
Home » Posts tagged 'atrial fibrillation'
Tag Archives: atrial fibrillation
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SULCARDINE SULPHATE
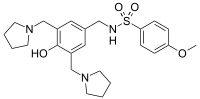
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
BMS 919373
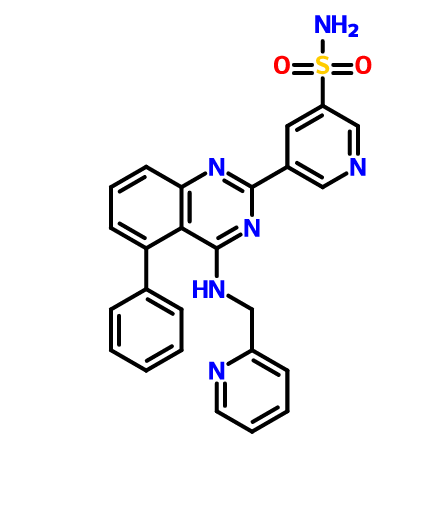
 .
.
Bethany Halford on Twitter: “BMS-919373, from $BMS for …https://twitter.com/beth_halford/status/634105343719682048
Aug 19, 2015 – BMS–919373, from $BMS for atrial fibrillation #ACSBoston MEDI 1st disclosures @bmsnews pic.twitter.com/y3D4Yv2U7M.
BMS 919373
- Phase IIParoxysmal atrial fibrillation
- Phase IAcute coronary syndromes; Atrial fibrillation
-
- 01 Oct 2014Phase-I clinical trials in Atrial fibrillation in Canada (PO) (NCT02153437)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in Canada (PO) (NCT02156076)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in USA (PO)
- https://clinicaltrials.gov/ct2/show/NCT02153437
- https://clinicaltrials.gov/ct2/show/NCT02156076
- CAS HCL SALT 1272356-77-0
| Latest Stage of Development | Phase I |
| Standard Indication | Fibrillation |
| Indication Details | Treat atrial fibrillation |
Synthesis
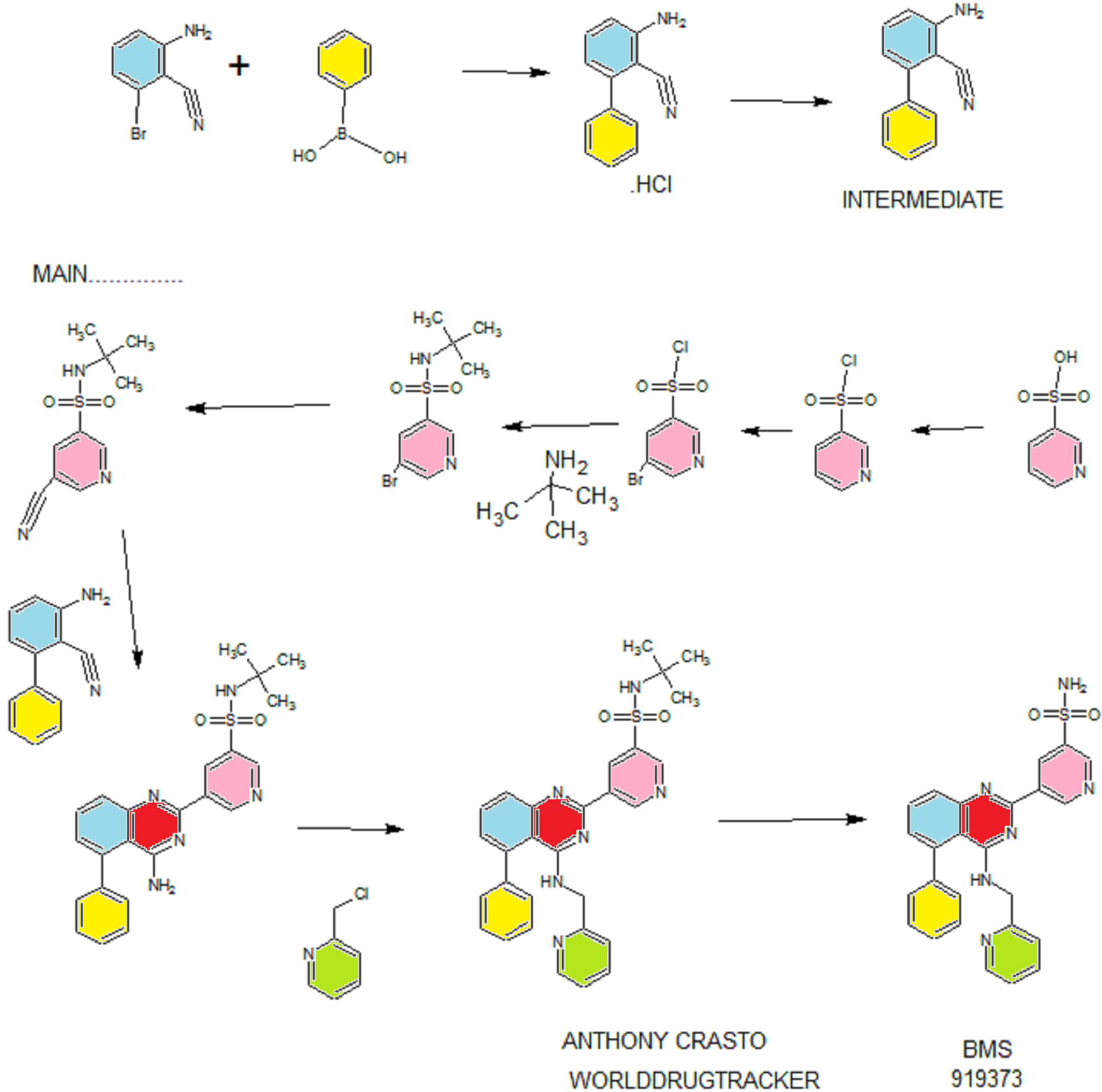
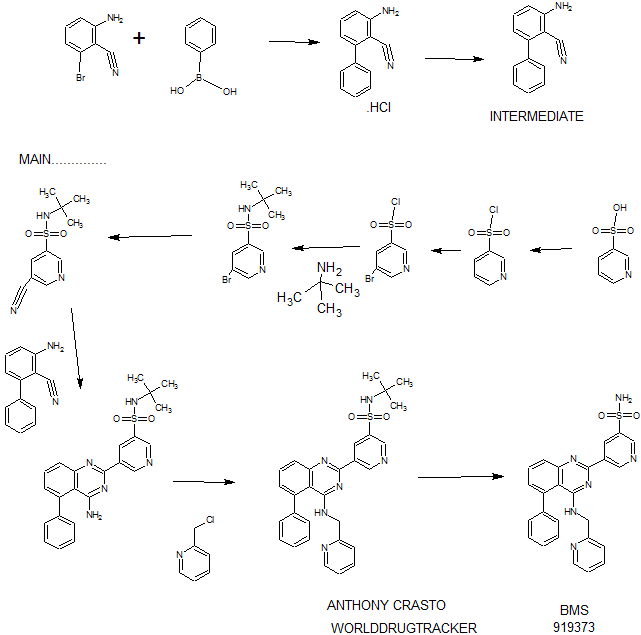
PATENT
WO 2011028741
http://www.google.co.in/patents/WO2011028741A1?cl=en
EXAMPLE 7
5-(5-Phenyl-4-(pyridin-2-ylmethylamino)quinazolin-2-yl)pyridine-3-sulfonamide

Step 1. Preparatio -Bromopyridine-3 -sulfonamide

See also U.S. Publication Nos. 2006/217387 and 2006/375834, and J. Org. Chem., 54:389 (1989). A mixture of pyridine-3 -sulfonic acid (10.3 g, 64.8 mmol), phosphorous pentachloride (20.82 g, 100 mmol) and phosphorous oxychloride (10 mL, 109 mmol) was heated to reflux where it stirred for 4h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was evaporated to dryness under reduced pressure to yield a residue. The residue was treated with bromine (6.00 mL, 1 16 mmol) and then heated to reflux where it stirred for 14h. After this time, the reaction mixture was cooled to 0 °C and then a saturated solution of NH4OH in ¾0 (40 mL) was slowly added. The resulting mixture was allowed to warm to room temperature where it stirred for 30 min. The reaction mixture was then filtered and the filter cake was washed with hexane to afford 5 -bromopyridine-3 -sulfonamide (6.0 g) as an off- white solid. The product was used without further purification. LCMS Method Q: retention time 0.75 min; [M+l] = 237.0.
Step 2. Preparation of pyridine-3-sulfonamide-5-ylboronic acid pinacol ester

See also WO2008/150827 Al and WO2008/144463. A mixture of 5- bromopyridine-3 -sulfonamide (1.5 g, 6.33 mmol), bis(pinacolato)diboron (2.41 g, 9.5 mmol) and potassium acetate (1.86 g, 19.0 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 15 min then (l, l’-bis(diphenylphosphino)- ferrocene)palladium (II) chloride dichloromethane complex (232 mg, 0.317 mmol) was added and the resulting mixture was degassed again with nitrogen for 10 min. At the conclusion of this period, the reaction mixture was heated in a microwave at 120 °C for 45 min. After this time, the reaction mixture was filtered through CELITE® and the filtrate was concentrated under reduced pressure to provide pyridine-3- sulfonamide-5-ylboronic acid pinacol ester (740 mg) as a brown solid. The product was used without further purification. XH NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.56-7.74 (bs, 2H), 1.17 (s, 12H).
Step 3. Example 7

To a solution of 2-chloro-5-phenyl-N-(pyridin-2-ylmethyl)quinazolin-4- amine (150 mg, 0.43 mmol) in 1,4-dioxane (6 mL) and ¾0 (1 mL) under nitrogen was added pyridine-3-sulfonamide-5-ylboronic acid pinacol ester (185 mg, 0.65 mmol), and potassium carbonate (119 mg, 0.86 mmol). Upon completion of addition, the mixture was degassed with nitrogen for 15 minutes and then (1, 1′- bis(diphenylphosphino)ferrocene)palladium (II) chloride dichloromethane complex (31 mg, 0.043 mmol) was added. The resulting mixture was again degassed with nitrogen for 10 min. After this time, the mixture was heated to 90 °C where it stirred for 16h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was quenched by the addition of water and then transferred to a separation funnel. The aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with water and saturated NaCl, dried over Na2S04, filtered and concentrated under reduced pressure. The resulting concentrate was purified by preparative TLC using 5% methanol in dichloromethane to afford Example 7 (50 mg) as a brown solid. ‘H NMR (400 MHz, DMSO-d6) δ (ppm): 9.81 (s, 1H), 9.17 (s, 1H), 9.09 (s, 1H), 8.24 (d, J= 4.4 Hz, 1H), 7.94 (d, J=7.2 Hz, 1H), 7.86 (t, J= 7.6 Hz, 1Η),7.75-7.72 (t, J= 7.6 Hz, 3H), 7.59-7.51 (m, 5H), 7.34 (d, J=7.2 Hz, 2H), 7.24 (t, J=6.4 Hz, 1H), 6.98 (t, J= 3.2 Hz, 1H), 4.77 (d, J= 4.0 Hz, 2H). LCMS Method Q: retention time 1.39 min; [M+l] = 469.0. HPLC Method B: purity 98.1%, retention time = 8.74 min. [00120] Alternatively, Example 7 can be synthesized as follows:
Step 1. Preparation of 5-Bromo-pyridine-3-sulfonyl chloride

PC15 (2.95 Kg, 14.16 moles) and POCl3 (2.45 Kg, 15.98 moles) were added into pyridine-3 -sulfonic acid (1.5 Kg, 9.42 mol) in 10 L RB flask equipped with mechanical stirrer under inert atmosphere. The reaction mass was heated to 120- 125°C where it stirred for 18 h. After this time, the reaction progress was monitored by HPLC, which indicated the reaction was complete. Excess POCI3 was removed under vacuum to give a residue. The residue was cooled to ambient temperature and bromine (1.2 Kg, 7.5 moles) was added. Upon completion of addition, the resulting mixture was heated to 120-125°C where it stirred for 5 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then poured into ice-water (10 L), and the resulting mixture was extracted with DCM (10.5 Lx2). The DCM extracts were combined and the solvent was removed under vacuum to yield crude product (1.8 Kg, 74.4% yield).
Step 2. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

Crude 5 -bromopyridine-3-sulfonyl chloride from step 1 above was dissolved in THF (14 L, 8 vol) and then transferred to a 20 L RB flask equipped with mechanical stirrer under inert atmosphere. The solution was cooled to 0-5°C and tert- butyl amine (1.95 Kg, 26.66 moles) was added at 0-5°C. Upon completion of addition, the reaction mixture was warmed to ambient temperature where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The solvent was evaporated under vacuum to give a thick residue. The residue was dissolved in ethyl acetate (18 L, 12 vol). The organic layer was separated, washed with water (9 L, 5 vol) and then concentrated under vacuum to yield a residue. Hexanes (9 L, 5 vol) were added to the residue and the product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.5 Kg, 54.28% overall yield). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 8.99 (d, J = 2Hz, 1H), 8.81 (d, J= 2 Hz, 1H), 8.29 (t, J= 2Hz, 1H). [M++l] = 293. Step 3. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

5 -Bromo-N-tert-butylpyridine-3 -sulfonamide (1.5 Kg, 5.11 moles) was dissolved in dimethylformamide (7.5 L, 5 vol) and the solution was added to a 20 L glass-lined reactor equipped with mechanical stirrer. The solution was degassed with nitrogen for 30 min. After this time, potassium ferrocyanide trihydrate (867 g, 2.05 moles), sodium carbonate (1.08 Kg, 10.189 moles), copper (I) iodide (73.2 g, 0.374 moles) and dichloro-bis (triphenylphosphine) palladium (II) (71.6 g, 0.102 moles) were added. Upon completion of addition, the reaction mixture was heated to 120- 125°C where it stirred for 4 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then filtered through a celite bed. Water (18 L, 12 vol) was added into the filtrate and the resulting mixture was extracted with ethyl acetate (7.5L*2). The organic layers were combined, washed with water and then concentrated to yield a thick residue. Hexanes (7.5 L, 5 vol) were added to the residue. The product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.0 Kg, 82.8% yield, 89% purity by HPLC). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 9.21 – 9.24 (d,d J= 7.2Hz, 3.2Hz, 2H), 8.70-8.71(m,lH), 7.98 (s, lH). [M++l] = 239.2.
Step 4. Preparation of 3-aminobiphenyl-2-carbonitrile

2-Amino-6-bromo-benzonitrile (1.0 Kg, 5.07 moles) and toluene (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer under inert atmosphere. Potassium acetate (996 g, 10.16 moles) and phenylboronic acid (866, 7.10 moles) were added into the solution and the solution was degassed with nitrogen for 30 min. After this time, dichloro-bis (triphenylphosphine) palladium (II) (17.8 g, 0.025 moles) was added to the reaction mixture at ambient temperature. The mixture was heated to 110°C, where it stirred for 17 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was completed. The reaction mixture was filtered through a celite bed. The filtrate was transferred back to the reactor and concentrated hydrochloric acid (-35%, 2 L, 2 vol) was charged to the reactor at ambient temperature. The HCl salt of the title compound precipitated out from the reaction and was collected by filtration. The HCl salt was transferred into the 20 L reactor and then made basic with 10% NaOH solution (pH 8-9). The resulting product was extracted with ethyl acetate (10 L, 10 vol). The ethyl acetate layer was washed with water (5 L, 5 vol) and then the solvent was evaporated under vacuum to give a residue. Hexanes (5 L, 5 vol) were added to the residue at 35-40°C, and the resulting slurry was cooled to ambient temperature. Once at the prescribed temperature, the product was collected by filtration to provide a pale yellow solid (802 g, 81.4%, 99% by HPLC). XH NMR (DMSO-D6, 400 MHz, δ ppm); 7.43-7.52 (m, 5H), 7.33-7.37 (m, 1H), 6.83 (d, J=8Hz, 1H), 6.62 (d, J=8Hz, 1H), 6.1 (s, 2H). ES-MS: [M++l] = 194.23.
Step 5. Preparation of 5-(4-amino-5-phenylquinazolin-2-yl)-N-tert-butylpyridine-3-

3-Aminobiphenyl-2-carbonitrile (1028 g, 5.30 moles), 5-bromo-N-tert- butylpyridine-3 -sulfonamide (1440 g, 5.55 moles) and 1,4-dioxane (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. Sodium tert-butoxide (1.275 Kg 12.870 moles) was added to the solution portion-wise at 20- 30°C. Upon completion of addition, the reaction mixture was heated to reflux where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 30-35°C and then poured into water (40 L, 40 vol). The resulting mixture was extracted with DCM (20 L*2). The DCM layers were combined, washed with water (10 L, 10 vol) and then dried over sodium sulfate. The solvent was evaporated under vacuum to give a residue. Isopropyl alcohol (1.2 L, 1.2 vol) was added to the residue at 40°C. The resulting precipitate slurry was cooled to 10-15°C and then stirred for 2 h. After this time, the precipitate was collected by filtration and dried at 50°C for 16 h to yield the product (1.9 Kg, 82.9% yield, 99% purity by HPLC). Ή NMR (DMSO-D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.11 (s, 2H), 7.83-7.94 (m, 4H), 7.49-7.60 (m, 5H), 7.31 (d,d /=6.8Hz,1.2Hz, 1H). ES-MS: [M++l] = 433.53.
Step 6. Preparation of N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide

2-(Chloromethyl) pyridine hydrochloride (564 g, 3.44 moles) and dimethyl acetamide (7L, 7 vol) were added to a 20 L RB flask- 1 equipped with mechanical stirrer under inert atmosphere. The resulting solution was cooled to 0- 5°C and triethylamine (346.3, 3.44 moles) was added at 0-5°C. 5-(4-Amino-5- phenylquinazolin-2-yl)-N-tert-butylpyridine-3-sulfonamide (1.0 Kg. 2.306 moles) and dimethylacetamide (4 L, 4 vol) were added to a separate 20 L RB flask-2 equipped with mechanical stirrer under inert atmosphere. This solution was cooled to 0-5°C and sodium tert-butoxide (884 g, 9.24 moles) was added at 0-5°C. The resulting solution was stirred to affect dissolution and then transferred to the RB flask- 1 at 0- 5°C. Upon completion of addition, the reaction mixture was stirred at 0-5°C for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The reaction mass was poured into water (60 L, 60 vol) with stirring. The crude product was collected by filtration and dried at 60°C for 12 h. After this time, the dried material was dissolved in THF (20 L, 20 vol). Upon dissolution, 6M HC1 in isopropyl alcohol (1 L, 1 vol) was added at 20-25°C. The crude HCL salt of the product was obtained a pale-yellow free flow solid (920 g, 71% yield, 93% purity by HPLC). The crude HC1 salt (1.345 Kg, 2.56moles), methanol (6.7 L, 5 vol) and dichloromethane (13.5 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The slurry was stirred for 20-30 min at 30°C. After this time, the solvent was distilled to 4 vol with respect to input under vacuum. The resulting slurry was cooled to 20-25°C, where stirred for 2 h. At the conclusion of this period, the slurry was filtered and dried at 50°C for 6 h to yield the product (1.1 Kg, 82% yield, 98% purity by HPLC). XH NMR (DMSO- D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.10-9.14 (m, 2H), 8.39 (s, 1H), 7.92-8.03 (m, 4H), 7.56-7.58 (m, 5H), 7.43-7.49 (m, 3H), 7.1 (bs, 1H), 4.88 (s, 2H), 1.17 (2, 9H).
Step 7. Example 7

N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide (1.0 Kg, 1.9 moles) and concentrated hydrochloric acid (7 L, 7 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The reaction mixture was heated to 90-100°C where it stirred for 1 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 5-10°C and the pH was adjusted to 1.7 to 2.0 using 12% aqueous sodium hydroxide solution. Once at the prescribed pH, the crude HC1 salt of the product was collected by filtration. The HC1 salt filter cake and ethanol (5 L, 5 vol) were added to 10 L glass-lined reactor equipped with a mechanical stirrer. The resulting mixture was made basic to pH 7-8 at 20-25°C using triethyl amine (2.25 Kg, 22.23 moles). Once at the prescribed pH, the basic mixture was stirred for 2 h. After this time, the free base of product was filtered and washed with water (10 L, 10 vol) followed by ethanol (2L, 2 vol). The resulting product was dried at 50-55°C for 8 h to yield Example 7 (644 g, 72% yield, 99.9% purity by HPLC).
XH NMR (DMSO-D6, 400 MHz, δ ppm); 9.81 (d, J=2.0Hz, 1H), 9.18 (t, J=2Hz, 1H), 9.1 1 (d, J=2Hz, 1H), 8.23 (d, J=4.4Hz, 1H), 7.92-7.94 (m, 1H), 7.83-7.87 (m, 1H), 7.78 (s, 2H), 7.70-7.72 (m, 1H), 7.50-7.59 (m, 5H), 7.31-7.34 (m, 2H), 7.22-7.25 (m, 1H), 6.95 (t, J=4Hz, 1H), 4.76 (d, J=4Hz, 2H). ES-MS: [M++l] = 469.
/////////atrial fibrillation, Potassium channel Kv1.5 (KCNA5) inhibitor, IKur antagonist, Bristol-Myers Squibb Co., BMS 919373, BMS-919373, PHASE 2
NS(=O)(=O)c1cc(cnc1)c4nc2cccc(c2c(NCc3ccccn3)n4)c5ccccc5
