
Home » Posts tagged 'VERTEX'
Tag Archives: VERTEX
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
VX-445, Elexacaftor, エレクサカフトル
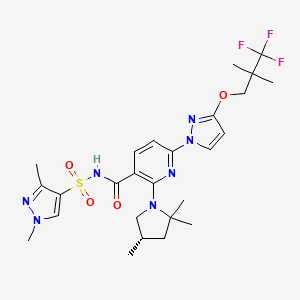

VX-445, Elexacaftor, エレクサカフトル
597.658 g/mol, C26H34F3N7O4S
N-[(1,3-Dimethyl-1H-pyrazol-4-yl)sulfonyl]-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-3-pyridinecarboxamide
3-Pyridinecarboxamide, N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)-2-((4S)-2,2,4-trimethyl-1-pyrrolidinyl)-
Cas 2216712-66-0
WHO 11180
Treatment of cystic fibrosis, CFTR modulator
Elexacaftor is under investigation in clinical trial NCT03525548 (A Study of VX-445 Combination Therapy in CF Subjects Homozygous for F508del (F/F)).
Cystic fibrosis transmembrane conductance regulator (CFTR) corrector designed to restore Phe508del CFTR protein function in patients with cystic fibrosis when administered with tezacaftor and ivacaftor.
VX-445 (elexacaftor), tezacaftor, and ivacaftor triple-drug combo
Vertex Pharmaceuticals (NASDAQ: VRTX) already claims a virtual monopoly in treating the underlying cause of cystic fibrosis (CF). The biotech’s current three CF drugs should generate combined sales of close to $3.5 billion this year. Another blockbuster is likely to join those three drugs on the market in 2020 — Vertex’s triple-drug CF combo featuring VX-445 (elexacaftor), tezacaftor, and ivacaftor.
EvaluatePharma projects that this triple-drug combo will rake in close to $4.3 billion by 2024. The market researcher pegs the net present value of the drug at nearly $20 billion, making it the most valuable pipeline asset in the biopharmaceutical industry right now.
PATENT
WO 2018107100
Also disclosed herein is Compound 1:
[0013] N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide.
Synthesis of Compound 1
[00256] Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00257] Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
[00258] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature.2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L).2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00259] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
[00260] Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00261] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00262] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
[00263] Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00264] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %).1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
[00265] Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00266] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by
vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
[00267] Part B: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
[00268] Preparation of starting materials:
[00269] 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol
[00270] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g). The estimated total amount of product is 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
[00271] tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate
[00272] A 50L Syrris controlled reactor was started and jacket set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise (exothermic), maintaining reaction temp <30 °C.
A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L), and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then distilled off 1-2 volumes of solvent. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C , 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse, crystalline solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
[00273] Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate
[00274] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced
pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
[00275] Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
[00276] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white waxy solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +;
Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
[00277] Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate
[00278] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
[00279] Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate
[00280] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and he mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
[00281] Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
[00282] tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room
temperature under stirring for 2.5 h. The solid was collected by filtration, washed with isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
[00283] Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide
[00284] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give the product as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +; Retention time: 0.68 minutes.
[00285] Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
[00286] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol) , and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
[00287] Alternative Steps F and G:
[00288] Alternative Step F: 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide
[00289]
[
[00291] To a suspension of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (20.0 g, 53.89 mmol) in THF (78.40 mL) was added solid carbonyldiimidazole (approximately 10.49 g, 64.67 mmol) portion wise and the resulting solution was stirred at room temperature (slight exotherm from 18-21 °C was observed). After 1 h, solid 1,3-dimethylpyrazole-4-sulfonamide
(approximately 11.33 g, 64.67 mmol) was added, followed by DBU (approximately 9.845 g, 9.671 mL, 64.67 mmol) in two equal portions over 1 min (exotherm from 19 to 35 °C). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl acetate (118 mL) and then HCl (approximately 107.8 mL of 2 M, 215.6 mmol). The phases were separated and the aqueous phase was extracted
with ethyl aceate (78 mL). The combined organics were washed with water (39.2 mL), then brine (40 mL), dried over sodium sulfate and concentrated. The resulting foam was crystallized from a 1:1 isopropanol:heptane mixture (80 mL) to afford 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide (26.1 g, 93%) as a white solid. ESI-MS m/z calc.520.0, found 520.9 (M+1) +; Retention time: 1.83 minutes.
[00292] Alternative Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
[
[00294] 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (20.0 g, 38.39 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (approximately 14.36 g, 95.98 mmol), and K2CO3 (approximately 26.54 g, 192.0 mmol) were combined in DMSO (80.00 mL) and 1,2-diethoxyethane (20.00 mL) in a 500-mL flask with reflux condenser. The reaction mixture was heated at 120 °C for 16 h then cooled to room temperature. The reaction was diluted with DCM (200.0 mL) and HCl (approximately 172.8 mL of 2 M, 345.5 mmol); aqueous pH ~1. The phases were separated, and the aqueous phase was extracted with DCM (100.0 mL). The organic phases were combined, washed with water (100.0 mL) (3 x), and dried (Na2SO4) to afford an amber solution. The solution was filtered through a DCM-packed silica gel bed (80 g; 4 g/g) and washed with 20% EtOAc/DCM (5 x 200 mL). The combined filtrate/washes were concentrated to afford 22.2 g of an off-white powder. The powder was slurried in MTBE (140 mL) for 30 min. The solid was collected by filtration (paper/sintered-glass) to afford 24 g after air-drying. The solid was transferred to a drying dish and vacuum-dried (40 °C/200 torr/N2 bleed) overnight to afford 20.70 g (90%) of a white powder. ESI-MS m/z calc.
597.2345, found 598.0 (M+1)+; Retention time: 2.18 minutes.
[00295] 1H NMR (400 MHz, Chloroform-d) δ 13.85 (s, 1H), 8.30 (d, J = 8.6 Hz, 1H), 8.23 (d, J = 2.8 Hz, 1H), 8.08 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 5.98 (d, J = 2.8 Hz, 1H), 4.24 (s, 2H), 3.86 (s, 3H), 3.44 (dd, J = 10.3, 8.4 Hz, 1H), 3.09 (dd, J = 10.3, 7.8 Hz, 1H), 2.67– 2.52 (m, 1H), 2.47 (s, 3H), 2.12 (dd, J = 12.3, 7.8 Hz, 1H), 1.70 (dd, J = 12.4, 9.6 Hz, 1H), 1.37 (s, 3H), 1.33 (s, 3H), 1.27 (s, 6H), 1.20 (d, 3H).
[00296] Alternative Synthesis of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
Step 1: Preparation of 3,3,3-trifluoro-2,2-dimethylpropan-1-ol
A reactor was loaded with toluene (300 mL) and 3,3,3-trifluoro-2,2-dimethylpropanoic acid (30 g, 192.2 mmol), capped, purged under nitrogen. The reaction was set to control the internal temperature to 40 °C. A solution of Vitride (65% in toluene. approximately 119.6 g of 65 %w/w, 115.4 mL of 65 %w/w, 384.4 mmol) was set up for addition via syringe, and addition was begun at 40 °C, with the target addition temperature between 40 and 50 °C. The reaction was stirred at 40 °C for 90 min. The reaction was cooled to 10 °C then the remaining Vitride was quenched with slow addition of water (6 mL). A solution of 15 % aq NaOH (30 mL) was added in portions, and solids precipitated half way through the base addition. Water (60.00 mL) was added. The mixture was warmed to 30 °C and held for at least 15 mins. The mixture was then cooled to 20 °C. The
aqueous layer was removed. The organic layer was washed with water (60 mL x 3), and then washed with brine (60 mL). The washed organic layer was dried under Na2SO4, followed with MgSO4. The mix was filtered through Celite, and the cake washed with toluene (60.00 mL) and pulled dry. The product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (22.5 g, 82%) was obtained as clear colorless solution.
Step 2: Preparation of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate
A reactor was charged with 3,3,3-trifluoro-2,2-dimethylpropan-1-ol (17.48 g, 123.0 mmol) solution in toluene (250g), 1-(tert-butyl) 4-ethyl 3-hydroxy-1H-pyrazole-1,4-dicarboxylate (30.0 g, 117.1 mmol), and PPh3 (35.33 g, 134.7 mmol). The reaction was heated to 40 °C. DIAD (26.09 mL, 134.7 mmol) was weighed and placed into a syringe and added over 10 minutes while maintaining an internal temperature ranging between 40 and 50 °C. The reaction was then heated to 100 °C over 30 minutes. After holding at 100 °C for 30 minutes, the reaction was complete, and the mixture was cooled to 70 °C over 15 minutes. Heptane (180.0 mL) was added, and the jacket was cooled to 15 °C over 1 hour. (TPPO began crystallizing at ~35 °C). The mixture stirring at 15 °C was filtered (fast), the cake was washed with a pre-mixed solution of toluene (60 mL) and heptane (60 mL) and then pulled dry. The clear solution was concentrated to a waxy solid (45 °C, vacuum, rotovap). Crude 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (53.49g) was obtained as a waxy solid, (~120% of theoretical mass recovered).
Step 3: Preparation of 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid
A solution of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (50.0 g, 131 mmol) in 2-methyltetrahydrofuran (500 mL) was prepared in a reactor and stirred at 40 °C. Portions of KOt-Bu (80.85 g, 720.5 mmol) were then added over 30 minutes. Addition was exothermic. After 2053.49g UPLC-MS showed complete removal of the Boc group, so water (3.53 g, 3.53 mL, 196 mmol) was added drop-wise addition via syringe over 20 min to keep the reaction temperature between 40-50 °C. The mixture was then stirred for 17 hours to complete the reaction. The mixture was then cooled to 20 °C and water (400 mL) was added. The stirring was stopped and the layers were separated. The desired product in the aqueous layer was returned to the reactor and the organic layer was discarded. The aqueous layer was washed with 2-Me-THF (200 mL). Isopropanol (50. mL) was added followed by dropwise addition of aqueous HCl (131 mL of 6.0 M, 786.0 mmol) to adjust the pH to ❤ while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.
[00297] Step 4: Preparation of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (1.0 equiv) was added to a reactor followed by DMF (6.0 vol, 2.6 wt equiv). The mixture was stirred at 18– 22 °C. DBU (0.2 equiv.) was charged to the reaction mixture at a rate of approximately 45 mL/min. The reaction temperature was then raised to 98– 102 °C over 45 minutes. The reaction mixture was stirred at 98– 102 °C for no less than 10 h. The reaction mixture was then cooled to -2°C to 2 °C over approximately 1 hour and was used without isolation to make ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate.
[00298] Alternate procedure for the preparation of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
[00299] Step 1. Ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate
[00300] A solution of ethyl 2,6-dichloronicotinate (256 g, 1.16 mol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (242 g, 1.16 mol) in DMF (1.53 L) was treated with potassium carbonate (209 g, 1.51 mol) and DABCO (19.6 g, 174 mmol). The resultant suspension was stirred allowed to exotherm from 14 to 25 °C and then maintained at 20– 25 °C with external cooling for 3 days. The suspension was cooled to below 10 °C when water (2.0 L) was added in a thin stream while maintaining the temperature below 25 °C. After the addition was complete, the suspension was stirred for an additional 1 h. The solid was collected by filtration (sintered-glass/polypad) and the filter-cake was washed with water (2 x 500-mL) and dried with suction for 2 h to afford water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (512 g; 113% yield) as white powder which was used without further steps in the subsequent reaction.
[00301] Step 2.2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid
[00302] The water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (455 g, 1.16 mol; assumed 100% yield from previous step) in EtOH (1.14 L) and THF (455 mL) was stirred at ambient temperature (17 °C) when 1 M NaOH (1.16 L, 1.16 mol) was added. The reaction mixture exothermed to 30 °C and was further warmed at 40 °C for 2 h. The solution was quenched with 1 M HCl (1.39 L, 1.39 mol) which resulted in an immediate precipitation which became thicker as the acid was added. The creamy suspension was allowed to cool to room temperature and was stirred overnight. The solid was collected by filtration (sintered-glass/poly pad). The filter-cake was washed with water (2 x 500-mL). The filter-cake was dried by suction for 1 h but remained wet. The damp solid was transferred to a 10-L Buchi flask for further drying (50 °C/20 torr), but was not effective. Further effort to dry by chasing with i-PrOH was also ineffective. Successful drying was accomplished after the damp solid was backfilled with i-PrOAc (3 L), the suspension was heated at 60 °C (homogenization), and re-concentrated to dryness (50 °C/20 torr) to afford dry 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid (408 g; 97% yield for two steps) as a fine, white powder. The product was further dried in a vacuum oven (50 °C/10 torr/N2 bleed) for 2 h but marginal weight loss was observed. 1H NMR (400 MHz, DMSO-d6) δ 13.64 (s, 1H), 8.49– 8.36 (m, 2H), 7.77 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.8 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).19F NMR (376 MHz, DMSO-d6) δ -75.2. KF analysis: 0.04% water.
2. Preparation of Form A of Compound 1
[00303] The crystalline Form A of Compound 1 was obtained as a result of the following synthesis. Combined 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide(108 g, 207.3 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (77.55 g, 518.2 mmol), was combined with K2CO3 (143.2 g, 1.036 mol) in DMSO (432.0 mL) and 1,2-
diethoxyethane (108.0 mL) in a 1-L RB flask with a reflux condenser. The resulting suspension was heated at 120°C and was stirred at temperature overnight. Then the reaction was diluted with DCM (1.080 L) and HCl (933.0 mL of 2 M, 1.866 mol) was slowly added. The liquid phases were separated, and the aqueous phase was extracted with DCM (540.0 mL).The organic phases were combined, washed with water (540.0 mL) (3 x), then dried with (Na2SO4) to afford an amber solution. Silica gel (25 g) was added and then the drying agent/silica gel was filtered off. The filter-bed was washed with DCM (3 x 50-mL). The organic phases were combined and concentrated (40 °C/40 torr) to afford crude N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (198.6 g, 160% theory) as an off-white solid. The solid was diluted with MTBE (750 mL), warmed at 60 °C (external temperature), and mixed to a homogenous suspension. The suspension was cooled to 30 °C with stirring and the solid was collected by filtration, air-dried, and vacuum-dried to afford Compound 1 (111.1 g; 90 %) as a fine, white powder.
[00304] The crystalline Form A of Compound 1 was also obtained through the following procedure. A suspension of Compound 1 (150.0 g, 228.1 mmol) in iPrOH (480 mL) and water (120 mL) was heated at 82 °C to obtain a solution. The solution was cooled with a J-Kem controller at a cooling rate of 10 °C/h. Once the temperature reached 74 °C, the solution was seeded with a sample of Compound 1 in crystalline Form A. Crystallization occurred immediately. The suspension was cooled to 20 °C. The solid was collected by filtration, washed with i-PrOH (2 x 75 mL), air-dried with suction, and vacuum-dried (55 °C/300 torr/N2 bleed) to afford Compound 1, Form A (103.3 g) as a white powder.. The sample was cooled to ~5 °C, let stir for 1 h, and then the solid was collected by filtration (sintered glass/paper). the filter-cake was washed with i-PrOH (75 mL) (2 x), air-dried with suction, air-dried in a drying dish (120.6 g mostly dried), vacuum-dried (55 °C/300 torr/N2 bleed) for 4 h, and then RT overnight. Overnight drying afforded 118.3 g (87% yield) of a white powder.
PATENT
WO-2019113476
Example 1: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
[00110] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L). 2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00111] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00112] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00113] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00114] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white solid (2.042 kg, 16.1 mol, 87 %). 1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00115] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
Example 2: Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
Example 2A
[00116] 2,2,6,6-tetramethylpiperidin-4-one (50.00 g, 305.983 mmol, 1.000 equiv), tributylmethyl ammonium chloride (2.89 g, 3.0 mL, 9.179 mmol, 0.030 equiv), chloroform (63.92 g, 43.2 mL, 535.470 mmol, 1.750 equiv), and DCM
(dichloromethane) (100.0 mL, 2.00 vol) were charged to a 1000 mL three-neck round bottom flask equipped with an overhead stirrer. The reaction mixture was stirred at 300 rpm, and 50 wt% NaOH (195.81 g, 133.2 mL, 2,447.863 mmol, 8.000 equiv) was added dropwise (via addition funnel) over 1.5 h while maintaining the temperature below 25 °C with intermittent ice/acetone bath. The reaction mixture was stirred at 500 rpm for 18 h, and monitored by GC (3% unreacted piperidinone after 18 h). The suspension was diluted with DCM (100.0 mL, 2.00 vol) and H2O (300.0 mL, 6.00 vol), and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol). The organic phases were combined and 3 M hydrochloric acid (16.73 g, 153.0 mL, 458.974 mmol, 1.500 equiv) was added. The mixture was stirred at 500 rpm for 2 h. The conversion was complete after approximately 1 h. The aqueous phase was saturated with NaCl, H2O (100.0 mL, 2.00 vol) was added to help reduce the emulsion, and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol) twice. H2O (100.0 mL, 2.00 vol) was added to help with emulsion separation. The organic phases were combined, dried (MgSO4), and concentrated to afford 32.6 g (85%) of crude 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) as a pale orange clumpy solid. The crude was recrystallized from hot (90°C) iPrOAc (71.7 mL, 2.2 vol. of crude), cooled to 80 °C, and ~50 mg of crystalline 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) was added for seeding. Crystallization started at 77 °C, the mixture was slowly cooled to ambient temperature, and aged for 2 h. The solid was collected by filtration, washed with 50/50 iPrOAc/heptane (20.0 mL, 0.40 vol) twice, and dried overnight in the vacuum oven at 40 °C to afford the desired product (23.70 g, 189.345 mmol, 62% yield) as a white sand colored crystalline solid.
1H NMR (400 MHz, CDCl3, 7.26 ppm) δ 7.33 (bs, 1H), 5.96– 5.95 (m, 1H), 5.31-5.30 (m, 1H), 2.6 (t, J = 2.5 Hz, 2H), 1.29 (s, 6H).
Example 2B
[00117] Step 1: Under a nitrogen atmosphere, 2,2,6,6-tetramethylpiperidin-4-one (257.4 kg, 1658.0 mol, 1.00 eq.), tri-butyl methyl ammonium chloride (14.86 kg, 63.0 mol, 0.038 eq.), chloroform (346.5 kg, 2901.5 mol, 1.75 eq.) and DCM (683.3 kg) were added to a 500 L enamel reactor. The reaction was stirred at 85 rpm and cooled to 15~17°C. The solution of 50wt% sodium hydroxide (1061.4 kg, 13264.0 mol, 8.00 eq.) was added dropwise over 40 h while maintaining the temperature between 15~25°C. The reaction mixture was stirred and monitored by GC.
[00118] Step 2: The suspension was diluted with DCM (683.3 kg) and water (1544.4 kg). The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg). The organic phases were combined, cooled to 10°C and then 3 M
hydrochloric acid (867.8 kg, 2559.0 mol, 1.5 eq.) was added. The mixture was stirred at 10~15 °C for 2 h. The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg x 2). The organic phases were combined, dried over Na2SO4 (145.0 kg) for 6 h. The solid was filtered off and washed with DCM (120.0 kg). The filtrate was stirred with active charcoal (55 kg) for 6 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure (30~40°C, -0.1MPa). Then isopropyl acetate (338 kg) was added and the mixture was heated to 87~91°C, stirred for 1 h. Then the solution was cooled to 15 °C in 18 h and stirred for 1 h at 15 °C. The solid was collected by filtration, washed with 50% isopropyl acetate/hexane (80.0 kg x 2) and dried overnight in the vacuum oven at 50 °C to afford 5,5-dimethyl-3-methylenepyrrolidin-2-one as an off white solid, 55% yield.
Example 3: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
Example 3A – Use of Rh Catalyst
Step 1 – Preparation of Rh Catalyst Formation:
[00119] In a 3 L Schlenk flask, 1.0 l of tetrahydrofurn (THF) was degassed with an argon stream. Mandyphos Ligand SL-M004-1 (1.89 g) and [Rh(nbd)Cl]2 (98%, 0.35 g) (chloronorbornadiene rhodium(I) dimer) were added. The resulting orange catalyst solution was stirred for 30 min at room temperature to form a catalyst solution.
Step 2:
[00120] A 50 L stainless steel autoclave was charged with 5,5-dimethyl-3-methylenepyrrolidin-2-one (6.0 kg) and THF (29 L). The autoclave was sealed and the resulting suspension was flushed with nitrogen (3 cycles at 10 bar), and then released of pressure. Next the catalyst solution from Step 1 was added. The autoclave was flushed with nitrogen without stirring (3 cycles at 5 bar) and hydrogen (3 cycles at 5 bar). The pressure was set to 5 bar and a 50 L reservoir was connected. After 1.5 h with stirring at 1000 rpm and no hydrogen uptake the reactor was flushed again with nitrogen (3 cycles at 10 bar) with stirring and additional catalyst solution was added. The autoclave was again flushed to hydrogen with the above described procedure (3 x 5 bar N2, 3 x 5 bar H2) and adjusted to 5 bar. After 2 h, the pressure was released, the autoclave was flushed with nitrogen (3 cycles at 5 bar) and the product solution was discharged into a 60 L inline barrel. The autoclave was charged again with THF (5 L) and stirred with 1200 rpm for 5 min. The wash solution was added to the reaction mixture.
Step 3:
[00121] The combined solutions were transferred into a 60 L reactor. The inline barrel was washed with 1 L THF which was also added into the reactor. 20 L THF were removed by evaporation at 170 mbar and 40°C.15 L heptane were added. The distillation was continued and the removed solvent was continuously replaced by heptane until the THF content in the residue was 1% w/w (determined by NMR). The reaction mixture was heated to 89°C (turbid solution) and slowly cooled down again (ramp: 14°C/h). Several heating and cooling cycles around 55 to 65°C were made. The off-white suspension was transferred to a stirred pressure filter and filtered (ECTFE-pad, d = 414 mm, 60 my, Filtration time = 5 min). 10 L of the mother liquor was transferred back into the reactor to wash the crystals from the reactor walls and the obtained slurry was also added to the filter. The collected solid was washed with 2 x 2.5 l heptane, discharged and let dry on the rotovap at 40°C and 4 mbar to obtain the product, (S)-3,5,5-trimethyl-pyrrolidin-2-one; 5.48Kg (91%), 98.0% ee.
Example 3B – Use of Ru Catalyst
[00122] The reaction was performed in a similar manner as described above in Example 3A except the use of a Ru catalyst instead of a Rh catalyst.
[00123] Compound (15) (300 g) was dissolved in THF (2640 g, 10 Vol) in a vessel. In a separate vessel, a solution of [RuCl(p-cymene){(R)-segphos}]Cl (0.439g, 0.0002 eq) in THF (660 g, 2.5 Vol) was prepared. The solutions were premixed in situ and passed through a Plug-flow reactor (PFR). The flow rate for the Compound (15) solution was at 1.555 mL/min and the Ru catalyst solution was at 0.287 mL/min. Residence time in the PFR was 4 hours at 30 °C, with hydrogen pressure of 4.5 MPa. After completion of reaction, the THF solvent was distilled off to give a crude residue. Heptane (1026 g, 5 vol) was added and the resulting mixture was heated to 90 °C. The mixture was seeded with 0.001 eq. of Compound 16S seeds. The mixture was cooled to -15 °C at 20 °C/h. After cooling, heptane (410 g, 2 vol) was added and the solid product was recovered by filtration. The resulting product was dried in a vacuum oven at 35 °C to give (S)-3,5,5-trimethyl-pyrrolidin-2-one (281.77 g, 98.2 % ee, 92 % yield).
Example 3C – Analytical Measurements
[00124] Analytical chiral HPLC method for the determination of the conversion, chemoselectivity, and enantiomeric excess of the products from Example 3A and 3B was made under the following conditions
Instrument: Agilent Chemstation 1100
Column: Phenomenex Lux 5u Cellulose-2, 4.6 mm x 250 mm x 5 um, LHS6247 Solvent: Heptane/iPrOH (90:10)
Flow: 1.0 ml/min
Detection: UV (210 nm)
Temperature: 25°C
Sample concentration: 30 μl of reaction solution evaporated, dissolved in 1 mL heptane/iPrOH (80/20)
Injection volume: 10.0 μL, Run time 20 min
Retention times:
5,5–‐dimethyl–3–methylenepyrrolidin–‐2–‐one: 13.8 min (S)-3,5,5-trimethyl-pyrrolidin-2-one: 10.6 min
(R)-3,5,5-trimethyl-pyrrolidin-2-one: 12.4 min
Example 4: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
[00125] Mandyphos (0.00479 mmol, 0.12 eq) was weighed into a GC vial. In a separate vial Ru(Me-allyl)2(COD) (16.87 mg, 0.0528 mmol) was weighed and dissolved in DCM (1328 µL). In another vial HBF4•Et2O (6.6 µL) and BF3 ^Et2O (2.0 µL) were dissolved in DCM (240 µL). To the GC vial containing the ligand was added, under a flow of argon, the Ru(Me-allyl)2(COD) solution (100 µL; 0.00399 mmol, 0.1eq) and the HBF4•Et2O / BF3 ^Et2O solution (20 µL; 1 eq HBF4 ^Et2O and catalytic BF3 ^Et2O). The resulting mixtures were stirred under a flow of argon for 30 minutes.
[00126] 5,5-dimethyl-3-methylenepyrrolidin-2-one (5 mg, 0.0399 mmol) in EtOH (1 mL) was added. The vials were placed in the hydrogenation apparatus. The apparatus was flushed with H2 (3×) and charged with 5 bar H2. After standing for 45 minutes, the apparatus was placed in an oil bath at temperature of 45°C. The reaction mixtures were stirred overnight under H2.200 µL of the reaction mixture was diluted with MeOH (800 µL) and analyzed for conversion and ee.
1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (ddd, J = 12.4, 8.6, 0.8 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Table 1: IPC method for Asymmetric Hydrogenation
Example 5. Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride from (S)- 3,5,5-trimethyl-pyrrolidin-2-one
Example 5A
[00127] Anhydrous THF (100ml) was charged to a dry 750ml reactor and the jacket temperature was set to 50°C. Once the vessel contents were at 50°C LiAlH4pellets (10g, 263mmol, 1.34 eq.) were added. The mixture was stirred for 10 minutes, then a solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) (25g, 197mmol) in anhydrous THF (100ml) was added dropwise over 45 minutes, maintaining the temperature between 50-60°C. Once the addition was complete the jacket temperature was increased to 68°C and the reaction stirred for 18.5hrs. The reaction mixture was cooled to 30°C then saturated sodium sulfate solution (20.9ml) was added dropwise over 30 minutes, keeping the temperature below 40°C. Vigorous evolution of hydrogen was observed and the reaction mixture thickened but remained mixable. The mixture thinned towards the end of the addition. The mixture was cooled to 20°C, diluted with iPrOAc (100ml) and stirred for an additional 10 minutes. The suspension was then drained and collected through the lower outlet valve, washing through with additional iPrOAc (50ml). The collected suspension was filtered through a celite pad on a sintered glass funnel under suction and washed with iPrOAc (2x50ml).
[00128] The filtrate was transferred back to the cleaned reactor and cooled to 0°C under nitrogen. 4M HCl in dioxane (49.1ml, 197mmol, 1eq.) was then added dropwise over 15 minutes, maintaining the temperature below 20°C. A white precipitate formed. The reactor was then reconfigured for distillation, the jacket temperature was increased to 100 °C, and distillation of solvent was carried out. Additional i-PrOAc (100 mL) was added during concentration, after >100 mL distillate had been collected. Distillation was continued until ~250 mL total distillate was collected, then a Dean-Stark trap was attached and reflux continued for 1 hour. No water was observed to collect. The reaction mixture was cooled to 20 °C and filtered under suction under nitrogen. The filtered solid was washed with i-PrOAc (100 mL), dried under suction in nitrogen, then transferred to a glass dish and dried in a vacuum oven at 40 °C with a nitrogen bleed. (S)-2,2,4-Trimethylpyrrolidine hydrochloride (17S•HCl) was obtained as a white solid (24.2g, 82%).
GC analysis (purity): >99.5%
GC chiral purity: 99.5%
Water content (by KF): 0.074%
Residual solvent (by 1H-NMR): 0.41%
Example 5B
[00129] To a glass lined 120 L reactor was charged LiAlH4 pellets (2.5 kg 66 mol, 1.2 equiv.) and dry THF (60 L) and warmed to 30 °C. To the resulting suspension was
charged (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C and sampled to check for completion, then cautiously quenched with the addition of EtOAc (1.0 L, 10 moles, 0.16 eq) followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq) then followed by a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 eq water with 1.4 eq sodium hydroxide relative to aluminum), followed by 7.5 L water (6 eq“Fieser” quench). After the addition was completed, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C.
[00130] The resultant solution was concentrated by vacuum distillation to a slurry in two equal part lots on the 20 L Buchi evaporator. Isopropanol (8 L) was charged and the solution reconcentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added and the product slurried by warming to about 50 °C. Distillation from Isopropanol continued until water content by KF is≤ 0.1 %. Methyl tertbutyl ether (6 L) was added and the slurry cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L methyl tert-butyl ether and pulled dry with a strong nitrogen flow and further dried in a vacuum oven (55 °C/300 torr/N2bleed) to afford (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, , 3H).
Example 5C
[00131] With efficient mechanical stirring, a suspension of LiAlH4 pellets (100 g 2.65 mol; 1.35 eq.) in THF (1 L; 4 vol. eq.) warmed at a temperature from 20 °C– 36 °C (heat of mixing). A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (250 g; 1.97 mol) in THF (1 L; 4 vol. eq.) was added to the suspension over 30 min. while allowing the reaction temperature to rise to ~60 °C. The reaction temperature was increased to near reflux (~68 °C) and maintained for about 16 h. The reaction mixture was cooled to below 40 °C and cautiously quenched with drop-wise addition of a saturated aqueous solution of Na2SO4 (209 mL) over 2 h. After the addition was completed, the reaction mixture was cooled to ambient temperature, diluted with i-PrOAc (1 L), and mixed thoroughly. The solid was removed by filtration (Celite pad) and washed with i-PrOAc (2 x 500 mL). With external cooling and N2 blanket, the filtrate and washings were combined and treated with drop-wise addition of anhydrous 4 M HCl in dioxane (492 mL; 2.95 mol; 1 equiv.) while maintaining the temperature below 20 °C. After the addition was completed (20 min), the resultant suspension was concentrated by heating at reflux (74– 85 °C) and removing the distillate. The suspension was backfilled with i-PrOAc (1 L) during concentration. After about 2.5 L of distillate was collected, a Dean-Stark trap was attached and any residual water was azeotropically removed. The suspension was cooled to below 30 °C when the solid was collected by filtration under a N2 blanket. The solid is dried under N2 suction and further dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford 261 g (89% yield) of (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H). 1H NMR (400 MHz, CDCl3) δ 9.55 (d, J = 44.9 Hz, 2H), 3.52 (ddt, J = 12.1, 8.7, 4.3 Hz, 1H), 2.94 (dq, J = 11.9, 5.9 Hz, 1H), 2.70– 2.51 (m, 1H), 2.02 (dd, J = 13.0, 7.5 Hz, 1H), 1.62 (s, 3H), 1.58– 1.47 (m, 4H), 1.15 (d, J = 6.7 Hz, 3H).
Example 5D
[00132] A 1L four-neck round bottom flask was degassed three times. A 2M solution of LiAlH4 in THF (100 mL) was charged via cannula transfer. (S)-3,5,5-trimethylpyrrolidin-2-one (19.0 g) in THF (150 mL) was added dropwise via an addition funnel over 1.5 hours at 50-60 °C, washing in with THF (19 mL). Upon completion of the addition, the reaction was stirred at 60 °C for 8 hours and allowed to cool to room temperature overnight. GC analysis showed <1% starting material remained.
[00133] Deionized water (7.6 mL) was added slowly to the reaction flask at 10-15 °C, followed by 15% potassium hydroxide (7.6 mL). Isopropyl acetate (76 mL) was added, the mixture was stirred for 15 minutes and filtered, washing through with isopropyl acetate (76 mL).
[00134] The filtrate was charged to a clean and dry 500 mL four neck round bottom flask and cooled to 0-5 °C. 36% Hydrochloric acid (15.1 g, 1.0 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (190 mL), was carried out to leave a residual volume of ~85 mL. Karl Fischer analysis = 0.11% w/w H2O. MTBE (methyl tertiary butyl ether) (19 mL) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (25 mL) and drying under vacuum at 40-45 °C to give crude (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (17.4 g, 78% yield). GC purity = 99.5%. Water content = 0.20% w/w. Chiral GC gave an ee of 99.0% (S).
Ruthenium content = 0.004 ppm. Lithium content = 0.07 ppm.
[00135] A portion of the dried crude (S)-2,2,4-trimethylpyrrolidine hydrochloride (14.3g) was charged to a clean and dry 250 mL four-neck round bottom flask with isopropanol (14.3 mL) and the mixture held at 80-85 °C (reflux) for 1 hour to give a clear solution. The solution was allowed to cool to 50 °C (solids precipitated on cooling) then MTBE (43 mL) was added and the suspension held at 50-55 °C (reflux) for 3 hours. The solids were filtered off at 10 °C, washing with MTBE (14 mL) and dried under vacuum at 40 °C to give recrystallised (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystallised solid (13.5 g, 94% yield on recrystallisation, 73% yield). GC purity = 99.9%. Water content = 0.11% w/w. Chiral GC gave an ee of 99.6 (S). Ruthenium content = 0.001 ppm. Lithium content = 0.02 ppm.
Example 5E:
[00136] A reactor was charged with lithium aluminum hydride (LAH) (1.20 equiv.) and 2-MeTHF (2-methyltetrahydrofuran) (4.0 vol), and heated to internal temperature of 60 °C while stirring to disperse the LAH. A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (1.0 equiv) in 2-MeTHF (6.0 vol) was prepared and stirred at 25 °C to fully dissolve the (S)-3,5,5-trimethylpyrrolidin-2-one. The (S)-3,5,5-trimethylpyrrolidin-2-one solution was added slowly to the reactor while keeping the off-gassing manageable, followed by rinsing the addition funnel with 2-MeTHF (1.0 vol) and adding it to the reactor. The reaction was stirred at an internal temperature of 60 ± 5 °C for no longer than 6 h. The internal temperature was set to 5 ± 5 °C and the agitation rate was increased. A solution of water (1.35 equiv.) in 2-MeTHF (4.0v) was prepared and added slowly to the reactor while the internal temperature was maintained at or below 25 °C. Additional water (1.35 equiv.) was charged slowly to the reactor while the internal temperature was maintained at or below 25 °C. Potassium hydroxide (0.16 equiv.) in water (0.40 vol) was added to the reactor over no less than 20 min while the temperature was maintained at or below 25 °C. The resulting solids were removed by filtration, and the reactor and cake were washed with 2-MeTHF (2 x 2.5 vol). The filtrate was transferred back to a jacketed
vessel, agitated, and the temperature was adjusted to 15 ± 5 °C. Concentrated aqueous HCl (35-37%, 1.05 equiv.) was added slowly to the filtrate while maintaining the temperature at or below 25 °C and was stirred no less than 30 min. Vacuum was applied and the solution was distilled down to a total of 4.0 volumes while maintaining the internal temperature at or below 55 °C, then 2-MeTHF (6.00 vol) was added to the vessel. The distillation was repeated until Karl Fischer analysis (KF) < 0.20% w/w H2O. Isopropanol was added (3.00 vol), and the temperature was adjusted to 70 °C (65– 75 °C) to achieve a homogenous solution, and stirred for no less than 30 minutes at 70 °C. The solution was cooled to 50 °C (47– 53 °C) over 1 hour and stirred for no less than 1 h, while the temperature was maintained at 50°C (47– 53 °C). The resulting slurry was cooled to -10 °C (-15 to -5°C) linearly over no less than 12 h. The slurry was stirred at -10 °C for no less than 2 h. The solids were isolated via filtration or centrifugation and were washed with a solution of 2-MeTHF (2.25 vol) and IPA (isopropanol) (0.75 vol). The solids were dried under vacuum at 45 ± 5 °C for not less than 6 h to yield (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl).
Example 6: Phase Transfer Catalyst (PTC) Screens for the Synthesis of 5,5- dimethyl-3-methylenepyrrolidin-2-one
[00137] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.05 eq.), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added dropwise over 2 min. The reaction mixture was stirred until completion as assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion and assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the
organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 2.
Table 2
Example 7: Solvent Screens for the Synthesis of 5,5-dimethyl-3- methylenepyrrolidin-2-one
[00138] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.), and solvent (2 vol. or 4 vol., as shown in Table 3 below) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL,
2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC.
Reaction results are summarized in Table 3.
Table 3
Example 8: Base Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin- 2-one
[00139] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of an amount wt% sodium hydroxide as shown in Table 4 below in water (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase is extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as
an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 4.
Table 4
Example 9: Various Amounts of Phase Transfer Catalyst (PTC) for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[00140] In this experiment, various amounts of PTCs were tested as described below: Tetrabutylammonium hydroxide (0.01 eq.), TBAB (0.01 eq.), Tributylmethylammonium chloride (0.01 eq.), Tetrabutylammonium hydroxide (0.02 eq.), TBAB (0.02 eq.), Tributylmethylammonium chloride (0.02 eq.), Tetrabutylammonium hydroxide (0.03 eq.), TBAB (0.03 eq.), Tributylmethylammonium chloride (0.03 eq.).
[00141] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion, assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by
HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. The reaction results are summarized in Table 5.
Table 5
Example 10: Preparation of 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl)
[00142] 2,2,6,6-tetramethyl-4-piperidinone (14) (30 g, 193.2 mmol, 1.0 eq) was charged to a 500 mL nitrogen purged three necked round bottomed flask equipped with condenser. IPA (300 mL, 10 vol) was added to the flask and the mixture heated to 60 °C until dissolved.
[00143] To the solution at 60 °C was added 5-6 M HCl in IPA (40 mL, 214.7 mmol, 1.1 eq) over 10 min and the resulting suspension stirred at 60 °C for 30 min then allowed to cool to ambient temperature. The suspension was stirred at ambient temperature overnight, then filtered under vacuum and washed with IPA (3 x 60 mL, 3 x 2 vol). The cream colored solid was dried on the filter under vacuum for 10 min.
[00144] The wet cake was charged to a 1 L nitrogen purged three necked round bottomed flask equipped with condenser. IPA (450 mL, 15 vol) was added to the flask and the suspension heated to 80 °C until dissolved. The mixture was allowed to cool slowly to ambient temperature over 3 h and the resulting suspension stirred overnight at ambient temperature.
[00145] The suspension was filtered under vacuum, washed with IPA (60 mL, 2 vol) and dried on the filter under vacuum for 30 min. The resulting product was dried in a vacuum oven at 40 °C over the weekend to give 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl) a white crystalline solid, 21.4 g, 64% yield.
Example 11: Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) from (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S)
[00146] Each reactor was charged with (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF, H2, and the catalyst shown in the below table. The reactor was heated to 200 °C and pressurized to 60 bar, and allowed to react for 12 hours. GC analysis showed that (S)-2,2,4-trimethylpyrrolidine was produced in the columns denoted by“+.”
[00147] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/SiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 130 °C under 80 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (74.8% yield, 96.1% ee).
Alternate synthesis
[00148] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 4% Pt-2%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 200 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (88.5% yield, 29.6% ee).
Alternate synthesis
[00149] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/TiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 150 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.9% yield, 98.0% ee).
Alternate synthesis
[00150] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.03 mL/min into a packed bed reactor prepacked with 2% Pt-8%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 40 mL/min. The reaction was carried out at 180 °C under 55 bar pressure with a residence time of 6 min. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.4% yield, 96.8% ee).
Example 12: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3- trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
Compound 1
I. Preparation of Starting Materials:
A. Synthesis of 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt:
Step 1: tert-Butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (28)
[00151] A 2L 3-necked round-bottom flask, equipped with a J-Kem thermocouple and an overhead stirrer, was purged with nitrogen for >20 minutes. Hexyllithium solution (2.3 M in hexanes; 1.05 equiv; 0.260 L, 597 mmol) was transferred into the flask via cannula. The flask was then cooled to–65°C in a dry ice/isopropyl alcohol bath and diisopropylamine (1.05 equiv; 0.842 L; 597mmol) was added via an addition funnel, and the internal temperature was maintained at–40 ±5 °C. Once the diisopropylamine addition was complete, tetrahydrofuran (THF) (0.423 L; 6.4 vol) was added to the reactor and the reaction was warmed to room temperature and stirred for 15 minutes. The solution was then cooled to–60 °C and ethyl isobutyrate (1.0 equiv; 0.754 L; 568 mmol) was added dropwise maintaining the temperature below–45 °C. 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) (0.9 equiv; 0.616 L; 511 mmol) was then added dropwise to the reaction flask and the temperature was maintained below–45 °C. In a separate flask, tert-butyldimethylsilyl chloride (TBSCl) (1.05 equiv; 89.9 g; 597 mmol) was dissolved in THF (2.2 vol w.r.t. TBSCl) and then added to the 2L reactor. The internal temperature was maintained at≤–30°C during the addition of the TBSCl solution. The resulting reaction mixture was allowed to warm to room
temperature and stirred overnight under inert atmosphere. The reaction solution was transferred to a 2L one-neck round-bottom flask. Additional THF (50 mL, x 2) was used to rinse and transfer. The solution was concentrated in vacuo to remove most of the THF. Hexanes were added to the concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (500 mL). The organic phase was washed with three times with water (500 mL x 3), to remove salts. The organic layer was dried over Na2SO4 (100 g). The solution was filtered and the waste cake washed with additional hexanes (100 mL). The resulting hexanes solution of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was concentrated in vacuo. A quantitative 1H-NMR assay was performed with benzyl benzoate as an internal standard. The quantitative NMR assay indicated that 108.6 grams of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (83% yield) was present, and that 1.2 mol% of ethyl isobutyrate relative to tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was also present. The resulting tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane solution was used without further purification for the photochemical reaction of Step 2. Step 2: 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt
[00152] Stock solution A: The concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1- yl)oxy)dimethylsilane (198 g; 0.86 mol) was dissolved in acetonitrile (895 g; 1.14 L; 5.8 vol) to give a cloudy, yellow solution that was then filtered. The density of the clear, filtered solution was measured to be 0.81 g/mL and the molar concentration was calculated to be 0.6 M. This is referred to as stock solution A (substrate).
[00153] Stock solution B: The catalyst and reagent solution was prepared by dissolving Ru(bpy)3Cl2 hexahydrate in acetonitrile, followed by adding ethanol and pyrrolidine to give a red-colored solution (density measured: 0.810 g/mL). The molar concentration of the catalyst was calculated to be 0.00172 M. The molar concentration of the solution with respect to EtOH/pyrrolidine was calculated to be ~2.3 M. See Table 6.
Table 6
(i) Photochemical Trifluoromethylation
[00154] CF3I gas was delivered to the reactor directly from the lecture bottle using a regulator and mass flow controller. Stock solutions A and B were pumped at 6.7 g/min and 2.07 g/min, respectively, to mix in a static mixer. The resulting solution was then combined with CF3I in a static mixer. The CF3I was metered into the reactor via a mass flow controller at 2.00 g/min (2 equiv). Liquid chromatography (LC) assay indicated that 1.0% of the tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was left unreacted. Details of the reaction parameters are shown in the table below. The reaction stream was passed through the 52 mL photoreactor while being irradiated with the 800 W 440-445 LED light source. The first 5 minutes of eluent was discarded. Thereafter the eluent was collected for a total of 3.05 hours. A total of ~2.3 L of solution was collected during the reaction (~1.06 mol). See Table 7.
Table 7
(ii) Saponification & Salt Formation
[00155] The saponication of the crude solution (4.1 L, from 1.60 mol tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane) was carried out in a 5L 4-necked round-bottomed flask in 2 roughly equal size batches using 15 wt% NaOH (aq) (total ~320g NaOH) at 50 °C for 2-4 h. Upon completion of the reaction determined by gas chromatography (GC) analysis, the re-combined batches were cooled to room
temperature and hexanes (500 mL) and toluene (500 mL) were added to give a clear phase separation. The top organic layer was washed with half-brine (1 L) and combined with the first portion of the product-containing aqueous solution (4.5 L). The combined aqueous stream was washed with hexanes (500 mL) and concentrated to 2-3 L to remove a majority of volatile acetonitrile. To the aqueous phase was added concentrated HCl (1 L, 12 N) and the resulting mixture was extracted with hexanes (4 x 1 L). The combined hexanes extracts were washed with half brine (2 x 500 mL) and concentrated to give an oil (216 g). The oil was dissolved in THF (580 mL), and morpholine (120 mL, 1.0 equiv) was added slowly via an addition funnel. Upon completion of addition, the batch was seeded (0.5-1 g) with morpholine salt, and the seeds were held and allowed to thicken over 30 min. Hexanes (1660 mL) were added over ~ 2 h, and the mixture was aged for another 3 h. The batch was filtered, washed with hexanes (~500 mL) in portions and dried under vacuum/dry air flush to give 3,3,3-trifluoro-2,2-dimethylpropionic acid, morpholine salt as a white solid (283 g, 73%).1H NMR (400 MHz, CD3OD) δ 3.84-3.86 (m, 4H), 3.15-3.18 (m, 4H), 1.33 (s, 6H); 19F NMR (376 MHz, CD3OD): δ -75.90 (s, 3F).
B. Synthesis of 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol (5)
[00156] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g), which corresponds to 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
C. Synthesis of tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate (22)
[00157] A 50L Syrris controlled reactor was started and the jacket was set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethylamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise, maintaining reaction temp <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L) and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then 1-2 volumes of solvent was distilled off. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
II. Preparation of Compound I
Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (23)
[00158] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-
isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (7)
[00159] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +; Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate (25)
[00160] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (26)
[00161] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and the mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (10)
[00162] tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room temperature under stirring for 2.5 h. The solid was collected by filtration, washed with
isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (13)
[00163] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +;
Retention time: 0.68 minutes.
Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
[00164] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol), and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
///////////VX-445, Elexacaftor, VX445, エレクサカフトル , PHASE 3, CYSTIC FIBRIOSIS, VX 445
C[C@@H]1CN(c2nc(ccc2C(=O)NS(=O)(=O)c3cn(C)nc3C)n4ccc(OCC(C)(C)C(F)(F)F)n4)C(C)(C)C1
CC1CC(N(C1)C2=C(C=CC(=N2)N3C=CC(=N3)OCC(C)(C)C(F)(F)F)C(=O)NS(=O)(=O)C4=CN(N=C4C)C)(C)C
VX-659, Bamocaftor potassium

VX-659, BAMOCAFTOR
N-(Benzenesulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
3-Pyridinecarboxamide, N-(phenylsulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-
N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
| CAS Number | 2204245-48-5 |
UNII: 8C7XEW3K7S |
BAMOCAFTOR |
| M. Wt | 591.65 |
| Formula | C28H32F3N5O4S |
Bamocaftor potassium
CAS 2204245-47-4
| Molecular Formula | C28 H31 F3 N5 O4 S . K |
| Molecular Weight | 629.735 |
VX-659
VX-659 potassium salt
VY7D8MTV72 (UNII code)
WHO 11167
3-Pyridinecarboxamide, N-(phenylsulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-, potassium salt (1:1)
Potassium (benzenesulfonyl)[6-(3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl)-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carbonyl]azanide
PHASE 2 CYSTIC FIBRIOSIS , VERTEX
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) (DeltaF508 Mutant) Correctors
Bamocaftor potassium is a CFTR channel (DeltaF508-CFTR Mutant) corrector in phase II clinical trials at Vertex, in patients with CF who are homozygous for the F508del mutation of the CF transmembrane conductance regulator (CFTR) gene, or who are heterozygous for the F508del mutation and a minimal function (MF) CFTR mutation not likely to respond to tezacaftor, ivacaftor, or tezacaftor/ivacaftor and also in combination with tezacaftor and VX-561 in F508del/MF in patients with cystic fibrosis.
The compound is also developed by the company as a fixed-dose combination of VX-659, tezacaftor and ivacaftor.
Vertex Pharmaceuticals is developing a combination regimen comprising VX-659, a next-generation cystic fibrosis transmembrane conductance regulator (CFTR) corrector, with tezacaftor and ivacaftor, as a triple fixed-dose combination tablet. In March 2019, Vertex planned to file an NDA in the US in 3Q19 concurrently in patients aged 12 years or older with one F508del mutation and one minimal function mutation and in patients with two F508del mutations for either the VX-659 or VX-445 triple combination regimen; the regimen selected for a regulatory filing would be based on final 24-week data.
PATENT
WO 2018064632
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018064632
Example 4: Synthesis of Compounds 1-65
[00229] Synthetic Example 1: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (Compound 1)
[00230] Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00231] Step 1: Synthesis of methyl-2,4-dimethyl-4-nitro-pentanoate
[00232] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg. 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L). 2 M HC1 (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible – a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HC1 (3 L). After separation, the HC1 washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. This was dried with MgSC and filtered to afford methyl-2,4-dimethyl-4-mtro-pentanoate as a clear green oil (3.16 kg, 99% yield). 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56 – 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J= 6.8 Hz, 3H). [00233] Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00234] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00235] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
[00236] Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00237] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at -2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (-1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to -1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %). 1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
[00238] Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00239] A glass lined 120 L reactor was charged with lithium aluminium hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and 1he product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the
slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4- trimethylpyrrolidine’HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, / = 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, J= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J= 6.6 Hz, 3H).
[00240] Part B: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
[00241] Synthesis of starting materials:
[00242] Synthesis of tert-Butyl 2,6-dichloropyridine-3-carboxylate
[00243] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and stirred overnight at room temperature. At this point, HC1 IN (400 mL) was added, and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL), and the combined organic layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert- butyl 2,6-dichloropyndine-3-carboxylate as a colorless oil. ESI-MS m/z calc. 247.02, found 248.1 (M+l) +; Retention time: 2.27 minutes. 1H NMR (300 MHz, CDC13) ppm 1.60 (s, 9H), 7.30 (d, .7=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
[00244] Synthesis of tert-Butyl 3-oxo-2,3-dihydro-lH-pyrazole-l-carboxylate
[00245] A 50L reactor was started, and the jacket was set to 20 °C, with stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added, and the reactor was capped. The reaction was heated to an internal temperature of 40 °C, and the system was set to hold jacket temperature at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 °C for 1 h. The reaction mixture was cooled to 20 °C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion-wise, maintaining reaction temperature <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L. 22.76 mol) in MeOH (2.860 L) was added portion-wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 °C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear, light amber oil. The resulting oil was transferred to the 50L reactor, stirred and water (7.150 L) and heptane (7.150 L) were added. The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container, and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol) and added dropwise. The jacket was set to 0 °C to absorb the quench exotherm. After the addition was complete (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L), and washed a second time with water (3.575 L). The crystalline solid was transferred into a 20L rotovap bulb, and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and 1-2 volumes of solvent were distilled off The slurry in the rotovap flask was filtered, and the solids were washed with heptane (3.575 L). The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-lH-pyrazole-2-carboxylate (2921 g, 71%) as a coarse, crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J= 2.9 Hz, 1H), 5.90 (d, J= 2.9 Hz, 1H), 1.54 (s, 9H).
[00246] Synthesis of 2-[l-(trifluoromethyl)cyclopropyl]ethanol
[00247] To a solution of lithium aluminum hydride (293 mg, 7.732 mmol) in THF (10.00 mL) in an ice-bath, 2-[l-(trifluoromethyl)cyclopropyl]acetic acid (1.002 g, 5.948 mmol) in THF (3.0 mL) was added dropwise over a period of 30 minutes keeping the reaction temperature below 20 ° C. The mixture was allowed to gradually warm to ambient temperature and was stirred for 18 h. The mixture was cooled with an ice-bath and sequentially quenched with water (294 mg, 295 μL, 16.36 mmol), NaOH (297 μL of 6 M, 1.784 mmol), and then water (884.0 μL, 49.07 mmol) to afford a granular solid in the mixture. The solid was filtered off using celite, and the precipitate was washed with ether. The filtrate was further dried with MgSO4 and filtered and concentrated in vacuo to afford the product with residual THF and ether. The mixture was taken directly into the next step without further purification.
[00248] Step 1: tert-Butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-1-carboxylate
[00249] rerf-Butyl 5-oxo-lH-pyrazole-2-carboxylate (1.043 g, 5.660 mmol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (916 mg, 5.943 mmol), and triphenyl phosphine (1.637 g, 6.243 mmol) were combined in THF (10.48 mL) and the reaction was cooled in an ice-bath. Diisopropyl azodicarboxylate (1.288 g, 1.254 mL, 6.368 mmol) was added dropwise to the reaction mixture, and the reaction was allowed to warm to room temperature for 16 hours. The mixture was evaporated, and the resulting material was partitioned between ethyl acetate (30 mL) and IN sodium hydroxide (30 mL). The organic layer was separated, washed with brine (30 mL), dried over sodium sulfate, and concentrated. The crude material was purified by silica gel chromatography eluting with a gradient of ethyl acetate in hexanes (0- 30%) to give tert-butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 57%). ESI-MS m/z calc. 320.13, found 321.1 (M+l) +; Retention time: 0.72 minutes.
[00250] Step 2: 3-[2-[l-(Trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole
[00251] terr-Butyl-3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 3.216 mmol) was dissolved in dichloromethane (10.30 mL) with trifluoroacetic acid (2.478 mL, 32.16 mmol), and the reaction was stirred at room temperature for 2 hours. The reaction was evaporated, and the resulting oil was partitioned between ethyl acetate (10 mL) and a saturated sodium bicarbonate solution.
The organic layer was separated, washed with brine, dried over sodium sulfate, and evaporated to give 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (612 mg, 86%). ESI-MS m/z calc. 220.08, found 221.0 (M+1) +; Retention time: 0.5 minutes. ¾ NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 7.50 (t, J = 2.1 Hz, 1H), 5.63 (t, J= 2.3 Hz, 1H), 4.14 (t, J= 7.1 Hz, 2H), 2.01 (t, J= 7.1 Hz, 2H), 0.96 – 0.88 (m, 2H), 0.88 -0.81 (m, 2H).
[00252] Step 3: tert- Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate
[00253] tert-Butyl 2,6-dichloropyridine-3-carboxylate (687 mg, 2.770 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (610 mg, 2.770 mmol), and freshly ground potassium carbonate (459 mg, 3.324 mmol) were combined in anhydrous DMSO (13.75 mL). l,4-diazabicyclo[2.2.2]octane (DABCO (1,4-diazabicyclo[2.2.2]octane), 62 mg, 0.5540 mmol) was added, and the mixture was stirred at room temperature under nitrogen for 16 hours. The reaction mixture was diluted with water (20 mL) and stirred for 15 minutes. The resulting solid was collected and washed with water. The solid was dissolved in dichloromethane and dried over magnesium sulfate. The mixture was filtered and concentrated to give ferf-butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 84%). ESI-MS m/z calc. 431.12, found 432.1 (M+1) +; Retention time: 0.88 minutes.
[00254] Step 4: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00255] tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 2.339 mmol) and trifluoroacetic acid (1.8 mL, 23.39 mmol) were combined in dichloromethane (10 mL) and heated at 40 °C for 3 h. The reaction was concentrated. Hexanes were added, and the mixture was concentrated again to give 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (873 mg, 99%) ESI-MS m/z calc. 375.06, found 376.1 (M+l)+; Retention time: 0.69 minutes.
[00256] Step 5: N-(Benzenesulfonyl)-2-chloro-6-[3- [2- [1-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide
[00257] A solution of 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (0.15 g, 0.3992 mmol) and carbonyl diimidazole (77 mg, 0,4790 mmol) in THF (2.0 mL) was stirred for one hour, and benzenesulfonamide (81 mg, 0.5190 mmol) and DBU (72 μL, 0.4790 mmol) were added. The reaction was stirred for 16 hours, acidified with 1 M aqueous citric acid, and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated. The residue was purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-5%) to give N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyndine-3-carboxamide (160 mg, 78%). ESI-MS m/z calc. 514.07, found 515.1 (M+l)+; Retention time: 0.74 minutes.
[00258] Step 6: N-(Benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy] pyrazol-l-yl] -2- [(4S)-2,2,4-trimethylpyrrolidin-l-yl] pyridine-3-carboxamide
[00259] A mixture of N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l -(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 0.3107 mmol), (4S)-2,2,4-trimethylpyrrolidine hydrochloride salt (139 mg, 0.9321 mmol), and potassium carbonate (258 mg, 1.864 mmol) in DMSO (1.5 mL) was stirred at 130 °C for 17 hours. The reaction mixture was acidified with 1 M aqueous citric acid and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated to yield a crude product that was purified by reverse-phase HPLC utilizing a gradient of 10-99% acetonitrile in 5 mM aqueous HCI to yield N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (87 mg, 47%). ESI-MS mJz calc. 591.21, found 592.3 (M+l) +; Retention time: 2.21 minutes. 1H NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.19 (d, J = 2.8 Hz, 1H), 8.04 – 7.96 (m, 2H), 7.81 (d, J= 8.2 Hz, 1H), 7.77 – 7.70 (m, 1H), 7.70 – 7.62 (m, 2H), 6.92 (d, J= 8.2 Hz, 1H), 6.10 (d, J= 2.8 Hz, 1H), 4.31 (t, J= 7.0 Hz, 2H), 2.42 (t, J = 10.5 Hz, 1H), 2.28 (dd, J = 10.2, 7.0 Hz, 1H), 2.17 – 2.01 (m, 3H), 1.82 (dd, J= 11.9, 5.5 Hz, 1H), 1.52 (d, .7= 9.4 Hz, 6H), 1.36 (t, J= 12.1 Hz, 1H), 1.01 – 0.92 (m, 2H), 0.92 – 0.85 (m, 2H), 0.65 (d, J = 6.3 Hz, 3H). pKa: 4.95±0.06.
Alternate synthesis of 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00263] Step 1: ethyl 3-hydroxy-lH-pyrazole-4-carboxylate
[00264] A mixture of EtOH (20.00 L, 10 vol) and diethyl 2-(ethoxymethylene)propanedioate (2000 g, 9.249 mol, 1.0 equiv) was added under nitrogen purge a to a 50 L reactor equipped with a reflux condenser (10 °C) and the jacket set to 40 °C. The mixture was stirred, and then hydrazine hydrate (538.9 g of 55 %w/w, 523.7 mL of 55 %w/w, 9.249 mol, 1.00 equiv) was added in portions via an addition funnel. Once the addition was complete, the reaction was heated to 75 °C for 22 h to afford a solution of ethy l 3-hydroxy-lH-pyrazole-4-carboxylate that was used directly in the next step.
[00265] Step 2: l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate
[00266] The solution of ethyl 3-hydroxy-lH-pyrazole-4-carboxylate was cooled from 75 °C to 40 °C, then triethylamine (TEA) (46.80 g, 64.46 mL, 462.5 mmol, 0.05 eq.) was added. A solution of Boc anhydride (2.119 kg, 9.711 mol 1.05 equiv) in EtOH (2.000 L, 1 equiv) was added to the reactor over 35 min. The mixture was stirred for 4 hours to complete the reaction; then water (10.00 L, 5.0 vol) was added over 15 mins. The resulting mixture was cooled to 20 °C to complete crystallization of the product. The crystals were allowed to age for 1 hour, then the mixture was filtered. The solid was washed with a mixture of EtOH (4.000 L, 2.0 vol) and water (2.000 L, 1 0 vol) The solid was then dried in vacuo to afford l-(tert-butyl)-4-ethyl-3-hydroxy-lH-pyrazole-1,4-dicarboxylate (1530 g, 65%) as colorless, fine needle, crystalline solid. ‘H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.40 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.56 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
[00267] Step 3: l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate
[00268] A 5L reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temperature and nitrogen purge. The vessel was charged with toluene (1.0L, 10.0 vol), 2-[l-(tnfluoromethyl)cyclopropyl]ethanol (lOO.Og, 648.8 mmol, 1.0 equiv), and l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate (166.3 g, 648.8 mmol), and the mixture was stirred. The reaction mixture was charged with triphenyl phosphine (195.7 g, 746.1 mmol, 1.15 equiv), then the reactor was set to maintain an internal temperature of 40 °C. Diisopropyl azoldicarboxylate (150.9 g, 746.1 mmol, 1.15 equiv) was added into an addition funnel and was added to the
reaction while maintaining the reaction temperature between 40 and 50 °C (addition was exothermic, exotherm addition controlled), and stirred for a total of 2.5 hours. Once the reaction was deemed complete by HPLC, heptane was added (400 mL, 4 vol), the solution was cooled to 20 °C over 60 minutes, and the bulk of tnphenylphosphine oxide-DIAD complex (TPPO-DIAD) crystallized out. Once at room temp, the mixture was filtered, and the solid was washed with heptane (400 mL, 4.0 vol) and pulled dry. The filtrate was used in the next step as a solution in toluene-heptane without further purification.
[00269] Step 4: ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate
[00270] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with a toluene solution consisting of approximately 160 mmol, 65.0 g of 1 -(tert-buty 1) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate in 3 vol of toluene (prepared by concentrating a 25% portion of filtrate from previous reaction down to 4 volumes in a rotovap). The reaction was set to maintain an internal temperature at 40 °C and KOH (33.1 g, 1.5 eq. of aqueous 45 % KOH solution) was added in one portion, resulting in a mild exothermic addition, while CO2 was generated upon removal of the protecting group. The reaction proceeded for 1.5 hr, monitored by HPLC, with the product partially crystallizing during the reaction. Heptane (160 mL, 2.5 vol) was added to the reaction mixture and the reaction was cooled to room temperature over 30 minutes. The resulting mixture was filtered, and the solid was washed with heptane (80.00 mL, 1.25 vol), pulled dry, then dried in vacuo (55 °C, vacuum). 52.3 g of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate was obtained as a crude, colorless solid that was used without further purification.
[00271] Step 5: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid
[00272] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with methanol (150.0 mL, 3.0 vol), a solution of ethyl 3-(2-(l-(triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate (50.0 g, 171.1 mmol, 1.0 equiv), and the reaction was stirred to suspend the solids. The reactor was set to maintain internal temperature at 40 °C. To the mixture was added KOH (96 g of aqueous 45 % KOH, 1.71 mol, 10.0 equiv) in portions maintaining the internal temperature <50 °C. Once addition was complete, the reaction was set to maintain temperature at 50 °C, and the reaction proceeded for 23 hours, monitored by HPLC. Once complete the reaction was cooled to 10 °C then partially concentrated on a rotary evaporator to remove most of the MeOH. The resulting solution was diluted with water (250 mL, 5.0 vol) and 2-Me-THF (150 mL, 3.0 vol), and transferred to the reactor, stirred at room temp, then stopped, and layers were allowed to separate. The layers were tested, with remaining TPPO-DIAD complex in the organic layer and product in the aqueous layer. The aqueous layer was washed again with 2-Me-THF (100 mL, 2.0 vol), the layers separated, and the aqueous layer returned to the reactor vessel. The stirrer was started and set to 450 rpm, and the reactor jacket was set to 0 °C. The pH was adjusted to pH acidic by addition of 6M aqueous HC1 (427mL, 15 equiv) portion wise, maintaining the internal temperature between 10 and 30 °C. The product began to crystallize close to pH neutral and was accompanied with strong off-gassing, and so the acid was added slowly, and then further added to reach pH 1 once the off-gassing had ended. To the resulting suspension was added 2-Me-THF (400 mL, 8.0 vol), and the product was allowed to dissolve into the organic layer. Stirring was stopped, the layers were separated, and the aqueous layer was returned to the reactor, stirred and re-extracted with 2-Me-THF (100 mL, 2.0 vol). The organic lay ers were combined in the reactor and stirred at room temperature, washed with brine (lOOmL, 2 vols), dried over Na2S04, filtered through celite, and the solid was washed with 2-Me-THF (50 mL, 1.0 vol). The filtrate was transferred to a clean rotovap flask, stirred, warmed to 50 °C and heptane (200 mL, 4.0 vol) added, and then partially concentrated with the addition of heptane (300 mL, 6.0 vol) and then seeded with 50mg of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid), and the product crystallized during solvent removal. The distillation was stopped when the bulk of the 2-Me-THF had distilled off. The bath heater was turned off, the vacuum removed, and the mixture was allowed to stir and cool to room temperature. The mixture was filtered (slow speed) and the solid was washed with heptane (100 mL, 2.0 vol), and the solid was collected and dried in vacuo (50 °C, rotovap). 22.47 g of 3-(2-(l-(triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid was obtained as an off-white solid. 1H NMR (400 MHz, DMSO-d) δ 12.45 (s, 2H), 8.01 (s, 1H), 4.26 (t, J = 7.0 Hz, 2H), 2.05 (t, J= 7.0 Hz, 2H), 0.92 (m, 4H).
[00273] Step 6: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole
[00274] A mixture of toluene (490.0 mL), 3-(2-(l- (triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid (70.0 g, 264.9 mmol), and DMSO (70.00 mL) was placed in a reactor and heated to 100 °C with stirring. DBU (approximately 20.16 g, 19.80 mL, 132.4 mmol) was added to the reactor over 15 min. The mixture was stirred for 20 h to complete the reaction and then cooled to 20 °C. The mixture was washed with water (350.0 mL), then 0.5N aq HC1 (280.0 mL), then water (2 x 140.0 mL), and lastly with bnne (210.0 mL). The organic layer was dried with Na2S04, and then activated charcoal (5 g, Darco 100 mesh) was added to the stirred slurry. The dried mixture was filtered through celite, and the solid was washed with toluene (140.0 mL) and then pulled dry. The filtrate was concentrated in a rotovap (50 °C, vac) to afford 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-
pyrazole (30.89 g, 53%) as an amber oil. 1H NMR (400 MHz, DMSO-4,) δ 11.87 (s, 1H), 7.50 (d, J= 2.4 Hz, 1H), 5.63 (d, 7= 2.4 Hz, 1H), 4.23 – 4.06 (m, 2H), 2.01 (t, J= 7.1 Hz, 2H), 1.00 – 0.77 (m, 4H).
[00275] Step 7: ethyl 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate
[00276] A mixture of DMF (180.0 mL), ethyl 2,6-dichloropyridine-3-carboxylate (approximately 29.97 g, 136.2 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.0 g, 136.2 mmol), and K2CO3, (325 mesh, approximately 24.48 g, 177.1 mmol) was added to a stirred reactor at 20 °C. DABCO (approximately 2.292 g, 20.43 mmol) was then added to the reactor, and the mixture was stirred at 20 °C for 1 hour, and then the temperature was increased to 30 °C, and the mixture stirred for 24 hours to complete the reaction. The mixture was cooled to 20 °C; then water (360 mL) was added slowly. The mixture was then drained from the reactor and the solid was isolated by filtration. The solid was then washed with water (2 x 150 mL), and then the solid was dried under vacuum at 55 °C to afford ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (51.37 g, 93%) as a fine, beige colored solid. 1H NMR (400 MHz, DMSO-c4) δ 8.44 (d, J= 2.9 Hz, 1H), 8.41 (d, J= 8.5 Hz, 1H), 7.75 (d, J= 8.5 Hz, 1H), 6.21 (d, J= 2.9 Hz, 1H), 4.34 (m, 4H), 2.09 (t, J= 7.1 Hz, 2H), 1.34 (t, J= 7.1 Hz, 3H), 1.00 – 0.84 (m, 4H).
[00277] Step 8: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00278] A solution of ethyl 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (50.0 g, 123.8 mmol) in THF (300.0 mL) was prepared in a reactor at 20 °C. EtOH (150.0 mL) was added, followed by aqueous NaOH (approximately 59.44 g of 10 %w/w, 148.6 mmol). The mixture was stirred for 1 hour to complete the reaction; then aq IN HCl (750.0 mL) was slowly added. The resulting suspension was stirred for 30 mm at 10 °C, and then the solid was isolated by filtration. The solid was washed with water (150 mL then 2 x 100 mL) and then pulled dry by vacuum. The solid was then further dried under vacuum with heating to afford 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (42.29 g, 91%). 1H NMR (400 MHz, DMSO-d 6) 5 13.63 (s, 1H), 8.48 – 8.35 (m, 2H), 7.73 (d, J= 8.4 Hz, 1H), 6.20 (d, J= 2.9 Hz, 1H), 4.35 (t, J = 7.1 Hz, 2H), 2.09 (t, J= 7.1 Hz, 2H), 1.01 – 0.82 (m, 4H).
PATENT
WO2018227049
Follows on from WO2018227049 , claiming a composition comprising this compound and at least one of tezacaftor, ivacaftor, deutivacaftor or lumacaftor, useful for treating CF.
PATENT
WO-2019079760
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019079760&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of the compound, the potassium salt of which is presumed to be VX-659 , Such as Forms A, B, C, D, E, H and M , processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating cystic fibrosis, and compositions comprising VX-659, ivacaftoR, lumacaftor and tezacaftor .
This application claims priority to U.S. Provisional Application No.
62/574,677, filed October 19, 2017; U.S. Provisional Application No. 62/574,670, filed October 19, 2017; and U.S. Provisional Application No. 62/650,057, filed March 29, 2018, the entire contents of each of which are expressly incorporated herein by reference in their respective entireties.
[0002] Disclosed herein are crystalline forms of Compound I and pharmaceutically acceptable salts thereof, which are modulators of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), compositions comprising the same, methods of using the same, and processes for making the same.
[0003] Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 70,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure.
[0004] In patients with CF, mutations in CFTR endogenously expressed in respiratory epithelia lead to reduced apical anion secretion causing an imbalance in ion and fluid transport. The resulting decrease in anion transport contributes to enhanced mucus accumulation in the lung and accompanying microbial infections that ultimately cause death in CF patients. In addition to respiratory disease, CF patients typically suffer from gastrointestinal problems and pancreatic insufficiency that, if left untreated, result in death. In addition, the majority of males with cystic fibrosis are infertile, and fertility is reduced among females with cystic fibrosis.
[0005] Sequence analysis of the CFTR gene has revealed a variety of disease-causing mutations (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61 :863 :870; and Kerem, B-S. et al. (1989) Science 245: 1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). To date, greater than 2000 mutations in the CF gene have been identified; currently, the CFTR2 database contains information on only 322 of these identified mutations, with sufficient evidence to define 281 mutations as disease causing. The most prevalent disease-causing mutation is a deletion of phenylalanine at position 508 of the CFTR amino acid sequence and is
commonly referred to as the F508del mutation. This mutation occurs in approximately 70% of the cases of cystic fibrosis and is associated with severe disease.
[0006] The deletion of residue 508 in CFTR prevents the nascent protein from folding correctly. This results in the inability of the mutant protein to exit the endoplasmic reticulum (ER) and traffic to the plasma membrane. As a result, the number of CFTR channels for anion transport present in the membrane is far less than observed in cells expressing wild-type CFTR, i.e., CFTR having no mutations. In addition to impaired trafficking, the mutation results in defective channel gating.
Together, the reduced number of channels in the membrane and the defective gating lead to reduced anion and fluid transport across epithelia. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). The channels that are defective because of the F508del mutation are still functional, albeit less functional than wild-type CFTR channels. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). In addition to F508del, other disease-causing mutations in CFTR that result in defective trafficking, synthesis, and/or channel gating could be up-or down-regulated to alter anion secretion and modify disease progression and/or severity.
[0007] CFTR is a cAMP/ATP-mediated anion channel that is expressed in a variety of cell types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. In epithelial cells, normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue. CFTR is composed of approximately 1480 amino acids that encode a protein which is made up of a tandem repeat of transmembrane domains, each containing six
transmembrane helices and a nucleotide binding domain. The two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple
phosphorylation sites that regulate channel activity and cellular trafficking.
[0008] Chloride transport takes place by the coordinated activity of ENaC and CFTR present on the apical membrane and the Na+-K+-ATPase pump and CI- channels expressed on the basolateral surface of the cell. Secondary active transport of chloride from the luminal side leads to the accumulation of intracellular chloride, which can then passively leave the cell via CI“ channels, resulting in a vectorial transport. Arrangement of Na+/2C17K+ co-transporter, Na+-K+– ATPase pump and the basolateral membrane K+ channels on the basolateral surface and CFTR on the luminal side coordinate the secretion of chloride via CFTR on the luminal side. Because water is probably never actively transported itself, its flow across epithelia depends on tiny transepithelial osmotic gradients generated by the bulk flow of sodium and chloride.
[0009] Compound I and pharmaceutically acceptable salts thereof are potent CFTR modulators. Compound I is N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl) cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide, and has the following structure:
Example 1: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl] ethoxy] pyrazol-l-yl]-2- [(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (Compound I)
Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
° THF, Base
N02 1 “* N02 | -k/ B) HC‘
Step 1: Synthesis of methyl-2,4-dimethyl-4-nitro-pentanoate
[00381] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl fert-butyl ether (MTBE) (14 L). 2 M HC1 (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible – a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HC1 (3 L). After separation, the HC1 washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. This was dried with MgS04 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield). ¾ MR (400 MHz, Chloroform-i ) δ 3.68 (s, 3H), 2.56 – 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J= 6.8 Hz, 3H).
Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00382] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00383] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was
switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CCb (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2,S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00384] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at -2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (-1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to -1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %). ¾ MR (400 MHz, Chloroform-i ) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00385] A glass lined 120 L reactor was charged with lithium aluminium hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (¾)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HC1 (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine»HCl as a white, crystalline solid (6.21 kg, 75% yield). ¾ NMR (400 MHz, DMSO-^6) δ 9.34 (br d, 2H), 3.33 (dd, J= 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, J= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J= 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J= 6.6 Hz, 3H).
Part B: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
HO‘ CF,
Synthesis of starting materials:
Synthesis of terf-Butyl 2,6-dichloropyridine-3-carboxylate
[00386] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and stirred overnight at room temperature. At this point, HCI IN (400 mL) was added, and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL), and the combined organic layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc. 247.02, found 248.1 (M+1) +; Retention time: 2.27 minutes. ¾ NMR (300 MHz, CDCh) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
Synthesis of terf-Butyl 3-oxo-2,3-dihydro-lH-pyrazole-l-carboxylate
[00387] A 50L reactor was started, and the jacket was set to 20 °C, with stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added, and the reactor was capped. The reaction was heated to an internal temperature of 40 °C, and the system was set to hold jacket temperature at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 °C for 1 h. The reaction mixture was cooled to 20 °C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion-wise, maintaining reaction
temperature <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion-wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 °C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear, light amber oil. The resulting oil was transferred to the 50L reactor, stirred and water (7.150 L) and heptane (7.150 L) were added. The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container, and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol) and added dropwise. The jacket was set to 0 °C to absorb the quench exotherm. After the addition was complete (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L) and washed a second time with water (3.575 L). The crystalline solid was transferred into a 20L rotovap bulb, and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and 1-2 volumes of solvent were distilled off. The slurry in the rotovap flask was filtered, and the solids were washed with heptane (3.575 L). The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-lH-pyrazole-2-carboxylate (2921 g, 71%) as a coarse, crystalline solid. ¾ MR
(400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J= 2.9 Hz, 1H), 5.90 (d, J
1H), 1.54 (s, 9H).
Synthesis of 2-[l-(trifluoromethyl)cyclopropyl]ethanol
[00388] To a solution of lithium aluminum hydride (293 mg, 7.732 mmol) in THF (10.00 mL) in an ice-bath, 2-[l-(trifluoromethyl)cyclopropyl]acetic acid (1.002 g, 5.948 mmol) in THF (3.0 mL) was added dropwise over a period of 30 minutes keeping the reaction temperature below 20 0 C. The mixture was allowed to gradually warm to ambient temperature and was stirred for 18 h. The mixture was cooled with an ice-bath and sequentially quenched with water (294 mg, 295 μΐ., 16.36 mmol), NaOH (297 μΐ. of 6 M, 1.784 mmol), and then water (884.0 μΐ., 49.07 mmol) to afford a granular solid in the mixture. The solid was filtered off using celite, and the precipitate was washed with ether. The filtrate was further dried with MgS04 and filtered and concentrated in vacuo to afford the product with residual THF and ether. The mixture was taken directly into the next step without further purification.
Step 1: tert-Butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate
[00389] tert-Butyl 5-oxo-lH-pyrazole-2-carboxylate (1.043 g, 5.660 mmol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (916 mg, 5.943 mmol), and triphenyl phosphine (1.637 g, 6.243 mmol) were combined in THF (10.48 mL) and the reaction was cooled in an ice-bath. Diisopropyl azodicarboxylate (1.288 g, 1.254 mL, 6.368 mmol) was added dropwise to the reaction mixture, and the reaction was allowed to warm to room temperature for 16 hours. The mixture was evaporated, and the resulting material was partitioned between ethyl acetate (30 mL) and IN sodium hydroxide (30 mL). The organic layer was separated, washed with brine (30 mL), dried over sodium sulfate, and concentrated. The crude material was purified by silica gel chromatography eluting with a gradient of ethyl acetate in hexanes (0- 30%) to give tert-butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 57%). ESI-MS m/z calc. 320.13, found 321.1 (M+1) +; Retention time: 0.72 minutes.
Step 2: 3-[2-[l-(Trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole
[00390] tert-Butyl-3 -[2-[ 1 -(trifluoromethyl)cyclopropyl]ethoxy]pyrazole- 1 -carboxylate (1.03 g, 3.216 mmol) was dissolved in dichloromethane (10.30 mL) with trifluoroacetic acid (2.478 mL, 32.16 mmol), and the reaction was stirred at room temperature for 2 hours. The reaction was evaporated, and the resulting oil was partitioned between ethyl acetate (10 mL) and a saturated sodium bicarbonate solution. The organic layer was separated, washed with brine, dried over sodium sulfate, and evaporated to give 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (612 mg, 86%). ESI-MS m/z calc. 220.08, found 221.0 (M+1) +; Retention time: 0.5 minutes. ¾ MR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 7.50 (t, J= 2.1 Hz, 1H), 5.63 (t, J= 2.3 Hz, 1H), 4.14 (t, J= 7.1 Hz, 2H), 2.01 (t, J= 7.1 Hz, 2H), 0.96 – 0.88 (m, 2H), 0.88 -0.81 (m, 2H).
Step 3: tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate
[00391] tert-Butyl 2,6-dichloropyridine-3-carboxylate (687 mg, 2.770 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (610 mg, 2.770 mmol), and freshly ground potassium carbonate (459 mg, 3.324 mmol) were combined in anhydrous DMSO (13.75 mL). l,4-diazabicyclo[2.2.2]octane (DAB CO (1,4-diazabicyclo[2.2.2]octane), 62 mg, 0.5540 mmol) was added, and the mixture was
stirred at room temperature under nitrogen for 16 hours. The reaction mixture was diluted with water (20 mL) and stirred for 15 minutes. The resulting solid was collected and washed with water. The solid was dissolved in dichloromethane and dried over magnesium sulfate. The mixture was filtered and concentrated to give tert-butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 84%). ESI-MS m/z calc. 431.12, found 432.1 (M+l) +; Retention time: 0.88 minutes.
Step 4: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00392] tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 2.339 mmol) and trifluoroacetic acid (1.8 mL, 23.39 mmol) were combined in dichloromethane (10 mL) and heated at 40 °C for 3 h. The reaction was concentrated. Hexanes were added, and the mixture was concentrated again to give 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (873 mg, 99%) ESI-MS m/z calc. 375.06, found 376.1 (M+l)+; Retention time: 0.69 minutes.
Step 5: N-(Benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide
[00393] A solution of 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]
ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (0.15 g, 0.3992 mmol) and carbonyl diimidazole (77 mg, 0.4790 mmol) in THF (2.0 mL) was stirred for one hour, and
benzenesulfonamide (81 mg, 0.5190 mmol) and DBU (72 μΐ^, 0.4790 mmol) were added. The reaction was stirred for 16 hours, acidified with 1 M aqueous citric acid, and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated. The residue was purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-5%) to give N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 78%). ESI-MS m/z calc. 514.07, found 515.1 (M+l)+; Retention time: 0.74 minutes.
Step 6: N-(Benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
[00394] A mixture of N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 0.3107 mmol), (4S)-2,2,4-trimethylpyrrolidine hydrochloride salt (139 mg, 0.9321 mmol), and potassium carbonate (258 mg, 1.864 mmol) in DMSO (1.5 mL) was stirred at 130 °C for 17 hours. The reaction mixture was acidified with 1 M aqueous citric acid and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated to yield a crude product that was purified by reverse-phase HPLC utilizing a gradient of 10-99%) acetonitrile in 5 mM aqueous HC1 to yield N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (87 mg, 47%). ESI-MS m/z calc. 591.21, found 592.3 (M+l) +; Retention time: 2.21 minutes. 1H MR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.19 (d, J= 2.8 Hz, 1H), 8.04 – 7.96 (m, 2H), 7.81 (d, J= 8.2 Hz, 1H), 7.77 – 7.70 (m, 1H), 7.70 – 7.62 (m, 2H), 6.92 (d, J= 8.2 Hz, 1H), 6.10 (d, J= 2.8 Hz, 1H), 4.31 (t, J= 7.0 Hz, 2H), 2.42 (t, J= 10.5 Hz, 1H), 2.28 (dd, J = 10.2, 7.0 Hz, 1H), 2.17 – 2.01 (m, 3H), 1.82 (dd, J= 11.9, 5.5 Hz, 1H), 1.52 (d, J = 9.4 Hz, 6H), 1.36 (t, J= 12.1 Hz, 1H), 1.01 – 0.92 (m, 2H), 0.92 – 0.85 (m, 2H), 0.65 (d, J = 6.3 Hz, 3H). pKa: 4.95±0.06.
Synthesis of sodium salt of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl) cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (sodium salt of Compound I)
[00395] N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (1000 mg, 1.679 mmol) was dissolved in ethanol (19.87 ml) under warming, filtered clear through a syringe filter (0.2 μπι), washed with warm ethanol (10 ml) and the warm solution was treated with 1M NaOH (1.679 ml, 1.679 mmol). The solution was evaporated at 30-35 °C, co-evaporated 3 times with ethanol (-20 ml), to give a solid, which was dried overnight under vacuum in a drying cabinet at 45 °C with a nitrogen bleed to give 951 mg of a cream colored solid. The solid was further dried under vacuum in a drying cabinet at 45 °C with a nitrogen bleed over the weekend. 930 mg (89%) of the sodium salt of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide was obtained as an off-white amorphous solid. ¾ NMR (400 MHz, DMSO-d) δ 8.15 (d, J= 2.7 Hz, 1H), 7.81 (dd, J= 6.7, 3.1 Hz, 2H), 7.61 (d, J= 7.9 Hz, 1H), 7.39 (dd, J= 4.9, 2.0 Hz, 3H), 6.74 (d, J= 7.9 Hz, 1H), 6.01 (d, J= 2.6 Hz, 1H), 4.29 (t, J= 7.0 Hz, 2H), 2.93 – 2.78 (m, 2H), 2.07 (t, J= 7.1 Hz, 3H), 1.78 (dd, J= 11.8, 5.6 Hz, 1H), 1.52 (d, J= 13.6 Hz, 6H), 1.33 (t, J= 12.0 Hz, 1H), 1.00 – 0.92 (m, 2H), 0.89 (q, J= 5.3, 4.6 Hz, 2H), 0.71 (d, J= 6.3 Hz, 3H). EST-MS m/z calc. 591.2127, found 592.0 (M+l)+; Retention time: 3.28 minutes. XRPD (see FIG. 16).
Alternate synthesis of 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy] pyrazol-l-yl] pyridine-3-carboxylic acid
Step 1: ethyl 3-hydroxy-lH-pyrazole-4-carboxylate
[00396] A mixture of EtOH (20.00 L, 10 vol) and diethyl 2-(ethoxymethylene) propanedioate (2000 g, 9.249 mol, 1.0 equiv) was added under nitrogen purge a to a 50 L reactor equipped with a reflux condenser (10 °C) and the jacket set to 40 °C. The mixture was stirred, and then hydrazine hydrate (538.9 g of 55 %w/w, 523.7 mL of 55 %w/w, 9.249 mol, 1.00 equiv) was added in portions via an addition funnel. Once the addition was complete, the reaction was heated to 75 °C for 22 h to afford a solution of ethyl 3-hydroxy-lH-pyrazole-4-carboxylate that was used directly in the next step.
Step 2: l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate
[00397] The solution of ethyl 3 -hydroxy- lH-pyrazole-4-carboxylate was cooled from 75 °C to 40 °C, then triethylamine (TEA) (46.80 g, 64.46 mL, 462.5 mmol, 0.05 eq.) was added. A solution of Boc anhydride (2.119 kg, 9.711 moll .05 equiv) in EtOH (2.000 L, 1 equiv) was added to the reactor over 35 min. The mixture was stirred for 4 hours to complete the reaction; then water (10.00 L, 5.0 vol) was added over 15 mins. The resulting mixture was cooled to 20 °C to complete crystallization of the product. The crystals were allowed to age for 1 hour, then the mixture was filtered. The solid was washed with a mixture of EtOH (4.000 L, 2.0 vol) and water (2.000 L, 1.0 vol). The solid was then dried in vacuo to afford l-(tert-butyl)-4-ethyl-3-hydroxy-lH-pyrazole-1,4-dicarboxylate (1530 g, 65%) as colorless, fine needle, crystalline solid. ¾ NMR (400 MHz, DMSO-de) δ 11.61 (s, 1H), 8.40 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.56 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
Step 3: l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-ΙΗ-pyr azole- 1 ,4-dicarboxylate
[00398] A 5L reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temperature and nitrogen purge. The vessel was charged with toluene (1.0L, 10.0 vol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (lOO.Og, 648.8 mmol, 1.0 equiv), and l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate (166.3 g, 648.8 mmol), and the mixture was stirred. The reaction mixture was charged with triphenyl phosphine (195.7 g, 746.1 mmol, 1.15 equiv), then the reactor was set to maintain an internal temperature of 40 °C. Diisopropyl azoldicarboxylate (150.9 g, 746.1 mmol, 1.15 equiv) was added into an addition funnel and was added to the reaction while maintaining the reaction temperature between 40 and 50 °C (addition was exothermic, exotherm addition controlled), and stirred for a total of 2.5 hours. Once the reaction was deemed complete by HPLC, heptane was added (400 mL, 4 vol), the solution was cooled to 20 °C over 60 minutes, and the bulk of triphenylphosphine oxide-DIAD complex (TPPO-DIAD) crystallized out. Once at room temp, the mixture was filtered, and the solid was washed with heptane (400 mL, 4.0 vol) and pulled dry. The filtrate was used in the next step as a solution in toluene-heptane without further purification.
Step 4: ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate
[00399] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with a toluene solution consisting of approximately 160 mmol, 65.0 g of l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate in 3 vol of toluene (prepared by concentrating a 25% portion of filtrate from previous reaction down to 4 volumes in a rotovap). The reaction was set to maintain an internal temperature at 40 °C and KOH (33.1 g, 1.5 eq. of aqueous 45 % KOH solution) was added in one portion, resulting in a mild exothermic addition, while CO2 was generated upon removal of the protecting group. The reaction proceeded for 1.5 hr, monitored by HPLC, with the product partially crystallizing during the reaction. Heptane (160 mL, 2.5 vol) was added to the reaction mixture and the reaction was cooled to room temperature over 30 minutes. The resulting mixture was filtered, and the solid was
washed with heptane (80.00 mL, 1.25 vol), pulled dry, then dried in vacuo (55 °C, vacuum). 52.3 g of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate was obtained as a crude, colorless solid that was used without further purification.
Step 5: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid
[00400] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with methanol (150.0 mL, 3.0 vol), a solution of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl) ethoxy)-lH-pyrazole-4-carboxylate (50.0 g, 171.1 mmol, 1.0 equiv), and the reaction was stirred to suspend the solids. The reactor was set to maintain internal temperature at 40 °C. To the mixture was added KOH (96 g of aqueous 45 % KOH, 1.71 mol, 10.0 equiv) in portions maintaining the internal temperature <50 °C. Once addition was complete, the reaction was set to maintain temperature at 50 °C, and the reaction proceeded for 23 hours, monitored by HPLC. Once complete the reaction was cooled to 10 °C then partially concentrated on a rotary evaporator to remove most of the MeOH. The resulting solution was diluted with water (250 mL, 5.0 vol) and 2-Me-THF (150 mL, 3.0 vol), and transferred to the reactor, stirred at room temp, then stopped, and layers were allowed to separate. The layers were tested, with remaining TPPO-DIAD complex in the organic layer and product in the aqueous layer. The aqueous layer was washed again with 2-Me-THF (100 mL, 2.0 vol), the layers separated, and the aqueous layer returned to the reactor vessel. The stirrer was started and set to 450 rpm, and the reactor jacket was set to 0 °C. The pH was adjusted to pH acidic by addition of 6M aqueous HC1 (427mL, 15 equiv) portion wise, maintaining the internal temperature between 10 and 30 °C. The product began to crystallize close to pH neutral and was accompanied with strong off-gassing, and so the acid was added slowly, and then further added to reach pH 1 once the off-gassing had ended. To the resulting suspension was added 2-Me-THF (400 mL, 8.0 vol), and the product was allowed to dissolve into
the organic layer. Stirring was stopped, the layers were separated, and the aqueous layer was returned to the reactor, stirred and re-extracted with 2-Me-THF (100 mL, 2.0 vol). The organic layers were combined in the reactor and stirred at room temperature, washed with brine (lOOmL, 2 vols), dried over Na2S04, filtered through celite, and the solid was washed with 2-Me-THF (50 mL, 1.0 vol). The filtrate was transferred to a clean rotovap flask, stirred, warmed to 50 °C and heptane (200 mL, 4.0 vol) added, and then partially concentrated with the addition of heptane (300 mL, 6.0 vol) and then seeded with 50mg of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid), and the product crystallized during solvent removal. The distillation was stopped when the bulk of the 2-Me-THF had distilled off. The bath heater was turned off, the vacuum removed, and the mixture was allowed to stir and cool to room temperature. The mixture was filtered (slow speed) and the solid was washed with heptane (100 mL, 2.0 vol), and the solid was collected and dried in vacuo (50 °C, rotovap). 22.47 g of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid was obtained as an off-white solid. ¾ MR (400 MHz, DMSO-de) δ
12.45 (s, 2H), 8.01 (s, 1H), 4.26 (t, J= 7.0 Hz, 2H), 2.05 (t, J= 7.0 Hz, 2H), 0.92 (m,
4H).
Step 6: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole
[00401] A mixture of toluene (490.0 mL), 3-(2-(l-(trifluoromethyl)cyclopropyl) ethoxy)-lH-pyrazole-4-carboxylic acid (70.0 g, 264.9 mmol), and DMSO (70.00 mL) was placed in a reactor and heated to 100 °C with stirring. DBU (approximately 20.16 g, 19.80 mL, 132.4 mmol) was added to the reactor over 15 min. The mixture was stirred for 20 h to complete the reaction and then cooled to 20 °C. The mixture was washed with water (350.0 mL), then 0.5N aq HC1 (280.0 mL), then water (2 x 140.0 mL), and lastly with brine (210.0 mL). The organic layer was dried with Na2S04, and then activated charcoal (5 g, Darco 100 mesh) was added to the stirred slurry. The dried mixture was filtered through celite, and the solid was washed with toluene (140.0 mL) and then pulled dry. The filtrate was concentrated in a rotovap (50 °C, vac) to afford 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.89 g, 53%) as an amber oil. 1H MR (400 MHz, DMSO-d) δ 11.87 (s, 1H), 7.50 (d, J= 2.4 Hz, 1H), 5.63 (d, J = 2.4 Hz, 1H), 4.23 – 4.06 (m, 2H), 2.01 (t, J= 7.1 Hz, 2H), 1.00 – 0.77 (m, 4H).
Step 7: ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy] pyrazol-l-yl]pyridine-3-carboxylate
[00402] A mixture of DMF (180.0 mL), ethyl 2,6-dichloropyridine-3-carboxylate (approximately 29.97 g, 136.2 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.0 g, 136.2 mmol), and K2CO3, (325 mesh, approximately 24.48 g, 177.1 mmol) was added to a stirred reactor at 20 °C. DABCO (approximately 2.292 g, 20.43 mmol) was then added to the reactor, and the mixture was stirred at 20 °C for 1 hour, and then the temperature was increased to 30 °C, and the mixture stirred for 24 hours to complete the reaction. The mixture was cooled to 20 °C; then water (360 mL) was added slowly. The mixture was then drained from the reactor and the solid was isolated by filtration. The solid was then washed with water (2 x 150 mL), and then the solid was dried under vacuum at 55 °C to afford ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (51.37 g, 93%) as a fine, beige colored solid. ¾ MR (400 MHz, DMSO-^e) δ 8.44 (d, J= 2.9 Hz, 1H), 8.41 (d, J= 8.5 Hz, 1H), 7.75 (d, J= 8.5 Hz, 1H), 6.21 (d, J= 2.9 Hz, 1H), 4.34 (m, 4H), 2.09 (t, J= 7.1 Hz, 2H), 1.34 (t, J= 7.1 Hz, 3H), 1.00 – 0.84 (m, 4H).
Step 8: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00403] A solution of ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (50.0 g, 123.8 mmol) in THF (300.0 mL) was prepared in a reactor at 20 °C. EtOH (150.0 mL) was added, followed by aqueous NaOH (approximately 59.44 g of 10 %w/w, 148.6 mmol). The mixture was stirred for 1 hour to complete the reaction; then aq IN HC1 (750.0 mL) was slowly added. The resulting suspension was stirred for 30 min at 10 °C, and then the solid was isolated by filtration. The solid was washed with water (150 mL then 2 x 100 mL) and then pulled dry by vacuum. The solid was then further dried under vacuum with heating to afford 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (42.29 g, 91%). ¾ NMR (400 MHz, DMSO-i¾) δ 13.63 (s, 1H), 8.48 -8.35 (m, 2H), 7.73 (d, J= 8.4 Hz, 1H), 6.20 (d, J= 2.9 Hz, 1H), 4.35 (t, J= 7.1 Hz, 2H), 2.09 (t, J= 7.1 Hz, 2H), 1.01 – 0.82 (m, 4H).
Example 2: Preparation of a Spray Dried Dispersion (SDD) of Compound I
[00404] A spray dried dispersion of Compound I (free form) was prepared using Buchi Mini Spray Dryer B290. HPMCAS-HG (6.0 grams) was dissolved in 200 mL of MeOH/DCM (1/1), and Compound I (6.0 grams) was added and stirred for 30 minutes forming a clear solution. The resulting solution was spray dried under the following conditions resulting in a 50 wt% Compound 1/50 wt% HPMCAS- HG spray dried dispersion (Yield: 80%, Solid load: 6%). FIG. 14 shows the XRPD spectrum of a SDD of 50% Compound I in HPMCAS-HG. FIG. 15 is spectrum showing modulated differential scanning calorimetry (MDSC) spectrum of a spray dried dispersion (SDD) of 50% Compound I in HPMCAS-HG.
Table 64 SDD of Compound I
Example 3: Synthesis of Compound II: (R)-l-(2,2-Difluorobenzo[d][l,3]dioxol-5- yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2- -2-yl)-lH-indol-5-yl)cyclopropanecarboxamide
Step 1: (R)-Benzyl 2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate and ((S)-2,2-Dimethyl-l,3-dioxolan-4-yl)methyl 2-(l-(((R)-2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate
[00405] Cesium carbonate (8.23 g, 25.3 mmol) was added to a mixture of benzyl 2-(6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate (3.0 g, 8.4 mmol) and (S)-(2,2-dimethyl-l,3-dioxolan-4-yl)methyl 4-methylbenzenesulfonate (7.23 g, 25.3 mmol) in DMF (N,N-dimethylformamide) (17 mL). The reaction was stirred at 80 °C for 46 hours under a nitrogen atmosphere. The mixture was then partitioned between ethyl acetate and water. The aqueous layer was extracted with ethyl acetate. The combined ethyl acetate layers were washed with brine, dried over MgS04, filtered and concentrated. The crude product, a viscous brown oil which contains both of the products shown above, was taken directly to the next step without further purification. (R)-Benzyl 2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate, ESI-MS m/z calc. 470.2, found 471.5 (M+l)+. Retention time 2.20 minutes. ((S)-2,2-Dimethyl-l,3-dioxolan-4-yl)methyl 2-(l-(((R)-2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate, ESI-MS m/z calc. 494.5, found 495.7 (M+l)+. Retention time 2.01 minutes.
Step 2: (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol
[00406] The crude reaction mixture obtained in step (A) was dissolved in THF (tetrahydrofuran) (42 mL) and cooled in an ice-water bath. LiAlH4 (16.8 mL of 1 M solution, 16.8 mmol) was added drop-wise. After the addition was complete, the
mixture was stirred for an additional 5 minutes. The reaction was quenched by adding water (1 mL), 15% NaOH solution (1 mL) and then water (3 mL). The mixture was filtered over Celite, and the solids were washed with THF and ethyl acetate. The filtrate was concentrated and purified by column chromatography (30-60% ethyl acetate-hexanes) to obtain (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol as a brown oil (2.68g, 87 % over 2 steps). ESI-MS m/z calc. 366.4, found 367.3 (M+l)+. Retention time 1.68 minutes. 1H MR (400 MHz, DMSO-^6) δ 8.34 (d, J = 7.6 Hz, 1H), 7.65 (d, J = 13.4 Hz, 1H), 6.57 (s, 1H), 4.94 (t, J = 5.4 Hz, 1H), 4.64 – 4.60 (m, 1H), 4.52 – 4.42(m, 2H), 4.16 – 4.14 (m, 1H), 3.76 – 3.74 (m, 1H), 3.63 – 3.53 (m, 2H), 1.42 (s, 3H), 1.38 – 1.36 (m, 6H) and 1.19 (s, 3H) ppm. (DMSO is dimethylsulfoxide).
Step 3: (R)-2-(5-amino-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-lH-indol-2-yl)-2-methylpropan-l-ol
[00407] (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol (2.5 g, 6.82 mmol) was dissolved in ethanol (70 mL) and the reaction was flushed with N2. Then Pd-C (250 mg, 5% wt) was added. The reaction was flushed with nitrogen again and then stirred under H2 (atm). After 2.5 hours only partial conversion to the product was observed by LCMS. The reaction was filtered through Celite and concentrated. The residue was re-subjected to the conditions above. After 2 hours LCMS indicated complete conversion to product. The reaction mixture was filtered through Celite. The filtrate was concentrated to yield the product (1.82 g, 79 %). ESI-MS m/z calc. 336.2, found 337.5 (M+l)+. Retention time 0.86 minutes. ¾ NMR (400 MHz, DMSO-^6) δ 7.17 (d, J = 12.6 Hz, 1H), 6.76 (d, J = 9.0 Hz, 1H), 6.03 (s, 1H), 4.79 – 4.76 (m, 1H), 4.46 (s, 2H), 4.37 – 4.31 (m, 3H),4.06 (dd, J = 6.1, 8.3 Hz, 1H), 3.70 – 3.67 (m, 1H), 3.55 – 3.52 (m, 2H), 1.41 (s, 3H), 1.32 (s, 6H) and 1.21 (s, 3H) ppm.
Step 4: (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide
[00408] DMF (3 drops) was added to a stirring mixture of l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarboxylic acid (1.87 g, 7.7 mmol) and thionyl chloride (1.30 mL, 17.9 mmol). After 1 hour a clear solution had formed. The
solution was concentrated under vacuum and then toluene (3 mL) was added and the mixture was concentrated again. The toluene step was repeated once more and the residue was placed on high vacuum for 10 minutes. The acid chloride was then dissolved in dichloromethane (10 mL) and added to a mixture of (R)-2-(5 -amino- 1-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-lH-indol-2-yl)-2-methylpropan-l-ol (1.8 g, 5.4 mmol) and triethylamine (2.24 mL, 16.1 mmol) in dichloromethane (45 mL). The reaction was stirred at room temperature for 1 hour. The reaction was washed with IN HC1 solution, saturated NaHCCb solution and brine, dried over MgSCb and concentrated to yield the product (3g, 100%). ESI-MS m/z calc. 560.6, found 561.7 (M+l)+. Retention time 2.05 minutes. ¾ NMR (400 MHz, DMSO-^6) δ 8.31 (s, 1H), 7.53 (s, 1H), 7.42 – 7.40 (m, 2H), 7.34 – 7.30 (m, 3H), 6.24 (s, 1H), 4.51 – 4.48 (m, 1H), 4.39 – 4.34 (m,2H), 4.08 (dd, J = 6.0, 8.3 Hz, 1H), 3.69 (t, J = 7.6 Hz, 1H), 3.58 – 3.51 (m, 2H), 1.48 – 1.45 (m, 2H), 1.39 (s, 3H), 1.34 – 1.33 (m, 6H), 1.18 (s, 3H) and 1.14 -1.12 (m, 2H) ppm
Step 5: (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide
[00409] (R)-l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-N-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-2-(l -hydroxy -2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide (3.0 g, 5.4 mmol) was dissolved in methanol (52 mL). Water (5.2 mL) was added followed by p-TsOH.H20 (p-toluenesulfonic acid hydrate) (204 mg, 1.1 mmol). The reaction was heated at 80 °C for 45 minutes. The solution was concentrated and then partitioned between ethyl acetate and saturated NaHCCb solution. The ethyl acetate layer was dried over MgS04 and concentrated. The residue was purified by column chromatography (50-100 % ethyl acetate – hexanes) to yield the product. (1.3 g, 47 %, ee >98% by SFC). ESI-MS m/z calc. 520.5, found 521.7 (M+l)+. Retention time 1.69 minutes. ¾ NMR (400 MHz, DMSC 6) δ 8.31 (s, 1H), 7.53 (s, 1H), 7.42 – 7.38 (m, 2H), 7.33 – 7.30 (m, 2H), 6.22 (s, 1H), 5.01 (d, J = 5.2 Hz, 1H), 4.90 (t, J = 5.5 Hz, 1H), 4.75 (t, J = 5.8 Hz, 1H), 4.40 (dd, J = 2.6, 15.1 Hz, 1H), 4.10 (dd, J = 8.7, 15.1 Hz, 1H), 3.90 (s, 1H), 3.65 – 3.54 (m, 2H), 3.48 – 3.33 (m, 2H), 1.48 -1.45 (m, 2H), 1.35 (s, 3H), 1.32 (s, 3H) and 1.14 – 1.11 (m, 2H) ppm.
Example 4: Synthesis of Compound III: N-(2,4-di-terf-butyl-5-hydroxyphi oxo-l,4-dihydroquinoline-3-carboxamide
Part A: Synthesis of 4-oxo-l,4-dihydroquinoline-3-carboxylic acid
Step 1: 2-Phenylaminomethylene-malonic acid diethyl ester
[00410] A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 °C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. ¾ MR (OMSO-de) δ 1 1.00 (d, 1H), 8.54 (d, J = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
Step 2: 4-Hydroxyquinoline-3-carboxylic acid ethyl ester
[00411] A I L three-necked flask fitted with a mechanical stirrer was charged with 2-phenylaminomethylene-malonic acid diethyl ester (26.3 g, 0.100 mol), polyphosphoric acid (270 g) and phosphoryl chloride (750 g). The mixture was heated to 70 °C and stirred for 4 h. The mixture was cooled to room temperature and filtered. The residue was treated with aqueous Na2CCb solution, filtered, washed with water and dried. 4-Hydroxyquinoline-3-carboxylic acid ethyl ester was obtained as a pale brown solid (15.2 g, 70%). The crude product was used in next step without further purification.
Step 3: 4-Oxo-l,4-dihydroquinoline-3-carboxylic acid
[00412] 4-Hydroxyquinoline-3-carboxylic acid ethyl ester (15 g, 69 mmol) was suspended in sodium hydroxide solution (2N, 150 mL) and stirred for 2 h at reflux. After cooling, the mixture was filtered, and the filtrate was acidified to pH 4 with 2N HCl. The resulting precipitate was collected via filtration, washed with water and dried under vacuum to give 4-oxo-l,4-dihydroquinoline-3-carboxylic acid as a pale white solid (10.5 g, 92 %). ¾ MR (DMSO-^e) δ 15.34 (s, 1 H), 13.42 (s, 1 H), 8.89 (s, 8.28 (d, J = 8.0 Hz, 1H), 7.88 (m, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.60 (m, 1H).
Part B: Synthesis of N-(2,4-di-terf-butyl-5-hydroxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide
Step 1: Carbonic acid 2,4-di-ferf-butyl-phenyl ester methyl ester
[00413] Methyl chloroformate (58 mL, 750 mmol) was added dropwise to a solution of 2,4-di-fert-butyl-phenol (103.2 g, 500 mmol), Et3N (139 mL, 1000 mmol) and DMAP (3.05 g, 25 mmol) in dichloromethane (400 mL) cooled in an ice-water bath to 0 °C. The mixture was allowed to warm to room temperature while stirring overnight, then filtered through silica gel (approx. 1L) using 10% ethyl acetate – hexanes (~ 4 L) as the eluent. The combined filtrates were concentrated to yield carbonic acid 2,4-di-tert-butyl-phenyl ester methyl ester as a yellow oil (132 g, quant.). ¾ MR (400 MHz, DMSO-i¾) δ 7.35 (d, J = 2.4 Hz, 1H), 7.29 (dd, J = 8.5, 2.4 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H), 1.30 (s, 9H), 1.29 (s, 9H).
Step 2: Carbonic acid 2,4-di-ferf-butyl-5-nitro-phenyl ester methyl ester and Carbonic acid 2,4-di-terf-butyl-6-nitro-phenyl ester methyl ester
[00414] To a stirring mixture of carbonic acid 2,4-di-tert-butyl-phenyl ester methyl ester (4.76 g, 180 mmol) in cone, sulfuric acid (2 mL), cooled in an ice-water bath, was added a cooled mixture of sulfuric acid (2 mL) and nitric acid (2 mL). The addition was done slowly so that the reaction temperature did not exceed 50 °C. The reaction was allowed to stir for 2 h while warming to room temperature. The reaction mixture was then added to ice-water and extracted into diethyl ether. The ether layer was dried (MgS04), concentrated and purified by column chromatography (0 – 10% ethyl acetate – hexanes) to yield a mixture of carbonic acid 2,4-di-tert-butyl-5-nitro-phenyl ester methyl ester and carbonic acid 2,4-di-tert-butyl-6-nitro-phenyl ester methyl ester as a pale yellow solid (4.28 g), which was used directly in the next step.
Step 3: 2,4-Di-terf-butyl-5-nitro-phenol and 2,4-Di-terf-butyl-6-nitro-phenol
[00415] The mixture of carbonic acid 2,4-di-tert-butyl-5-nitro-phenyl ester methyl ester and carbonic acid 2,4-di-tert-butyl-6-nitro-phenyl ester methyl ester (4.2 g, 14.0 mmol) was dissolved in MeOH (65 mL) before KOH (2.0 g, 36 mmol) was added. The mixture was stirred at room temperature for 2 h. The reaction mixture was then made acidic (pH 2-3) by adding cone. HC1 and partitioned between water and diethyl ether. The ether layer was dried (MgS04), concentrated and purified by column
chromatography (0 – 5 % ethyl acetate – hexanes) to provide 2,4-di-tert-butyl-5-nitro-phenol (1.31 g, 29% over 2 steps) and 2,4-di-tert-butyl-6-nitro-phenol. 2,4-Oi-tert-butyl-5-nitro-phenol: ¾ MR (400 MHz, DMSO-i¾) δ 10.14 (s, 1H, OH), 7.34 (s, 1H), 6.83 (s, 1H), 1.36 (s, 9H), 1.30 (s, 9H). 2,4-Di-tert-butyl-6-nitro-phenol: ¾ MR (400 MHz, CDCh) δ 11.48 (s, 1H), 7.98 (d, J = 2.5 Hz, 1H), 7.66 (d, J = 2.4 Hz, 1H), 1.47 (s, 9H), 1.34 (s, 9H).
Step 4: 5-Amino-2,4-di-terf-butyl-phenol
[00416] To a refluxing solution of 2,4-di-tert-butyl-5-nitro-phenol (1.86 g, 7.40 mmol) and ammonium formate (1.86 g) in ethanol (75 mL) was added Pd-5% wt. on activated carbon (900 mg). The reaction mixture was stirred at reflux for 2 h, cooled to room temperature and filtered through Celite. The Celite was washed with methanol and the combined filtrates were concentrated to yield 5-amino-2,4-di-tert-butyl-phenol as a grey solid (1.66 g, quant.). ¾ MR (400 MHz, DMSO-^e) δ 8.64 (s, 1H, OH), 6.84 (s, 1H), 6.08 (s, 1H), 4.39 (s, 2H, H2), 1.27 (m, 18H); HPLC ret. time 2.72 min, 10-99 % CftCN, 5 min run; ESI-MS 222.4 m/z [M+H]+.
Step 5: N-(5-hydroxy-2,4-di-ieri-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide
[00417] To a suspension of 4-oxo-l,4-dihydroquinolin-3-carboxylic acid (35.5 g, 188 mmol) and HBTU (85.7 g, 226 mmol) in DMF (280 mL) was added Et3N (63.0 mL, 451 mmol) at ambient temperature. The mixture became homogeneous and was allowed to stir for 10 min before 5-amino-2,4-di-tert-butyl-phenol (50.0 g, 226 mmol) was added in small portions. The mixture was allowed to stir overnight at ambient temperature. The mixture became heterogeneous over the course of the reaction. After all of the acid was consumed (LC-MS analysis, MH+ 190, 1.71 min), the solvent was removed in vacuo. EtOH (ethyl alcohol) was added to the orange solid material to produce a slurry. The mixture was stirred on a rotovap (bath temperature 65 °C) for 15 min without placing the system under vacuum. The mixture was filtered and the captured solid was washed with hexanes to provide a white solid that was the EtOH crystalate. Et20
(diethyl ether) was added to the solid obtained above until a slurry was formed. The mixture was stirred on a rotovapor (bath temperature 25 °C) for 15 min without placing the system under vacuum. The mixture was filtered and the solid captured. This procedure was performed a total of five times. The solid obtained after the fifth precipitation was placed under vacuum overnight to provide N-(5-hydroxy-2,4-di-tert-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide (38 g, 52%). HPLC ret. time 3.45 min, 10-99% CftCN, 5 min run; 1H MR (400 MHz, DMSO-i¾) δ 12.88 (s, 1H), 11.83 (s, 1H), 9.20 (s, 1H), 8.87 (s, 1H), 8.33 (dd, J = 8.2, 1.0 Hz, 1H), 7.83-7.79 (m, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.54-7.50 (m, 1H), 7.17 (s, 1H), 7.10 (s, 1H), 1.38 (s, 9H), 1.37 (s, 9H); ESI-MS m/z calc’d 392.21; found 393.3 [M+H]+.
PAPER
The New England journal of medicine (2018), 379(17), 1599-1611
https://www.nejm.org/doi/10.1056/NEJMoa1807119
////////////VX-659, VX 659, VX659, PHASE 2, CYSTIC FIBRIOSIS , VERTEX, Bamocaftor potassium
[K+].C[C@@H]1CN(c2nc(ccc2C(=O)[N-]S(=O)(=O)c3ccccc3)n4ccc(OCCC5(CC5)C(F)(F)F)n4)C(C)(C)C1
C[C@@H]1CN(c2nc(ccc2C(=O)NS(=O)(=O)c3ccccc3)n4ccc(OCCC5(CC5)C(F)(F)F)n4)C(C)(C)C1
TEZACAFTOR, VX 661 for treatment of cystic fibrosis disease.

TEZACAFTOR, VX 661
CAS : 1152311-62-0;
- Molecular FormulaC26H27F3N2O6
- Average mass520.498 Da
l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide).
(R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl]-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)cyclopropanecarboxamide
Vertex (INNOVATOR)
UNII: 8RW88Y506K
![]()
In July 2016, this combination was reported to be in phase 3 clinical development.
Update
Symdeko (tezacaftor/ivacaftor) ; Vertex; For the treatment of cystic fibrosis , Approved February 2018
Urology
Tezacaftor, also known asVX-661, is CFTR modulator. VX-661 is potentially useful for treatment of cystic fibrosis disease. Cystic fibrosis (CF) is a genetic disease caused by defects in the CF transmembrane regulator (CFTR) gene, which encodes an epithelial chloride channel. The most common mutation, Δ508CFTR, produces a protein that is misfolded and does not reach the cell membrane. VX-661 can correct trafficking of Δ508CFTR and partially restore chloride channel activity. VX-661 is currently under Phase III clinical trial.
VX-661 is an orally available deltaF508-CFTR corrector in phase III clinical trials at Vertex for the treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
Novel deuterated analogs of a cyclopropanecarboxamide ie tezacaftor (VX-661), as modulators of cystic fibrosis transmembrane conductance regulator (CFTR) proteins, useful for treating a CFTR-mediated disorder eg cystic fibrosis.
VX-661 (CAS #: 1152311-62-0; l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide). VX-661 is a cystic fibrosis transmembrane conductance regulator modulator. VX-661 is currently under investigation for the treatment of cystic fibrosis. VX-661 has also shown promise in treating sarcoglycanopathies, Brody’s disease, cathecolaminergic polymorphic ventricular tachycardia, limb girdle muscular dystrophy, asthma, smoke induced chronic obstructive pulmonary disorder, chronic bronchitis, rhinosinusitis, constipation, pancreatitis, pancreatic insufficiency, male infertility caused by congenital bilateral absence of the vas deferens (CBAVD), mild pulmonary disease, idiopathic pancreatitis, allergic bronchopulmonary aspergillosis (ABPA), liver disease, hereditary emphysema, hereditary hemochromatosis, coagulation-fibrinolysis deficiencies, such as protein C deficiency, type 1 hereditary angioedema, lipid processing deficiencies, such as familial hypercholesterolemia, type 1 chylomicronemia, abetalipoproteinemia, lysosomal storage diseases, such as I-cell disease/pseudo-Hurler, mucopolysaccharidoses, Sandhof/Tay-Sachs, Crigler-Najjar type II, polyendocrinopathy/hyperinsulinemia, diabetes mellitus, Laron dwarfism, myeloperoxidase deficiency, primary hypoparathyroidism, melanoma, glycanosis CDG type 1, congenital hyperthyroidism, osteogenesis imperfecta, hereditary hypofibrinogenemia, ACT deficiency, diabetes insipidus (DI), neurohypophyseal DI, nephrogenic DI, Charcot-Marie tooth syndrome, Pelizaeus-Merzbacher disease, neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, progressive supranuclear palsy, Pick’s disease, polyglutamine neurological disorders such as Huntington’s, spinocerebellar ataxia type I, spinal and bulbar muscular atrophy, dentatombral pallidoluysian, and myotonic dystrophy, as well as spongifiorm encephalopathies, such as hereditary Creutzfeldt- Jakob disease (due to prion protein processing defect), Fabry disease, Gerstrnarm-Straussler-Scheinker syndrome, chronic obstructive pulmonary disorder, dry-eye disease, or Sjogren’s disease, osteoporosis, osteopenia, bone healing and bone growth (including bone repair, bone regeneration, reducing bone resorption and increasing bone deposition), Gorham’s Syndrome, chloride channelopathies such as myotonia congenita (Thomson and Becker forms), Bartter’s
syndrome type III, Dent’s disease, hyperekplexia, epilepsy, lysosomal storage disease, Angelman syndrome, and primary ciliary dyskinesia (PCD), a term for inherited disorders of the structure and/or function of cilia, including PCD with situs inversus (also known as Kartagener syndrome), PCD without situs inversus, and ciliary aplasia. WO 2014086687; WO2013185112.

VX-661

VX-661 is likely subject to extensive CYP45o-mediated oxidative metabolism. These, as well as other metabolic transformations, occur in part through polymorphically-expressed enzymes, exacerbating interpatient variability. Additionally, some metabolites of VX-661 may have undesirable side effects. In order to overcome its short half-life, the drug likely must be taken several times per day, which increases the probability of patient incompliance and discontinuance.Deuterium Kinetic Isotope Effect
PATENT
Scheme I


EXAMPLE 1
(R)-l-(2,2-difluorobenzo[dl[l,31dioxol-5-vn-N-(l-q,3-dihvdroxypropyn-6-fluoro-2-(l- hvdroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cvclopropanecarboxamide
(VX-661)

![]()
Methyl 2.2-difluorobenzo[dl [1.31dioxole-5-carboxylate: To a 200 mL pressure tank reactor (10 atm. in CO), was placed 5-bromo-2,2-difluoro-2H-l,3-benzodioxole (20.0 g, 84.4 mmol, 1.00 equiv), methanol (40 mL), triethylamine (42.6 g, 5.00 equiv.), Pd2(dba)3 (1.74 g, 1.69 mmol, 0.02 equiv), Pd(dppf)Cl2 (1.4 g, 1.69 mmol, 0.02 equiv.). The resulting solution was stirred at 85 °C under an atmosphere of CO overnight and the reaction progress was monitored by GCMS. The reaction mixture was cooled. The solids were filtered out. The organic phase was concentrated under vacuum to afford 17.5 g of methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate as a crude solid, which was used directly in the next step. Step 2
![]()
2 step 2 3
(2.2-difluorobenzo[dl [ 1.31 dioxol-5 -vDmethanol : To a 500mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen were placed methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate (17.5 g, 81.01 mmol, 1.00 equiv.), tetrahydrofuran (200 mL). This was followed by the addition of L1AIH4 (6.81 mg, 162.02 mmol, 2.00 equiv.) at 0 °C. The resulting solution was stirred for 1 h at 25 °C and monitored by GCMS. The reaction mixture was cooled to 0 °C until GCMS indicated the completion of the reaction. The pH value of the solution was adjusted to 8 with sodium hydroxide (1 mol/L). The solids were filtered out. The organic layer combined and concentrated under vacuum to afford 13.25 g (87%) of (2,2-difluoro-2H-l,3-benzodioxol-5-yl)methanol as yellow oil.
Step 3
![]()
step 3
5-(chloromethyl)-2.2-difluorobenzo[diri.31dioxole: (2.2-difluoro-2H-1.3-benzodioxol-5-yl)methanol (13.25 g, 70.4 mmol, 1.00 equiv.) was dissolved in DCM (200 mL). Thionyl chloride (10.02 g, 1.20 equiv.) was added to this solution. The resulting mixture was stirred at room temperature for 4 hours and then concentrated under vacuum. The residue was then diluted with DCM (500 mL) and washed with 2 x 200 mL of sodium bicarbonate and 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate, filtered and evaporated to afford 12.36 g (85%) of 5-(chloromethyl)-2,2-difluoro-2H-l ,3-benzodioxole as yellow oil.
Step 4
![]()
step 4 5
[00160] 2-(2.2-difluorobenzordi ri .31dioxol-5-yl)acetonitrile: 5-(chloromethyl)-2,2-difluoro-2H-l,3-benzodioxole (12.36 g, 60 mmol, 1.00 equiv.) was dissolved in DMSO (120 mL). This was followed by the addition of NaCN (4.41 g, 1.50 equiv.) with the inert temperature below 40 °C. The resulting solution was stirred for 2 hours at room temperature. The reaction progress was monitored by GCMS. The reaction was then quenched by the addition of 300 mL of water/ice. The resulting solution was extracted with 3 x 100 mL of ethyl acetate. The organic layers combined and washed with 3 x 100 mL brine dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.84 g (92%) of 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile as brown oil.
Step 5

l -(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l -carbonitrile: To a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, were placed 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile (10.84 g, 55 mmol, 1.00 equiv.),
NaOH (50%) in water), 1 -bromo-2-chloroethane (11.92g, 82.5 mmol, 1.50 equiv.), BmNBr
(361 mg, 1.1 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 °C. The reaction progress was monitored by GCMS. The reaction mixture was cooled. The resulting solution was extracted with 3 x 200 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.12g of 1 -(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile as brown oil.
Step 6

[00162] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 250-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile (10.12 g, 45.38 mmol, 1.00 equiv), 6 N NaOH (61 mL) and EtOH (60 mL). The resulting solution was stirred for 3 h at 100 °C. The reaction mixture was cooled and the pH value of the solution was adjusted to 2 with hydrogen chloride (1 mol/L) until LCMS indicated the completion of the reaction. The solids were collected by filtration to afford 9.68 g (88%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid as a light yellow solid.
Step 7

[00163] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carbonyl chloride; To a solution of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid (687 mg, 2.84 mmol, 1.00 equiv.) in toluene (5 mL) was added thionyl chloride (1.67 g, 5.00 equiv.). The resulting solution was stirred for 3h at 65 °C. The reaction mixture was cooled and concentrated under vacuum to afford 738 mg (99%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride as a yellow solid.
Step 8
![]()
9 STEP 8 10
2-methyl-4-(trimethylsilyl)but-3-vn-2-ol: To a solution of ethynyltrimethylsilane (20 g, 203.63 mmol, 1.00 equiv) in THF (100 mL) was added n-BuLi (81 mL, 2.5M in THF)
dropwise with stirring at -78 °C. Then the resulting mixture was warmed to 0 °C for 1 h with stirring and then cooled to -78 °C. Propan-2-one (11.6 g, 199.73 mmol, 1.00 equiv.) was added dropwise with the inert temperature below -78 °C. The resulting solution was stirred at -78 °C for 3 h. The reaction was then quenched by the addition of 100 mL of water and extracted with 3 x 100 mL of MTBE. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 28 g (90%) of 2-methyl-4-(trimethylsilyl)but-3-yn-2-ol as an off-white solid. ¾ NMR (400 MHz, CDCh) δ: 1.50 (s, 6H), 1.16-1.14 (m, 9H).
Step 9
![]()
step 9
10
(3-chloro-3-methylbut-l-vnvntrimethylsilane: To a lOOmL round-bottom flask, was placed 2-methyl-4-(trimethylsilyl) but-3-yn-2-ol (14 g, 89.57 mmol, 1.00 equiv.), cone. HC1 (60 mL, 6.00 equiv.). The resulting solution was stirred for 16 h at 0 °C. The resulting solution was extracted with 3 x 100 mL of hexane. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 8 g (51%) of (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 1.84 (s, 6H), 1.18-1.16 (m, 9H).
Step 10
![]()
step 10
11 12
(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)trimethylsilane: Magnesium turnings (1.32 g, 1.20 equiv) were charged to a 250-mL 3-necked round-bottom flask and then suspended in THF (50 mL). The resulting mixture was cooled to 0 °C and maintained with an inert atmosphere of nitrogen. (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane (8 g, 45.78 mmol, 1.00 equiv.) was dissolved in THF (50 mL) and then added dropwise to this mixture with the inert temperature between 33-37 °C. The resulting solution was stirred at room temperature for an addition 1 h before BnOCH2Cl (6.45 g, 41.33 mmol, 0.90 equiv.) was added dropwise with the temperature below 10 °C. Then the resulting solution was stirred for 16 h at room temperature. The reaction was then quenched by the addition of 50 mL of water and extracted with 3 x 100 mL of hexane. The combined organic layers was dried over
anhydrous sodium sulfate and concentrated under vacuum to afford 10 g (84%) of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 7.37-7.35 (m, 5H), 4.62 (s, 2H), 3.34 (s, 2H), 1.24 (s, 6H), 0.17-0.14 (m, 9H).
Step 11
![]()
((2.2-dimethylbut-3-vnyloxy)methyl)benzene: To a solution of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane (10 g, 38.40 mmol, 1.00 equiv) in methanol (100 mL) was added potassium hydroxide (2.53 g, 38.33 mmol, 1.30 equiv). The resulting solution was stirred for 16 h at room temperature. The resulting solution was diluted with 200 mL of water and extracted with 3 x 100 mL of hexane. The organic layers combined and washed with 1 x 100 mL of water and then dried over anhydrous sodium sulfate and concentrated under vacuum to afford 5 g (69%) of [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene as light yellow oil. ¾ NMR (300 MHz, D20) δ: 7.41-7.28 (m, 5H) , 4.62 (s, 2H), 3.34 (s, 2H), 2.14 (s, 1H), 1.32-1.23 (m, 9H).
Step 12
![]()
14 15
methyl 2.2-difluorobenzo[d1[1.31dioxole-5-carboxylate: To a solution of 3-fluoro-4-nitroaniline (6.5 g, 41.64 mmol, 1.00 equiv) in chloroform (25 mL) and AcOH (80 mL) was added Bn (6.58 g, 41.17 mmol, 1.00 equiv.) dropwise with stirring at 0 °C in 20 min. The resulting solution was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 150 mL of water/ice. The pH value of the solution was adjusted to 9 with sodium hydroxide (10 %). The resulting solution was extracted with 3 x 50 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was re-crystallized from PE/EA (10: 1) to afford 6 g (61%) of 2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 13

(R)-l-(benzyloxy)-3-(2-bromo-5-fluoro-4-nitrophenylamino)propan-2-ol: 2-bromo-5-fluoro-4-nitroaniline (6.00 g, 25.56 mmol, 1.00 equiv.), Zn(C104)2 (1.90 g, 5.1 mmol, 0.20 equiv.), 4A Molecular Sieves (3 g), toluene (60 mL) was stirred at room temperature for 2 h and maintain with an inert atmosphere of N2 until (2R)-2-[(benzyloxy)methyl]oxirane (1.37 g, 8.34 mmol, 2.00 equiv.) was added. Then the resulting mixture was stirred for 15 h at 85 °C. The reaction progress was monitored by LCMS. The solids were filtered out and the resulting solution was diluted with 20 mL of ethyl acetate. The resulting mixture was washed with 2 x 20 mL of Sat. NH4CI and 1 x 20 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :5) to afford 7.5 g (70%) of N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 14

[00170] (R)-l-(4-amino-2-bromo-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 250-mL round-bottom flask, was placed N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline (7.5 g, 18.84 mmol, 1.00 equiv.), ethanol (80 mL), water (16 mL), NH4CI (10 g, 189 mmol, 10.00 equiv.), Zn (6.11 g, 18.84 mmol, 5.00 equiv.). The resulting solution was stirred for 4 h at 85 °C. The solids were filtered out and the resulting solution was concentrated under vacuum and diluted with 200 mL of ethyl acetate. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :3) to afford 4.16 g (60%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine as light yellow oil.
Step 15
TsO

(R)-4-(3-(benzyloxy)-2-hvdroxypropylamino)-5-bromo-2-fluorobenzenaminium 4-methylbenzenesulfonate: l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine (2 g, 5.42 mmol, 1.00 equiv.) was dissolved in dichloromethane (40 mL) followed by the addition of TsOH (1 g, 5.81 mmol, 1.10 equiv.). The resulting mixture was stirred for 16 h at room temperature and then concentrated under vacuum to afford 2.8 g (95%) of 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate as an off-white solid.
Step 16

(R)-l-(4-amino-2-(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 100-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate (2.9 g, 5.36 mmol, 1.00 equiv.), [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene (1.2 g, 6.37 mmol, 1.20 equiv.), Pd(OAc)2 (48 mg, 0.21 mmol, 0.04 equiv.), dppb (138 mg, 0.32 mmol, 0.06 equiv.), potassium carbonate (2.2 g, 15.92 mmol, 3.00 equiv.) and MeCN (50 mL). The resulting solution was stirred for 16 h at 80 °C. The solids were filtered out and the resulting mixture was concentrated under vacuum until LCMS indicated the completion of the reaction. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :4) to afford 2.2 g (86%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l ,4-diamine as a light brown solid.
Step 17

l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 40-mL vial purged and maintained with an inert atmosphere of nitrogen, was placed 1-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l,4-diamine (1 g, 2.1 mmol, 1.00 equiv.), MeCN (10 mL), Pd(MeCN)2Cl2 (82 mg, 0.32 mmol, 0.15 equiv.). The resulting solution was stirred for 12 h at 85 °C. The reaction progress was monitored by LCMS. The resulting mixture was concentrated under vacuum to afford 900 mg (crude) of (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol as a brown solid, which was used for next step without further purification.
Step 18

(R)-N-(l-(3-(benzyloxy)-2-hvdroxypropyl)-2-(l-(benzyloxy)-2-methylpropan-2-yl)-6-fluoro- lH-indol-5-yl)- 1 -(2.2-difluorobenzo[dl [ 1.31 dioxol-5-vDcvclopropanecarboxamide: To a 40 mL vial purged and maintained with an inert atmosphere of nitrogen, was placed (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol (800 mg, 1.68 mmol, 1.00 equiv.), dichloromethane (20 mL), TEA (508 mg, 5.04 mmol, 3.00 equiv.). l-(2,2-difiuoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride (524 mg, 2 mmol, 1.20 equiv.) was added to this mixture at 0 °C. The resulting solution was stirred for 2 h at 25 °C. The reaction progress was monitored by LCMS. The resulting solution was diluted with 20 mL of DCM and washed with 3 xlO mL of brine. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1:5) to afford 400 mg (30%) of N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide as a light yellow solid.
Step 19

(R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide: To a 100-mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of H2, were placed N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide (400 mg, 0.77 mmol, 1.00 equiv.) dry Pd/C (300 mg) and MeOH (5 Ml, 6M HC1). The resulting mixture was stirred at room temperature for 2 h until LCMS indicated the completion of the reaction. The solids were filtered out and the resulting mixture was concentrated under vacuum. The residue was purified by prep-HPLC with the following conditions: Column, XBridge Prep C18 OBD Column 19 x 150 mm, 5um; mobile phase and Gradient, Phase A: Waters (0.1%FA ), Phase B: ACN; Detector, UV 254 nm to afford 126.1 mg (42.4%) of (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide as a light yellow solid.
¾ NMR (400 MHz, OMSO-de) δ: 8.32 (s, 1H), 7.54 (s, 1H), 7.41-7.38 (m, 2H), 7.34-7.31 (m, 2H), 6.22 (s, 1H), 5.03-5.02 (m, 1H), 4.93-4.90 (m, 1H), 4.77-4.75 (m, 1H), 4.42-4.39 (m, 1H), 4.14-4.08 (m, 1H), 3.91 (brs, 1H) , 3.64-3.57 (m, 2H), 3.47-3.40 (m, 2H), 1.48-1.46 (m, 2H), 1.36-1.32 (m, 6H), 1.14-1.12 (m, 2H).
LCMS: m/z = 521.2[M+H]+.
PATENT
WO 2015160787
https://www.google.com/patents/WO2015160787A1?cl=en
PATENT
WO 2014014841
https://www.google.com/patents/WO2014014841A1?cl=en
All tautomeric forms of the Compound 1 are included herein. For example, Compound 1 may exist as tautomers, both of which are included herein:

Methods of Preparing Compound 1 Amorphous Form and Compound 1 Form A
Compound 1 is the starting point and in one embodiment can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

Toluene, H20, 70 °C

Bu4NBr
1. NaOH
2. HC1
Scheme 2. Synthesis of acid chloride moiety – alternative synthesis.

1. NaCN
2. H20

SOC1,

Scheme 3. Synthesis of the amine moiety.



Scheme 4. Formation of Compound 1.

Compound 1
Methods of Preparing Compound 1 Amorphous Form
Starting from Compound 1 , or even a crystalline form of Compound 1 , Compound 1 Amorphous Form may be prepared by rotary evaporation or by spray dry methods.
Dissolving Compound 1 in an appropriate solvent like methanol and rotary evaporating the methanol to leave a foam produces Compound 1 Amorphous Form. In some embodiments, a warm water bath is used to expedite the evaporation.
Compound 1 Amorphous Form may also be prepared from Compound 1 using spray dry methods. Spray drying is a process that converts a liquid feed to a dried particulate form. Optionally, a secondary drying process such as fluidized bed drying or vacuum drying, may be used to reduce residual solvents to pharmaceutically acceptable levels. Typically, spray drying involves contacting a highly dispersed liquid suspension or solution, and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. The preparation to be spray dried can be any solution, coarse suspension, slurry, colloidal dispersion, or paste that may be atomized using the selected spray drying apparatus. In a standard procedure, the preparation is sprayed into a current of warm filtered air that evaporates the solvent and conveys the dried product to a collector (e.g. a cyclone). The spent air is then exhausted with the solvent, or alternatively the spent air is sent to a condenser to capture and potentially recycle the solvent. Commercially available types of apparatus may be used to conduct the spray drying. For example, commercial spray dryers are manufactured by Buchi Ltd. And Niro (e.g., the PSD line of spray driers manufactured by Niro) (see, US 2004/0105820; US 2003/0144257).
Spray drying typically employs solid loads of material from about 3% to about 30% by weight, (i.e., drug and excipients), for example about 4% to about 20% by weight, preferably at least about 10%. In general, the upper limit of solid loads is governed by the viscosity of (e.g., the ability to pump) the resulting solution and the solubility of the components in the solution. Generally, the viscosity of the solution can determine the size of the particle in the resulting powder product.
Techniques and methods for spray drying may be found in Perry’s Chemical
Engineering Handbook, 6th Ed., R. H. Perry, D. W. Green & J. O. Maloney, eds.), McGraw-Hill book co. (1984); and Marshall “Atomization and Spray-Drying” 50, Chem. Eng. Prog. Monogr. Series 2 (1954). In general, the spray drying is conducted with an inlet temperature of from about 60 °C to about 200 °C, for example, from about 95 °C to about 185 °C, from about 110 °C to about 182 °C, from about 96 °C to about 180 °C, e.g., about 145 °C. The spray drying is generally conducted with an outlet temperature of from about 30 °C to about 90 °C, for example from about 40 °C to about 80 °C, about 45 °C to about 80 °C e.g., about 75 °C. The atomization flow rate is generally from about 4 kg h to about 12 kg/h, for example, from about 4.3 kg/h to about 10.5 kg h, e.g., about 6 kg/h or about 10.5 kg/h. The feed flow rate is generally from about 3 kg/h to about 10 kg/h, for example, from about 3.5 kg/h to about 9.0 kg/h, e.g., about 8 kg/h or about 7.1 kg/h. The atomization ratio is generally from about 0.3 to 1.7, e.g., from about 0.5 to 1.5, e.g., about 0.8 or about 1.5.
Removal of the solvent may require a subsequent drying step, such as tray drying, fluid bed drying (e.g., from about room temperature to about 100 °C), vacuum drying, microwave drying, rotary drum drying or biconical vacuum drying (e.g., from about room temperature to about 200 °C).
Synthesis of Compound 1
Acid Chloride Moiety
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile

ouene, 2 , CN
A reactor was purged with nitrogen and charged with 900 mL of toluene. The solvent was degassed via nitrogen sparge for no less than 16 h. To the reactor was then charged Na3P04 (155.7 g, 949.5 mmol), followed by bis(dibenzylideneacetone) palladium (0) (7.28 g, 12.66 mmol). A 10% w/w solution of tert-butylphosphine in hexanes (51.23 g, 25.32 mmol) was charged over 10 min at 23 °C from a nitrogen purged addition funnel. The mixture was allowed to stir for 50 min, at which time 5-bromo-2,2-difluoro-l,3-benzodioxole (75 g, 316.5 mmol) was added over 1 min. After stirring for an additional 50 min, the mixture was charged with ethyl cyanoacetate (71.6 g, 633.0 mmol) over 5 min followed by water (4.5 mL) in one portion. The mixture was heated to 70 °C over 40 min and analyzed by HPLC every 1 – 2 h for the percent conversion of the reactant to the product. After complete conversion was observed (typically 100% conversion after 5 – 8 h), the mixture was cooled to 20 – 25 °C and filtered through a celite pad. The celite pad was rinsed with toluene (2 X 450 mL) and the combined organics were concentrated to 300 mL under vacuum at 60 – 65 °C. The concentrate was charged with 225mL DMSO and concentrated under vacuum at 70 – 80 °C until active distillation of the solvent ceased. The solution was cooled to 20 – 25 °C and diluted to 900 mL with DMSO in preparation for Step 2. Ή NMR (500 MHz, CDC13) δ 7.16 – 7.10 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t, J= 7.1 Hz, 3H).
Synthesis of (2,2-difluoro-l^-benzodioxol-5-yl)-acetonitrile.

[00311] The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature < 40 °C. The mixture was then heated to 75°C over 1 h and analyzed by HPLC every 1 – 2 h for % conversion. When a conversion of > 99% was observed (typically after 5 – 6 h), the reaction was cooled to 20 – 25 °C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 – 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60°C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 – 130 °C (oven temperature) and 1.5 – 2.0 Torr. (2,2-Difluoro-l,3- benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2- difluoro-l,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). Ή NMR (500 MHz, DMSO) 6 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H). Synthesis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cycIopropanecarbonitrUe.

MTBE
A stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h. An appropriate amount of MTBE was similarly degassed for several hours. To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol). The volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing. To the stirring mixture at 23.0 °C was charged degassed 50% w/w NaOH (143 mL) over 10 min followed by l-bromo-2-chloroethane (44.7 g, 311.6 mmol) over 30 min. The reaction was analyzed by HPLC in 1 h intervals for % conversion. Before sampling, stirring was stopped and the phases allowed to separate. The top organic phase was sampled for analysis. When a % conversion > 99 % was observed (typically after 2.5 – 3 h), the reaction mixture was cooled to 10 °C and was charged with water (461 mL) at such a rate as to maintain a temperature < 25 °C. The temperature was adjusted to 20 – 25 °C and the phases separated. Note: sufficient time should be allowed for complete phase separation. The aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HC1 (163mL) and 5% NaCl (163 mL). The solution of (2,2-difluoro- 1,3 -benzodioxol-5-yl)- cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40 – 50 °C. The solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50 – 60 °C. Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50 – 60 °C. The resulting mixture was cooled to 20 – 25 °C and diluted with ethanol to 266 mL in preparation for the next step. lH NMR (500 MHz, DMSO) 6 7.43 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 1.9 Hz, 1H), 7.30 (dd, J= 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H). [00314] Synthesis of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid.

The solution of (2,2-difluoro-l ,3-benzodioxol-5-yl)-cyclopropanecarbonitrile in ethanol from the previous step was charged with 6 N NaOH (277 mL) over 20 min and heated to an internal temperature of 77 – 78 °C over 45 min. The reaction progress was monitored by HPLC after 16 h. Note: the consumption of both (2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarbonitrile and the primary amide resulting from partial hydrolysis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile were monitored. When a % conversion > 99 % was observed (typically 100% conversion after 16 h), the reaction mixture was cooled to 25 °C and charged with ethanol (41 mL) and DCM (164 mL). The solution was cooled to 10 °C and charged with 6 N HC1 (290 mL) at such a rate as to maintain a temperature < 25 °C. After warming to 20 – 25 °C, the phases were allowed to separate. The bottom organic phase was collected and the top aqueous phase was back extracted with DCM (164 mL). Note: the aqueous phase was somewhat cloudy before and after the extraction due to a high concentration of inorganic salts. The organics were combined and concentrated under vacuum to 164 mL. Toluene (328 mL) was charged and the mixture condensed to 164 mL at 70 – 75 °C. The mixture was cooled to 45 °C, charged with MTBE (364 mL) and stirred at 60 °C for 20 min. The solution was cooled to 25 °C and polish filtered to remove residual inorganic salts. MTBE (123 mL) was used to rinse the reactor and the collected solids. The combined organics were transferred to a clean reactor in preparation for the next step.
Isolation of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecar boxy lie acid.

The solution of l-(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid from the previous step is concentrated under vacuum to 164 mL, charged with toluene (328 mL) and concentrated to 164 mL at 70 – 75 °C. The mixture was then heated to 100 – 105 °C to give a homogeneous solution. After stirring at that temperature for 30 min, the solution was cooled to 5 °C over 2 hours and maintained at 5 °C for 3 hours. The mixture was then filtered and the reactor and collected solid washed with cold 1 :1 toluene/n-heptane (2 X 123 mL). The material was dried under vacuum at 55 °C for 17 hours to provide l-(2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarboxylic acid as an off-white crystalline solid. l-(2,2-difluoro-l,3-benzodioxol- 5-yl)-cyclopropanecarboxylic acid was isolated in 79% yield from (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (3 steps including isolation) and with an HPLC purity of 99.0% AUC. ESI-MS m/z calc. 242.04, found 241.58 (M+l)+; Ή NMR (500 MHz, DMSO) δ 12.40 (s, 1H), 7.40 (d, J= 1.6 Hz, 1H), 7.30 (d, J= 8.3 Hz, 1H), 7.17 (dd, J= 8.3, 1.7 Hz, 1H), 1.46 (m, 2H), 1.17 (m, 2H).
Alternative Synthesis of the Acid Chloride Moiety [00319] Synthesis of (2,2-ditluoro-l,3-benzodioxol-5-yl)-methanol.
1. Vitride (2 equiv)
PhCH3 (10 vol)

[00320] Commercially available 2,2-difluoro-l,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-methanol that is used directly in the next step.
Synthesis of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole.
1. SOCl2 (1.5 equiv)
DMAP (0.01 equiv)

(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-methanol ( 1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and S0C12 (1.2 eq) is added via addition funnel. The S0C12 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO_ , filtered, and concentrated to afford crude 5-chloromethyl- 2,2-difluoro-l,3-benzodioxole that is used directly in the next step.
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile.

A solution of 5-chloromethyl-2,2-difluoro- 1 ,3-benzodioxole ( 1 eq) in DMSO ( 1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1,8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
The remaining steps are the same as described above for the synthesis of the acid moiety.
Amine Moiety
Synthesis of 2-bromo-5-fluoro-4-ntroaniline.

Synthesis of benzyIglycoIated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt.
1) l ^OBn
cat. Zn(C104)2-2H20 ®

DCM
A thoroughly dried flask under N2 was charged with the following: Activated powdered 4A molecular sieves (50 wt% based on 2-bromo-5-fluoro-4-nitroaniline), 2-Bromo-5- fluoro-4-nitroaniline (1.0 equiv), zinc perchlorate dihydrate (20 mol%), and toluene (8 vol). The mixture was stirred at room temperature for NMT 30 min. Lastly, (R)-benzyl glycidyl ether (2.0 equiv) in toluene (2 vol) was added in a steady stream. The reaction was heated to 80 °C (internal temperature) and stirred for approximately 7 hours or until 2-Bromo-5-fluoro-4-nitroaniline was <5%AUC.
The reaction was cooled to room temperature and Celite (50 wt%) was added, followed by ethyl acetate (10 vol). The resulting mixture was filtered to remove Celite and sieves and washed with ethyl acetate (2 vol). The filtrate was washed with ammonium chloride solution (4 vol, 20% w/v). The organic layer was washed with sodium bicarbonate solution (4 vol x 2.5% w/v). The organic layer was concentrated in vacuo on a rotovap. The resulting slurry was dissolved in isopropyl acetate (10 vol) and this solution was transferred to a Buchi hydrogenator.
The hydrogenator was charged with 5wt% Pt(S)/C (1.5 mol%) and the mixture was stirred under N2 at 30 °C (internal temperature). The reaction was flushed with N2 followed by hydrogen. The hydrogenator pressure was adjusted to 1 Bar of hydrogen and the mixture was stirred rapidly (>1200 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with dichloromethane (10 vol). The filtrate was concentrated in vacuo. Any remaining isopropyl acetate was chased with dichloromethane (2 vol) and concentrated on a rotavap to dryness.
The resulting residue was dissolved in dichloromethane (10 vol). jP-Toluenesulfonic acid monohydrate (1.2 equiv) was added and stirred overnight. The product was filtered and washed with dichloromethane (2 vol) and suction dried. The wetcake was transferred to drying trays and into a vacuum oven and dried at 45 °C with N2 bleed until constant weight was achieved. Benzylglycolated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt was isolated as an off-white solid.
Chiral purity was determined to be >97%ee.
[00334] Synthesis of (3-Chloro-3-methylbut-l-ynyl)trimethylsilane.

[00335] Propargyl alcohol (1.0 equiv) was charged to a vessel. Aqueous hydrochloric acid (37%, 3.75 vol) was added and stirring begun. During dissolution of the solid alcohol, a modest endotherm (5-6 °C) is observed. The resulting mixture was stirred overnight (16 h), slowly becoming dark red. A 30 L jacketed vessel is charged with water (5 vol) which is then cooled to 10 °C. The reaction mixture is transferred slowly into the water by vacuum, maintaining the internal temperature of the mixture below 25 °C. Hexanes (3 vol) is added and the resulting mixture is stirred for 0.5 h. The phases were settled and the aqueous phase (pH < 1) was drained off and discarded. The organic phase was concentrated in vacuo using a rotary evaporator, furnishing the product as red oil. [00336] Synthesis of (4-(Benzyloxy)-3,3-dimethylbut-l-yttyl)trimethylsiIane.

[00337] Method A
[00338] All equivalent and volume descriptors in this part are based on a 250g reaction.
Magnesium turnings (69.5 g, 2.86 mol, 2.0 equiv) were charged to a 3 L 4-neck reactor and stirred with a magnetic stirrer under nitrogen for 0.5 h. The reactor was immersed in an ice- water bath. A solution of the propargyl chloride (250 g, 1.43 mol, 1.0 equiv) in THF (1.8 L, 7.2 vol) was added slowly to the reactor, with stirring, until an initial exotherm (-10 °C) was observed. The Grignard reagent formation was confirmed by IPC usingΉ-NMR spectroscopy. Once the exotherm subsided, the remainder of the solution was added slowly, maintaining the batch temperature <15 °C. The addition required ~3.5 h. The resulting dark green mixture was decanted into a 2 L capped bottle.
[00339] All equivalent and volume descriptors in this part are based on a 500g reaction. A 22 L reactor was charged with a solution of benzyl chloromethyl ether (95%, 375 g, 2.31 mol, 0.8 equiv) in THF (1.5 L, 3 vol). The reactor was cooled in an ice-water bath. Two Grignard reagent batches prepared as described above were combined and then added slowly to the benzyl chloromethyl ether solution via an addition funnel, maintaining the batch temperature below 25 °C. The addition required 1.5 h. The reaction mixture was stirred overnight (16 h).
[00340] All equivalent and volume descriptors in this part are based on a 1 kg reaction. A solution of 15%» ammonium chloride was prepared in a 30 L jacketed reactor (1.5 kg in 8.5 kg of water, 10 vol). The solution was cooled to 5 °C. Two Grignard reaction mixtures prepared as described above were combined and then transferred into the ammonium chloride solution via a header vessel. An exotherm was observed in this quench, which was carried out at a rate such as to keep the internal temperature below 25 °C. Once the transfer was complete, the vessel jacket temperature was set to 25 °C. Hexanes (8 L, 8 vol) was added and the mixture was stirred for 0.5 h. After settling the phases, the aqueous phase (pH 9) was drained off and discarded. The remaining organic phase was washed with water (2 L, 2 vol). The organic phase was concentrated in vacuo using a 22 L rotary evaporator, providing the crude product as an orange oil.
[00341] Method B
[00342] Magnesium turnings (106 g, 4.35 mol, 1.0 eq) were charged to a 22 L reactor and then suspended in THF (760 mL, 1 vol). The vessel was cooled in an ice-water bath such that the batch temperature reached 2 °C. A solution of the propargyl chloride (760 g, 4.35 mol, 1.0 equiv) in THF (4.5 L, 6 vol) was added slowly to the reactor. After 100 mL was added, the addition was stopped and the mixture stirred until a 13 °C exotherm was observed, indicating the Grignard reagent initiation. Once the exotherm subsided, another 500 mL of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. The remainder of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The addition required -1.5 h. The resulting dark green solution was stirred for 0.5 h. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. Neat benzyl
chloromethyl ether was charged to the reactor addition funnel and then added dropwise into the reactor, maintaining the batch temperature below 25 °C. The addition required 1.0 h. The reaction mixture was stirred overnight. The aqueous work-up and concentration was carried out using the same procedure and relative amounts of materials as in Method A to give the product as an orange oil.
[00343] Syntheisis of 4-Benzyloxy-3,3-dimethylbut-l-yne.

2 steps
[00344] A 30 L jacketed reactor was charged with methanol (6 vol) which was then cooled to 5 °C. Potassium hydroxide (85%, 1.3 equiv) was added to the reactor. A 15-20 °C exotherm was observed as the potassium hydroxide dissolved. The jacket temperature was set to 25 °C. A solution of 4-benzyloxy-3,3-dimethyl-l-trimethylsilylbut-l-yne (1.0 equiv) in methanol (2 vol) was added and the resulting mixture was stirred until reaction completion, as monitored by HPLC. Typical reaction time at 25 °C is 3-4 h. The reaction mixture is diluted with water (8 vol) and then stirred for 0.5 h. Hexanes (6 vol) was added and the resulting mixture was stirred for 0.5 h. The phases were allowed to settle and then the aqueous phase (pH 10-11) was drained off and discarded. The organic phase was washed with a solution of KOH (85%, 0.4 equiv) in water (8 vol) followed by water (8 vol). The organic phase was then concentrated down using a rotary evaporator, yielding the title material as a yellow-orange oil. Typical purity of this material is in the 80% range with primarily a single impurity present. Ή NMR (400 MHz, C6D6) δ 7.28 (d, 2 H, J = 7.4 Hz), 7.18 (t, 2 H, J= 7.2 Hz), 7.10 (d, 1H, J= 7.2 Hz), 4.35 (s, 2 H), 3.24 (s, 2 H), 1.91 (s, 1 H), 1.25 (s, 6 H).
[00345] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.
[00346] Method A
[00347] Synthesis of Benzylglycolated 4-Amino-2-(4-benzyloxy-3,3-dimethyIbut- l-ynyl)-5- fluoroaniline.

[00348] Benzylglycolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt was freebased by stirring the solid in EtOAc (5 vol) and saturated NaHCC>3 solution (5 vol) until clear organic layer was achieved. The resulting layers were separated and the organic layer was washed with saturated NaHC03 solution (5 vol) followed by brine and concentrated in vacuo to obtain benzylglocolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt as an oil.
[00349] Then, a flask was charged with benzylglycolated 4-ammonium-2-bromo-5- flouroaniline tosylate salt (freebase, 1.0 equiv), Pd(OAc) (4.0 mol%), dppb (6.0 mol%) and powdered K2CO3 (3.0 equiv) and stirred with acetonitrile (6 vol) at room temperature. The resulting reaction mixture was degassed for approximately 30 min by bubbling in N2 with vent. Then 4-benzyloxy-3,3-dimethylbut-l-yne (1.1 equiv) dissolved in acetonitrile (2 vol) was added in a fast stream and heated to 80 °C and stirred until complete consumption of 4-ammonium-2- bromo-5-flouroaniline tosylate salt was achieved. The reaction slurry was cooled to room temperature and filtered through a pad of Celite and washed with acetonitrile (2 vol). Filtrate was concentrated in vacuo and the residue was redissolved in EtOAc (6 vol). The organic layer was washed twice with NH4CI solution (20% w/v, 4 vol) and brine (6 vol). The resulting organic layer was concentrated to yield brown oil and used as is in the next reaction.
[00350] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.

[00351] Crude oil of benzylglycolated 4-amino-2-(4-benzyloxy-3,3-dimethylbut-l-ynyl)-5- fluoroaniline was dissolved in acetonitrile (6 vol) and added (MeCN)2PdCl2 (15 mol%) at room temperature. The resulting mixture was degassed using N2 with vent for approximately 30 min. Then the reaction mixture was stirred at 80 °C under N2 blanket overnight. The reaction mixture was cooled to room temperature and filtered through a pad of Celite and washed the cake with acetonitrile (1 vol). The resulting filtrate was concentrated in vacuo and redissolved in EtOAc (5 vol). Deloxane-II THP (5 wt% based on the theoretical yield of N-benzylglycolated-5-amino-2- (2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole) was added and stirred at room temperature overnight. The mixture was then filtered through a pad of silica (2.5 inch depth, 6 inch diameter filter) and washed with EtOAc (4 vol). The filtrate was concentrated down to a dark brown residue, and used as is in the next reaction.
[00352] Repurification of crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1,1- dimethylethyl)-6-fluoroindole:
[00353] The crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1 , l-dimethylethyl)-6- fluoroindole was dissolved in dichloromethane (~1.5 vol) and filtered through a pad of silica initially using 30% EtOAc/heptane where impurities were discarded. Then the silica pad was washed with 50% EtO Ac/heptane to isolate N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l- dimethylethyl)-6-fluoroindole until faint color was observed in the filtrate. This filtrate was concentrated in vacuo to afford brown oil which crystallized on standing at room temperature. Ή NMR (400 MHz, DMSO) 6 7.38-7.34 (m, 4 H), 7.32-7.23 (m, 6 H), 7.21 (d, 1 H, J= 12.8 Hz), 6.77 (d, 1H, J= 9.0 Hz), 6.06 (s, 1 H), 5.13 (d, 1H, J = 4.9 Hz), 4.54 (s, 2 H), 4.46 (br. s, 2 H), 4.45 (s, 2 H), 4.33 (d, 1 H, J= 12.4 Hz), 4.09-4.04 (m, 2 H), 3.63 (d, 1H, J= 9.2 Hz), 3.56 (d, 1H, J= 9.2 Hz), 3.49 (dd, 1H, J= 9.8, 4.4 Hz), 3.43 (dd, 1H, J= 9.8, 5.7 Hz), 1.40 (s, 6 H).
[00354] Synthesis of N-benzyIglycolated-5-amino-2-(2-benzyIoxy-l,l-diniethylethyl)-6- fluoroindole.
[00355] Method B

2. (MeCN)2PdCl2
MeCN, 80 <€
3. Silica gel filtration
[00356] Palladium acetate (33 g, 0.04 eq), dppb (94 g, 0.06 eq), and potassium carbonate (1.5 kg, 3.0 eq) are charged to a reactor. The free based oil benzylglocolated 4-ammonium-2-bromo- 5-flouroaniline (1.5 kg, 1.0 eq) was dissolved in acetonitrile (8.2 L, 4.1 vol) and then added to the reactor. The mixture was sparged with nitrogen gas for NLT 1 h. A solution of 4-benzyloxy- 3,3-dimethylbut-l-yne (70%), 1.1 kg, 1.05 eq) in acetonitrile was added to the mixture which was then sparged with nitrogen gas for NLT 1 h. The mixture was heated to 80 °C and then stirred overnight. IPC by HPLC is carried out and the reaction is determined to be complete after 16 h. The mixture was cooled to ambient temperature and then filtered through a pad of Celite (228 g). The reactor and Celite pad were washed with acetonitrile (2 x 2 L, 2 vol). The combined phases are concentrated on a 22 L rotary evaporator until 8 L of solvent have been collected, leaving the crude product in 7 L (3.5 vol) of acetonitrile. [00357] 5 s-acetonitriledichloropalladium ( 144 g, 0.15 eq) was charged to the reactor. The crude solution was transferred back into the reactor and the roto-vap bulb was washed with acetonitrile (4 L, 2 vol). The combined solutions were sparged with nitrogen gas for NLT 1 h. The reaction mixture was heated to 80 °C for NLT 16 h. In process control by HPLC shows complete consumption of starting material. The reaction mixture was filtered through Celite (300 g). The reactor and filter cake were washed with acetonitrile (3 L, 1.5 vol). The combined filtrates were concentrated to an oil by rotary evaporation. The oil was dissolved in ethyl acetate (8.8 L, 4.4 vol). The solution was washed with 20% ammonium chloride (5 L, 2.5 vol) followed by 5% brine (5 L, 2.5 vol). Silica gel (3.5 kg, 1.8 wt. eq.) of silica gel was added to the organic phase, which was stirred overnight. Deloxan THP II metal scavenger (358 g) and heptane (17.6 L) were added and the resulting mixture was stirred for NLT 3 h. The mixture was filtered through a sintered glass funnel. The filter cake was washed with 30% ethyl acetate in heptane (25 L). The combined filtrates were concentrated under reduced pressure to give N- benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole as a brown paste ( 1.4 kgl.Svnthesis of Compound 1
[00358] Synthesis of benzyl protected Compound 1.



[00359] 1 -(2,2-difluoro- 1 ,3 -benzodioxol-5-yl)-cyclopropanecarboxylic acid (1.3 equiv) was slurried in toluene (2.5 vol, based on l-(2,2-difluoro-l,3-benzodioxol-5-yi)- cyclopropanecarboxylic acid) and the mixture was heated to 60 °C. SOCl2 (1.7 equiv) was added via addition runnel. The resulting mixture was stirred for 2 hr. The toluene and the excess
SOCI2 were distilled off using rotavop. Additional toluene (2.5 vol, based on l-(2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid) was added and distilled again. The crude acid chloride was dissolved in dichloromethane (2 vol) and added via addition funnel to a mixture of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole (1.0 equiv), and triethylamine (2.0 equiv) in dichloromethane (7 vol) while maintaining 0-3 °C (internal temperature). The resulting mixture was stirred at 0 °C for 4 hrs and then warmed to room temperature overnight. Distilled water (5 vol) was added to the reaction mixture and stirred for NLT 30 min and the layers were separated. The organic phase was washed with 20 wt% K2CO3 (4 vol x 2) followed by a brine wash (4 vol) and concentrated to afford crude benzyl protected Compound 1 as a thick brown oil, which was purified further using silica pad filtration.
[00360] Silica gel pad filtration: Crude benzyl protected Compound 1 was dissolved in ethyl acetate (3 vol) in the presence of activated carbon Darco-G (10wt%, based on theoretical yield of benzyl protected Compound 1) and stirred at room temperature overnight. To this mixture was added heptane (3 vol) and filtered through a pad of silica gel (2x weight of crude benzyl protected Compound 1). The silica pad was washed with ethyl acetate/heptane (1:1, 6 vol) or until little color was detected in the filtrate. The filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish brown oil, and used directly in the next step.
[00361] Repurification: Benzyl protected Compound 1 was redissolved in dichloromethane (1 vol, based on theoretical yield of benzyl protected Compound 1) and loaded onto a silica gel pad (2x weight of crude benzyl protected Compound 1). The silica pad was washed with
dichloromethane (2 vol, based on theoretical yield of benzyl protected Compound 1) and the filtrate was discarded. The silica pad was washed with 30% ethyl acetate/heptane (5 vol) and the filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish orange oil, and used directly in the next step. [00362] Synthesis of Compound 1.

OBn 4 steps

[00363] Method A
[00364] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, ~ 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 °C.
[00365] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank.
[00366] After 2 full days of reaction, more Pd / C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [00367] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg.
[00368] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1 :1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1 :1 ethyl acetate-heptane. The silica pad was flushed first with 1 :1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC.
[00369] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of Compound 1 as an off-white solid.
[00370] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1 :1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g).
[00371] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of -6% methanol. The thermostat was set to 48 °C (below the boiling temp of the MTBE-methanol azeotrope, which is 52 °C). The mixture was cooled to 20 °C over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air- dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
[00372] Method B
[00373] Benzyl protected Compound 1 was dissolved in THF (3 vol) and then stripped to dryness to remove any residual solvent. Benzyl protected Compound 1 was redissolved in THF (4 vol) and added to the hydrogenator containing 5 wt% Pd/C (2.5 mol%, 60% wet, Degussa E5 El 01 N /W). The internal temperature of the reaction was adjusted to 50 °C, and flushed with N2 (x5) followed by hydrogen (x3). The hydrogenator pressure was adjusted to 3 Bar of hydrogen and the mixture was stirred rapidly (>1100 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with THF (1 vol). The filtrate was concentrated in vacuo to obtain a brown foamy residue. The resulting residue was dissolved in MTBE (5 vol) and 0.5N HC1 solution (2 vol) and distilled water (1 vol) were added. The mixture was stirred for NLT 30 min and the resulting layers were separated. The organic phase was washed with 10wt% K2CO3 solution (2 vol x2) followed by a brine wash. The organic layer was added to a flask containing silica gel (25 wt%), Deloxan-THP II (5wt%, 75% wet), and
Na2S04 and stirred overnight. The resulting mixture was filtered through a pad of Celite and washed with 10%THF/MTBE (3 vol). The filtrate was concentrated in vacuo to afford crude Compound 1 as pale tan foam.
[00374] Compound 1 recovery from the mother liquor: Option A.
[00375] Silica gel pad filtration: The mother liquor was concentrated in vacuo to obtain a brown foam, dissolved in dichloromethane (2 vol), and filtered through a pad of silica (3x weight of the crude Compound 1). The silica pad was washed with ethyl acetate/heptane (1 :1, 13 vol) and the filtrate was discarded. The silica pad was washed with 10% THF/ethyl acetate (10 vol) and the filtrate was coiicentraied in vacuo to afford Compound 1 as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{00376] Compound 1 recovery from the mother liquor: Option B,
[00377] Silica gel column chromatography: After chromatography on silica gel (50% ethyl acetate/hexaties to 100% ethyl acetate), the desired compound was isolated as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{003781 Additional Recrystaliization of Compound 1
[ 0379‘j Solid Compound 1 (135 kg) was suspended in IPA (5.4 L, 4 vol) and then heated to 82 °C. Upon complete dissolution (visual), heptane (540 mL, 0.4 vol) was added slowly. The mixture was cooled to 58 °C The mixture was then cooled slowly to 51 °C, during which time crystallization occurs. The heat source was shut down and the recrystalfeation mixture was allowed to cool naturally overnight. The mixture was filtered using a benchtop Buclmer funnel and the filter cake was washed with IPA (2.7 L, 2 vol). The filler cake was dried in the tunnel under air flow for 8 h and then was oven-dried in vacuo at 45-50 °C overnight to give 1.02 kg of recrystallized Compound 1 ,
100380] Compound 1 may also be prepared by one of several synthetic routes disclosed in US published patent application U S20090131 92, incorporated herein by reference.
{003811 Table 6 below recites analytical data for Compound 1.
Table 6.

Synthesis of Compound 1 Amorphous Form [00383] Spray-Dried Method
[00384] 9.95g of Hydroxypropylmethylcellulose acetate succinate HG grade (HPMCAS-HG) was weighed into a 500 ml beaker, along with 50 mg of sodium lauryl sulfate (SLS). MeOH (200 ml) was mixed with the solid. The material was allowed to stir for 4 h. To insure maximum dissolution, after 2 h of stirring the solution was sonicated for 5 mins, then allowed to continue stirring for the remaining 2 h. A very fin suspension of HPMCAS remained in solution. However, visual observation determined that no gummy portions remained on the walls of the vessel or stuck to the bottom after tilting the vessel.
[00385] Compound 1 (1 Og) was poured into the 500 ml beaker, and the system was allowed to continue stirring. The solution was spray dried using the following parameters:
Formulation Description: Compound 1 Form A/HPMCAS/SLS (50/49.5/0.5)
Buchi Mini Spray Dryer
T inlet (setpoint) 145 °C
T outlet (start) 75 °C
T outlet (end) 55 °C
Nitrogen Pressure 75 psi
Aspirator 100 %
Pump 35 %
Rotometer 40 mm
Filter Pressure 65 mbar
Condenser Temp -3 °C
Run Time l h
REFERENCES
1: Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med. 2014 Jul 23;6(246):246ra97. doi: 10.1126/scitranslmed.3008889. PubMed PMID: 25101887.
2: Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014 Jul;39(7):500-11. PubMed PMID: 25083129; PubMed Central PMCID: PMC4103577.
3: Norman P. Novel picolinamide-based cystic fibrosis transmembrane regulator modulators: evaluation of WO2013038373, WO2013038376, WO2013038381, WO2013038386 and WO2013038390. Expert Opin Ther Pat. 2014 Jul;24(7):829-37. doi: 10.1517/13543776.2014.876412. Epub 2014 Jan 7. PubMed PMID: 24392786.
//////TEZACAFTOR, VX 661, PHASE 3, 1152311-62-0, UNII: 8RW88Y506K, deltaF508-CFTR corrector, Vertex, treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
CC(C)(CO)C1=CC2=CC(=C(C=C2N1CC(CO)O)F)NC(=O)C3(CC3)C4=CC5=C(C=C4)OC(O5)(F)F
CC(C)(CO)c1cc2cc(c(cc2n1C[C@H](CO)O)F)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F
VX-? , an Azaindolyl-Pyrimidine Inhibitor of Influenza Virus Replication from Vertex
VX-?
An Azaindolyl-Pyrimidine Inhibitor of Influenza Virus Replication from Vertex
SYNTHESIS COMING……..
Specific Rotation
Influenza spreads around the world in seasonal epidemics, resulting in the deaths of hundreds of thousands annually – millions in pandemic years. For example, three influenza pandemics occurred in the 20th century and killed tens of millions of people, with each of these pandemics being caused by the appearance of a new strain of the virus in humans. Often, these new strains result from the spread of an existing influenza virus to humans from other animal species.
Influenza is primarily transmitted from person to person via large virus-laden droplets that are generated when infected persons cough or sneeze; these large droplets can then settle on the mucosal surfaces of the upper respiratory tracts of susceptible individuals who are near (e.g. within about 6 feet) infected persons. Transmission might also occur through direct contact or indirect contact with respiratory secretions, such as touching surfaces contaminated with influenza virus and then touching the eyes, nose or mouth. Adults might be able to spread influenza to others from 1 day before getting symptoms to approximately 5 days after symptoms start. Young children and persons with weakened immune systems might be infectious for 10 or more days after onset of symptoms. [00103] Influenza viruses are RNA viruses of the family Orthomyxoviridae, which comprises five genera: Influenza virus A, Influenza virus B, Influenza virus C, Isavirus and Thogoto virus.
The Influenza virus A genus has one species, influenza A virus. Wild aquatic birds are the natural hosts for a large variety of influenza A. Occasionally, viruses are transmitted to other species and may then cause devastating outbreaks in domestic poultry or give rise to human influenza pandemics. The type A viruses are the most virulent human pathogens among the three influenza types and cause the most severe disease. The influenza A virus can be subdivided into different serotypes based on the antibody response to these viruses. The serotypes that have been confirmed in humans, ordered by the number of known human pandemic deaths, are: HlNl (which caused Spanish influenza in 1918), H2N2 (which caused Asian Influenza in 1957), H3N2 (which caused Hong Kong Flu in 1968), H5N1 (a pandemic threat in the 2007-08 influenza season), H7N7 (which has unusual zoonotic potential), H1N2 (endemic in humans and pigs), H9N2, H7N2 , H7N3 and H10N7. [00105] The Influenza virus B genus has one species, influenza B virus. Influenza B almost exclusively infects humans and is less common than influenza A. The only other animal known to be susceptible to influenza B infection is the seal. This type of influenza mutates at a rate 2-3 times slower than type A and consequently is less genetically diverse, with only one influenza B serotype. As a result of this lack of antigenic diversity, a degree of immunity to influenza B is usually acquired at an early age. However, influenza B mutates enough that lasting immunity is not possible. This reduced rate of antigenic change, combined with its limited host range (inhibiting cross species antigenic shift), ensures that pandemics of influenza B do not occur.
The Influenza virus C genus has one species, influenza C virus, which infects humans and pigs and can cause severe illness and local epidemics. However, influenza C is less common than the other types and usually seems to cause mild disease in children. [00107] Influenza A, B and C viruses are very similar in structure. The virus particle is 80-120 nanometers in diameter and usually roughly spherical, although filamentous forms can occur. Unusually for a virus, its genome is not a single piece of nucleic acid; instead, it contains seven or eight pieces of segmented negative-sense RNA. The Influenza A genome encodes 11 proteins: hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), Ml, M2, NSl, NS2(NEP), PA, PBl, PB1-F2 and PB2.
HA and NA are large glycoproteins on the outside of the viral particles. HA is a lectin that mediates binding of the virus to target cells and entry of the viral genome into the target cell, while NA is involved in the release of progeny virus from infected cells, by cleaving sugars that bind the mature viral particles. Thus, these proteins have been targets for antiviral drugs. Furthermore, they are antigens to which antibodies can be raised. Influenza A viruses are classified into subtypes based on antibody responses to HA and NA, forming the basis of the H and N distinctions (vide supra) in, for example, H5N1. [00109] Influenza produces direct costs due to lost productivity and associated medical treatment, as well as indirect costs of preventative measures. In the United States, influenza is responsible for a total cost of over $10 billion per year, while it has been estimated that a future pandemic could cause hundreds of billions of dollars in direct and indirect costs. Preventative costs are also high. Governments worldwide have spent billions of U.S. dollars preparing and planning for a potential H5N1 avian influenza pandemic, with costs associated with purchasing drugs and vaccines as well as developing disaster drills and strategies for improved border controls.
Current treatment options for influenza include vaccination, and chemotherapy or chemoprophylaxis with anti-viral medications. Vaccination against influenza with an influenza vaccine is often recommended for high-risk groups, such as children and the elderly, or in people that have asthma, diabetes, or heart disease. However, it is possible to get vaccinated and still get influenza. The vaccine is reformulated each season for a few specific influenza strains but cannot possibly include all the strains actively infecting people in the world for that season. It takes about six months for the manufacturers to formulate and produce the millions of doses required to deal with the seasonal epidemics; occasionally, a new or overlooked strain becomes prominent during that time and infects people although they have been vaccinated (as by the H3N2 Fujian flu in the 2003-2004 influenza season). It is also possible to get infected just before vaccination and get sick with the very strain that the vaccine is supposed to prevent, as the vaccine takes about two weeks to become effective. [00111] Further, the effectiveness of these influenza vaccines is variable. Due to the high mutation rate of the virus, a particular influenza vaccine usually confers protection for no more than a few years. A vaccine formulated for one year may be ineffective in the following year, since the influenza virus changes rapidly over time, and different strains become dominant.
Also, because of the absence of RNA proofreading enzymes, the RNA- dependent RNA polymerase of influenza vRNA makes a single nucleotide insertion error roughly every 10 thousand nucleotides, which is the approximate length of the influenza vRNA. Hence, nearly every newly-manufactured influenza virus is a mutant — antigenic drift. The separation of the genome into eight separate segments of vRNA allows mixing or reassortment of vRNAs if more than one viral line has infected a single cell. The resulting rapid change in viral genetics produces antigenic shifts and allows the virus to infect new host species and quickly overcome protective immunity.
Antiviral drugs can also be used to treat influenza, with neuraminidase inhibitors being particularly effective, but viruses can develop resistance to the standard antiviral drugs.
PAPER
http://pubs.acs.org/doi/full/10.1021/acs.oprd.6b00063
Development of a Scalable Synthesis of an Azaindolyl-Pyrimidine Inhibitor of Influenza Virus Replication

A scalable, asymmetric route for the synthesis of the influenza virus replication inhibitor 2 is presented. The key steps include an enzymatic desymmetrization of cis-1,3-cyclohexanediester in 99% yield and 96% ee, SNAr displacement of a methanesulfinylpyrimidine, and a Curtius rearrangement to form a morpholinyl urea. This high-yielding route allowed us to rapidly synthesize hundreds of grams of 2 in 99% purity to support in vivo studies.

About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
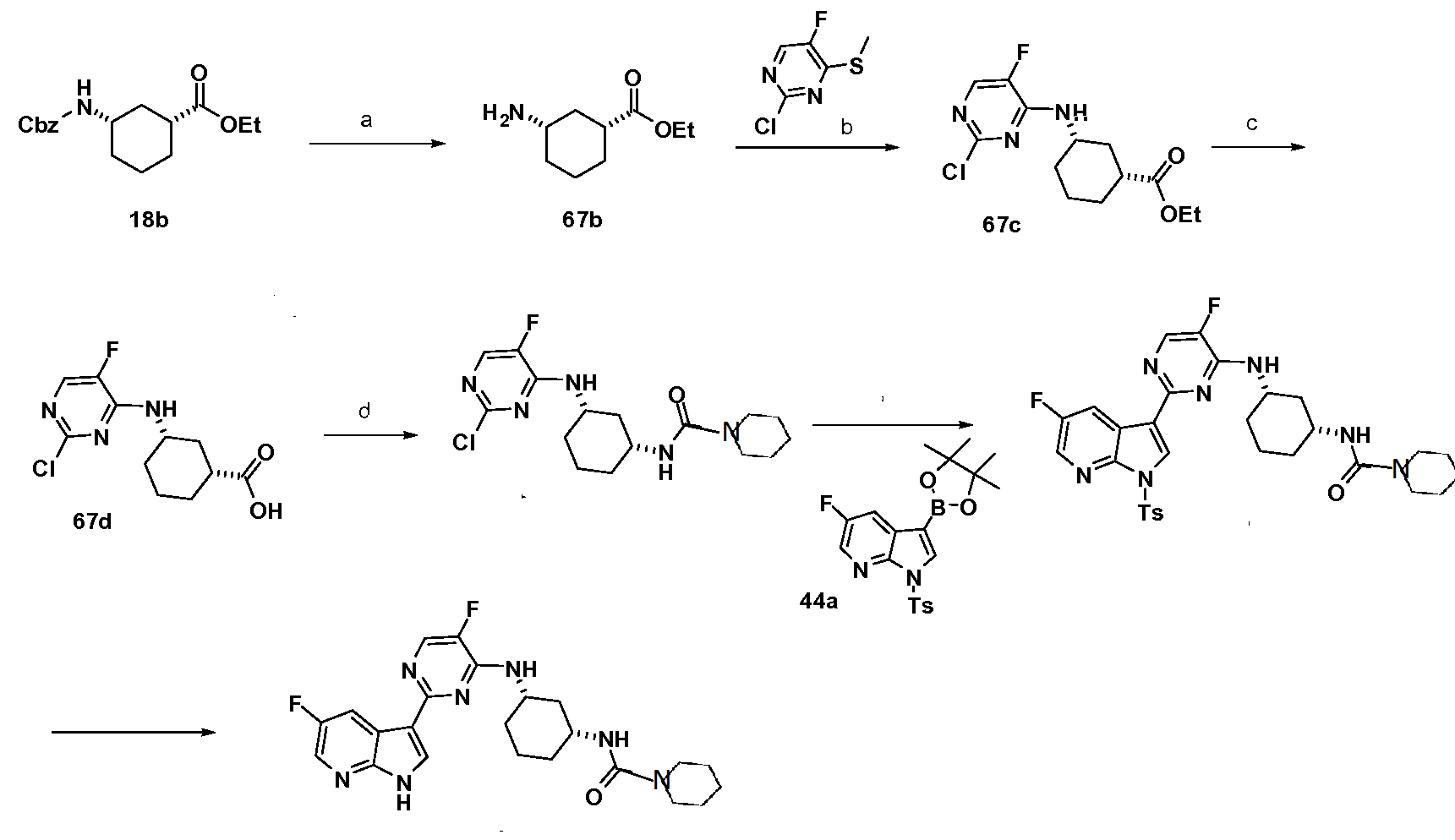
SYNTHESIS COMING
WO-2010148197
http://www.google.co.in/patents/WO2010148197A1?cl=en
General Scheme 44 SIMILAR TO A POINT BUT NOT SAME

(a) Pd(PPh3)4 sodium carbonate, DME/water, reflux (b) meta-chloroperbenzoic acid, dichloromethane, rt. (c) 20a, tetrahydrofuran, 5O°C (d) trifluoroacetic acid, dichloromethane, rt.
SIMILAR NOT SAME
(e) morpholιne-4-carbonyl chloride, dimethylformamide, rt (f) sodium methoxide, methanol, rt.
Formation of 5-fluoro-3-[5-fluoro-4-(methylthio)pyrimidin-2-yl]-1-tosyl-lΗ- pyrrolo[2,3-b]pyridine (44b)
2-Chloro-5-fluoro-4-methylsulfanyl-pyrimidine (34.1 g, 191.0 mmol) , 5-fluoro-1-(p- tolylsulfonyl)-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyrrolo[2,3-b]pyridine, 44a, (53.0 g, 127.3 mmol) and Na2Cθ3 (40.5 g, 381.9 mmol) were dissolved in a mixture of DME (795 mL) and water (159 mL). The mixture was purged with nitrogen for 20 minutes and treated with Pd(PPh3 )4 (7.4 g, 6.6 mmol). After purging with nitrogen for another 20 minutes, the reaction was heated to reflux overnight, cooled to room temperature and diluted with water (60OmL). The resulting suspension was stirred at room temperature for 30 minutes and the precipitate was then collected by filtration, washed with water and acetonitrile and dried at 50 °C to afford 48.2 g of 5-fluoro-3-[5-fluoro-4-(methylthio)pyrimidin-2-yl]-1-tosyl-1H- pyrrolo[2,3-b]pyridine as a white solid.
1H NMR (300 MHz, OMSO-d6) δ 8.70 – 8.58 (m, 2H), 8.54 – 8.41 (m, 2H), 8.09 (d, J = 8.4 Hz, 2H), 7.45 (d, J= 8.2 Hz, 2H), 2.76 (s, 3H), 2.36 (s, 3H).
Formation of 5-fluoro-3-[5-fluoro-4-(methylsulfinyl)pyrimidin-2-yl]-1- tosyl-1H-pyrrolo[2,3-b]pyridine (44c)
5-fluoro-3 – [5 -fluoro-4-(methylthio)pyrimidin-2-yl] – 1 -tosyl- 1 H-pyrrolo [2,3 – b]pyridine, 44b, (48.2 g, 111.5 mmol) was dissolved in dichloromethane (2.3 L) and treated portionwise with m-CPBA (27.5 g, 122.6 mmol) while keeping the temperature below 20 °C. After addition was complete, the reaction was stirred at room temperature for 2 hours, then treated with another portion of m-CPBA (1.9 g) and stirred for another hour. The reaction mixture was washed with 12% aqueuous K2CO3 (2 x 1.0 L) and the organic layer was dried on Na2SO4 and concentrated in vacuo to provide 50 g of 5-fluoro-3-[5-fluoro-4- (methylsulfinyl)pyrimidin-2-yl]-1-tosyl-1H-pyrrolo[2,3-b]pyridine as a yellow solid.
1H NMR (300 MHz, DMSO-rf<5) δ 9.11 (d, J= 1.5 Hz, 1H), 8.69 (s, 1H), 8.65 (dd, J = 9.0, 2.9 Hz, 1H), 8.52 (dd, J= 2.8, 1.2 Hz, 1H), 8.11 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 3.05 (s, 3H), 2.36 (s, 3H).
[001057] Formation of tert-butyl N-[(IR, 3S)-3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo [2,3-b] pyridin-3-yl]pyrimidin-4-yl] amino] cyclohexyl] carbamate (44d)
5-fluoro-3-(5-fluoro-4-methylsulfinyl-pyrimidin-2-yl)-1-(p-tolylsulfonyl)pyrrolo[2,3- b]pyridine, 44c, (5.9 g, 10.5 mmol) and tert-butyl N-[(IR, 35*)-3-aminocyclohexyl]carbamate (3 g, 12.60 mmol) were dissolved in THF (100 mL). The reaction mixture was heated to 50 °C for 6 hours, then cooled to room temperature. C6 lite was added and the solvent was removed under reduced pressure. The C6 lite-supported residue was purified by silica gel chromatography (20-80% EtOAc/hexanes gradient to provide 3.7 g of tert-butyl N-[(IR, 3S)- 3-[[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4- yl]amino]cyclohexyl]carbamate.
1H NMR (300 MHz, CDCl3) δ 8.51 (s, 1H), 8.46 – 8.41 (m, 1H), 8.29 (d, J = 1.6 Hz, 1H), 8.11 (s, 1H), 8.08 (s, 1H), 8.06 (d, J= 3.2 Hz, 1H), 7.27 (d, J= 8.4 Hz, 2H), 4.91 (d, J = 8.0 Hz, 1H), 4.41 (s, 1H), 4.29 – 4.01 (m, 1H), 3.64 (s, 1H), 2.47 (d, J= 11.5 Hz, 1H), 2.36 (s, 3H), 2.24 (d, J = 13.1 Hz, 1H), 2.08 (d, J= 10.9 Hz, 1H), 1.91 (d, J= 13.8 Hz, 1H), 1.43 (s, 9H), 1.30 – 1.03 (m, 4H).
Formation of (IS, SΛHVHS-fluoro^-β-fluoro-1-Cp- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]cyclohexane-1,3-diamine (44e) tert-Butyl N-[(IR, 3S>3-[[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3- b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl]carbamate, 44d, (3.7 g, 6.2 mmol) was dissolved in dichloromethane (105 mL) and treated with trifluoroacetic acid (31 mL). After 5 minutes, the volatiles were evaporated under reduced pressure, and the resulting residue was treated with IN NaOH (75 mL). The resulting precipitate was collected by filtration, washed with water (3 x 30 mL) and vacuum dried to provide 2.7 g of (IS, 3R)-Nl -[5-fluoro-2-[5- fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]cyclohexane-l,3-diamine as a white solid.
1H NMR (300 MHz, MeOD) d 8.56 (dd, J = 8.0, 3.9 Hz, 2H), 8.35 – 8.26 (m, 1H), 8.12 (dd, J= 10.3, 6.1 Hz, 3H), 7.43 (d, J= 8.4 Hz, 2H), 4.36 – 4.21 (m, 1H), 3.28 – 3.13 (m, 1H), 2.48 (d, J= 12.3 Hz, 1H), 2.46 (s, 3H), 2.25 – 1.97 (m, J= 17.3, 10.6, 4.1 Hz, 4H), 1.76 – 1.28 (m, 3H).
Formation of N-[(IR, 3S>3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl] morpholine- 4-carboxamide (44f)
(15, 3R)-M-[5-fluoro-2-[5-fluoro-1-(p-tolylsulfonyl)pyrrolo[2,3-b]pyridin-3- yl]pyrimidin-4-yl]cyclohexane- 1,3 -diamine, 44e, (2.3 g, 4.6 mmol) was dissolved in DMF (5OmL) and treated with morpholine-4-carbonyl chloride (2.1 g, 13.8 mmol) and DIPEA (4.2 g, 5.6 mL, 32.3 mmol). After one hour, the resulting solution was diluted with water (400 mL) and stirred for an additional two hours. The resulting precipitate was collected by filtration, washed with water (3 x 50 mL) and dried to provide the crude product. This material was purified by flash chromatography on a 4Og column using EtOAc/DCM 20- 100%, to provide 2.0 g of N-[(1R, 35)-3-[[5-fluoro-2-[5-fluoro-1-(p- tolylsulfonyl)pyrrolo[2,3-b]pyridin-3-yl]pyrimidin-4-yl]amino]cyclohexyl]morpholine-4- carboxamide as a white solid.
1H NMR (300 MHz, DMSO-Λ5) δ 8.53 – 8.43 (m, J = 11.9, 2.7 Hz, 3H), 8.22 (d, J = 3.9 Hz, 1H), 8.07 (d, J= 8.4 Hz, 2H), 7.44 (d, J= 8.3 Hz, 2H), 6.32 (d, J= 7.5 Hz, 1H), 4.05 (s, J= 19.4 Hz, 1H), 3.62 (s, 1H), 3.58 – 3.45 (m, 4H), 3.27 – 3.18 (m, 4H), 2.36 (s, 3H), 2.12 (d, J= 11.7 Hz, 1H), 1.99 (d, J= 9.5 Hz, 1H), 1.83 (d, J= 10.3 Hz, 2H), 1.53 – 1.11 (m, J = 32.3, 22.8, 10.9 Hz, 4H).
ormation of N-[(IR, 3S>3-[[5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-yl] amino] cyclohexyl]morpholine-4-carboxamide (706)
N- [( IR, 35)-3 – [ [5 -fluoro-2- [5 -fluoro- 1 -(p-tolylsulfonyl)pyrrolo [2,3 -b]pyridin-3 – yl]pyrimidin-4-yl]amino]cyclohexyl]morpholine-4-carboxamide, 44f, (2.0 g, 3.2 mmol) was suspended in methanol (50 mL) and treated with 25% sodium methoxide in methanol (19.9 mL, 92.3 mmol) . After stirring for 1 hour, the solvent was evaporated under reduced pressure, and the residue was partitioned between water (100 mL) and ethyl acetate (100 mL). The organic layer was collected, dried on Νa2SO4 and concentrated to provide the crude product as a yellow solid. This material was purified by silica gel chromatography on a 4Og column, using DCM/MeOH 1-6%. The purified fractions were treated with 2N HCl in ether and concentrated to provide 1.5 g of N-[(1R, 35)-3-[[5-fluoro-2-(5-fluoro-1H- pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]amino]cyclohexyl]-morpholine-4-carboxamide as a white solid.
HCI D DCM

44e
Formation of (IS, S^-M-^-fluoro-S-CS-fluoro-1H-pyrrolo^S-^pyridin- 3-yl)phenyl)cyclohexane-1,3-diamine (44e)
To a solution of tert-butyl (IR, 35)-3-(2-fluoro-5-(5-fluoro-1-tosyl-lH-pyrrolo-[2,3- &]pyridin-3-yl)phenylamino)cyclohexylcarbamate, 44d, (0.65 g, 1.09 mmol) in methylene chloride (22 mL) was added hydrogen chloride (2.71 mL of 4M solution in 1,4-dioxane, 10.86 mmol). The reaction was heated to 50 °C and stirred for 6 hours. The mixture was cooled to room temperature and concentrated in vacuo, producing a yellow solid. The crude residue was purified via silica gel chromatography (25-50% Ethyl Acetate/hexanes gradient). Desired fractions were combined and concentrated in vacuo to produce 350 mg of 44e as a yellow powder.
General Scheme 67 SIMILAR TO A POINT BUT NOT SAME

(a) Pd/C (wet, Degussa), hydrogen, EtOH (b) 2,4-dichloro-5-fluoropyrimidine, 1Pr2NEt, THF, reflux (c) LiOH, THF/water, 5O°C
SIMILAR BUT NOT SAME
(d) DPPA, Et3N, THF, 85 °C (e) 5-fluoro-3-(4,4,5,5-tetramethyl-1,3 ,2-dioxaborolan-2-yl)-1- tosyl-l//-pyrrolo[2,3-i]pyridine, XPhos, Pd2(dba)3, K3PO4, 2-methylTHF, water, 125 °C (f)
Formation (IR, 35)-ethyl 3-aminocyclohexanecarboxylate (67b)
To a solution of (IR, 35)-ethyl 3-(benzyloxycarbonylamino)cyclohexane-carboxylate, 18b, (14.0 g, 45.9 mmol) in ethanol (3 mL) was added Pd/C (wet, Degussa (2.4 g, 2.3 mmol). The mixture was evacuated and then stirred under atmosphere of nitrogen at room temperature overnight. The reaction mixture was filtered through a pad of celite and the resulting filtrate concentrated in vacuo to provide an oil that was used without further purification.
Formation (IR, SS^-ethyl 3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexane- carboxylate (67c)
To a solution of (IR, 3«S)-ethyl S-aminocyclohexanecarboxylate, 67b, (5.1 g, 24.1 mmol) and 2,4-dichloro-5,-fluoropyrimidine (6.0 g, 36.0 mmol) in THF (60 mL) was added diisopropylethylamine (9.6 mL, 55.4 mmol). The mixture was heated to reflux overnight. The reaction was cooled to room temperature and concentrated in vacuo. The residue was diluted with water and extracted twice with ethyl acetate. The combined organic phases were dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (0-40% EtOAc/hexanes gradient) to provide 6.7 g of (IR, 35*)-ethyl 3-(2- chloro-5-fluoropyrimidin-4-ylamino)cyclohexane-carboxylate as a white solid: LCMS RT = 3.1 (M+H) 302.2.
Formation (IR, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexanecarboxylic acid (67d)
To a solution of (IR, 35*)-ethyl 3-(2-chloro-5-fluoropyrimidin-4- ylamino)cyclohexane-carboxylate, 67c, (20.0 g, 66.3 mmol) in THF (150 mL) was added added a solution of LiOH hydrate (8.3 g, 198.8 mmol) in 100ml water. The reaction mixture was stirred at 50 °C overnight, To the reaction mixture was added HCl (16.6 mL of 12 M solution, 198.8 mmol) and EtOAc. The organic phase was washed with brine and dried over MgSO4 and the solvent was removed under reduced pressure to afford 17.5 g of product that was used without further purification: 1H NMR (300 MHz, CDC13) δ 7.91 (d, J = 2.7 Hz, 2H), 5.24 (d, J = 7.3 Hz, 2H), 4.19 – 4.03 (m, 3H), 3.84 – 3.68 (m, 3H), 2.59 (ddd, J= 11.5, 8.2, 3.6 Hz, 2H), 2.38 (d, J = 12.4 Hz, 2H), 2.08 (d, J = 9.6 Hz, 6H), 1.99 – 1.76 (m, 5H), 1.63 – 1.34 (m, 6H), 1.32 – 1.15 (m, 4H).
Formation N-((1R, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexyl)- pyrrolidine-1-carboxamide (67e)
A solution of (IR, 35)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexane- carboxylic acid, 67d, (8.2 g, 30.0 mmol), (azido(phenoxy)phosphoryl)oxybenzene (9.7 mL, 45.0 mmol) and triethylamine (5.8 mL, 42.0 mmol) in THF (200 mL) was degassed under nitrogen for 15 minutes. The reaction mixture was heated at 85 °C for 30 minutes until LC/MS indicated complete consumption of carboxylic acid, 67d. To the reaction mixture was added pyrrolidine (7.5 mL, 90.0 mmol) and the reaction was heated at 85 °C for an additional 15 min. The mixture was diluted into brine and extracted with EtOAc. The organic phase was separated, dried over MgSO4. The product was isolated (6.25 g) by filtration after partial removal of solvent in vacuo: 1H NMR (300 MHz, CDC13) δ 7.87 (d, J = 2.8 Hz, 2H), 5.04 (d, J = 8.1 Hz, 2H), 4.09 (ddd, J = 26.9, 13.4, 5.6 Hz, 4H), 3.91 – 3.71 (m, 2H), 3.32 (t, J= 6.5 Hz, 7H), 2.45 (d, J= 11.5 Hz, 2H), 2.08 (dd, J= 22.1, 12.0 Hz, 4H), 1.96- 1.82 (m, 9H), 1.54 (dd, J= 18.6, 8.5 Hz, 2H), 1.22 – 1.01 (m, 6H).
Formation N-((IR, 3S>3-(5-fluoro-2-(5-fluoro-1-tosyl-1H-pyrrolo[2,3-b]pyridm-3- yl)pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide (67f)
A solution of N-((1R, 3«S)-3-(2-chloro-5-fluoropyrimidin-4-ylamino)cyclohexyl)- pyrrolidine-1-carboxamide, 67e, (6.8 g, 20.0 mmol), 5-fluoro-1-(p-tolylsulfonyl)-3-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)pyrrolo[2,3-b]pyridine, 44a, (12.5 g, 30.0 mmol) and K3PO4 (17.0 g, 80.0 mmol) in 2-methyl TΗF (180 mL) and water (20 mL) was degassed under nitrogen for 30 min. To the mixture was added dicyclohexyl-[2-(2,4,6- triisopropylphenyl)phenyl]phosphane (XPhos) (1.1 g, 2.4 mmol) and Pd2(dba)3 (0.5 g, 0.5 mmol). The reaction mixture was heated in a pressure bottle at 125 °C for 2.5 hr. The reaction mixture was filtered through celite, the solvent was removed under reduced pressure. The resulting residue was purified by silica gel chromatography (8%MeOΗ/CΗ2Cl2) to afford 11.5 g of the desired product: 1H ΝMR (300 MHz, CDC13) δ 8.54 (s, 1H), 8.49 (dd, J= 9.0, 2.8 Hz, 1H), 8.32 (d, J= 2.1 Hz, 1H), 8.13 (d, J= 8.3 Hz, 2H), 8.07 (d, J= 3.2 Hz, 1H), 7.30 (d, J = 8.5 Hz, 2H), 4.98 (d, J = 6.3 Hz, 1H), 4.37 – 4.16 (m, 1H), 4.08 (d, J = 7.3 Hz, 1H), 3.99 – 3.80 (m, 1H), 3.33 (t, J= 6.5 Hz, 4H), 2.52 (d, J= 11.6 Hz, 1H), 2.39 (s, 3H), 2.29 (d, J= 11.3 Hz, 1H), 2.12 (d, J= 11.1 Hz, 1H), 1.99 – 1.81 (m, 5H), 1.70 – 1.55 (m, 1H), 1.22 – 1.08 (m, 2H).
Formation N-((IR, 3S>3-(5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)- pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide (895)
A solution of N-((1R, 35)-3-(5-fluoro-2-(5-fluoro-1-tosyl-lH-pyrrolo[2,3-b]pyridin-3- yl)pyrimidin-4-ylamino)cyclohexyl)pyrrolidine-1-carboxamide, 67f, (11.5 g, 19.3 mmol) in TΗF (150 mL) was added sodium methoxide (4.173 g, 19.31 mmol). After stirring the reaction mixture for 2 minutes, the mixture was poured into an aqueous saturated solution of NaHCO3. The organic phase was washed with brine, dried over MgSO4 and the solvent was removed under reduced pressure. The resulting residue was purified by silica gel chromatography (10%MeOH/CH2Cl2) to afford 6.5g of the desired product. The product was converted to an HCl salt by dissolving in MeOH (100 mL) and adding 2.4 mL of 12M HCl solution at room temperature. The solution was stirred at for lhour and the HCl salt precipitated out and filtered to provide 7.05g of the HCl salt: 1H NMR (300 MHz, DMSO) δ 9.36 (s, 2H), 9.05 (d, J= 3.0 Hz, 2H), 8.49 (d, J= 5.6 Hz, 2H), 8.41 (dd, J= 2.6, 1.4 Hz, 2H), 8.31 (d, J= 9.5 Hz, 2H), 5.92 (s, 3H), 4.24 (s, 3H), 3.64 (s, 2H), 3.18 (t, J= 6.6 Hz, 7H), 2.07 (dt, J = 22.7, 11.5 Hz, 4H), 1.87 (t, J = 12.6 Hz, 4H), 1.77 (dd, J = 8.0, 5.3 Hz, 7H), 1.65 – 1.13 (m, 8H).
PATENT
INHIBITORS OF INFLUENZA VIRUSES REPLICATION
/////////VX-? , an Azaindolyl-Pyrimidine Inhibitor, Influenza Virus Replication, Vertex, preclinical, 1259498-06-0
O=C(NC1CCC[C@@H](C1)Nc2nc(ncc2F)\C\4=C\N=C3\N\C=C(\F)/C=C3/4)N5CCCCC5
VX 787, PIMODIVIR, for Avian influenza

VRT-0928787
VX-787
vx 787
| Vertex Pharmaceuticals |
Janssen Pharmaceuticals, under license from Vertex Pharmaceuticals, is developing VX-787 and its back-up compound VX-353, an influenza A viral replication inhibitor, for treating influenza A virus infection, including pandemic and avian influenza strains. In May 2015, VX-787 was in phase II clinical trial.

Useful for treating influenza virus infection. For concurrent filing see WO2015073476 (claiming the polymorphic forms of VX-787) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
Polymorphic forms of hydrochloride (A,F and D) and tosylate salts (form A) of VX-787 are claimed. , useful for treating influenza virus infection. For concurrent filing see WO2015073481 (claiming the processes for the synthesis of VX-787 ) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
WO2010148197
http://www.google.com/patents/WO2010148197A1?cl=en
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1070 was made in a similar fashion as described above for compounds 946 and 947.
………………….
WO 2013019828
http://www.google.com/patents/WO2013019828A1?cl=en
WO 2012083122
http://www.google.co.in/patents/WO2012083122A1?cl=en
Synthetic Scheme 1

(a) CHC13; (b) NaOMe, MeOH; (c) DPPA, Et3N, BnOH; (d) H2, Pd/C;
Synthetic Scheme 2

(a) Et3N, CH3CN; (b) cone. H2S04; (c) 9M H2S04; (d) Ag2C03, HOAc, DMSO, 100 °C; (e) X- phos, Pd2(dba)3, K3PO4, 2-methyl THF, H20, 120 °C (f) LiOH, THF, MeOH, 70 °C
Synthetic Scheme 3

(a) Et3N, THF; (b) chiral SFC separation; (c) 5-fluoro- l -(p-tolylsulfonyl)-3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-
………………………
See new patents
WO2015073491
……………………………..
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Vertex Pharmaceuticals Inc

Vertex Licenses VX-787 to Janssen Pharmaceuticals for the Treatment of Influenza
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that it has entered into a licensing agreement with Janssen Pharmaceuticals, Inc. for the worldwide development and commercialization of VX-787, a novel medicine discovered by Vertex for the treatment of influenza. As part of the agreement, Vertex will receive an up-front payment of $30 million from Janssen and has the potential to receive additional development and commercial milestone payments as well as royalties on future product sales. Vertex completed a Phase 2a study of VX-787 in 2013 that showed statistically significant improvements in viral and clinical measurements of influenza infection. VX-787 is designed to directly inhibit replication of the influenza virus.
“With a deep history in developing new medicines for viral infections and diseases, Janssen is well-positioned to advance the global development of VX-787 for the treatment of influenza,” said Jeffrey Leiden, M.D., Ph.D., Chairman, President and Chief Executive Officer of Vertex. “This collaboration provides important support for the continued development of VX-787 in influenza and contributes to our financial strength to enable continued investment in our key development programs for cystic fibrosis and in research aimed at discovering new medicines.”
About the Collaboration
Under the terms of the collaboration, Janssen will have full global development and commercialization rights to VX-787. Vertex will receive a $30 million up-front payment from Janssen and could receive additional development and commercial milestone payments as well as royalties on future product sales. The collaboration, and the related $30 million up-front payment, is subject to the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act.
About VX-787
VX-787 is an investigational medicine that is designed to directly inhibit replication of influenza A, including recent H1 (pandemic) and H5 (avian) influenza strains, based on in-vitro data. VX-787’s mechanism represents a new class of potential medicines for the treatment of influenza, distinct from neuraminidase inhibitors, the current standard of care for the treatment of influenza. VX-787 is intended to provide a rapid onset of action and an expanded treatment window.
In a Phase 2a influenza challenge study, statistically significant improvements in viral and clinical measurements of influenza infection were observed after treatment with VX-787. The study met its primary endpoint and showed a statistically significant decrease in the amount of virus in nasal secretions (viral shedding) over the seven-day study period. In addition, at the highest dosing regimen evaluated in the study, there was a statistically significant reduction in the severity and duration of influenza-like symptoms. In this study, VX-787 was generally well-tolerated, with no adverse events leading to discontinuation. Those who took part in the study volunteered to be experimentally exposed to an attenuated form of live H3N2 influenza A virus. H3N2 is a common type of influenza virus and was the most common type observed in the 2012/2013 influenza season in the United States.
VX-787 was discovered by Vertex scientists.
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
| WO2002024705A1 | 13 Sep 2001 | 28 Mar 2002 | Charles Jackson Barnett | Stereoselective process for preparing cyclohexyl amine derivatives |
| WO2003015798A1 | 13 Aug 2002 | 27 Feb 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | 30 Mar 2005 | 13 Oct 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006069258A1 * | 20 Dec 2005 | 29 Jun 2006 | Amgen Inc | Substituted heterocyclic compounds and methods of use |
| WO2007084557A2 | 17 Jan 2007 | 26 Jul 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2008079346A1 | 21 Dec 2007 | 3 Jul 2008 | Vertex Pharma | 5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009073300A1 | 31 Oct 2008 | 11 Jun 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | 17 Jun 2010 | 23 Dec 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2011008915A1 * | 15 Jul 2010 | 20 Jan 2011 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| US20100038988 | 12 Aug 2008 | 18 Feb 2010 | Gannon Ramy | Stator and Method of Making the Same |
| WO2003015798A1 | Aug 13, 2002 | Feb 27, 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2009073300A1 | Oct 31, 2008 | Jun 11, 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | Jun 17, 2010 | Dec 23, 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US20100038988 | Aug 12, 2008 | Feb 18, 2010 | Gannon Ramy | Stator and Method of Making the Same |
……
.

Vertex Pharmaceuticals’ Boston Campus, United States of America
Lynette Hopkinson VP Commercial Regulatory Affairs, Global Regulatory Affairs Vertex Pharmaceuticals Incorporated, United States

swati Patel, a lead analyst, shared a toast with Mir Hussain, a systems engineer, at Vertex Pharmaceuticals during the Friday beer hour, which features beer and chips for employees.
On Fridays around 5 o’clock, after a hard week of work, Frank Holland likes to unwind with a beer. And he doesn’t have to leave work to get one.
Holland is a research scientist at Vertex Pharmaceuticals, which every Friday rings in “beer hour,” offering free adult beverages and munchies to its 1,300 Boston employees.
For Holland, the weekly ritual is a chance to escape the bubble of his chemistry lab and bump into colleagues from other departments — as well as Vertex’s top executives, who regularly attend. For those who prefer grapes to hops, there is also wine.
“Some of the other companies I worked at, you really had to go out of your way to meet people,” said Holland, 32. “At Vertex all you have to do is show up in the cafeteria on a Friday afternoon.”
Sure, free beer is common at hip tech offices; some even have their own bars. But Vertex, best known for its treatment for cystic fibrosis, was doing this way before it was cool. The beer-hour tradition goes back to the company’s founding days, in 1989. Back then, it was just two dozen people in a small office in Cambridge. Someone went to a corner store, bought a case of beer and some chips, and beer hour was born.

Virginia Carden Carnahan
Vice President, New Product Planning and Strategy, Vertex Pharmaceuticals

A scientist works in the lab at Boston-based Vertex Pharmaceuticals.
Vertex Pharmaceuticals Headquarters Lobby
…………

Vertex Pharmaceuticals: Another FDA Orphan Drug Designation For Cystic Fibrosis
| FDA Orphan Drug Designation Database Record | |
| Generic Name: | (R)-1-(2,2-difluorobenzo [d][1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl) cyclopropanecarboxamide |
| Trade Name: | n/a |
| Date Designated: | 04-24-2014 |
| Orphan Designation: | Treatment of cystic fibrosis |
| Orphan Designation Status: | Designated |
| FDA Orphan Approval Status: | Not FDA Approved for Orphan Indication |
| Sponsor: | Vertex Pharmaceuticals Inc. 50 Northern Avenue Boston, MA 02210-1862 The sponsor address listed is the last reported by the sponsor to OOPD. |
.
VX-661 is a Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) corrector. VX-661 is being studied in combination with Kalydeco (Ivacaftor) for patients who have the F508del mutation. VX-661 is currently recruiting participants for a Phase II clinical trial to evaluate the safety and efficacy of VX-661 in combination with Kalydeco in subjects with CF who are homozygous (have 2 copies) for the F508del CFTR…
View original post 173 more words
