT
WO 2015160787
https://www.google.com/patents/WO2015160787A1?cl=en
PATENT
WO 2014014841
https://www.google.com/patents/WO2014014841A1?cl=en
All tautomeric forms of the Compound 1 are included herein. For example, Compound 1 may exist as tautomers, both of which are included herein:

Methods of Preparing Compound 1 Amorphous Form and Compound 1 Form A
Compound 1 is the starting point and in one embodiment can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

Toluene, H20, 70 °C

Bu4NBr
1. NaOH
2. HC1
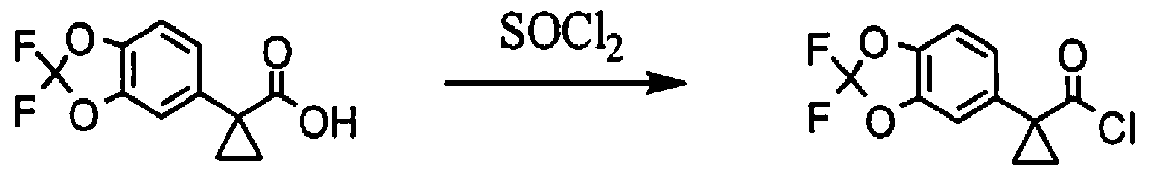
Scheme 2. Synthesis of acid chloride moiety – alternative synthesis.

1. NaCN
2. H20

SOC1,

Scheme 3. Synthesis of the amine moiety.



Scheme 4. Formation of Compound 1.

Compound 1
Methods of Preparing Compound 1 Amorphous Form
Starting from Compound 1 , or even a crystalline form of Compound 1 , Compound 1 Amorphous Form may be prepared by rotary evaporation or by spray dry methods.
Dissolving Compound 1 in an appropriate solvent like methanol and rotary evaporating the methanol to leave a foam produces Compound 1 Amorphous Form. In some embodiments, a warm water bath is used to expedite the evaporation.
Compound 1 Amorphous Form may also be prepared from Compound 1 using spray dry methods. Spray drying is a process that converts a liquid feed to a dried particulate form. Optionally, a secondary drying process such as fluidized bed drying or vacuum drying, may be used to reduce residual solvents to pharmaceutically acceptable levels. Typically, spray drying involves contacting a highly dispersed liquid suspension or solution, and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. The preparation to be spray dried can be any solution, coarse suspension, slurry, colloidal dispersion, or paste that may be atomized using the selected spray drying apparatus. In a standard procedure, the preparation is sprayed into a current of warm filtered air that evaporates the solvent and conveys the dried product to a collector (e.g. a cyclone). The spent air is then exhausted with the solvent, or alternatively the spent air is sent to a condenser to capture and potentially recycle the solvent. Commercially available types of apparatus may be used to conduct the spray drying. For example, commercial spray dryers are manufactured by Buchi Ltd. And Niro (e.g., the PSD line of spray driers manufactured by Niro) (see, US 2004/0105820; US 2003/0144257).
Spray drying typically employs solid loads of material from about 3% to about 30% by weight, (i.e., drug and excipients), for example about 4% to about 20% by weight, preferably at least about 10%. In general, the upper limit of solid loads is governed by the viscosity of (e.g., the ability to pump) the resulting solution and the solubility of the components in the solution. Generally, the viscosity of the solution can determine the size of the particle in the resulting powder product.
Techniques and methods for spray drying may be found in Perry’s Chemical
Engineering Handbook, 6th Ed., R. H. Perry, D. W. Green & J. O. Maloney, eds.), McGraw-Hill book co. (1984); and Marshall “Atomization and Spray-Drying” 50, Chem. Eng. Prog. Monogr. Series 2 (1954). In general, the spray drying is conducted with an inlet temperature of from about 60 °C to about 200 °C, for example, from about 95 °C to about 185 °C, from about 110 °C to about 182 °C, from about 96 °C to about 180 °C, e.g., about 145 °C. The spray drying is generally conducted with an outlet temperature of from about 30 °C to about 90 °C, for example from about 40 °C to about 80 °C, about 45 °C to about 80 °C e.g., about 75 °C. The atomization flow rate is generally from about 4 kg h to about 12 kg/h, for example, from about 4.3 kg/h to about 10.5 kg h, e.g., about 6 kg/h or about 10.5 kg/h. The feed flow rate is generally from about 3 kg/h to about 10 kg/h, for example, from about 3.5 kg/h to about 9.0 kg/h, e.g., about 8 kg/h or about 7.1 kg/h. The atomization ratio is generally from about 0.3 to 1.7, e.g., from about 0.5 to 1.5, e.g., about 0.8 or about 1.5.
Removal of the solvent may require a subsequent drying step, such as tray drying, fluid bed drying (e.g., from about room temperature to about 100 °C), vacuum drying, microwave drying, rotary drum drying or biconical vacuum drying (e.g., from about room temperature to about 200 °C).
Synthesis of Compound 1
Acid Chloride Moiety
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile

ouene, 2 , CN
A reactor was purged with nitrogen and charged with 900 mL of toluene. The solvent was degassed via nitrogen sparge for no less than 16 h. To the reactor was then charged Na3P04 (155.7 g, 949.5 mmol), followed by bis(dibenzylideneacetone) palladium (0) (7.28 g, 12.66 mmol). A 10% w/w solution of tert-butylphosphine in hexanes (51.23 g, 25.32 mmol) was charged over 10 min at 23 °C from a nitrogen purged addition funnel. The mixture was allowed to stir for 50 min, at which time 5-bromo-2,2-difluoro-l,3-benzodioxole (75 g, 316.5 mmol) was added over 1 min. After stirring for an additional 50 min, the mixture was charged with ethyl cyanoacetate (71.6 g, 633.0 mmol) over 5 min followed by water (4.5 mL) in one portion. The mixture was heated to 70 °C over 40 min and analyzed by HPLC every 1 – 2 h for the percent conversion of the reactant to the product. After complete conversion was observed (typically 100% conversion after 5 – 8 h), the mixture was cooled to 20 – 25 °C and filtered through a celite pad. The celite pad was rinsed with toluene (2 X 450 mL) and the combined organics were concentrated to 300 mL under vacuum at 60 – 65 °C. The concentrate was charged with 225mL DMSO and concentrated under vacuum at 70 – 80 °C until active distillation of the solvent ceased. The solution was cooled to 20 – 25 °C and diluted to 900 mL with DMSO in preparation for Step 2. Ή NMR (500 MHz, CDC13) δ 7.16 – 7.10 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t, J= 7.1 Hz, 3H).
Synthesis of (2,2-difluoro-l^-benzodioxol-5-yl)-acetonitrile.

[00311] The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature < 40 °C. The mixture was then heated to 75°C over 1 h and analyzed by HPLC every 1 – 2 h for % conversion. When a conversion of > 99% was observed (typically after 5 – 6 h), the reaction was cooled to 20 – 25 °C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 – 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60°C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 – 130 °C (oven temperature) and 1.5 – 2.0 Torr. (2,2-Difluoro-l,3- benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2- difluoro-l,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). Ή NMR (500 MHz, DMSO) 6 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H). Synthesis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cycIopropanecarbonitrUe.

MTBE
A stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h. An appropriate amount of MTBE was similarly degassed for several hours. To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol). The volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing. To the stirring mixture at 23.0 °C was charged degassed 50% w/w NaOH (143 mL) over 10 min followed by l-bromo-2-chloroethane (44.7 g, 311.6 mmol) over 30 min. The reaction was analyzed by HPLC in 1 h intervals for % conversion. Before sampling, stirring was stopped and the phases allowed to separate. The top organic phase was sampled for analysis. When a % conversion > 99 % was observed (typically after 2.5 – 3 h), the reaction mixture was cooled to 10 °C and was charged with water (461 mL) at such a rate as to maintain a temperature < 25 °C. The temperature was adjusted to 20 – 25 °C and the phases separated. Note: sufficient time should be allowed for complete phase separation. The aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HC1 (163mL) and 5% NaCl (163 mL). The solution of (2,2-difluoro- 1,3 -benzodioxol-5-yl)- cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40 – 50 °C. The solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50 – 60 °C. Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50 – 60 °C. The resulting mixture was cooled to 20 – 25 °C and diluted with ethanol to 266 mL in preparation for the next step. lH NMR (500 MHz, DMSO) 6 7.43 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 1.9 Hz, 1H), 7.30 (dd, J= 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H). [00314] Synthesis of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid.

The solution of (2,2-difluoro-l ,3-benzodioxol-5-yl)-cyclopropanecarbonitrile in ethanol from the previous step was charged with 6 N NaOH (277 mL) over 20 min and heated to an internal temperature of 77 – 78 °C over 45 min. The reaction progress was monitored by HPLC after 16 h. Note: the consumption of both (2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarbonitrile and the primary amide resulting from partial hydrolysis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile were monitored. When a % conversion > 99 % was observed (typically 100% conversion after 16 h), the reaction mixture was cooled to 25 °C and charged with ethanol (41 mL) and DCM (164 mL). The solution was cooled to 10 °C and charged with 6 N HC1 (290 mL) at such a rate as to maintain a temperature < 25 °C. After warming to 20 – 25 °C, the phases were allowed to separate. The bottom organic phase was collected and the top aqueous phase was back extracted with DCM (164 mL). Note: the aqueous phase was somewhat cloudy before and after the extraction due to a high concentration of inorganic salts. The organics were combined and concentrated under vacuum to 164 mL. Toluene (328 mL) was charged and the mixture condensed to 164 mL at 70 – 75 °C. The mixture was cooled to 45 °C, charged with MTBE (364 mL) and stirred at 60 °C for 20 min. The solution was cooled to 25 °C and polish filtered to remove residual inorganic salts. MTBE (123 mL) was used to rinse the reactor and the collected solids. The combined organics were transferred to a clean reactor in preparation for the next step.
Isolation of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecar boxy lie acid.

The solution of l-(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid from the previous step is concentrated under vacuum to 164 mL, charged with toluene (328 mL) and concentrated to 164 mL at 70 – 75 °C. The mixture was then heated to 100 – 105 °C to give a homogeneous solution. After stirring at that temperature for 30 min, the solution was cooled to 5 °C over 2 hours and maintained at 5 °C for 3 hours. The mixture was then filtered and the reactor and collected solid washed with cold 1 :1 toluene/n-heptane (2 X 123 mL). The material was dried under vacuum at 55 °C for 17 hours to provide l-(2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarboxylic acid as an off-white crystalline solid. l-(2,2-difluoro-l,3-benzodioxol- 5-yl)-cyclopropanecarboxylic acid was isolated in 79% yield from (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (3 steps including isolation) and with an HPLC purity of 99.0% AUC. ESI-MS m/z calc. 242.04, found 241.58 (M+l)+; Ή NMR (500 MHz, DMSO) δ 12.40 (s, 1H), 7.40 (d, J= 1.6 Hz, 1H), 7.30 (d, J= 8.3 Hz, 1H), 7.17 (dd, J= 8.3, 1.7 Hz, 1H), 1.46 (m, 2H), 1.17 (m, 2H).
Alternative Synthesis of the Acid Chloride Moiety [00319] Synthesis of (2,2-ditluoro-l,3-benzodioxol-5-yl)-methanol.
1. Vitride (2 equiv)
PhCH3 (10 vol)

[00320] Commercially available 2,2-difluoro-l,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-methanol that is used directly in the next step.
Synthesis of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole.
1. SOCl2 (1.5 equiv)
DMAP (0.01 equiv)

(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-methanol ( 1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and S0C12 (1.2 eq) is added via addition funnel. The S0C12 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO_ , filtered, and concentrated to afford crude 5-chloromethyl- 2,2-difluoro-l,3-benzodioxole that is used directly in the next step.
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile.

A solution of 5-chloromethyl-2,2-difluoro- 1 ,3-benzodioxole ( 1 eq) in DMSO ( 1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1,8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
The remaining steps are the same as described above for the synthesis of the acid moiety.
Amine Moiety
Synthesis of 2-bromo-5-fluoro-4-ntroaniline.

Synthesis of benzyIglycoIated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt.
1) l ^OBn
cat. Zn(C104)2-2H20 ®

DCM
A thoroughly dried flask under N2 was charged with the following: Activated powdered 4A molecular sieves (50 wt% based on 2-bromo-5-fluoro-4-nitroaniline), 2-Bromo-5- fluoro-4-nitroaniline (1.0 equiv), zinc perchlorate dihydrate (20 mol%), and toluene (8 vol). The mixture was stirred at room temperature for NMT 30 min. Lastly, (R)-benzyl glycidyl ether (2.0 equiv) in toluene (2 vol) was added in a steady stream. The reaction was heated to 80 °C (internal temperature) and stirred for approximately 7 hours or until 2-Bromo-5-fluoro-4-nitroaniline was <5%AUC.
The reaction was cooled to room temperature and Celite (50 wt%) was added, followed by ethyl acetate (10 vol). The resulting mixture was filtered to remove Celite and sieves and washed with ethyl acetate (2 vol). The filtrate was washed with ammonium chloride solution (4 vol, 20% w/v). The organic layer was washed with sodium bicarbonate solution (4 vol x 2.5% w/v). The organic layer was concentrated in vacuo on a rotovap. The resulting slurry was dissolved in isopropyl acetate (10 vol) and this solution was transferred to a Buchi hydrogenator.
The hydrogenator was charged with 5wt% Pt(S)/C (1.5 mol%) and the mixture was stirred under N2 at 30 °C (internal temperature). The reaction was flushed with N2 followed by hydrogen. The hydrogenator pressure was adjusted to 1 Bar of hydrogen and the mixture was stirred rapidly (>1200 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with dichloromethane (10 vol). The filtrate was concentrated in vacuo. Any remaining isopropyl acetate was chased with dichloromethane (2 vol) and concentrated on a rotavap to dryness.
The resulting residue was dissolved in dichloromethane (10 vol). jP-Toluenesulfonic acid monohydrate (1.2 equiv) was added and stirred overnight. The product was filtered and washed with dichloromethane (2 vol) and suction dried. The wetcake was transferred to drying trays and into a vacuum oven and dried at 45 °C with N2 bleed until constant weight was achieved. Benzylglycolated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt was isolated as an off-white solid.
Chiral purity was determined to be >97%ee.
[00334] Synthesis of (3-Chloro-3-methylbut-l-ynyl)trimethylsilane.

[00335] Propargyl alcohol (1.0 equiv) was charged to a vessel. Aqueous hydrochloric acid (37%, 3.75 vol) was added and stirring begun. During dissolution of the solid alcohol, a modest endotherm (5-6 °C) is observed. The resulting mixture was stirred overnight (16 h), slowly becoming dark red. A 30 L jacketed vessel is charged with water (5 vol) which is then cooled to 10 °C. The reaction mixture is transferred slowly into the water by vacuum, maintaining the internal temperature of the mixture below 25 °C. Hexanes (3 vol) is added and the resulting mixture is stirred for 0.5 h. The phases were settled and the aqueous phase (pH < 1) was drained off and discarded. The organic phase was concentrated in vacuo using a rotary evaporator, furnishing the product as red oil. [00336] Synthesis of (4-(Benzyloxy)-3,3-dimethylbut-l-yttyl)trimethylsiIane.

[00337] Method A
[00338] All equivalent and volume descriptors in this part are based on a 250g reaction.
Magnesium turnings (69.5 g, 2.86 mol, 2.0 equiv) were charged to a 3 L 4-neck reactor and stirred with a magnetic stirrer under nitrogen for 0.5 h. The reactor was immersed in an ice- water bath. A solution of the propargyl chloride (250 g, 1.43 mol, 1.0 equiv) in THF (1.8 L, 7.2 vol) was added slowly to the reactor, with stirring, until an initial exotherm (-10 °C) was observed. The Grignard reagent formation was confirmed by IPC usingΉ-NMR spectroscopy. Once the exotherm subsided, the remainder of the solution was added slowly, maintaining the batch temperature <15 °C. The addition required ~3.5 h. The resulting dark green mixture was decanted into a 2 L capped bottle.
[00339] All equivalent and volume descriptors in this part are based on a 500g reaction. A 22 L reactor was charged with a solution of benzyl chloromethyl ether (95%, 375 g, 2.31 mol, 0.8 equiv) in THF (1.5 L, 3 vol). The reactor was cooled in an ice-water bath. Two Grignard reagent batches prepared as described above were combined and then added slowly to the benzyl chloromethyl ether solution via an addition funnel, maintaining the batch temperature below 25 °C. The addition required 1.5 h. The reaction mixture was stirred overnight (16 h).
[00340] All equivalent and volume descriptors in this part are based on a 1 kg reaction. A solution of 15%» ammonium chloride was prepared in a 30 L jacketed reactor (1.5 kg in 8.5 kg of water, 10 vol). The solution was cooled to 5 °C. Two Grignard reaction mixtures prepared as described above were combined and then transferred into the ammonium chloride solution via a header vessel. An exotherm was observed in this quench, which was carried out at a rate such as to keep the internal temperature below 25 °C. Once the transfer was complete, the vessel jacket temperature was set to 25 °C. Hexanes (8 L, 8 vol) was added and the mixture was stirred for 0.5 h. After settling the phases, the aqueous phase (pH 9) was drained off and discarded. The remaining organic phase was washed with water (2 L, 2 vol). The organic phase was concentrated in vacuo using a 22 L rotary evaporator, providing the crude product as an orange oil.
[00341] Method B
[00342] Magnesium turnings (106 g, 4.35 mol, 1.0 eq) were charged to a 22 L reactor and then suspended in THF (760 mL, 1 vol). The vessel was cooled in an ice-water bath such that the batch temperature reached 2 °C. A solution of the propargyl chloride (760 g, 4.35 mol, 1.0 equiv) in THF (4.5 L, 6 vol) was added slowly to the reactor. After 100 mL was added, the addition was stopped and the mixture stirred until a 13 °C exotherm was observed, indicating the Grignard reagent initiation. Once the exotherm subsided, another 500 mL of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. The remainder of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The addition required -1.5 h. The resulting dark green solution was stirred for 0.5 h. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. Neat benzyl
chloromethyl ether was charged to the reactor addition funnel and then added dropwise into the reactor, maintaining the batch temperature below 25 °C. The addition required 1.0 h. The reaction mixture was stirred overnight. The aqueous work-up and concentration was carried out using the same procedure and relative amounts of materials as in Method A to give the product as an orange oil.
[00343] Syntheisis of 4-Benzyloxy-3,3-dimethylbut-l-yne.

2 steps
[00344] A 30 L jacketed reactor was charged with methanol (6 vol) which was then cooled to 5 °C. Potassium hydroxide (85%, 1.3 equiv) was added to the reactor. A 15-20 °C exotherm was observed as the potassium hydroxide dissolved. The jacket temperature was set to 25 °C. A solution of 4-benzyloxy-3,3-dimethyl-l-trimethylsilylbut-l-yne (1.0 equiv) in methanol (2 vol) was added and the resulting mixture was stirred until reaction completion, as monitored by HPLC. Typical reaction time at 25 °C is 3-4 h. The reaction mixture is diluted with water (8 vol) and then stirred for 0.5 h. Hexanes (6 vol) was added and the resulting mixture was stirred for 0.5 h. The phases were allowed to settle and then the aqueous phase (pH 10-11) was drained off and discarded. The organic phase was washed with a solution of KOH (85%, 0.4 equiv) in water (8 vol) followed by water (8 vol). The organic phase was then concentrated down using a rotary evaporator, yielding the title material as a yellow-orange oil. Typical purity of this material is in the 80% range with primarily a single impurity present. Ή NMR (400 MHz, C6D6) δ 7.28 (d, 2 H, J = 7.4 Hz), 7.18 (t, 2 H, J= 7.2 Hz), 7.10 (d, 1H, J= 7.2 Hz), 4.35 (s, 2 H), 3.24 (s, 2 H), 1.91 (s, 1 H), 1.25 (s, 6 H).
[00345] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.
[00346] Method A
[00347] Synthesis of Benzylglycolated 4-Amino-2-(4-benzyloxy-3,3-dimethyIbut- l-ynyl)-5- fluoroaniline.

[00348] Benzylglycolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt was freebased by stirring the solid in EtOAc (5 vol) and saturated NaHCC>3 solution (5 vol) until clear organic layer was achieved. The resulting layers were separated and the organic layer was washed with saturated NaHC03 solution (5 vol) followed by brine and concentrated in vacuo to obtain benzylglocolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt as an oil.
[00349] Then, a flask was charged with benzylglycolated 4-ammonium-2-bromo-5- flouroaniline tosylate salt (freebase, 1.0 equiv), Pd(OAc) (4.0 mol%), dppb (6.0 mol%) and powdered K2CO3 (3.0 equiv) and stirred with acetonitrile (6 vol) at room temperature. The resulting reaction mixture was degassed for approximately 30 min by bubbling in N2 with vent. Then 4-benzyloxy-3,3-dimethylbut-l-yne (1.1 equiv) dissolved in acetonitrile (2 vol) was added in a fast stream and heated to 80 °C and stirred until complete consumption of 4-ammonium-2- bromo-5-flouroaniline tosylate salt was achieved. The reaction slurry was cooled to room temperature and filtered through a pad of Celite and washed with acetonitrile (2 vol). Filtrate was concentrated in vacuo and the residue was redissolved in EtOAc (6 vol). The organic layer was washed twice with NH4CI solution (20% w/v, 4 vol) and brine (6 vol). The resulting organic layer was concentrated to yield brown oil and used as is in the next reaction.
[00350] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.

[00351] Crude oil of benzylglycolated 4-amino-2-(4-benzyloxy-3,3-dimethylbut-l-ynyl)-5- fluoroaniline was dissolved in acetonitrile (6 vol) and added (MeCN)2PdCl2 (15 mol%) at room temperature. The resulting mixture was degassed using N2 with vent for approximately 30 min. Then the reaction mixture was stirred at 80 °C under N2 blanket overnight. The reaction mixture was cooled to room temperature and filtered through a pad of Celite and washed the cake with acetonitrile (1 vol). The resulting filtrate was concentrated in vacuo and redissolved in EtOAc (5 vol). Deloxane-II THP (5 wt% based on the theoretical yield of N-benzylglycolated-5-amino-2- (2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole) was added and stirred at room temperature overnight. The mixture was then filtered through a pad of silica (2.5 inch depth, 6 inch diameter filter) and washed with EtOAc (4 vol). The filtrate was concentrated down to a dark brown residue, and used as is in the next reaction.
[00352] Repurification of crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1,1- dimethylethyl)-6-fluoroindole:
[00353] The crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1 , l-dimethylethyl)-6- fluoroindole was dissolved in dichloromethane (~1.5 vol) and filtered through a pad of silica initially using 30% EtOAc/heptane where impurities were discarded. Then the silica pad was washed with 50% EtO Ac/heptane to isolate N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l- dimethylethyl)-6-fluoroindole until faint color was observed in the filtrate. This filtrate was concentrated in vacuo to afford brown oil which crystallized on standing at room temperature. Ή NMR (400 MHz, DMSO) 6 7.38-7.34 (m, 4 H), 7.32-7.23 (m, 6 H), 7.21 (d, 1 H, J= 12.8 Hz), 6.77 (d, 1H, J= 9.0 Hz), 6.06 (s, 1 H), 5.13 (d, 1H, J = 4.9 Hz), 4.54 (s, 2 H), 4.46 (br. s, 2 H), 4.45 (s, 2 H), 4.33 (d, 1 H, J= 12.4 Hz), 4.09-4.04 (m, 2 H), 3.63 (d, 1H, J= 9.2 Hz), 3.56 (d, 1H, J= 9.2 Hz), 3.49 (dd, 1H, J= 9.8, 4.4 Hz), 3.43 (dd, 1H, J= 9.8, 5.7 Hz), 1.40 (s, 6 H).
[00354] Synthesis of N-benzyIglycolated-5-amino-2-(2-benzyIoxy-l,l-diniethylethyl)-6- fluoroindole.
[00355] Method B

2. (MeCN)2PdCl2
MeCN, 80 <€
3. Silica gel filtration
[00356] Palladium acetate (33 g, 0.04 eq), dppb (94 g, 0.06 eq), and potassium carbonate (1.5 kg, 3.0 eq) are charged to a reactor. The free based oil benzylglocolated 4-ammonium-2-bromo- 5-flouroaniline (1.5 kg, 1.0 eq) was dissolved in acetonitrile (8.2 L, 4.1 vol) and then added to the reactor. The mixture was sparged with nitrogen gas for NLT 1 h. A solution of 4-benzyloxy- 3,3-dimethylbut-l-yne (70%), 1.1 kg, 1.05 eq) in acetonitrile was added to the mixture which was then sparged with nitrogen gas for NLT 1 h. The mixture was heated to 80 °C and then stirred overnight. IPC by HPLC is carried out and the reaction is determined to be complete after 16 h. The mixture was cooled to ambient temperature and then filtered through a pad of Celite (228 g). The reactor and Celite pad were washed with acetonitrile (2 x 2 L, 2 vol). The combined phases are concentrated on a 22 L rotary evaporator until 8 L of solvent have been collected, leaving the crude product in 7 L (3.5 vol) of acetonitrile. [00357] 5 s-acetonitriledichloropalladium ( 144 g, 0.15 eq) was charged to the reactor. The crude solution was transferred back into the reactor and the roto-vap bulb was washed with acetonitrile (4 L, 2 vol). The combined solutions were sparged with nitrogen gas for NLT 1 h. The reaction mixture was heated to 80 °C for NLT 16 h. In process control by HPLC shows complete consumption of starting material. The reaction mixture was filtered through Celite (300 g). The reactor and filter cake were washed with acetonitrile (3 L, 1.5 vol). The combined filtrates were concentrated to an oil by rotary evaporation. The oil was dissolved in ethyl acetate (8.8 L, 4.4 vol). The solution was washed with 20% ammonium chloride (5 L, 2.5 vol) followed by 5% brine (5 L, 2.5 vol). Silica gel (3.5 kg, 1.8 wt. eq.) of silica gel was added to the organic phase, which was stirred overnight. Deloxan THP II metal scavenger (358 g) and heptane (17.6 L) were added and the resulting mixture was stirred for NLT 3 h. The mixture was filtered through a sintered glass funnel. The filter cake was washed with 30% ethyl acetate in heptane (25 L). The combined filtrates were concentrated under reduced pressure to give N- benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole as a brown paste ( 1.4 kgl.Svnthesis of Compound 1
[00358] Synthesis of benzyl protected Compound 1.



[00359] 1 -(2,2-difluoro- 1 ,3 -benzodioxol-5-yl)-cyclopropanecarboxylic acid (1.3 equiv) was slurried in toluene (2.5 vol, based on l-(2,2-difluoro-l,3-benzodioxol-5-yi)- cyclopropanecarboxylic acid) and the mixture was heated to 60 °C. SOCl2 (1.7 equiv) was added via addition runnel. The resulting mixture was stirred for 2 hr. The toluene and the excess
SOCI2 were distilled off using rotavop. Additional toluene (2.5 vol, based on l-(2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid) was added and distilled again. The crude acid chloride was dissolved in dichloromethane (2 vol) and added via addition funnel to a mixture of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole (1.0 equiv), and triethylamine (2.0 equiv) in dichloromethane (7 vol) while maintaining 0-3 °C (internal temperature). The resulting mixture was stirred at 0 °C for 4 hrs and then warmed to room temperature overnight. Distilled water (5 vol) was added to the reaction mixture and stirred for NLT 30 min and the layers were separated. The organic phase was washed with 20 wt% K2CO3 (4 vol x 2) followed by a brine wash (4 vol) and concentrated to afford crude benzyl protected Compound 1 as a thick brown oil, which was purified further using silica pad filtration.
[00360] Silica gel pad filtration: Crude benzyl protected Compound 1 was dissolved in ethyl acetate (3 vol) in the presence of activated carbon Darco-G (10wt%, based on theoretical yield of benzyl protected Compound 1) and stirred at room temperature overnight. To this mixture was added heptane (3 vol) and filtered through a pad of silica gel (2x weight of crude benzyl protected Compound 1). The silica pad was washed with ethyl acetate/heptane (1:1, 6 vol) or until little color was detected in the filtrate. The filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish brown oil, and used directly in the next step.
[00361] Repurification: Benzyl protected Compound 1 was redissolved in dichloromethane (1 vol, based on theoretical yield of benzyl protected Compound 1) and loaded onto a silica gel pad (2x weight of crude benzyl protected Compound 1). The silica pad was washed with
dichloromethane (2 vol, based on theoretical yield of benzyl protected Compound 1) and the filtrate was discarded. The silica pad was washed with 30% ethyl acetate/heptane (5 vol) and the filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish orange oil, and used directly in the next step. [00362] Synthesis of Compound 1.

OBn 4 steps

[00363] Method A
[00364] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, ~ 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 °C.
[00365] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank.
[00366] After 2 full days of reaction, more Pd / C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [00367] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg.
[00368] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1 :1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1 :1 ethyl acetate-heptane. The silica pad was flushed first with 1 :1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC.
[00369] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of Compound 1 as an off-white solid.
[00370] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1 :1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g).
[00371] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of -6% methanol. The thermostat was set to 48 °C (below the boiling temp of the MTBE-methanol azeotrope, which is 52 °C). The mixture was cooled to 20 °C over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air- dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
[00372] Method B
[00373] Benzyl protected Compound 1 was dissolved in THF (3 vol) and then stripped to dryness to remove any residual solvent. Benzyl protected Compound 1 was redissolved in THF (4 vol) and added to the hydrogenator containing 5 wt% Pd/C (2.5 mol%, 60% wet, Degussa E5 El 01 N /W). The internal temperature of the reaction was adjusted to 50 °C, and flushed with N2 (x5) followed by hydrogen (x3). The hydrogenator pressure was adjusted to 3 Bar of hydrogen and the mixture was stirred rapidly (>1100 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with THF (1 vol). The filtrate was concentrated in vacuo to obtain a brown foamy residue. The resulting residue was dissolved in MTBE (5 vol) and 0.5N HC1 solution (2 vol) and distilled water (1 vol) were added. The mixture was stirred for NLT 30 min and the resulting layers were separated. The organic phase was washed with 10wt% K2CO3 solution (2 vol x2) followed by a brine wash. The organic layer was added to a flask containing silica gel (25 wt%), Deloxan-THP II (5wt%, 75% wet), and
Na2S04 and stirred overnight. The resulting mixture was filtered through a pad of Celite and washed with 10%THF/MTBE (3 vol). The filtrate was concentrated in vacuo to afford crude Compound 1 as pale tan foam.
[00374] Compound 1 recovery from the mother liquor: Option A.
[00375] Silica gel pad filtration: The mother liquor was concentrated in vacuo to obtain a brown foam, dissolved in dichloromethane (2 vol), and filtered through a pad of silica (3x weight of the crude Compound 1). The silica pad was washed with ethyl acetate/heptane (1 :1, 13 vol) and the filtrate was discarded. The silica pad was washed with 10% THF/ethyl acetate (10 vol) and the filtrate was coiicentraied in vacuo to afford Compound 1 as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{00376] Compound 1 recovery from the mother liquor: Option B,
[00377] Silica gel column chromatography: After chromatography on silica gel (50% ethyl acetate/hexaties to 100% ethyl acetate), the desired compound was isolated as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{003781 Additional Recrystaliization of Compound 1
[ 0379‘j Solid Compound 1 (135 kg) was suspended in IPA (5.4 L, 4 vol) and then heated to 82 °C. Upon complete dissolution (visual), heptane (540 mL, 0.4 vol) was added slowly. The mixture was cooled to 58 °C The mixture was then cooled slowly to 51 °C, during which time crystallization occurs. The heat source was shut down and the recrystalfeation mixture was allowed to cool naturally overnight. The mixture was filtered using a benchtop Buclmer funnel and the filter cake was washed with IPA (2.7 L, 2 vol). The filler cake was dried in the tunnel under air flow for 8 h and then was oven-dried in vacuo at 45-50 °C overnight to give 1.02 kg of recrystallized Compound 1 ,
100380] Compound 1 may also be prepared by one of several synthetic routes disclosed in US published patent application U S20090131 92, incorporated herein by reference.
{003811 Table 6 below recites analytical data for Compound 1.
Table 6.

Synthesis of Compound 1 Amorphous Form [00383] Spray-Dried Method
[00384] 9.95g of Hydroxypropylmethylcellulose acetate succinate HG grade (HPMCAS-HG) was weighed into a 500 ml beaker, along with 50 mg of sodium lauryl sulfate (SLS). MeOH (200 ml) was mixed with the solid. The material was allowed to stir for 4 h. To insure maximum dissolution, after 2 h of stirring the solution was sonicated for 5 mins, then allowed to continue stirring for the remaining 2 h. A very fin suspension of HPMCAS remained in solution. However, visual observation determined that no gummy portions remained on the walls of the vessel or stuck to the bottom after tilting the vessel.
[00385] Compound 1 (1 Og) was poured into the 500 ml beaker, and the system was allowed to continue stirring. The solution was spray dried using the following parameters:
Formulation Description: Compound 1 Form A/HPMCAS/SLS (50/49.5/0.5)
Buchi Mini Spray Dryer
T inlet (setpoint) 145 °C
T outlet (start) 75 °C
T outlet (end) 55 °C
Nitrogen Pressure 75 psi
Aspirator 100 %
Pump 35 %
Rotometer 40 mm
Filter Pressure 65 mbar
Condenser Temp -3 °C
Run Time l h
REFERENCES
1: Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med. 2014 Jul 23;6(246):246ra97. doi: 10.1126/scitranslmed.3008889. PubMed PMID: 25101887.
2: Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014 Jul;39(7):500-11. PubMed PMID: 25083129; PubMed Central PMCID: PMC4103577.
3: Norman P. Novel picolinamide-based cystic fibrosis transmembrane regulator modulators: evaluation of WO2013038373, WO2013038376, WO2013038381, WO2013038386 and WO2013038390. Expert Opin Ther Pat. 2014 Jul;24(7):829-37. doi: 10.1517/13543776.2014.876412. Epub 2014 Jan 7. PubMed PMID: 24392786.
//////TEZACAFTOR, VX 661, PHASE 3, 1152311-62-0, UNII: 8RW88Y506K, deltaF508-CFTR corrector, Vertex, treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
CC(C)(CO)C1=CC2=CC(=C(C=C2N1CC(CO)O)F)NC(=O)C3(CC3)C4=CC5=C(C=C4)OC(O5)(F)F
CC(C)(CO)c1cc2cc(c(cc2n1C[C@H](CO)O)F)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F

