 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

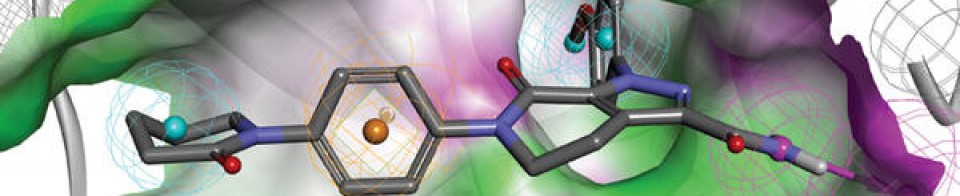
((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, Carbamic acid, methyl ester
Carbamic acid, N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, methyl ester
CAS 1374883-22-3, 819.87, C41 H48 F3 N9 O6
CAS of DIHCl 1480448-59-6
CAS of DIHCl, H2O 1480448-63-2
Theravance, Inc. INNOVATOR
To treat hepatitis C virus infection

-
((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (compound 1):
-
 Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds
Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds

SYNTHESIS
CLICK ON IMAGES FOR CLEAR VIEW

………………………..

……………………………….



PATENT
WO-2013/165796
https://www.google.co.in/patents/WO2013165796A1?cl=en
PATENT
http://www.google.com/patents/US20130295048
crystalline compound 1 is advantageously prepared directly from the crude product of the final step of the synthesis of compound 1, illustrated in the following scheme, without purification of the amorphous form.

As described in Example 3 below, ((S)-1-{(S)-2-[4-(4′-{[6-(2R,5S)-2,5-dimethyl-piperazin-1-yl)-pyridine-3-carbonyl}-amino]-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (2) is reacted with methylaminoformyl chloride to provide a crude product, which is recovered by conventional extraction and drying. The reaction is typically performed in the presence of an excess of base, in an inert diluent such as dichloromethane. Next, methanol is added to the crude product followed by the slow addition of water in a ratio of methanol:water of about 2.5:1 to about 2.7:1 to form a crystallization mixture. Seeds of crystalline compound 1 are added about halfway through the water addition. The crystallization mixture is stirred for a period of several days to form crystalline compound 1. To increase purity, the product can be recrystallized by a similar process: the crystalline compound is dissolved in methanol, water and seeds are added, such that the ratio of methanol to water in the mixture is about 2.5:1, and the mixture is stirred for a period of at least 12 hours to provide crystalline compound 1, which is recovered conventionally
-

-
To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1:4 ethanol:water (2×25 mL) to give the title intermediate as a white solid (3.87 g). Analytical HPLC: Retention time=21.3 min.
- Preparation 1: (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester(a) N-(4-Bromo-3-trifluoromethoxy-phenyl)-6-fluoro-nicotinamide

(b) (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N-diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120° C. for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4Cl, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83% yield). Analytical HPLC: Retention time=21.1 min.

Preparation 2: (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-Methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of ((S)-1-{(S)-2-[4-(4-bromo-phenyl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1.00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90° C. overnight.
-
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol), and (2S,5R)-4-[5-(4-bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (4.35 g, 7.13 mmol). The reaction mixture was stirred at 95° C. overnight.
-
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100% EtOAc/hexane) to provide the title intermediate (5.3 g, 90% yield). Analytical HPLC: Retention time=14.7 min.

Preparation 3: ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-

-
Acetyl chloride (63.2 mL, 888 mmol) was added to ethanol (360 mL) and stirred at RT for 1 h. To the resulting HCl solution was added a solution of (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (73 g, 84 mmol) in ethanol (360 mL). The reaction mixture was stirred at RT overnight.
-
The reaction mixture was concentrated to dryness (124 g crude). Water 500 mL) was added and the mixture was extracted with EtOAc (2×500 mL). The aqueous layer was adjusted to pH 4 with 1:1 NaOH:water. Ethyl acetate (400 mL) and sat. aq. Na2CO3 (100 mL) were added and the layers were separated. The organic layer was dried over Na2SO4 and evaporated to give the title intermediate (62.8 g; 88% yield). Analytical HPLC: Retention time=10.0 min.

Example 1Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

(a) ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester tri-HCl
-
Acetyl chloride (0.71 mL, 10.0 mmol) was added to ethanol (7 mL) and stirred at RT for 1 h. The resulting HCl solution was added to a solution of (2S,5R)-4-[5-(4′-{2-[(5)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (1.55 g, 1.8 mmol) in ethanol (7 mL). The reaction mixture was warmed to 35° C. and stirred overnight. The mixture was concentrated to dryness, and chased with DCM to provide the crude tri-HCl salt of the title intermediate (1.57 g) which was used directly in the next step. HPLC method C: Retention time=10.0 min.
(b) Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-
To a solution of the product of the previous step (1.57 g crude, ca. 1.80 mmol) and N,N-diisopropylethylamine (3.14 mL, 18.0 mmol) in DCM (24 mL) was slowly added 1 M methylaminoformyl chloride in DMA (1.8 mL). The reaction mixture stirred at RT for 1 h, and then 1 M methylaminoformyl chloride in DMA (1.8 mL) was added. The reaction was quenched with sat. aq. NaHCO3 and the reaction mixture was stirred for 20 min. The layers were separated and the organic layer was dried and evaporated to give a residue. To the residue was added methanol (15 mL) followed by 2 N LiOH/water (3 mL). The reaction mixture was stirred at RT for 1 h, diluted with water, extracted with DCM (80 mL), dried, and evaporated to give a crude product which was purified by silica gel chromatography (40 g silica, 2-8% MeOH/DCM) to provide the title compound (0.93 g, 63% yield). Analytical HPLC: Retention time=11.0 min.
HPLC
- Analytical HPLC Method
-
-
- Column: Zorbax Bonus-RP 3.5 μm. 4.6×150 mm
- Column temperature: 35° C.
- Flow rate: 1.0 mL/min
- Mobile Phases: A=Water/ACN (98:2)+0.1% TFA
- B=Water/ACN (10:90)+0.1% TFA,
- Injection volume: 100-1500 μL
- Detector wavelength: 214 nm
- Sample preparation: Dissolve in 1:1 ACN:water
- Gradient: 29 min total (time (min)/% B): 0.5/10, 24/90, 25/90, 26/10, 29/10
-
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| WO2012061552A1 * | 3 Nov 2011 | 10 May 2012 | Theravance, Inc. | Novel inhibitors of hepatitis c virus |
| US201113288216 |
About Theravance Biopharma
The mission of Theravance Biopharma (NASDAQ: TBPH) is to create value from a unique and diverse set of assets: an approved product; a development pipeline of late-stage assets; and a productive research platform designed for long-term growth.
Our pipeline of internally discovered product candidates includes potential best-in-class opportunities in underserved markets in the acute care setting, representing multiple opportunities for value creation. VIBATIV® (telavancin), our first commercial product, is a once-daily dual-mechanism antibiotic approved in the U.S., Europe and certain other countries for certain difficult-to-treat infections. Revefenacin (TD-4208) is an investigational long-acting muscarinic antagonist (LAMA) being developed as a potential once-daily, nebulized treatment for COPD. Axelopran (TD-1211) is an investigational potential once-daily, oral treatment for opioid-induced constipation (OIC). Our earlier-stage clinical assets represent novel approaches for potentially treating diseases of the lung and gastrointestinal tract and infectious disease. In addition, we have an economic interest in future payments that may be made by GlaxoSmithKline plc pursuant to its agreements with Theravance, Inc. relating to certain drug development programs, including the combination of fluticasone furoate, umeclidinium and vilanterol (the “Closed Triple”).
With our successful drug discovery and development track record, commercial infrastructure, experienced management team and efficient corporate structure, we believe that we are well positioned to create value for our shareholders and make a difference in the lives of patients.
For more information, please visit www.theravance.com.
THERAVANCE®, the Cross/Star logo, MEDICINES THAT MAKE A DIFFERENCE® and VIBATIV® are registered trademarks of the Theravance Biopharma group of companies.
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672……….

(e)Thalladi, V. R.; Nzerem, J.; Huang, X.; Zhang, W. Crystalline form of a pyridyl-piperazinyl hepatitis C virus inhibitor. World Patent Application WO-2013/165796, November 7, 2013.
PATENT
WO 2012061552
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 28: ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-piperazin-l- yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2- yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

A mixture of [(5)-2-methyl-l-((5)-2- {4-[4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-phenyl]-lH-imidazol-2-yl}-pyrrolidine-l-carbonyl)-propyl]- carbamic acid methyl ester (86 mg, 0.17 mmol) and (25′,5R)-4-[5-(4-bromo-3- trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-l-carboxylic acid tert-butyl ester (100 mg, 0.2 mmol, Preparation 27) was dissolved in 1,4-dioxane (1.8 mL, 23 mmol) and water (0.25 mL, 14 mmol). Cesium carbonate (170 mg, 0.52 mmol) was added. The reaction mixture was sparged with nitrogen and then
tetrakis(triphenylphosphine)palladium(0) (12.1 mg, 0.011 mmol) was added. The reaction mixture was sealed under nitrogen and heated at 95 °C overnight. The reaction mixture was extracted with ethyl acetate/water, the organic layer was dried over sodium sulfate and concentrated to produce an orange oil.
The oil from the previous step was treated with 4 M HCl in 1,4-dioxane (2 mL, 7 mmol) and stirred at room temperature for 1 h. The reaction mixture was concentrated and evaporated with ethyl acetate (2 x) to produce the HCl salt of the title compound as a yellow solid which was purified by preparative HPLC to provide the tri-TFA salt of the title compound (150 mg, 30 % overall yield), (m/z): [M+H] calcd for
763.35 found 763.7.
Example 29 ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-4- methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy- biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)- carbamic a

To a solution of ((5)- l – {(5)-2-[4-(4′- { [6-((2R,55)-2,5-dimethyl-piperazin- l-yl)- pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]- pyrrolidine- l-carbonyl} -2-methyl-propyl)-carbamic acid methyl ester tri-TFA (1 1.4 mg, 0.01 1 mmol; Preparation 28) and N,N-diisopropylethylamine (18 uL, 0.1 1 mmol) dissolved in DMA (0.4 mL, 4 mmol) was added 1.0 M methyl isocyanate in toluene (10 uL, 0.01 mmol). The reaction mixture was stirred at RT overnight, concentrated, dissolved in 1 : 1 acetic acid:water (1.5 mL) and purified by preparative HPLC to provide the di-TFA salt of the title compound (7.1 mg). (m/z): [M+H]+ calcd for C41H48F3N906 820.37 found 820.5.
Alternative synthesis of ((5)-1-{(5)-2-[ -(4′-{[6-((2Λ,ί» 2,5- Dimethyl-4-methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′- trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2- methyl-propyl)-carbamic acid methyl ester
(a) N-(‘4-Bromo-3-trifluoromethoxy-phenyl -6-fluoro-nicotinamide

To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1 :4 ethanohwater (2 x 25 mL) to give the title intermediate as a white solid (3.87 g). HPLC method C: Retention time = 21.3 min.
(b) (25,,5R)-4-r5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl) -pyridin-2-yl1-2,5- dimethyl-piperazin – 1 -carboxylic acid fe/t-butyl ester

The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl- piperazine-1 -carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N- diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120 °C for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4C1, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83 % yield). HPLC method C: Retention time = 21.1 min (c) (2 .5R)-4 5-(4′-{2 ffl -((5f)-2-Methoxycarbonylamino-3-methyl-butyrvn- pyiTolidin-2-yl1-lH-imidazol-4-yl}-2-tri
pyridin-2- -2,5-dimethyl-piperazine-l-carboxylic acid fert-butyl ester

To a solution of ((5′)-l-{(5,)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol;), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1,00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and l,l’-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90 °C overnight.
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol). The reaction mixture was stirred at 95°C overnight.
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100 % EtOAc/hexane) to provide the title intermediate (5.3 g, 90 % yield). HPLC method C: Retention time = 14.7 min.
(d) (ffl ffl-2 4-(4′ r6-((2R,5^-2,5-Dimethyl-piperazin-l-vn-pyridine-3-carbonyl1- amino}-2′ rifluoromethoxy-biphenyl-4-yl -lH-imidazol-2-yl1-pyrrolidine-l- carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

Acetyl chloride (2.57 mL, 36.2 mmol) was added to ethanol (18 mL) and stirred at RT for 1 h. To the resulting HQ solution was added a solution of the product of the previous step (3.90 g, 4.5 mmol) in ethanol (18 mL). The reaction mixture was warmed to 35 °C and stirred overnight. Acetyl chloride (1.28 mL, 18.1 mmol) was added to ethanol (7.8 mL) and stirred for 30 min. The resulting HC1 solution was added to the reaction mixture at 35 °C. The temperature was raised to 40 °C. The mixture was concentrated to dryness chased by dichloromethane to provide the crude tri-HCl salt of the title intermediate (5.4 g) which was used directly in the next step. HPLC method C: Retention time = 10.1 min.
(e) ((^-l- {(^-2-r4-(4′- {r6-((2R.5^-2.5-Dimethyl-4-methylcarbamoyl-piperazin-l-yl)- pyridine-3-carbonyl1-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl1- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

To a solution of the product of the previous step (5.4 g crude, ca. 3.96 mmol) and N,N-diisopropylethylamine (6.89 mL, 39.6 mmol) in DCM (52 mL) was slowly added 1 M methylaminoformyl chloride in DMA (4.3 mL). The reaction mixture stirred at room temperature for 1 h, and then water (50 mL) was added. The organic layer was washed with saturated NH4C1 and then brine, dried over Na2S04 and evaporated to give 5.2 g crude product, which was purified by silica gel chromatography (133 g silica, 2 to 8 % methanol/DCM for 15 min then 8 % methanol/DCM for 40 min) to provide the title compound (2.4 g, 74 % yield). HPLC method C: Retention time 1 1.2 min
Synthesis of intermediates
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 1: 4-(4-bro -phenyl)-2-(S)-pyrrolidin-2-yl-lH-imidazole

(a) 2-Bromo-l-(4-bromo-phenyl)-ethanone
Bromine (80 g, 500 mmol) was added dropwise to a solution of l-(4-bromo- phenyl)-ethanone (100 g, 500 mmol) in dichloromethane (1500 mL) at ambient temperature. The reaction mixture was stirred for 3 h and then concentrated. The residue was washed with dichloromethane (100 mL) to give the crude title compound (120 g, 86
% yield) as a white solid. XH NMR (CDC13, 400 MHz) δ (ppm): 7.78 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 4.32 (s, 2H).
(b) (^-pyrrolidine- 1 ,2-dicarboxylic acid 2-r2-(4-bromo-phenyl)-2-oxo-ethyl1 ester \-tert- butyl ester
Diisopropylethylamine (67 g, 518 mmol) was added dropwise to a solution of the product of the previous step (120 g, 432 mmol) and (5)-pyrrolidine-l,2-dicarboxylic acid 1-tert-butyl ester ( -Boc proline) (102 g, 475 mmol) in acetonitrile (2 L) at room temperature. The reaction mixture was stirred overnight and concentrated to dryness. The residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated to give crude title compound (178 g, 100 % yield).
(c) (5f)-2-r4-(4-bromo-phenyl -lH-imidazol-2-yl1-pyrrolidine-l-carboxylic acid fe/t-butyl ester
A solution of the product of the previous step (178 g, 432 mmol) and ammonium acetate (500 g, 6.5 mol) in toluene (2 L) was heated at reflux overnight. The solvent was removed and the residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1:3 petroleum ether: ethyl acetate to give the title compound (120 g, 71 % yield) as a yellow solid. ¾ NMR (CDC13, 400 MHz) δ (ppm): 7.56 (s, 1H), 7.39 (d, J=8.0 Hz, 2H), 7.24 (m, 1H), 7.14 (s, 1H), 4.88 (m, 1H), 3.33 (m, 2H), 2.94 (s, 1H), 2.07 (m, 2H), 1.88 (m, 1H), 1.42 (s, 9H).
(d) 4-(4-bromo-phenyl)-2-(5f)-pyrrolidin-2-yl-lH-imidazole
To a solution of (5)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]-pyrrolidine-l- carboxylic acid tert-butyl ester (3 g, 7.6 mmol) in methanol (3 mL) was added 4N HQ in methanol (60 mL) at 0 °C. The reaction mixture was stirred for 2 h and then concentrated to give crude hydrochloride salt of the title compound (2.51 g 100 % yield) as a yellow solid.
Preparation 2: (5)-2-Methoxycarbonylamino-3-methyl-butyric acid

A mixture of (5)-2-amino-3 -methyl-butyric acid (10 g, 85 mmol), NaOH (10.3 g, 255 mmol) in water (100 mL) was treated with methylchloridocarbonate (8 g, 85 mmol) at 0 0 C. The reaction mixture was stirred for 24 h at room temperature and then 5 N aqueous HC1 was added to the reaction mixture to adjust pH to 4. The mixture was filtered through a pad of Celite to give the product (10 g, 67% yield) as a white solid. ¾ NMR (CH3OD, 400 MHz) δ (ppm) 4.05(d, 1H), 3.65(s, 3H), 2.14(m, 1H), 0.95(m, 6H). Preparation 3: ((S)-l-{(S)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-met methyl ester

Triethylamine (2.3 g, 11.4 mmol) was added to a solution of 4-(4-bromo-phenyl)- 2-(5)-pyrrolidin-2-yl-lH- imidazole hydrochloride (2 g, 11.4 mol), (5)-2- methoxycarbonylamino-3-methyl-butyric acid (2.5 g, 7.6 mmol), and HATU (4.3 g, 11.4 mmol) in dimethylformamide (50 mL) at 0 °C under nitrogen. The reaction mixture was stirred at room temperature overnight and treated with ethyl acetate (100 mL) and water (1000 mL). The organic layer was washed with water (2 x 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1 : 1 petroleum ether: ethyl acetate to give the title compound (2.5 g 74 % yield) as a yellow solid. XH NMR (i¾-DMSO, 400 MHz) δ (ppm) 7.63 (d, J=8.8 Hz, 2H), 7.54 (m, IH), 7.47 (m, 2H), 7.26 (d, J=8.4 Hz, IH), 5.03 (m, IH), 4.02 (t, J =8.4 Hz, IH), 3.76 (m, 2H), 3.51 (s, 3H), 2.10 (m, 2H), 1.93 (m, 3H), 0.85 (d, J=6.8 Hz, 3H), 0.81 (d, J=6.8 Hz, 3H).
I AM NOT SURE OF BELOW DATA, It is a cut paste for TD 6450 , NOT ABLE TO CONNECT CAS 1374883-22-3 WITH TD 6450
IF YOU HAVE A STRUCTURE PIC FOR THE SAME MAIL ME amcrasto@gmail.com, call +919323115463
POSTER
50th Annu Meet Eur Assoc Study Liver (EASL) (April 22-26, Vienna) 2015, Abst P0898
http://ilc-congress.eu/abstract_25_04/ILC2015-abstract-book-25-04-Saturday.pdf
P0898
TD-6450,
A NEXT GENERATION ONCE-DAILY NS5A INHIBITOR, HAS POTENT ANTIVIRAL ACTIVITY FOLLOWING A 3-DAY MONOTHERAPY STUDY IN GENOTYPE 1 HCV INFECTION
E. Lawitz1, M. Rodriguez-Torres2, R. Kohler3, A. Amrite3, C. Barnes3, M.L.C. Pecoraro3, J. Budman3, M. McKinnell3, C.B. Washington3. 1Texas Liver Institute, University of Texas Health Science Center, San Antonio, TX, United States; 2Fundacion de Investigacion, San Juan, Puerto Rico; 3Theravance Biopharma, South San Francisco, CA, United States
E-mail: cwashington@theravance.com
Background and Aims: TD-6450 is a next generation HCV NS5A inhibitor with superior in vitro potency against resistanceassociated variants (RAVs) encountered with first-generation NS5A inhibitors. This study evaluated the safety, pharmacokinetics (PK) and antiviral activity of TD-6450 following multiple oral doses in HCV patients
Theravance Biopharma Announces Positive Results From Phase 1 Proof-of-Concept Study of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
240 mg Achieved a Median Maximal Viral Load Decline of 4.9 Log10 IU/mL Following Three Daily Doses in Genotype 1a Patients
SOUTH SAN FRANCISCO, CA — (Marketwired) — 11/03/14 — Theravance Biopharma (NASDAQ: TBPH), through its U.S. operating subsidiary, Theravance Biopharma US, Inc., today announced positive results from the first three cohorts of Study 0110, a Phase 1 proof-of-concept study of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus infection (HCV).
TD-6450 was evaluated in three cohorts of eight genotype 1a (GT-1a) patients each at doses of 60, 120 and 240 mg, administered once-daily for three days. TD-6450 demonstrated dose-dependent antiviral activity with median maximal declines of HCV RNA of 3.87, 4.63 and 4.89 log10 IU/mL for doses of 60, 120 and 240 mg, respectively.
In the 120 and 240 mg dose groups, three days of once-daily oral treatment resulted in levels of serum HCV RNA below the limit of detection (LOD) in 43% (3/7) and 57% (4/7) of patients treated with TD-6450, respectively. Three of the seven LOD patients went on to show no measurable virus at Day 14, and two of these patients still had no measurable virus at Day 28. At a two-month time point in a long-term follow-up study, the viral load in these two patients was measurable, but both remained more than three logs below their baseline.
None of the patients in the three dose groups had virologic breakthrough during their three-day treatment course, and 100% of the treated GT-1a patients in the study achieved at least a three log10 IU/mL reduction of HCV RNA. At the 120 and 240 mg doses, 71% (5/7) and 86% (6/7) of treated patients achieved at least a four log10 IU/mL reduction in HCV RNA, respectively.
All doses of TD-6450 were generally well tolerated after three doses and for the 28-day observation period. There were no serious adverse events and no patient discontinuations. There was no pattern of clinical adverse events or laboratory abnormalities related to treatment.
“We see diverse responses to direct antivirals in genotype 1 populations. Despite recent advances in HCV therapy, significant treatment challenges remain, including the required length of drug therapy. The robust activity of TD-6450 in genotype 1a patients suggests that this potentially best-in-class NS5A inhibitor could be a component of short and highly active combination therapy regimens,” said Eric Lawitz, MD, Vice President of Scientific and Research Development at the Texas Liver Institute and Clinical Professor of Medicine, The University of Texas Health Science Center San Antonio, and one of the principal investigators on the Phase 1 study.
“TD-6450, created using the principles of multivalent design, has a heterodimeric structure distinct from other NS5A inhibitors. We believe this unique structure allows it to bind asymmetrically across the NS5A protein interface, providing high in vitro potency against clinically encountered resistance-associated variants. We believe the potency of TD-6450 against both wild type virus and these resistance-associated variants enables the robust antiviral activity that we reported today,” said Mathai Mammen, MD, Senior Vice President, Research and Development, Theravance Biopharma. “We look forward to analyzing the full set of results from this Phase 1 study and evaluating the next steps in the development strategy for TD-6450.”
About the Phase 1 Proof-of-Concept Study (Study 0110)
This Phase 1 study is a double-blind, randomized, placebo-controlled, multiple-dose study to evaluate the safety, tolerability, pharmacokinetics and antiviral activity of orally administered TD-6450 in non-cirrhotic, treatment-naive patients with GT-1, 2, or 3 chronic HCV infection. The study includes seven cohorts. The first three cohorts enrolled eight GT-1a patients each (7 active; 1 placebo) and tested once-daily oral doses of 60, 120 or 240 mg, respectively. Patients were dosed for three days and followed for up to 28 days for viral load quantification. The limit of detection for the viral load quantification assay is 15 IU/mL.
Safety evaluations include monitoring for adverse events, routine laboratory assessments, vital signs and 12-lead ECG tracings.
In cohorts 4 through 6, patients with GT-1b, GT-2 and GT-3 are dosed once-daily at 240 mg. An additional cohort (cohort 7) of GT-1a patients is dosed twice daily with 240 mg. Data generation and analysis of results for cohorts 4 through 7 is ongoing. An interim analysis of those cohorts showed antiviral activity for GT-1b similar to that for GT-1a, but minimal antiviral activity for GT-2 and GT-3.
The Company anticipates presenting further data on all cohorts at a future scientific conference.
About TD-6450
TD-6450 is an internally discovered multivalent NS5A inhibitor designed to have improved antiviral activity against GT-1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450’s heterodimeric structure permits an asymmetric binding mode to NS5A relative to structurally symmetric inhibitors. TD-6450 has demonstrated additive activity with other classes of anti-HCV agents in replicon assays, and no cross-resistance with RAVs that confer resistance to other anti-HCV agents. The Company believes that the antiviral activity of TD-6450, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients.
TD-6450 was previously evaluated in a single-ascending dose and a 14-day multiple-ascending dose study in healthy subjects (study 0094). This randomized, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacokinetics of TD-6450. Single doses (up to 500 mg) and multiple doses of TD-6450 (up to 240 mg daily for 14 days) were evaluated in healthy subjects. Following single and multiple doses, TD-6450 was generally well-tolerated and no subjects discontinued due to adverse events. Headache was the most commonly reported adverse event following multiple doses (n=4). TD-6450 pharmacokinetics were linear up to 240 mg following single and multiple doses and its long half-life supports once-daily dosing.
About Hepatitis C and the NS5A Inhibitor Class
Hepatitis C is an infectious disease of the liver. Worldwide, health experts estimate that 130 – 150 million people have chronic hepatitis C, with as many as four million of those cases in the United States. Hepatitis C, like all forms of hepatitis, can damage the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The hepatitis C non-structural 5A (NS5A) protein of HCV has emerged as an attractive drug target and inhibitors of NS5A have a central role in all-oral HCV therapy. The multi-functional NS5A protein is required for ribonucleic acid (RNA) replication and virion assembly, and a number of investigational and approved NS5A inhibitors have shown antiviral efficacy in HCV-infected patients.
Theravance Biopharma and Trek Therapeutics Announce Initiation of Phase 2a Trial of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
Study Being Conducted by Trek Therapeutics Following Licensing of Worldwide Rights to Drug Candidate From Theravance Biopharma
DUBLIN, IRELAND and CAMBRIDGE, MA — (Marketwired) — 10/27/15 — Theravance Biopharma, Inc. (NASDAQ: TBPH) (“Theravance Biopharma”) and Trek Therapeutics (“TREKtx”) today announced that TREKtx has initiated a Phase 2a clinical trial of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus (HCV). Theravance Biopharma recently granted TREKtx an exclusive worldwide license for the development, manufacturing, use, marketing and sale of TD-6450 as a component in combination HCV products. Other terms of the transaction have not been disclosed.
The Phase 2a clinical trial will evaluate faldaprevir (FDV), an HCV protease inhibitor, combined with TD-6450 and ribavirin (RBV) in patients infected with HCV genotype 4. The trial is being conducted in the United States.
Mathai Mammen, M.D., Ph.D., Senior Vice President of Research and Development at Theravance Biopharma commented, “We are pleased to see the initiation of this Phase 2a clinical trial with TD-6450. This NS5A inhibitor has shown robust antiviral activity in a Phase 1 trial in patients with HCV genotype 1, as well as preclinical potency against both wild type HCV and resistance-associated variants. We believe that its antiviral activity, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients. We are especially pleased to collaborate with TREKtx and support their commitment to delivering novel and accessible combination HCV treatments to patients worldwide.”
“We are very excited about dosing our first genotype 4 patients in this combination study. If safety and efficacy are demonstrated, the goal is to initiate clinical trials in Egyptnext year, where the need is enormous,” said Dr. Robert Hindes, Chief Medical Officer of Trek Therapeutics.
About TD-6450
Theravance Biopharma discovered TD-6450, a multivalent NS5A inhibitor designed to have improved antiviral activity against genotype 1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450 has successfully completed Phase 1 studies in both healthy volunteers and HCV patients.
About Faldaprevir
Faldaprevir is a protease inhibitor that TREKtx acquired from Boehringer Ingelheim. FDV has completed Phase 3 studies in combination with pegylated interferon and RBV.
About HCV
Hepatitis C is an infectious disease of the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The U.S. Centers for Disease Control and Prevention estimates 2.7 million individuals in the United States have active hepatitis C virus (HCV) infection, most of whom are “baby boomers.” In the United States, chronic HCV infection is the leading cause of cirrhosis and liver cancer and the most common reason for liver transplantation. Worldwide, more than 135 million people have chronic HCV infection and most are undiagnosed.
About Trek Therapeutics
TREKtx is a private, clinical stage public benefit corporation developing treatments for serious infections. Its mission is to profitably develop affordable and accessible medicines to treat infectious diseases and to commercialize them for global populations. The company’s founders collectively participated in the development of seven approved antiviral drugs.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| US201161492267 |
| WO2013067267A1 * | 2 Nov 2012 | 10 May 2013 | Theravance, Inc. | Rod -like hepatitis c virus inhibitors containing the fragement {2- [4- (bi phenyl – 4 – yl) – 1h – imidazo – 2 – yl] pyrrolidine – 1 – carbonlymethyl} amine |
| WO2013163270A1 * | 24 Apr 2013 | 31 Oct 2013 | Theravance, Inc. | Hepatitis c virus inhibitors |
| WO2013165796A1 * | 25 Apr 2013 | 7 Nov 2013 | Theravance, Inc. | Crystalline form of a pyridyl-piperazinyl hepatitis c virus inhibitor |
//////////////


Reblogged this on Leaders in Pharmaceutical Business Intelligence and commented:
The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.
Thanks