
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
 .
.
Bethany Halford on Twitter: “BMS-919373, from $BMS for …https://twitter.com/beth_halford/status/634105343719682048
Aug 19, 2015 – BMS–919373, from $BMS for atrial fibrillation #ACSBoston MEDI 1st disclosures @bmsnews pic.twitter.com/y3D4Yv2U7M.
BMS 919373
- Phase IIParoxysmal atrial fibrillation
- Phase IAcute coronary syndromes; Atrial fibrillation
-
- 01 Oct 2014Phase-I clinical trials in Atrial fibrillation in Canada (PO) (NCT02153437)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in Canada (PO) (NCT02156076)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in USA (PO)
- https://clinicaltrials.gov/ct2/show/NCT02153437
- https://clinicaltrials.gov/ct2/show/NCT02156076
- CAS HCL SALT 1272356-77-0
| Latest Stage of Development | Phase I |
| Standard Indication | Fibrillation |
| Indication Details | Treat atrial fibrillation |
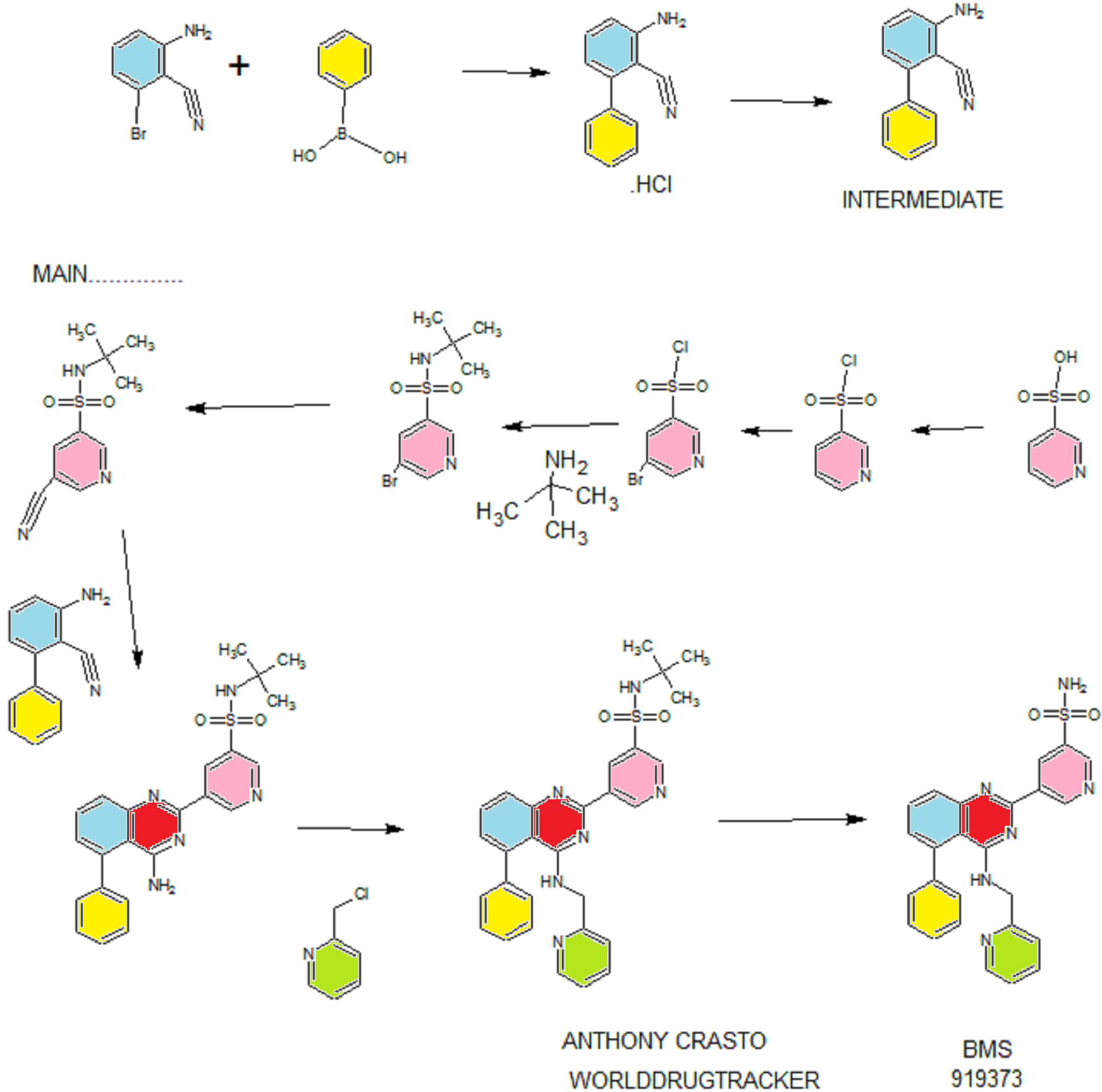
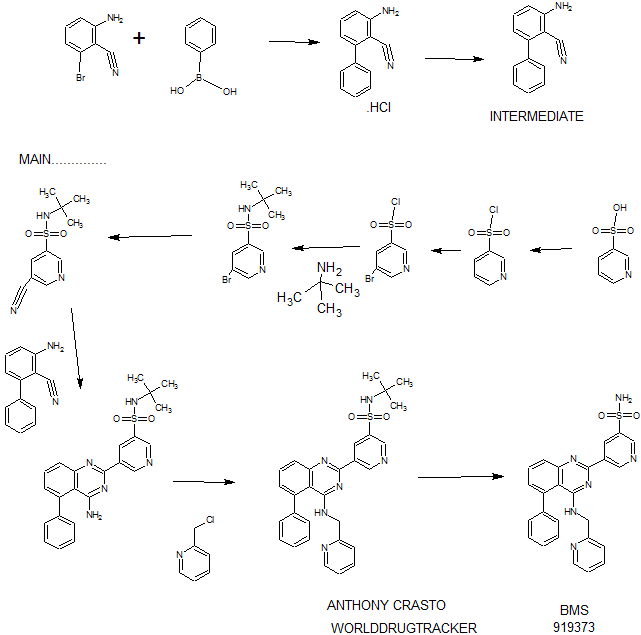
Synthesis
PATENT
WO 2011028741
http://www.google.co.in/patents/WO2011028741A1?cl=en
EXAMPLE 7
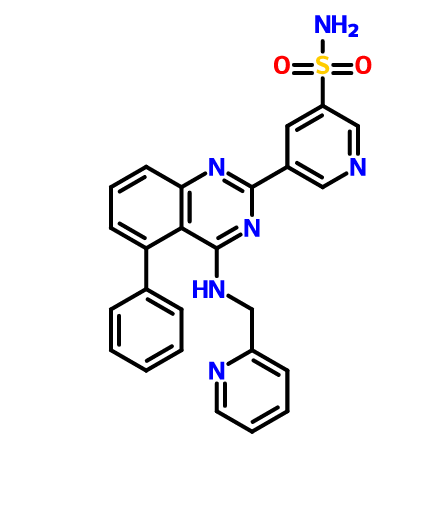
5-(5-Phenyl-4-(pyridin-2-ylmethylamino)quinazolin-2-yl)pyridine-3-sulfonamide

Step 1. Preparatio -Bromopyridine-3 -sulfonamide

See also U.S. Publication Nos. 2006/217387 and 2006/375834, and J. Org. Chem., 54:389 (1989). A mixture of pyridine-3 -sulfonic acid (10.3 g, 64.8 mmol), phosphorous pentachloride (20.82 g, 100 mmol) and phosphorous oxychloride (10 mL, 109 mmol) was heated to reflux where it stirred for 4h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was evaporated to dryness under reduced pressure to yield a residue. The residue was treated with bromine (6.00 mL, 1 16 mmol) and then heated to reflux where it stirred for 14h. After this time, the reaction mixture was cooled to 0 °C and then a saturated solution of NH4OH in ¾0 (40 mL) was slowly added. The resulting mixture was allowed to warm to room temperature where it stirred for 30 min. The reaction mixture was then filtered and the filter cake was washed with hexane to afford 5 -bromopyridine-3 -sulfonamide (6.0 g) as an off- white solid. The product was used without further purification. LCMS Method Q: retention time 0.75 min; [M+l] = 237.0.
Step 2. Preparation of pyridine-3-sulfonamide-5-ylboronic acid pinacol ester

See also WO2008/150827 Al and WO2008/144463. A mixture of 5- bromopyridine-3 -sulfonamide (1.5 g, 6.33 mmol), bis(pinacolato)diboron (2.41 g, 9.5 mmol) and potassium acetate (1.86 g, 19.0 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 15 min then (l, l’-bis(diphenylphosphino)- ferrocene)palladium (II) chloride dichloromethane complex (232 mg, 0.317 mmol) was added and the resulting mixture was degassed again with nitrogen for 10 min. At the conclusion of this period, the reaction mixture was heated in a microwave at 120 °C for 45 min. After this time, the reaction mixture was filtered through CELITE® and the filtrate was concentrated under reduced pressure to provide pyridine-3- sulfonamide-5-ylboronic acid pinacol ester (740 mg) as a brown solid. The product was used without further purification. XH NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.56-7.74 (bs, 2H), 1.17 (s, 12H).
Step 3. Example 7

To a solution of 2-chloro-5-phenyl-N-(pyridin-2-ylmethyl)quinazolin-4- amine (150 mg, 0.43 mmol) in 1,4-dioxane (6 mL) and ¾0 (1 mL) under nitrogen was added pyridine-3-sulfonamide-5-ylboronic acid pinacol ester (185 mg, 0.65 mmol), and potassium carbonate (119 mg, 0.86 mmol). Upon completion of addition, the mixture was degassed with nitrogen for 15 minutes and then (1, 1′- bis(diphenylphosphino)ferrocene)palladium (II) chloride dichloromethane complex (31 mg, 0.043 mmol) was added. The resulting mixture was again degassed with nitrogen for 10 min. After this time, the mixture was heated to 90 °C where it stirred for 16h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was quenched by the addition of water and then transferred to a separation funnel. The aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with water and saturated NaCl, dried over Na2S04, filtered and concentrated under reduced pressure. The resulting concentrate was purified by preparative TLC using 5% methanol in dichloromethane to afford Example 7 (50 mg) as a brown solid. ‘H NMR (400 MHz, DMSO-d6) δ (ppm): 9.81 (s, 1H), 9.17 (s, 1H), 9.09 (s, 1H), 8.24 (d, J= 4.4 Hz, 1H), 7.94 (d, J=7.2 Hz, 1H), 7.86 (t, J= 7.6 Hz, 1Η),7.75-7.72 (t, J= 7.6 Hz, 3H), 7.59-7.51 (m, 5H), 7.34 (d, J=7.2 Hz, 2H), 7.24 (t, J=6.4 Hz, 1H), 6.98 (t, J= 3.2 Hz, 1H), 4.77 (d, J= 4.0 Hz, 2H). LCMS Method Q: retention time 1.39 min; [M+l] = 469.0. HPLC Method B: purity 98.1%, retention time = 8.74 min. [00120] Alternatively, Example 7 can be synthesized as follows:
Step 1. Preparation of 5-Bromo-pyridine-3-sulfonyl chloride

PC15 (2.95 Kg, 14.16 moles) and POCl3 (2.45 Kg, 15.98 moles) were added into pyridine-3 -sulfonic acid (1.5 Kg, 9.42 mol) in 10 L RB flask equipped with mechanical stirrer under inert atmosphere. The reaction mass was heated to 120- 125°C where it stirred for 18 h. After this time, the reaction progress was monitored by HPLC, which indicated the reaction was complete. Excess POCI3 was removed under vacuum to give a residue. The residue was cooled to ambient temperature and bromine (1.2 Kg, 7.5 moles) was added. Upon completion of addition, the resulting mixture was heated to 120-125°C where it stirred for 5 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then poured into ice-water (10 L), and the resulting mixture was extracted with DCM (10.5 Lx2). The DCM extracts were combined and the solvent was removed under vacuum to yield crude product (1.8 Kg, 74.4% yield).
Step 2. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

Crude 5 -bromopyridine-3-sulfonyl chloride from step 1 above was dissolved in THF (14 L, 8 vol) and then transferred to a 20 L RB flask equipped with mechanical stirrer under inert atmosphere. The solution was cooled to 0-5°C and tert- butyl amine (1.95 Kg, 26.66 moles) was added at 0-5°C. Upon completion of addition, the reaction mixture was warmed to ambient temperature where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The solvent was evaporated under vacuum to give a thick residue. The residue was dissolved in ethyl acetate (18 L, 12 vol). The organic layer was separated, washed with water (9 L, 5 vol) and then concentrated under vacuum to yield a residue. Hexanes (9 L, 5 vol) were added to the residue and the product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.5 Kg, 54.28% overall yield). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 8.99 (d, J = 2Hz, 1H), 8.81 (d, J= 2 Hz, 1H), 8.29 (t, J= 2Hz, 1H). [M++l] = 293. Step 3. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

5 -Bromo-N-tert-butylpyridine-3 -sulfonamide (1.5 Kg, 5.11 moles) was dissolved in dimethylformamide (7.5 L, 5 vol) and the solution was added to a 20 L glass-lined reactor equipped with mechanical stirrer. The solution was degassed with nitrogen for 30 min. After this time, potassium ferrocyanide trihydrate (867 g, 2.05 moles), sodium carbonate (1.08 Kg, 10.189 moles), copper (I) iodide (73.2 g, 0.374 moles) and dichloro-bis (triphenylphosphine) palladium (II) (71.6 g, 0.102 moles) were added. Upon completion of addition, the reaction mixture was heated to 120- 125°C where it stirred for 4 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then filtered through a celite bed. Water (18 L, 12 vol) was added into the filtrate and the resulting mixture was extracted with ethyl acetate (7.5L*2). The organic layers were combined, washed with water and then concentrated to yield a thick residue. Hexanes (7.5 L, 5 vol) were added to the residue. The product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.0 Kg, 82.8% yield, 89% purity by HPLC). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 9.21 – 9.24 (d,d J= 7.2Hz, 3.2Hz, 2H), 8.70-8.71(m,lH), 7.98 (s, lH). [M++l] = 239.2.
Step 4. Preparation of 3-aminobiphenyl-2-carbonitrile

2-Amino-6-bromo-benzonitrile (1.0 Kg, 5.07 moles) and toluene (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer under inert atmosphere. Potassium acetate (996 g, 10.16 moles) and phenylboronic acid (866, 7.10 moles) were added into the solution and the solution was degassed with nitrogen for 30 min. After this time, dichloro-bis (triphenylphosphine) palladium (II) (17.8 g, 0.025 moles) was added to the reaction mixture at ambient temperature. The mixture was heated to 110°C, where it stirred for 17 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was completed. The reaction mixture was filtered through a celite bed. The filtrate was transferred back to the reactor and concentrated hydrochloric acid (-35%, 2 L, 2 vol) was charged to the reactor at ambient temperature. The HCl salt of the title compound precipitated out from the reaction and was collected by filtration. The HCl salt was transferred into the 20 L reactor and then made basic with 10% NaOH solution (pH 8-9). The resulting product was extracted with ethyl acetate (10 L, 10 vol). The ethyl acetate layer was washed with water (5 L, 5 vol) and then the solvent was evaporated under vacuum to give a residue. Hexanes (5 L, 5 vol) were added to the residue at 35-40°C, and the resulting slurry was cooled to ambient temperature. Once at the prescribed temperature, the product was collected by filtration to provide a pale yellow solid (802 g, 81.4%, 99% by HPLC). XH NMR (DMSO-D6, 400 MHz, δ ppm); 7.43-7.52 (m, 5H), 7.33-7.37 (m, 1H), 6.83 (d, J=8Hz, 1H), 6.62 (d, J=8Hz, 1H), 6.1 (s, 2H). ES-MS: [M++l] = 194.23.
Step 5. Preparation of 5-(4-amino-5-phenylquinazolin-2-yl)-N-tert-butylpyridine-3-

3-Aminobiphenyl-2-carbonitrile (1028 g, 5.30 moles), 5-bromo-N-tert- butylpyridine-3 -sulfonamide (1440 g, 5.55 moles) and 1,4-dioxane (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. Sodium tert-butoxide (1.275 Kg 12.870 moles) was added to the solution portion-wise at 20- 30°C. Upon completion of addition, the reaction mixture was heated to reflux where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 30-35°C and then poured into water (40 L, 40 vol). The resulting mixture was extracted with DCM (20 L*2). The DCM layers were combined, washed with water (10 L, 10 vol) and then dried over sodium sulfate. The solvent was evaporated under vacuum to give a residue. Isopropyl alcohol (1.2 L, 1.2 vol) was added to the residue at 40°C. The resulting precipitate slurry was cooled to 10-15°C and then stirred for 2 h. After this time, the precipitate was collected by filtration and dried at 50°C for 16 h to yield the product (1.9 Kg, 82.9% yield, 99% purity by HPLC). Ή NMR (DMSO-D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.11 (s, 2H), 7.83-7.94 (m, 4H), 7.49-7.60 (m, 5H), 7.31 (d,d /=6.8Hz,1.2Hz, 1H). ES-MS: [M++l] = 433.53.
Step 6. Preparation of N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide

2-(Chloromethyl) pyridine hydrochloride (564 g, 3.44 moles) and dimethyl acetamide (7L, 7 vol) were added to a 20 L RB flask- 1 equipped with mechanical stirrer under inert atmosphere. The resulting solution was cooled to 0- 5°C and triethylamine (346.3, 3.44 moles) was added at 0-5°C. 5-(4-Amino-5- phenylquinazolin-2-yl)-N-tert-butylpyridine-3-sulfonamide (1.0 Kg. 2.306 moles) and dimethylacetamide (4 L, 4 vol) were added to a separate 20 L RB flask-2 equipped with mechanical stirrer under inert atmosphere. This solution was cooled to 0-5°C and sodium tert-butoxide (884 g, 9.24 moles) was added at 0-5°C. The resulting solution was stirred to affect dissolution and then transferred to the RB flask- 1 at 0- 5°C. Upon completion of addition, the reaction mixture was stirred at 0-5°C for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The reaction mass was poured into water (60 L, 60 vol) with stirring. The crude product was collected by filtration and dried at 60°C for 12 h. After this time, the dried material was dissolved in THF (20 L, 20 vol). Upon dissolution, 6M HC1 in isopropyl alcohol (1 L, 1 vol) was added at 20-25°C. The crude HCL salt of the product was obtained a pale-yellow free flow solid (920 g, 71% yield, 93% purity by HPLC). The crude HC1 salt (1.345 Kg, 2.56moles), methanol (6.7 L, 5 vol) and dichloromethane (13.5 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The slurry was stirred for 20-30 min at 30°C. After this time, the solvent was distilled to 4 vol with respect to input under vacuum. The resulting slurry was cooled to 20-25°C, where stirred for 2 h. At the conclusion of this period, the slurry was filtered and dried at 50°C for 6 h to yield the product (1.1 Kg, 82% yield, 98% purity by HPLC). XH NMR (DMSO- D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.10-9.14 (m, 2H), 8.39 (s, 1H), 7.92-8.03 (m, 4H), 7.56-7.58 (m, 5H), 7.43-7.49 (m, 3H), 7.1 (bs, 1H), 4.88 (s, 2H), 1.17 (2, 9H).
Step 7. Example 7

N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide (1.0 Kg, 1.9 moles) and concentrated hydrochloric acid (7 L, 7 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The reaction mixture was heated to 90-100°C where it stirred for 1 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 5-10°C and the pH was adjusted to 1.7 to 2.0 using 12% aqueous sodium hydroxide solution. Once at the prescribed pH, the crude HC1 salt of the product was collected by filtration. The HC1 salt filter cake and ethanol (5 L, 5 vol) were added to 10 L glass-lined reactor equipped with a mechanical stirrer. The resulting mixture was made basic to pH 7-8 at 20-25°C using triethyl amine (2.25 Kg, 22.23 moles). Once at the prescribed pH, the basic mixture was stirred for 2 h. After this time, the free base of product was filtered and washed with water (10 L, 10 vol) followed by ethanol (2L, 2 vol). The resulting product was dried at 50-55°C for 8 h to yield Example 7 (644 g, 72% yield, 99.9% purity by HPLC).
XH NMR (DMSO-D6, 400 MHz, δ ppm); 9.81 (d, J=2.0Hz, 1H), 9.18 (t, J=2Hz, 1H), 9.1 1 (d, J=2Hz, 1H), 8.23 (d, J=4.4Hz, 1H), 7.92-7.94 (m, 1H), 7.83-7.87 (m, 1H), 7.78 (s, 2H), 7.70-7.72 (m, 1H), 7.50-7.59 (m, 5H), 7.31-7.34 (m, 2H), 7.22-7.25 (m, 1H), 6.95 (t, J=4Hz, 1H), 4.76 (d, J=4Hz, 2H). ES-MS: [M++l] = 469.
/////////atrial fibrillation, Potassium channel Kv1.5 (KCNA5) inhibitor, IKur antagonist, Bristol-Myers Squibb Co., BMS 919373, BMS-919373, PHASE 2
NS(=O)(=O)c1cc(cnc1)c4nc2cccc(c2c(NCc3ccccn3)n4)c5ccccc5

