
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

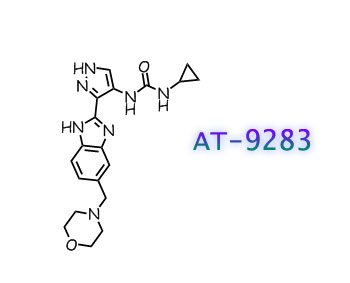
AT9283, AT 9283
N-cyclopropyl-N’-[3-[6-(4-morpholinylmethyl)-1H-benzimidazol-2-yl]-1H-pyrazol-4-yl]urea
1-cyclopropyl-3-[(3Z)-3-[5-(morpholin-4-ylmethyl)benzimidazol-2-ylidene]-1,2-dihydropyrazol-4-yl]urea
| 896466-04-9 | |
| Molecular Weight | 381.43 |
| Molecular Formula | C19H23N7O2 |
CAS
896466-04-9, 896466-57-2 ((±)-Lactic acid), 896466-61-8 (HCl), 896466-55-0 (methanesulfonate)
MolFormulaC22H29N7O5
MolWeight471.5096
CAS 896466-76-5 L LACTATE
(2S)-2-Hydroxypropanoic acid compd. with N-cyclopropyl-N’-[3-[6-(4-morpholinylmethyl)-1H-benzimidazol-2-yl]-1H-pyrazol-4-yl]urea
Astex Therapeutics Ltd, INNOVATOR

AT-9283 is a potent AuroraA/AuroraB and multi-kinase inhibitor. AT-9283 has shown to inhibit growth and survival of multiple solid tumor cell lines and is efficacious in mouse xenograft models.
AT 9283 is a substance being studied in the treatment of some types of cancer. It is small molecule a multi-targeted c-ABL, JAK2, Aurora A and B inhibition with 4, 1.2, 1.1 ad approximate 3 nM for Bcr-Abl (T3151), Jak2 and Jak3 aurora A and B, respectively. It blocks enzymes (Aurora kinases) involved in cell division and may kill cancer cells
WO2006070195 to Astex Therapeuitcs discloses pyrazole compounds of the general structure shown below as kinase inhibitors.

The compound AT9283 is in phase II clinical trials for treating advanced or metastatic solid tumors or Non-Hodgkin’s Lymphoma. AT9283 is shown below.


a Reagents and conditions:
(a) SOCl2, THF, DMF; (b) morpholine, THF, Et3N; ………FORMATION OOF ACID CHLORIDE AND COUPLING WITH MORPHOLINE
(c) NaBH4, BF3.OEt2, THF; …………..KETO TO CH2
(d) 10% Pd-C, H2, EtOH; TWO NITRO GPS TO TWO AMINO , REDN
(e) EDC, HOBt, DMF; (f) AcOH, reflux;COUPLING WITH 4-Nitro-lH-pyrazole-3-carboxylic acid
(g) 10%Pd-C, H2, DMF; NITRO GP TO AMINO
(h) standard amide and urea coupling methods
WO2006070195
https://www.google.co.in/patents/WO2006070195A1?cl=en
Stage 10: Synthesis of l-cvclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- beiizoimidazol-2-ylV 1 H-pyrazol-4-yli -urea.

To a mixture of 7-morpholin-4-ylmethyl-2,4-dihydro- 1 ,2,4,5a, 10- pentaaza- cyclopenta[a]fluoren-5-one (10.7 g, 32.9 mmol) in NMP (65 mL) was added cyclopropylamine (6.9 mL, 99 mmol). The mixture was heated at 100 0C for 5 h. LC/MS analysis indicated -75% conversion to product, therefore a further portion of cyclopropylamine (2.3 mL, 33 mmol) was added, the mixture heated at 100 0C for 4 h and then cooled to ambient. The mixture was diluted with water (100 mL) and extracted with EtOAc (100 niL). The organic portion was washed with sat. aq. NH4Cl (2 x 50 mL) and brine (50 rnL) and then the aqueous portions re-extracted with EtOAc (3 x 100 mL). The combined organic portions were dried over MgSO4 and reduced in vacuo to give l-cycloρropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea as an orange glassy solid (9.10 g).
Stage 11: Synthesis of l-cvclopropyl-S-P-fS-morpholin^-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yll-urea, L-lactate salt

To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea (9.10 g, 24 mmol) in EtOAc-iPrOH (1 :1, 90 mL) was added L-lactic acid (2.25 g, 25 mmol). The mixture was stirred at ambient temperature for 24 h then reduced in vacuo. The residue was given consecutive slurries using toluene (100 mL) and Et2O (100 mL) and the resultant solid collected and dried (8.04 g).
This solid was purified by recrystallisation from boiling iPrOH (200 mL) to give after drying l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)- lH-pyrazol-4-yl]-urea, L-lactate salt (5.7 g) as a beige solid.
EXAMPLE 66
Stage 1: Preparation of (3,4-dinitrophenyl)-morpholin-4-yl-methanone

3,4-Dinitrobenzoic acid (1.000Kg, 4.71mol, l.Owt), tetiuhydrofuran (10.00L5 lO.Ovol), and dimethylformamide (0.010L, O.Olvol) were charged to a flask under nitrogen. Thionyl chloride (0.450L, 6.16mol, 0.45vol) was added at 20 to 3O0C and the reaction mixture was heated to 65 to 7O0C. Reaction completion was determined by 1H NMR analysis (d6-DMSO), typically in 3 hours. The reaction mixture was cooled to 0 to 50C and triethylamine (1.25L, 8.97mol, 1.25vol) was added at 0 to 100C. Morpholine (0.62L, 7.07mol, 0.62vol) was charged to the reaction mixture at 0 to 1O0C and the slurry was stirred for 30 minutes at 0 to 1O0C. Reaction completion was determined by H NMR analysis (d6-DMSO). The reaction mixture was warmed to 15 to 2O0C and water (4.00L, 4.0vol) was added. This mixture was then charged to a 4OL flange flask containing water (21.0OL, 21.0vol) at 15 to 250C to precipitate the product. The flask contents were cooled to and aged at 0 to 50C for 1 hour and the solids were collected by filtration. The filter-cake was washed with water (4x 5.00L, 4x 5.0vol) and the pH of the final wash was found to be pH 7. The wet filter-cake was analysed by H NMR for the presence of triethylamine hydrochloride. The filter-cake was dried at 40 to 450C under vacuum until the water content by KF <0.2%w/w, to yield (3,4-dinitrophenyl)-morpholin-4-yl-methanone (1.286Kg, 97.0%, KF 0.069%w/w) as a yellow solid.
Stage 2: Preparation of 4-(3,4-dinitro-benzyl)-morpholine

C11H11N3O6 C11H13N3O5
FW:281.22 FW:267.24
(3,4-DinitiOphenyl)-morpholin-4-yl-methanone (0.750Kg, 2.67mol, l.Owt) and tetrahydrofuran (7.50L, lO.Ovol) were charged to a flask under nitrogen and cooled to 0 to 50C. Borontrifluoride etherate (0.713L, 5.63mol, 0.95vol) was added at 0 to 50C and the suspension was stirred at this temperature for 15 to 30 minutes. Sodium borohydride (0.212Kg, 5.60mol, 0.282wt) was added in 6 equal portions over 90 to 120 minutes. (A delayed exotherm was noted 10 to 15 minutes after addition of the first portion. Once this had started and the reaction mixture had been re-cooled, further portions were added at 10 to 15 minute intervals, allowing the reaction to cool between additions). The reaction mixture was stirred at 0 to 50C for 30 minutes. Reaction completion was determined by 1H NMR analysis (d6-DMSO). Methanol (6.30L, 8.4vol) was added drop wise at 0 to 1O0C to quench the reaction mixture (rapid gas evolution, some foaming). The quenched reaction mixture was stirred at 0 to 1O0C for 25 to 35 minutes then warmed to and stirred at 20 to 3O0C (exotherm, gas/ether evolution on dissolution of solid) until gas evolution had slowed. The mixture was heated to and stirred at 65 to 7O0C for 1 hour. The mixture was cooled to 30 to 4O0C and concentrated under vacuum at 40 to 450C to give crude 4-(3,4-dinitro-benzyl)-morpholine (0.702Kg, 98.4%) as a yellow/orange solid.
4-(3,4-Dinitro-benzyl)-niorpholme (2.815kg, 10.53mol, l.Owt) and methanol (12.00L, 4.3vol) were charged to a flask under nitrogen and heated to 65 to 7O0C. The temperature was maintained until complete dissolution. The mixture was then cooled to and aged at 0 to 50C for 1 hour. The solids were isolated by filtration. The filter-cake was washed with methanol (2x 1.50L, 2x 0.5vol) and dried under vacuum at 35 to 45°C to give 4-(3,4-dinitro-benzyl)-morpholine (2.353Kg, 83.5% based on input Stage 2, 82.5% overall yield based on total input Stage 1 material,) as a yellow solid.
Stage 3: Preparation of 4-morpholin-4-yl-methyl-benzene-L2-diamine

C11H13N3O5 C11H17N3O
FW:267.24 FW:207.27
4-(3,4-Dinitro-benzyl)-morρholine (0.800Kg, 2.99mol, l.Owt), and ethanol (11.20L, 14.0vol) were charged to a suitable flask and stirred at 15 to 250C and a vacuum / nitrogen purge cycle was performed three times. 10% Palladium on carbon (10%Pd/C, 50%wet paste, 0.040Kg, 0.05wt wet weight) was slurried in ethanol (0.80L, l.Ovol) and added to the reaction. The mixture was cooled to 10 to 2O0C and a vacuum / nitrogen purge cycle was performed three times. A vacuum / hydrogen purge cycle was performed three times and the reaction was stirred under a hydrogen atmosphere at 10 to 2O0C. Reaction completion was determined by 1H NMR analysis (d6-DMSO), typically 14 to 20 hours. A vacuum / nitrogen purge cycle was performed three times and the reaction mixture was filtered through glass microfibre paper under nitrogen. The filter-cake was washed with ethanol (3x 0.80L, 3x l.Ovol) and the combined filtrate and washes were concentrated to dryness under vacuum at 35 to 450C to give 4-morpholin-4-yl-methyl-benzene-l,2- diamine (0.61 IKg 98.6%) as a brown solid.
Stage 4: Preparation of 4-nitiO-lH-pyrazole-3-carboxγlic acid methyl ester

C4H3N3O4 C5H5N3O4
FW: 157.09 FW: 171.11
4-Nitro-lH-pyrazole-3-carboxylic acid (1.00kg, 6.37mol, l.Owt) and methanol (8.00L, 8.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. The suspension was cooled to 0 to 5°C under nitrogen and thionyl chloride (0.52L, 7.12mol, 0.52vol) was added at this temperature. The mixture was warmed to 15 to 25°C over 16 to 24 hours. Reaction completion was determined by 1H NMR analysis (d6-DMSO). The mixture was concentrated under vacuum at 35 to 45°C. Toluene (2.00L, 2.0vol) was charged to the residue and removed under vacuum at 35 to 450C. The azeotrope was repeated twice using toluene (2.00L, 2.0vol) to give 4-nitro-lH-pyrazole-3-carboxylic acid methyl ester (1.071Kg, 98.3%) as an off white solid.
Stage 5: Preparation of 4-amino-lH-pyrazole-3-carboxylic acid methyl ester. O2Me

C5H 5N3O4 C5H7N3O2 FW: 171.11 FW: 141.13
A suspension of 4-nitro-lH-pyrazole-3-carboxylic acid methyl ester (1.084Kg, 6.33mol, l.Owt) and ethanol (10.84L, lO.Ovol) was heated to and maintained at 30 to 35°C until complete dissolution occurred. 10% Palladium on carbon (10% Pd/C wet paste, 0.152Kg, 0.14wt) was charged to a separate flask under nitrogen and a vacuum / nitrogen purge cycle was performed three times. The solution of 4-nitro- lH-pyrazole-3-carboxylic acid methyl ester in ethanol was charged to the catalyst and a vacuum / nitrogen purge cycle was performed three times. A vacuum / hydrogen purge cycle was performed three times and the reaction was placed under an atmosphere of hydrogen. The reaction mixture was stirred at 28 to 30°C until deemed complete by 1H NMR analysis (d6-DMSO). The mixture was filtered under nitrogen and concentrated under vacuum at 35 to 450C to give 4-amino-lH- pyrazole-3-carboxylic acid methyl ester (0.883Kg, 98.9%) as a purple solid.
Stage 6: Preparation of 4-fert-butoxycarbonylamino-lH-pyrazole-3-carboxylic acid

C5H7N3O2 C9H13N3O4
FW: 141.13 FW:227.22
4-Amino-lH-pyrazole-3-carboxylic acid methyl ester (1.024Kg, 7.16mol, l.Owt) and dioxane (10.24L, lO.Ovol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. 2M aq. Sodium hydroxide solution (4.36L, 8.72mol, 4.26vol) was charged at 15 to 250C and the mixture was heated to 45 to 550C. The temperature was maintained at 45 to 550C until reaction completion, as determined by 1H NMR analysis (d6-DMSO). Di-te/Y-butyl dicarbonate (Boc anhydride, 1.667Kg, 7.64mol, 1.628wt) was added at 45 to 55°C and the mixture was stirred for 55 to 65 minutes. 1H NMR IPC analysis (d6-DMSO) indicated the presence of 9% unreacted intermediate. Additional di-fert-butyl dicarbonate (Boc anhydride, 0.141Kg, 0.64mol, 0.14wt) was added at 55°C and the mixture was stirred for 55 to 65 minutes. Reaction completion was determined by 1H NMR analysis (d6-DMSO). The dioxane was removed under vacuum at 35 to 450C and water (17.60L, 20.0vol) was added to the residue. The pH was adjusted to pH 2 with 2M aq. hydrochloric acid (4.30L, 4.20vol) and the mixture was filtered. The filter-cake was slurried with water (10.00L3 9.7vol) for 20 to 30 minutes and the mixture was filtered. The filter-cake was washed with heptanes (4.10L, 4.0vol) and pulled dry on the pad for 16 to 20 hours. The solid was azeodried with toluene (5x 4.00L, 5x 4.6vol) then dried under vacuum at 35 to 45°C to give 4-tert- butoxycarbonylamino-lH-pyrazole-3-carboxylic acid (1.389Kg, 85.4%) as a purple solid.
Stage 7: Preparation of [3-(2-amino-4-moipholin-4-ylmetliyl-phenylcarbamoviy lH-pyrazol-4-yl]-carbamic acid tert-butyl ester

C9H13N3O4 C11H17N3O C20H28N6O4
FW: 227.22 FW: 207.27 FW: 416.48
+ regioisomer
4-førf-Butoxycarbonylamino-lH-pyrazole-3-carboxylic acid (0.750Kg, 3.30 mol, l.Owt), 4-morpholin-4yl-methyl-benzene-l,2-diamine (0.752Kg, 3.63mol, l.Owt) and N,N’-dimethylformamide (11.25L, 15.0vol) were charged under nitrogen to a flange flask equipped with a mechanical stirrer and thermometer. 1- Hydroxybenzotriazole (HOBT, 0.540Kg, 3.96mol, 0.72wt) was added at 15 to 250C. N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide (EDC, 0.759Kg, 3.96mol, 1.01 wt) was added at 15 to 250C and the mixture was stirred at this temperature for 16 to 24 hours. Reaction completion was determined by 1H NMR analysis. The reaction mixture was concentrated under vacuum at 35 to 45°C. The residue was partitioned between ethyl acetate (7.50L, lO.Ovol) and sat. aq. sodium hydrogen carbonate solution (8.03L, 10.7vol) and the layers were separated. The organic phase was washed with brine (3.75L, 5.0vol), dried over magnesium sulfate (1.00Kg, 1.33wt) and filtered. The filter-cake was washed with ethyl acetate (1.50L, 2.0vol). The combined filtrate and wash were concentrated under vacuum at 35 to 450C to give [3-(2-amino-4-morpholin-4-ylmethyl-phenylcarbamoyl)-lH-pyrazol- 4-yl]-carbamic acid tert-butyl ester (1.217Kg, 88.6%) as a dark brown solid.
Stage 8 : Preparation of 3 -f 5-morpholin-4-ylmethyl- 1 H-benzoimidazol-2-ylV 1 H- pyrazol-4-ylamme

C15H19N6O

FW: 298.35
As a mixture of two regioisomers
[3-(2-Amino-4-morpholin-4-ylmethyl-phenylcarbamoyl)-lH-pyrazol-4-yl]- carbamic acid tert-butyl ester (1.350Kg, 3.24 mol, l.Owt) and ethanol (6.75L, 5.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. Cone. aq. hydrochloric acid (1.10L, 13.2 mol, 0.80vol) was added at 15 to 3O0C under nitrogen and the contents were then heated to 70 to 😯0C and maintained at this temperature for 16 to 24 hours. A second portion of hydrochloric acid (0.1 IL, 1.32 mol, O.OSOvol) was added at 70 to 😯0C and the reaction was heated for a further 4 hours. Reaction completion was determined by HPLC analysis. The reaction mixture was cooled to 10 to 200C and potassium carbonate (1.355Kg, 9.08mol, l.Owt) was charged portionwise at this temperature. The suspension was stirred until gas evolution ceased and was then filtered. The filter-cake was washed with ethanol (1.35L, l.Ovol) and the filtrates retained. The filter-cake was slurried with ethanol (4.00L, 3.0vol) at 15 to 250C for 20 to 40 minutes and the mixture was filtered. The filter-cake was washed with ethanol (1.35L3 1.Ovol) and the total combined filtrates were concentrated under vacuum at 35 to 450C. Ethanol (4.00L, 3. Ovol) was charged to the residue and removed under vacuum at 35 to 450C. Tetrahydrofuran (5.90L, 4.4vol) was added to the residue and stirred for 10 to 20 minutes at 15 to 25°C. The resulting solution was filtered, the filter-cake was washed with tetrahydrofuran (1.35L, l.Ovol) and the combined filtrates were concentrated under vacuum at 35 to 450C. Tetrahydrofuran (5.40L, 4. Ovol) was charged to the concentrate and removed under vacuum at 35 to 450C. Tetrahydrofuran (5.40L, 4. Ovol) was charged to the concentrate and removed under vacuum at 35 to 45°C to give the desired product, 3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-ylamine (0.924Kg, 95.5%, 82.84% by HPLC area) as a purple foam.
Stage 9: Preparation of 7-morpholin-4-ylmethyl-2,4-dihydro- 1,2,4,5a ,10-pentaaza- cyclopentaFal fluoren-5 -one

C15H18N6O C16H16N6O2 FW: 298.35 FW: 324.34
As a mixture of two regioisomers
3-(5-Morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)-lH-pyrazol-4-ylamine (0.993Kg, 3.33 mol, l.Owt) and tetrahydrofuran (14.0L, 15.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. The contents were stirred under nitrogen at 15 to 25°C and l,l ‘-carbonyldiimidazole (0.596Kg, 3.67 mol, O.όOwt) was added. The contents were then heated to 60 to 700C and stirred at this temperature for 16 to 24 hours. Reaction completion was determined by TLC analysis. The mixture was cooled to 15 to 200C and filtered. The filter-cake was washed with tetrahydrofuran (4.00L, 4. Ovol) and pulled dry for 15 to 30 minutes. The solid was dried under vacuum at 35 to 450C to yield 7- morpholin-4-ylmethyl-2,4-dihydro- 1 ,2,4,5a, 10-pentaaza-cyclopenta[a]fluoren-5- one (0.810Kg, 75.0%th, 92.19% by HPLC area) as a purple solid. Stage 10: Preparation of l-cvclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-vD- 1 H-pyrazol-4-yll -urea

C16H16N6O2 C19H23N7O2
FW: 324.34 FW: 381.44
As a mixture of two regioisomers
7-Morpholin-4-ylmethyl-254-dihydro-l,2,4,5a,10-pentaaza-cyclopenta[a]fluoren-5- one (0.797Kg, 2.46mol, l.Owt) and l-methyl-2-pyrrolidinone (2.40L, 3.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. Cyclopropylamine (0.279Kg, 4.88mol, 0.35 lwt) was added at 15 to 30°C under nitrogen. The contents were heated to 95 to 105°C and stirred at this temperature for 16 to 24 hours. Reaction completion was determined by 1H NMR analysis. The reaction mixture was cooled to 10 to 200C and ethyl acetate (8.00L, lO.Ovol) and sat. aq. sodium chloride (2.50L, 3.0vol) were charged, the mixture was stirred for 2 to 5 minutes and the layers separated. The organic phase was stirred with sat. aq. sodium chloride (5.00L, ό.Ovol) for 25 to 35 minutes, the mixture filtered and the filter-cake washed with ethyl acetate (0.40L, 0.5vol). The filter-cake was retained and the filtrates were transferred to a separating funnel and the layers separated. The procedure was repeated a further 3 times and the retained solids were combined with the organic phase and the mixture concentrated to dryness under vacuum at 35 to 450C. The concentrate was dissolved in propan-2-ol (8.00L, lO.Ovol) at 45 to 55°C and activated carbon (0.080Kg5 O.lwt) was charged. The mixture was stirred at 45 to 550C for 30 to 40 minutes and then hot filtered at 45 to 55°C. The filter-cake was washed with propan-2-ol (0.40L, 0.5vol). Activated carbon (0.080L, O.lwt) was charged to the combined filtrates and wash and the mixture stirred at 45 to 550C for 30 to 40 minutes. The mixture was hot filtered at 45 to 550C and the filter-cake washed with propan-2-ol (0.40L, 0.5vol). The filtrates and wash were concentrated under vacuum at 35 to 450C. Ethyl acetate (8.00, lO.Ovol) and water (2.20L, 3.0vol) were charged to the concentrate at 25 to 350C and the mixture stirred for 1 to 2 minutes. The layers were separated and the organic phase was concentrated under vacuum at 35 to 45°C. Ethyl acetate (4.00L, 5.0vol) was charged to the residue and concentrated under vacuum at 35 to 450C. Ethyl acetate (4.00L, 5.0vol) was charged to the residue and the mixture was stirred for 2 to 20 hours at 15 to 250C. The mixture was cooled to and aged at 0 to 5°C for 90 to 120 minutes and then filtered. The filter-cake was washed with ethyl acetate (0.80L, l.Ovol) and pulled dry for 15 to 30 minutes. The solid was dried under vacuum at 35 to 450C to yield l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea (0.533Kg, 56.8%, 93.20% by HPLC area) as a brown solid.
Several batches of Stage 9 product were processed in this way and the details of the quantities of starting material and product for each batch are set out in Table IA.
Table IA – Yields from urea formation step – Stage 10

Stage 11 : Preparation of l-cyclopiOpyl-3-r3-(5-moipholin-4-ylmethyl-lH- benzoimidazol-2-yls)-lH-pyrazol-4-yll-urea £-lactic acid salt L-Lactic acid


acid
C19H23N7O2 C22H29N7O5
FW: 381.44 FW: 471.52 l-Cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)-lH-ρyrazol- 4-yl]-urea (1.859Kg, 4.872mol, l.Owt), propan-2-ol (9.00L5 5.0vol) and ethyl acetate (8.0OL, 4.5vol) were charged to a flange flask equipped with a mechanical stirrer and thermometer. The contents were stirred under nitrogen and L-lactic acid (0.504Kg, 5.59mol, 0.269wt) was added at 15 to 25°C followed by a line rinse of ethyl acetate (0.90L, 0.5vol). The mixture was stirred at 15 to 25°C for 120 to 140 minutes. The solid was isolated by filtration, the filter-cake washed with ethyl acetate (2x 2.00L, 2x l.Ovol) and pulled dry for 20 to 40 minutes. The filter-cake was dissolved in ethanol (33.00L, 17.7vol) at 75 to 850C, cooled to 65 to 700C and the solution clarified through glass microfibre paper. The filtrates were cooled to and aged at 15 to 250C for 2 to 3 hours. The crystallised solid was isolated by filtration, the filter-cake washed with ethanol (2x 1.00L, 2x 0.5vol) and pulled dry for at least 30 minutes. The solid was dried under vacuum at 35 to 45°C to yield 1- cyclopropyl-3 – [3-(5 -morpholin-4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4- yl]-urea l-lactic acid salt (1.386Kg, 58.7%th, 99.47% by HPLC area,) as a dark pink uniform solid.
The infra-red spectrum of the lactate salt (KBr disc method) included characteristic peaks at 3229, 2972 and 1660 cm“1.
Without wishing to be bound by any theory, it is believed that the infra red peaks can be assigned to structural components of the salt as follow:
Peak: Due to:
3229 cm“1 N-H
2972 cm“1 aliphatic C-H
1660 cm“1 urea C=O EXAMPLE 67
Synthesis of Crystalline Free Base And Crystalline Salt Forms Of l-Cyclopropyl-3-
[3-(5-Morpholin-4-ylmethyl-lH-Benzoimidazol-2-vπ-lH-Pyrazol-4-yll-Urea
A. Preparation of l-Cvclopropyl-3-[3-f5-Moφholm-4-ylmethyl-lH- Benzoimidazol-2-yl)-lH-Pyrazol-4-yll-Urea free base
A sample of crude l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-lH-pyrazol-4-yl]-urea free base was prepared as outlined in Example 60 and initially purified by column chromatography on silica gel, eluting with EtOAc- MeOH (98:2 – 80:20). A sample of the free base obtained was then recrystallised from hot methanol to give crystalline material of l-cyclopropyl-3-[3-(5-morpholin- 4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4-yl] -urea free base.
B. Preparation of l-Cyclopropyl-S-rS-fS-Morpholin^-ylmethyl-lH-Benzoimidazol- 2-yl)-lH-Pyrazol-4-yl]-Urea free base dihydrate
A sample of crude l-cyclopropyl-3-[3-(5-moφholm-4-ylmethyl-lH-benzoimidazol- 2-yl)-l H-pyrazol-4-yl] -urea free base was dissolved in THF and then concentrated in vacuo to a minimum volume (~4 volumes). To the solution was added water dropwise (2 – 4 volumes) until the solution became turbid. A small amount of THF was added to re-establish solution clarity and the mixture left to stand overnight to give a crystalline material which was air-dried to give l-cyclopropyl-3-[3-(5- morpholin-4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4-yl] -urea free base dihydrate.
C. Preparation of l-Cyclopl^pyl-3-[3-(5-Morpholm-4-ylmethyl-lH-Benzoimidazol- 2-ylVlH-Pyrazol-4-yl]-Urea hydrochloride salt
A sample of crude l-cyclopropyl-3-[3-(5-moφholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-l H-pyrazol-4-yl] -urea free base was dissolved in the minimum amount of MeOH and then diluted with EtOAc. To the solution at 0 °C was slowly added 1.1 equivalents of HCl (4M solution in dioxane). Following addition, solid precipitated from solution which was collected by filtration. To the solid was added MeOH and the mixture reduced in vacuo. To remove traces of residual MeOH the residue was evaporated from water and then dried at 60 0C/ 0.1 mbar to give the hydrochloride salt.
D. Preparation of l-Cyclopropyl-3-[3-(‘5-Morpholm-4-ylmethyl-lH- Benzoimidazol-2-yiyiH-Pyrazol-4-yl1-Urea ethanesulfonate salt
To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea free base in MeOH-EtOAc was added 1 equivalent of ethanesulfonic acid. The mixture was stirred at ambient temperature and then reduced in vacuo. The residue was taken up in MeOH and to the solution was added Et2O. Mixture left to stand for 72 h and the solid formed collected by filtration and dried to give l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea ethanesulfonate salt.
E. Preparation of l-Cvclopropyl-3-[3-(‘5-Morpholm-4-ylmethyl-lH-Benzoimidazol- 2-yl)-lH-Pyrazol-4-yl]-Urea methanesulfonate salt
To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea free base (394 mg) in MeOH-EtOAc was added 1 equivalent of methanesulfonic acid (67 μl). A solid was formed which was collected by filtration, washing with EtOAc. The solid was dissolved in the minimum amount of hot MeOH, allowed to cool and then triturated with Et2O. The solid was left to stand for 72 h and then collected by filtration, washing with MeOH, to give l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea methanesulfonate salt.
EXAMPLE 68
Characterisation of l-Cvclopropyl-3-[3-(5-Morpholin-4-ylmethyl-lH-
Benzoimidazol-2-yl)-lH-Pyrazol-4-yll-Urea Free Base and Salts
Various forms of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-lH-pyrazol-4-yl]-urea were characterised. The forms selected for characterisation were identified from studies which primarily investigated extent of polymorphism and salt stability. The salts selected for further characterisation were the L-lactate salt, Free base dihydrate, Esylate salt, Free base and Hydrochloride salt.

Paper
Fragment-Based Discovery of the Pyrazol-4-yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent Aurora Kinase Activity†
†
Coordinates of the protein complexes with compounds 5, 7, 9, 10, and 16 have been deposited in the Protein Data Bank under accession codes 2w1d, 2w1f, 2w1c, 2w1e, 2w1g (Aurora A), 2w1h (CDK2), and 2w1i (JAK2).
, * To whom correspondence should be addressed. Phone: +44 (0)1223 226209. Fax: +44 (0)1223 226201. E-mail: s.howard@astex-therapeutics.com.
Abstract

Here, we describe the identification of a clinical candidate via structure-based optimization of a ligand efficient pyrazole-benzimidazole fragment. Aurora kinases play a key role in the regulation of mitosis and in recent years have become attractive targets for the treatment of cancer. X-ray crystallographic structures were generated using a novel soakable form of Aurora A and were used to drive the optimization toward potent (IC50 ≈ 3 nM) dual Aurora A/Aurora B inhibitors. These compounds inhibited growth and survival of HCT116 cells and produced the polyploid cellular phenotype typically associated with Aurora B kinase inhibition. Optimization of cellular activity and physicochemical properties ultimately led to the identification of compound16 (AT9283). In addition to Aurora A and Aurora B, compound 16 was also found to inhibit a number of other kinases including JAK2 and Abl (T315I). This compound demonstrated in vivo efficacy in mouse xenograft models and is currently under evaluation in phase I clinical trials.
1-Cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-1H-benzoimidazol-2-yl)-1H-pyrazol-4-yl]urea (16), Hydrochloride Salt
///////////
C1CC1NC(=O)NC2=CNNC2=C3N=C4C=CC(=CC4=N3)CN5CCOCC5


“Jiangsu Elitech Biochem Limited is a professional and renowned name among 2-Methyl-3 5-Dinitrobenzoic Acid suppliers. Since its inception in 2012 We have a mission to produce and provide customers with consistent and the best quality chemicals.”2-Methyl-3 5-Dinitrobenzoic Acid
LikeLike