
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Marbofloxacin
- Molecular FormulaC17H19FN4O4
- Average mass362.356 Da
115550-35-1[RN]
2,3-Dihydro-9-fluoro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[3,2,1-ij][4,1,2]benzoxadiazine-6-carboxylic Acid
6807
7H-1,3,4-Oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid, 9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-
8X09WU898T
марбофлоксацин
ماربوفلوكساسين
马波沙星
Marbofloxacin is a carboxylic acid derivative third generation fluoroquinolone antibiotic. It is used in veterinary medicine under the trade names Marbocyl, Forcyl, Marbo vet and Zeniquin. A formulation of marbofloxacin combined with clotrimazole and dexamethasone is available under the name Aurizon (CAS number 115550-35-1).

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
CN 107383058,
https://patents.google.com/patent/CN107383058B/enMarbofloxacin (Marbofloxacin) is fluoroquinolone antibacterial agent for animals, the entitled fluoro- 3- methyl-1 0- of 9- of chemistry (4- methylpiperazine-1-yl) -7- oxo -2,3- dihydro -7H- pyridine [3,2,1-ij] [4,1,2] benzo oxadiazines -6- carboxylic acid, It is developed by Roche Holding Ag, and is further developed by French Vetoquinol (method national strength and prestige are grand) company earliest, in nineteen ninety-five in Europe Listing.Marbofloxacin is after Enrofloxacin (Enrofloxacin), Danofloxacin (Danofloxacin), sarafloxacin (Sarafloxacin) etc. another third generation carbostyril family antibacterial drugs after, the drug have extensive antibacterial activity simultaneously With very good dynamic characteristic, sterilizing power is strong, absorbs fastly, widely distributed in vivo, with other antimicrobials without crossing drug resistant Property, easy to use, adverse reaction is small.Pharmacokinetic is studies have shown that Marbofloxacin removes long half time in animal body, biology Availability, almost without residual in the blood of animal, excrement and tissue, is well suited for clinically to antibiosis for animals close to 100% The requirement of element, structural formula are as follows:
Structure is complicated for Marbofloxacin, not only contains methyl piperazine substituent group, but also aromatic moieties contain pyridine benzo evil two Piperazine skeleton has had many documents and patent report at present and has reviewed its synthetic method, such as patent US4801584, ZL94190968.9, EP2010/067828, CN101619068, CN102060860, CN102617595, document J.Org. Chem., 1992,57 (2), 744-766, ” chemical reagent ” 2007,29 (11), 701-703., ” Chinese Journal of Pharmaceuticals ” 2002,33 (1), 1358-1363 etc..Patent US4801584 reports fluoro- via the fluoro- 4,8- dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis- preparation 6,7- bis- The method of 8- hydroxyl -1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester, this method are related to using valuableness And commercialization is not easy amination reagent O- (2, the 4- dinitrophenyl) oxyammonia largely purchased in 1 upper amino, by multistep reaction After complete the preparation of fluoro- 8- hydroxyl -1- (the methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis-, passed through after It crosses and paraformaldehyde, N methyl piperazine reacts the preparation for realizing Marbofloxacin.Correlated response formula is as follows:
The patent literature reports such as patent ZL94190968.9 are that raw material prepares Ma Bosha from 2,3,4,5 tetra fluoro benzoic acid The synthetic route of star, this method are not only related to the multisteps hazardous reactions such as carboxylic acyloxy chlorination, Grignard Reagent preparation reaction, synthesize road Wire length, and 3- (the N- methyl formyl hydrazono-) ethyl acrylate for being difficult to prepare is used, and yield is low, be not suitable for industrially putting Mass production, correlated response formula are as follows:
Patent CN101619068 is condensed using 2,3,4,5- phenyl tetrafluoride carbamoylalkyl esters and inferior amine salt, obtained N- bis- Methyl substituted enamine derivates react the enamine for preparing the substitution of N- methyl-N- acyl group under organic acid catalysis with N- methylhydrazide Derivative, then 6,7,8- tri- fluoro- 1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline-are completed in cyclization and hydrolysis under alkaline condition The preparation of 3- carboxylic acid realizes Ma Bosha finally by with N methyl piperazine, dimethyl formal (or diethyl formal) reaction The preparation of star.The technique uses the dimethyl suflfate and the height hazardous reaction reagent such as sodium hydride or alkalide of severe toxicity, because And it is subject to certain restrictions in commercial process.Correlated response formula is as follows:
In conclusion there are various deficiencies, such as chemistry examinations in the synthetic route of existing synthesis Marbofloxacin The defects of agent is expensive, reaction route is too long, using the chemical reagent for being unfavorable for industrialized production, the present inventor are real after study It tests, invents a kind of new method for preparing Marbofloxacin.The preparation of embodiment 1:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketone(E) -1,1,1- tri- chloro-4-methoxy butyl- 3- alkene -2- ketone (Formulas I, R=Me) (10.18g, 50mmol), 1- methyl The mixture of piperazine (6.0g, 60mmol) and mesitylene (50mL) is heated to reflux temperature and stirs 6 hours, and system is natural Be cooled to room temperature, remove organic solvent under high vacuum reduced pressure, residue (14.2g, crude product do not purify) without further purification, directly It connects for reacting in next step.Embodiment 2:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) urethanes (Formula VII) preparationUnder nitrogen protection, the product (14.2g is not purified, is directly used) of embodiment 1 is dissolved in toluene (120mL), then body Triethylamine (72mL, 514mmol) is added in system, system is heated to reflux temperature.Under reflux temperature, slowly dripped into reaction system Add toluene (60mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (16g, 75.3mmol).Rear system reflux is added dropwise 30min, then system slow cooling is to 60 DEG C, heat filtering.Filtrate is transferred in 500ml reaction flask, and carbazic acid second is then added Ester (Formula V, R2=Et) (6.25g, 60mmol).System is reacted 12 hours at a temperature of 60-65 DEG C after addition.To reaction H is slowly added in system2O (150mL) quenching reaction, system are naturally cooling to room temperature.Filtering, obtains solid, and solid uses heptan Alkane/ethyl acetate system mashing processing, obtains solid (Formula VII, R2=Et) (21.2g).Embodiment 3:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)2 obtained solid of embodiment (21.2g) is placed in 200ml reaction flask, ethyl alcohol (50mL) is added into reaction system With water (50mL), system is heated to flowing back.The aqueous solution (30mL) of KOH (7.0g) is slowly added under counterflow condition to system, is dripped System maintains the reflux for state response 96 hours after adding.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50ml) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (12.4g) after removing organic solvent.The preparation of embodiment 4:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketoneSequentially added in reaction flask the chloro- 4- ethyoxyl butyl- 3- alkene -2- ketone (Formulas I, R=Et) of (E) -1,1,1- three (14.1g, 65mmol) and 1- methyl piperazine (7.0g, 70mmol).Then system is heated to 130-155 DEG C and is stirred to react 5 hours.System is cold But to room temperature, the complete raw material of a little unreacted of high vacuum removed under reduced pressure, residue (16.8g, crude product do not purify) is without pure Change, is directly used in and reacts in next step.Embodiment 5:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) t-butyl carbamate (Formula VII, R2=tBu) preparationUnder nitrogen protection, the product (16.0g is not purified, is directly used) of embodiment 4 is dissolved in toluene (125mL), then N is added in system, N- diisopropylethylamine (104.5mL, 600mmol), system is heated to reflux temperature.Under reflux temperature, to Toluene (70mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (18.8g, 88mmol) is slowly added dropwise in reaction system.It is added dropwise Starting material Formula II is tracked to HPLC within system reflux 1 hour afterwards to disappear.Then system slow cooling is to 60 DEG C or so, hot mistake Filter.Filtrate is transferred in 500mL reaction flask, and tert-butyl carbazate (Formula V, R is then added2=tBu)(9.3g,70mmol).It is added After system reacted 48 hours at a temperature of 60-65 DEG C.H is slowly added into reaction system2O (150mL) quenching reaction, body System is naturally cooling to room temperature.Filtering obtains solid, and solid is handled using heptane/ethyl acetate system mashing, obtains solid (formula VII,R2=tBu) (19.3g) is directly used in next step without further purification.Embodiment 6:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)By 5 obtained solid of embodiment (19.0g) as in 200mL reaction flask, methanol (55mL) is added into reaction system With water (55mL), system is heated to flowing back.The aqueous solution (30mL) of CsOH (13.5g) is slowly added under counterflow condition to system, Rear system is added dropwise and maintains the reflux for state response 96 hours.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50mL) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (8.8g) after removing organic solvent.Embodiment 7: the preparation of Marbofloxacin1- amino-6- fluoro- 8- hydroxyl-7- (4- methylpiperazine-1-yl) oxo-1-4- is sequentially added in 100mL reaction flask, 4- dihydroquinoline -3- carboxylic acid (Formula VIII, 6.0g), 85% formic acid (30mL) and 36.5% formalin (6.0mL). System is carefully slowly heated to 75 DEG C or so reactions 1 hour after addition.Then system is cooled to 10 DEG C hereinafter, being carefully added into 25% ammonium hydroxide (25mL), stir 0.5 hour.Then activated carbon (1g) is added into system, mistake after 1 hour is sufficiently stirred Filter, filtrate methylene chloride extract 2 times (2 × 100mL).Merge organic phase, anhydrous sodium sulfate dries, filters, organic phase high vacuum Removed under reduced pressure solvent obtains Marbofloxacin crude product (5.4g).H is added in the crude product2In O (50mL), first acid for adjusting pH value is slowly added dropwise To 3.2 (pH meter detections), 4 hours are stood, filtering, filtrate added drop-wise sodium bicarbonate aqueous solution adjusting pH value to 6.2 (pH meter detections), A large amount of solids are precipitated, and ice salt bath cooling system stirs 1 hour to 0 DEG C or so, filtering, obtain Marbofloxacin after product drying (4.72g)。

Patent
Publication numberPriority datePublication dateAssigneeTitleUS4801584A *1986-09-121989-01-31Hoffmann-La Roche Inc.Pyrido(3,2,1-IJ)-1,3,4 benzoxadiazine derivativesCN1116849A *1993-01-231996-02-14辉瑞大药厂Process for the manufacture of a tricyclic compoundCN102060860A *2011-01-072011-05-18安徽美诺华药物化学有限公司Preparation method of MarbofloxacinCN102617595A *2012-03-232012-08-01江西华士药业有限公司Preparation method of fluoroquinolone antibacterial medicament marbofloxacinCN102712598A *2009-11-192012-10-03新梅斯托克尔卡·托瓦纳·兹德拉维尔公司A process for a preparation of marbofloxacin and intermediate thereof
CN110283186A *2019-07-192019-09-27海门慧聚药业有限公司A kind of crystal form of Marbofloxacin and preparation method thereof
PATENT
CN 107522718
PATENT
CN 102617595,
PATENT
Indian Pat. Appl., 2009CH00164,
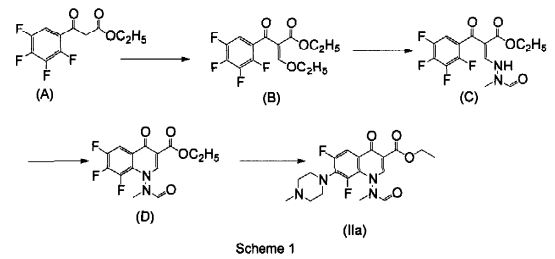

Example 2: Preparation of ethyl 6,8-difluoro-1-(N~methylfomnamido)-7-(4-methyl-1- piperazinyl)-4-oxo-4H-quinoline-3-carboxylate hydrochloride of Formula (Ilia)
STR IIIA
Water (400 ml) and the compound of Formula (IIa) (200 g) are charged into a round bottom flask at 28°C and concentrated HCI (124 ml) is added slowly at a temperature below 40°C, and the mass is heated to 95-1OO0C. 300 ml of water and ethanol are distilled under vacuum at 1004C. The mass is cooled to 25-30°C. Acetone (400 ml) Is added and the mass is cooled to 0-5°C. The mass is maintained at 0-58C for 30-60 minutes and the product is filtered. The product is washed with pre-chilled acetone (200 ml) and dried under vacuum at 70-75°C for 12-15 hours to obtain the title compound. Yield: 181.0 g (95%). Example 3: Preparation of marbofloxacin from the compound of Formula (Ilia) Ethylene glycol (100 ml) and potassium hydroxide (17.3 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (10 g) is added and the mass is heated to 120-130’C, and then maintained for 24 hours. The mass is cooled to 30°C and water (15 ml) is added. Hydrochloric acid (36%, 18 ml) is slowly added below 404C.rformic acid (6 ml) is slowly added below 40°C and the mass is stirred for 20-30 minutes. Formaldehyde (5 ml) is added and the mass is then heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to 15-20°C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled demineralized water (5 ml). The material is suction dried for 2-3 hours. Methanol (50 ml), demineralized water (15 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes.
Ammonia solution (25%, 7.5 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (5 ml) at 25- 35°C. The water and methanol are distilled at 60-70°C under vacuum until 20 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid is filtered at 0-5°C and the wet cake is washed with methanol (10 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 6.51 g (70%). Example 4: preparation of marbofloxacin from a compound of Formula (Ilia) Ethylene glycol (150 ml) and potassium hydroxide (72.2 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 115-1256C, and then is maintained for 10-12 hours at 115— 125°C. The mass is cooled to 25-35°C and water (150 ml) is added. Formic acid (98%, 100 m!) is slowly added below 45°C and the mass is stirred for 30-60 minutes. Formaldehyde (37-41%, 35 ml) is added to the mass, which is then heated to 70- 75°C and maintained for 1-2 hours. The mass is slowly cooled to 0-5°C and stirred for 1-2 hours. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (50 ml). The material is suction dried for 1 hour and washed with pre-chilled acetone (50 ml) and suction dried for 1 hour. Methanol (250 ml), water (100 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 40 mi) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (50 ml) at 25-35°C. The filtrate is distilled at 60-70°C under vacuum until 75-100 ml remain. The mass is cooled to 10-15’C and maintained for 30-60 minutes. The solid free base is filtered at 10-15°C and the wet cake is washed with chilled methanol (50 ml). The material is suction dried for 30-^60 minutes and the product is dried at 60-70°C under vacuum for 10-12 hours. Yield: 33.0 g (70.8%). Example 5: Preparation of marbofloxacin from a compound of Formula (Ilia) Water (350 ml) and potassium hydroxide (86.6 g) are stirred for 10 minutes. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 100-104°C. The mass is maintained for 105-110 hours at 100-1040C, then is copied to 25-35°C and water (65 ml) is added. Hydrochloric acid (36%, 125 ml) is slowly added below 40°C and the mass is stirred for 30 minutes. Formaldehyde (37%, 19 ml) is added and the mass is heated to 70-756C. The mass is maintained for 1-2 hours at 70-75 0C and then is slowly cooled to 0-5°C and maintained for 30-60 minutes. The obtained solid hydrochloride salt is filtered and the bed is washed with pre-chilled water (25 ml) at 0-5°C. The material is suction dried. Ethanol (250 ml), water (75 ml), ammonia solution (25%, 38 ml) and the wet cake are charged into a round-bottom flask and stirred for 1-2 hours at 25-35° C. The turbid solution is filtered and the bed is washed with ethanol (50 ml). The filtrate is distilled at 65-70°C under vacuum until 100 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered and the wet cake is washed with pre-chilled ethanol (50 ml). The product is dried under vacuum at 60-70°C for 15-^20 hours. Yield: 23.3 g (50%).
Example 6: Preparation of marbofloxacin from a compound of Formula (IIa) Ethylene glycol (60 ml) and potassium hydroxide (28.05 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (IId) (20 g) is added. The mass is heated to 120-135°C and maintained for 4-6 hours. The mass is cooled to 30°C and water (60 ml) is added. Formic acid (98-100%, 40 ml) is slowly added below 40°C and stirred for 20-30 minutes. Formaldehyde (37-41%, 12 ml) is added to the mass, which is heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to O-S6C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (20 ml). The material is suction dried for 2-3 hours. Methanol (100 ml), water (30 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 20 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (10 ml) at 25-35°C. The water and methanol are distilled at 60-70°C under vacuum until 40 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered at 0-5°C and the wet cake is washed with methanol (20 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 12.6 g (71%)
Example 7: Purification of marbofloxacin To crude marbofloxacin (25 g) is added methanol (125 ml) and ammonia (18.75 ml). Half of the volume of the methanol and ammonia solution is removed by azeotropic distillation. The mass is slowly cooled and maintained for 1 hour. The product is filtered and washed with chilled methanol (25 ml). The product is suction dried for 30 minutes and dried under vacuum for 12 hours, to yield pure marbofloxacin of a purity 99.80%. XRD pattern, DSC thermogram, TGA1 and IR are substantially in accordance with Figs. 1, 2, 3, and 4, respectively. Yield: 22 g (88.0%),
PATENT
Indian Pat. Appl., 2009CH00163,
PATENT
WO 2011061292
PATENT
CN 102060860,
PATENT
CN 101619068,
PATENT
https://patents.google.com/patent/EP2332916A2/en
- Marbofloxacin is the common name for 9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyridol(3,2,1-ij)(4,2,1)benzoxadiazin-6-carboxylic acid, of the formula :
- [0003]
Marbofloxacin is a potent antibiotic of the fluoroquinolone group. - [0004]
EP 259804 describes marbofloxacin as well as a synthesis for the preparation thereof by a multistep process which is unpractical for a large scale manufacture, since it requires high temperatures and reagents not suitable for large-scale production, resulting in low over-all yields. The process for the preparation is disclosed in the reaction scheme 1. - [0005]
EP 680482 discloses an alternative approach for the preparation of marbofloxacin, wherein hydroxy group is introduced into molecule by means of reaction of intermediate with alkali metal hydroxide in aqueous media. The starting material used is 2,3,4,5-tetrafluorobenzoic acid. Disadvantages of this process are relatively high excess of alkali metal hydroxide and lengthy procedure. The process for the synthesis according to this patent is shown in the reaction scheme 2. - [0006]
Research Disclosure No. 291, 1988, pages 548-551 discloses an alternative route of synthesis also starting from 2,3,4,5-tetrafluorobenzoic acid. Later steps of the process are shown in the reaction scheme 3. - [0007]
IT 1313683 relates to a process for preparation of marbofloxacin by a process via benzyl ether. Ether was debenzylated in aqueous solution by hydrogenating over 5% Pd/BaSO4 and the obtained product is cyclized using HCOOH/HCOH. - [0008]
In view of the prior art there still exists a need for an improved method for preparation of marbofloxacin and intermediates thereof suitable for a large-scale production.
Examples
- [0068]
A high resolution HPLC method is used to determine an amount and purity compounds of formula I, II and IV. The tests are carried out in X-Bridge C18, 150 x 4.6mm, 3.5µm column. The mobile phase is gradient of A) 5mM NH4COOCH3 pH=7.0 B) acetonitrile. Gradient: 0’=10%B, 10’=20%B, 25′-30’=90%B, 32’=10%B. - [0069]
The chromatograph is equipped with a UV detector set at 250 nm and 315nm, the flow rate is 1.0 ml per minute at 30°C.
Example 1a) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt
- [0070]
- [0071]
4.137g of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (10.14mmol) was put into 40mL of 10% H2SO4 and stirred at 100°C for 7 hours. Reaction mixture was cooled and crystals were formed. Mixture was cooled to 4°C and filtered with suction. Filter cake was washed with a mixture of H2O/EtOH/THF (1/1/5) and dried. 3.260g of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid as yellow crystals were obtained (91%). - [0072]
In case the sodium salt is desired the product obtained in previous step was put into 5mL of EtOH and 10mL of CH2Cl2 and 1.20g of NaOH dissolved in 2mL of water was added. Solution was stirred at room temperature. for 1h, dried with Na2SO4 and evaporated. 2.90g of pure title product was isolated (yellow powder, 7.71mmol, 76%).
b) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- [0073]
- [0074]
400mg of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (0, 979mmol) was put into 2mL of 10% H2SO4 and stirred at 100°C for 2 hours. Reaction mixture was cooled and crystals were formed. To this mixture 1,7mL of 25% aq. NH3 was slowly added. At first very dense suspension was formed that dissolves with further addition of ammonia solution. At the end clear solution formed with pH of 9. Ammonium sulphate was precipitated by the addition of 10mL of EtOH , filtered off and washed with 5mL of H2O/EtOH (1/2). Mother liquor was dried on the rotary evaporator and 10 mL of EtOH/H2O mixture (7/3) was added to precipitate residual inorganic salt, which was again filtered off. Remaining yellow solution was dried on a rotary evaporator to obtain 321mg of yellow powder (0.912 mmol, 93%).
Example 26-fluoro-8-hydroxy-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- [0075]
- [0076]
178 mg of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.470mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 0.10mL of HCOOH was added to neutralize hydroxide. 5mL of EtOH is added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 90mg of the product was obtained.
Example 39-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
- [0077]
- [0078]
180 mg of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.481mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 3 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1 mL of HCOOH was added followed by addition of 0.4 mL of 37% aq. solution of HCHO and stirred at 70°C for additional hour. Reaction mixture was cooled to room temperature and 5mL of EtOH was added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 111 mg of grey powder was obtained.
Example 49-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
- [0079]
- [0080]
1.14g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 3.06g of Me4NOH.5H2O (16.96mmol) and stirred at 100°C for 5 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1.44 mL of HCOOH (85% aq. sol) was added followed by addition of 0.5 mL of 37o aq. solution of HCHO and the flask was cooled on the water bath at 22°C. Another 1.44mL of 85% HCOOH was added and the reaction mixture was warmed to 70°C for 30min and after cooling 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.23g of grayish powder was obtained (90%) .
Example 59-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0081]
- [0082]
1.145g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 2.72g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved. After stirring at 70°C for 30min (precipitate was formed again after 5min) reaction flask was cooled to room temperature and 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.165g of grayish powder was obtained (85%), with a purity of 97.11% (HPLC). - [0083]
Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallized in a mixture of 26mL of EtOH and 14mL H2O. 0.673g of powder was obtained (61%) with a purity of 98.75% (HPLC).
Example 69-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0084]
- [0085]
1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 2.72g of Me4NOH.5H2O (15.01mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and 20mL of H2O was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.022g of greyish powder was obtained (75%). with a purity of 97.11% (HPLC). - [0086]
Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallised in a mixture of 20mL of EtOH and 6mL CHCl3. 0.771g of yellow powder was obtained (71%) with a purity of 99.50% as determined by HPLC.
Example 79-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0087]
- [0088]
1.142 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.26g of Me4NOH.5H2O (18.01mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.147g of white powder was obtained (84%). - [0089]
Crude reaction product was mixed with 5mL of water and 2mL of 25% aqueous solution of NH3 and clear solution was obtained. To this solution, 7mL of EtOH was added and dried under reduced pressure. Product was crystallized in a mixture of 15mL of EtOH and 10mL CHCl3 to obtain 0.4321g of white powder (41%) with a purity of 98.63% as determined by HPLC
Example 89-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0090]
- [0091]
1.136 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (2.98mmol) was mixed with 2.73g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 7 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.039g of grey powder was obtained (77%). - [0092]
Crude reaction product was neutralized with 2mL of 25% aqueous solution of NH3 and clear solution was diluted with 15mL of EtOH and 9mL of H2O. Solution was partially dried under reduced pressure until the formation of precipitate. At this point mixture was cooled in a refrigerator and precipitate was isolated by filtration under reduced pressure to obtain 0.675g of powder (65%) with a purity of 98.84% as determined by HPLC.
Example 99-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0093]
- [0094]
1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.30g of Me4NOH.5H2O (18.20mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure to obtain 0.847g of solid, while mother liquid was diluted with EtOH and concentrated under reduced pressure until precipitate forms, which was filtered again to obtain additional 0.208g of solid. The yield of combined solid material is 1.055g, 77%. Crude reaction product (formate salt) was crystallized in H2O/EtOH (25mL/10mL) to obtain 0.722g (53%) of yellow powder. Formate salt was put in 20mL of EtOH/CH2Cl2 mixture (1/1) and 0.5mL of 25%aq. NH3 was added to obtain clear solution. Solution was dried with Na2SO4 and solvent evaporated under reduced pressure to obtain 0.580g of yellow powder (53%).
Example 109-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0095]
- [0096]
100 mL reactor with a rotary stirrer was charged with 10,16g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (28,83mmol) and 26,50g of Me4NOH˙5H2O (146,25mmol) that was previously mixed together. Temperature of the heating jacket was set to 100°C and stirring to 100s-1, while water was allowed to evaporate out of the reactor during the reaction. Reaction was stirred at specified temperature for 5 hours and homogenous dark brown oil was obtained. Temperature of reactor was cooled to 20°C, 30mL of HCOOH was added and stirred well so that all oil is transformed into brown suspension. 4,5mL of 37% aq. HCHO was added drop-wise and heated at 70°C for 30min. Reaction mixture was cooled to 20°C and 20mL of water added to precipitate the product in the form of formate complex. Suspension was cooled to 0°C and filtered under reduced pressure and washed the filter cake with additional 10mL of cold water to obtain 8,38g of white powder. Mother liquor was partially evaporated under reduced pressure and when solid started to precipitate it was filtered again to obtain additional 0.80g of powder. 50mL of EtOH was added into the mother liquor to precipitate the product and after filtration at reduced pressure further 0.80g of white powder was obtained. Product was collected and 9,98g of white powder was suspended in a mixture of 50mL of EtOH and 50mL of CH2Cl2. Into the suspension 25% aq. NH3 was added to neutralize the formate complex and after addition of 12mL of NH3 all product was dissolved and small amount of solid material is formed. 5g of anhydrous Na2SO4 was added to dry the organic solution and it was filtered off and solvent evaporated under reduced pressure. 8.99g of slightly yellow powder was obtained in 86% yield.
Example 11Crystallization from ethanol/toluene/water 2:1:1
- [0097]
8.4g of crude marbofloxacin was suspended in a mixture of 83 ml of ethanol, 41ml of toluene and 41 ml of water and heated to reflux. From the clear yellow solution formed 83 ml of solvent mixture was distilled off, whereby the temperature rose from 74 to about 79°C, and a yellow precipitate was formed. The suspension was cooled to 20° – 25°C, stirred for 1 hour, filtered, and the filter cake was washed with 3 portions of 6 ml of ethanol to yield after drying in vacuum dryer the product in more than 95% yield.
Example 12Crystallization of marbofloxacin starting from marbofloxacin formate
- [0098]
26g of marbofloxacin formate was suspended in a mixture of 65ml of ethanol and 27ml of water. Under stirring a solution of 25% ammonia in ethanol (20ml 25%NH3/10ml EtOH) is slowly (about 30 minutes) added by drops until the substance is dissolved and pH value of 7-9 is reached. The reaction was stirred for about 15 minutes and filtered. The filtrate was evaporated at 110°C until about 60ml of the solvent was distilled off and marbofloxacin started to precipitate. After distillation the suspension was cooled and stirred for 0.5 to 1 hour at 0-5°C, filtered, to yield after drying at 40°C/50mbar for 3 to 5 hours the product in 100%yield.
Example 13Crystallization from ethanol
- [0099]
1g of marbofloxacin was dissolved under heating to reflux in 160ml of ethanol, after filtration, the solution is cooled and the crystallized product is recovered in more than 90% yield.
Example 146,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0100]
- [0101]
10mmol of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester was put in the round bottomed flask. 20mL of 10% H2SO4 was added and stirred with the temperature of the sand bath of 100°C for the time periods specified in the following table. Reaction mixture was cooled down to 4°C, filtered and the cake washed with water and the conversion an yield were determined. - [0102]
The experiment was repeated but starting compound was mixed with 1.0mL of solvent (EtOH, AcOH or MeCN as specified in the following table) before adding the 10% H2SO4. - [0103]
The starting compound is insoluble in aqueous phase. By mixing the starting compound with a small amount of polar solvent (EtOH, MeCN, AcOH) a film is formed around the crystals which improves wetting of the crystals with the aqueous acid. Without addition of polar solvent prior to adding the aqueous acid solution wetting of the crystals is impaired and the reaction is slower.Exp.Reaction time (solvent)Conversion (yield)14.016h65%14.027h60%14.0324h100%14.0424h100% (94%)14.056h (0.1mL AcOH per mmol)91%14.066h (0.1mL EtOH per mmol)89%14.0721h (0.1mL MeCN per mmol)100% (97%)14.0821h (0.1mL MeCN per mmol)100% (96%)
Example 156,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0104]
3.30g of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (10.054 mmol) was put into the round-bottomed flask equipped with the magnetic stirrer. 1mL of MeCN was added and stirred for a minute. 20mL of 10% H2SO4 was added and stirred. The flask was put into the sand bath (T = 100°C) and stirred for 21h. Suspension was cooled down to 4°C and filtered under suction. Yellow powder was washed twice with cold water and dried. 2.646g of yellow powder was obtained (9.721 mmol, 96.7%) and identified by NMR spectroscopy to be title compound.
Example 166,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0105]
6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (6.868g, 20.92 mmol) was mixed with 1mL of EtOH (to decrease the hydrophobicity of the substrate). Next, 40mL of 10% aqueous H2SO4 solution was added and the mixture was stirred at the temperature of the bath of 100°C for 12h. A white suspension formed which was cooled to 0°C and filtered under reduced pressure. The white powder was washed with cold water and cold EtOH and dried. 5.135g of yellow powder was obtained and identified as title compound by 19F and 1H NMR spectroscopy. The yield of hydrolysis was 90%.
Example 176,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0106]
- [0107]
6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (272mg, 1.0mmol, obtained as described in Example 16, and 400 mg of N-methylpiperazine (4.0mmol) were mixed with 1mL of EtOH and stirred under reflux temperature (jacket temperature Tj=100°C). After two hours of reaction clear solution formed, afterwards the product precipitated and a very dense suspension was formed. Reaction was stopped after three hours of stirring at Tj=100°C. A sample was put directly to the NMR analysis and only two signals were observed indicating reaction was quantitative. Crude reaction product was diluted with EtOH and neutralized by addition of aqueous solution of NH3 until pH of 8 was reached. Suspension was cooled to 0°C and product isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 138mg (39%) of product was obtained.
Example 186,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0108]
6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (1.087g, 3.993mmol), 484mg of N-methylpiperazine (4.83mmol) and 484 mg of Et3N (4.78mmol) were mixed with 8mL of EtOH and stirred under reflux temperature (Tj=100°C). After 19h of reflux yellow solution and white precipitate are formed in the reaction flask. Solvent was evaporated under reduced pressure and put directly to the NMR analysis. Crude reaction product was mixed with 20mL of EtOH and suspension cooled in the refrigerator. The product (white precipitate) was isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 1.178g of white powder was obtained (3.375 mmol, 800).
Example 196,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0109]
In accordance with examples 17 and 18 additional experiments were carried out using different reaction conditions for the conversion of 6,7,8-trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid into 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid. The experiments were performed according to the following general procedure: 1.0mm of starting compound was put in the round bottomed flask and N-methylpiperazine (NMP), base and solvent were added according to the following table. Reaction mixture was stirred at the corresponding temperature. Solvent was evaporated and crude reaction mixture analyzed directly by NMR (1H and 19F).
Example 206,8-Difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester
- [0111]
- [0112]
Substitution: 6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (220mg) and 400mg Et3N stirred for three hours at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of the reaction (that is the reason for higher amount of TEA). After 3 hours of stirring was reaction mixture cooled to room temperature and analyzed by NMR spectroscopy. - [0113]
Substitution: The above reaction was repeated but Et3N was replaced by 1 equivalent of DABCO. - [0114]
In both cases, substitution was quantitative and analysis of the crude reaction mixtures showed that there was some hydrolysis of the ethyl ester (EE) to the free carboxylic acid (CA) group resulting in a product mixture. The results are summarized in the following table. Ethyl ester is readily soluble in water.Exp.Reaction conditionsConversion (yield)20.012.5 NMP, 1 DABCO, 100°C, 3h100% (48% EE, 52% CA)20.022.5 NMP, 4 Et3N, 100°C, 3h100% (58% EE, 42% CA)
Example 216,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid (one-pot reaction)
- [0115]
- [0116]
6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (200mg) and stirred for one hour at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of reaction. After one hour of reaction the reaction mixture was cooled to room temperature and 10% aqueous H2SO4 (5mL) was added and stirred again at 100°C for two hours. Yellow solution was cooled to 0°C so that product precipitated. It was isolated by filtration under reduced pressure. Pure 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid in the form of sulfate salt was obtained (as determined by NMR) as slightly yellow powder (279mg, 58%).
Example 22Synthesis of 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3, 7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid (Marbofloxacin, MBX)
- [0117]
13.5 g of 6,8-Difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride and ca. 63 g of tetramethylammonium hydroxide water solution 25 % were charged into a reactor and slowly heated to 100°C. When this temperature was reached, water was removed by distillation at reduced pressure (between 0.8 to 0.3 bar) in such a manner that ca. 25 to 32 ml of water were removed in 3 hours. The reaction mixture was stirred for another 3 hours and after completion of the conversion, the reaction mixture was cooled to 0 – 10 °C and ca. 40.5 ml of formic acid were slowly added with violent agitation. The temperature was maintained below 20°C, preferable between 0 – 10°C. Then ca. 6.1 ml of formaldehyde were slowly added. After addition the reaction mixture was heated to 70°C and maintained at this temperature for about 30 minutes. - [0118]
The reaction mixture was cooled to room temperature (20 – 30°C), ca. 27 ml of purified water were added and the mixture was stirred for 30 minutes. Then the reaction mixture was cooled to 0 – 5°C and stirred at this temperature for at least 2 hours. The product marbofloxacin formate (MBXBZ) was centrifuged and washed with 10 – 15 g of cooled (0 – 5°C) purified water. The product was spun dried and collected. - [0119]
Wet product MBXBZ was added to the mixture of 67 ml of ethanol, 67 ml of methylene chloride and 16.2 ml of ammonia solution (ca. 25 0). If phases did not separate, additional 63 ml of methylene chloride and 33 ml of purified water were added. The pH of the water phase was adjusted to be between 7 and 9.5, preferable between 7.5 and 8.5. The mixture was agitated for approximately 15 minutes to 1 hour and then the layers were separated and both phases were subjected to in process control (IPC) analysis. - [0120]
If IPC results showed that extraction was not complete, ca. 63 ml of methylene chloride were added to the water layer and the extraction was repeated until the IPC specification was met. - [0121]
The organic phases were combined and ca. 6.8 mg of sodium sulphate anhydrous and optionally 0.4 mg of activated charcoal were added. The mixture was mixed for at least 30 minutes and filtered, then organic solvent was distilled off to obtain crude marbofloxacin.
Purification of the crude Marbofloxacin
- [0122]
In an inert atmosphere 5 g of purified water, 12 g of ethanol 96 % and 4.3 g of toluene (ratio between the solvents was within the following ranges: ethanol : toluene : water : 1.8 – 2.8 : 1 : 1.1 – 1.2) were charged into a reactor and wet crude marbofloxacin (MBXCA) from the previous step was added under nitrogen. The mixture was slowly heated to reflux (70 – 80°C) until a clear solution was obtained. The solution was stirred for 0.5 hour under this temperature and then one half of the azeotrope solvent mixture (toluene : water : ethanol = 51 % : 6 % : 43 %) was evaporated. Then the remaining mixture was cooled slowly to 5°C (allowed interval is between 0 and 25 °C) with agitation (optionally 1 % mass of product of disodium-EDTA can be added). The mixture was mixed for 1 to 3 hours and the product was then isolated by centrifugation, washed with 13 g of ethanol, spun dry and collected. The product was dried at temperature 40 – 45°C, p < 100 mbar for 8 hour.
Example 23Purification of Marbofloxacin
- [0123]
Marbofloxacin was dissolved in 20 parts by weight of water by addition of acetic acid. Marbofloxacin was completely dissolved at pH of 5.3. Active charcoal was added and the mixture was stirred overnight. The mixture was then filtered using activated charcoal filter. The pH of the filtrate was adjusted to 7.2 by use of KOH, the obtained suspension was stirred for 1 hour at room temperature and then the precipitated product was recovered. Marbofloxacin with a purity of 99,9% (HPLC area) was obtained. - [0124]
HPLC analysis was performed on a pentafluorophenyl propyl (PFP) column (type Luna® PFP, 150 x 4.6mm, 3µm, Phenomenex, USA); detector: UV315 nm; flow rate: 0.8 ml/min; injection volume: 5 µl; mobile phase: A: 0.02M NaH2PO4xH2O+0,1% TEA, pH2.5; B: acetonitrile : methanol = 5:95 (v/v) ; gradient: 0’=10B, 25’=100B, 30’= 100B, 32’=10B. The HPLC chromatogram of marbofloxacin prior to purification is shown in Figure 1, the HPLC chromatogram after purification is shown in Figure 2. As evident from the chromatograms all products with retention time above 24min were successfully eliminated.
Mechanism of action
Its mechanism of action is not thoroughly understood, but it is believed to be similar to the other fluoroquinolones by impairing the bacterial DNA gyrase which results in rapid bactericidal activity.[1] The other proposed mechanisms include that it acts against nondividing bacteria and does not require protein and RNA synthesis, which block protein and RNA synthesis respectively.[2]
Activity
Marbofloxacin is a synthetic, broad spectrum bactericidal agent. The bactericidal activity of marbofloxacin is concentration dependent, with susceptible bacteria cell death occurring within 20–30 minutes of exposure. Like other fluoroquinolones, marbofloxacin has demonstrated a significant post-antibiotic effect for both gram– and + bacteria and is active in both stationary and growth phases of bacterial replication.[3]
It has good activity against many gram-negative bacilli and cocci, is effective against:
- Aeromonas
- Brucella
- Campylobacter
- Chlamydia trachomatis
- Enterobacter
- Escherichia coli
- Haemophilus
- Klebsiella spp
- Mycobacterium
- Mycoplasma
- Proteus
- Pseudomonas aeruginosa
- Salmonella
- Serratia
- Shigella
- Staphylococci (including penicillinase-producing and methicillin-resistant strains)
- Vibrio
- Yersinia
Application
Marbofloxacin can be used both orally and topically. It is particularly used for infections of the skin, respiratory system and mammary glands in dogs and cats, as well as with urinary tract infections. For dogs, a dose ranges from 2.75 – 5.5 mg/kg once a day. The duration of treatment is usually at least five days, longer if there is a concurrent fungal or yeast infection.[4] Maximum duration of treatment is 30 days.[3]
Contraindications and side effects
Marbofloxacin should usually be avoided in young animals because of potential cartilage abnormalities. In rare occasion, it can cause central nervous system (CNS) stimulation and should be used with caution in patients with seizure disorders.[3] Under certain conditions it can cause discomfort such as cramps, treatable with diazepam. Other adverse effects are usually limited to gastrointestinal tract (GI) distress (vomiting, anorexia, soft stools, diarrhoea) and decreased activity.[3]
References
- ^ Boothe, D.M. (2001) Antimicrobial drugs. In Small Animal ClinicalPharmacology and Therapeutics, pp. 150–173. W. B. Saunders Co., Philadelphia, PA.
- ^ Hunter RP, Koch DE, Coke RL, Carpenter JW, Isaza R. Identification and comparison of marbofloxacin metabolites from the plasma of ball pythons (Python regius) and blue and gold macaws (Ara ararauna). J Vet Pharmacol Ther. 2007 Jun;30(3):257-62.
- ^ Jump up to:a b c d Plumb DC (ed). Plumb’s Veterinary Handbook, 7th ed. Ames, IA: Wiley-Blackwell Publishing, 2011.
- ^ Rougier S, Borell D, Pheulpin S, Woehrlé F, Boisramé B (October 2005). “A comparative study of two antimicrobial/anti-inflammatory formulations in the treatment of canine otitis externa”. Veterinary Dermatology. 16 (5): 299–307. doi:10.1111/j.1365-3164.2005.00465.x. PMID 16238809. Archived from the original on 2013-01-05.
External links
| Clinical data | |
|---|---|
| Trade names | XeniQuin bolus & Injection (Opsonin Agrovet BD) |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATCvet code | QJ01MA93 (WHO) |
| Legal status | |
| Legal status | Veterinary use only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 115550-35-1 |
| ChemSpider | 54663 |
| UNII | 8X09WU898T |
| ChEMBL | ChEMBL478120 |
| CompTox Dashboard (EPA) | DTXSID4046600 |
| ECHA InfoCard | 100.168.181 |
| Chemical and physical data | |
| Formula | C17H19FN4O4 |
| Molar mass | 362.356 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////Marbofloxacin, марбофлоксацин , ماربوفلوكساسين , 马波沙星 ,

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}