
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

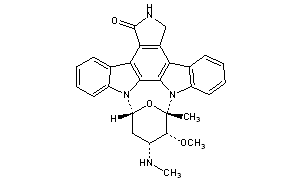
STAUROSPORINE
(+)-Staurosporine
- Molecular FormulaC28H26N4O3
- Average mass466.531 Da
(2S,3R,4R,6R)-3-Methoxy-2-methyl-4-(methylamino)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14,19,21,23,25,27-nonaen-16-one
6,10-Epoxy-6H,16H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-16-one, 7,8,9,10,17,18-hexahydro-7-methoxy-6-methyl-8-(methylamino)-, (6S,7R,8R,10R)-
62996-74-1[RN]
AM-2282
Antibiotic 230
antibiotic am 2282
StaurosporineCAS Registry Number: 62996-74-1
CAS Name: (9S,10R,11R,13R)- 2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3¢,2¢,1¢-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one
Manufacturers’ Codes: AM-2282; CGP-39360
Molecular Formula: C28H26N4O3, Molecular Weight: 466.53
Percent Composition: C 72.09%, H 5.62%, N 12.01%, O 10.29%
Literature References: Protein kinase C inhibitor; alkaloid isolated from Streptomyces staurosporeus. Isoln: S. Omura et al., J. Antibiot.30, 275 (1977). Crystal and molecular structure: A. Furusaki et al., J. Chem. Soc. Chem. Commun.1978, 800; eidem,Bull. Chem. Soc. Jpn.55, 3681 (1982). Corrected stereochemistry: N. Funato et al., Tetrahedron Lett.35, 1251 (1994). Total synthesis: J. T. Link et al., J. Am. Chem. Soc.117, 552 (1995); idem et al., ibid.118, 2825 (1996). Biosynthetic studies: D. Meksuriyen, G. A. Cordell, J. Nat. Prod.51, 884, 893 (1988); S.-W. Yang et al., ibid.62 1551 (1999). HPLC determn in blood and pharmacokinetics in rats: L. R. Gurley et al., J. Chromatogr. B712, 211 (1998). Inhibition of protein kinase C: T. Tamaoki et al., Biochem. Biophys. Res. Commun.135, 397 (1986); of other protein kinases: U. T. Rüegg, G. M. Burgess, Trends Pharmacol. Sci.10, 218 (1989). Induction of apoptosis: E. Falcieri et al., Biochem. Biophys. Res. Commun.193, 19 (1993); R. Bertrand et al., Exp. Cell Res.211, 314 (1994); of tyrosine phosphorylation: D. Rasouly, P. Lazarovici, Eur. J. Pharmacol.269, 255 (1994).
Properties: Pale yellow needles from chloroform-methanol as the methanol solvate, mp 270° (dec) (Omura). Also reported as yellow crystals from methanol, mp 288-291° (Meksuriyen, Cordell). [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol). uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85). Sol in DMSO, DMF. Slightly sol in chloroform, methanol.
Melting point: mp 270° (dec); mp 288-291° (Meksuriyen, Cordell)
Optical Rotation: [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol)
Absorption maximum: uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85)
Derivative Type: Hydrochloride
Molecular Formula: C28H26N4O3.HCl, Molecular Weight: 502.99
Percent Composition: C 66.86%, H 5.41%, N 11.14%, O 9.54%, Cl 7.05%
Properties: LD50 in mice (mg/kg): 6.6 i.p. (Omura).
Toxicity data: LD50 in mice (mg/kg): 6.6 i.p. (Omura)
Use: Pharmacological tool to study signal transduction pathways, tyrosine phosphorylation and to induce apoptosis.
An indolocarbazole that is a potent protein kinase C inhibitor which enhances cAMP-mediated responses in human neuroblastoma cells. (Biochem Biophys Res Commun 1995;214(3):1114-20)
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus.[1] It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.[2]
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive.[3] The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Chikara Murakata, Toshimitsu Takiguchi, Shigeo Katsumata, Akira Mihara, Keiichi Takahashi, Hiromitsu Saito, Shiro Akinaga, Masami Okabe, Yutaka Saito, “Process for producing staurosporine derivatives.” U.S. Patent US5344926, issued December, 1990.
SYN
CN 113122591
WO 2021127275
CN 110642872
WO 2020200945
CN 107603922
WO2006002422
PAPER
Journal of Antibiotics, 51(7), 679-682; 1998
PAPER
Journal of the American Chemical Society, 117(1), 552-3; 1995

PATENT
WO2006002422
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006002422
Preparation of Staurosporine Analogs
|00501 ] As will become apparent to a skilled artisan, many of the bridged epoxy diindolopyrrolo-hexahydrobenzodiazocines are commercially available as final compounds or modifiable intermediates. Staurosporine was originally isolated from the bacterium Streptomyces staurosporeus. (S.Omura et al. J.Antibiotics, 30, 275
1977).
[00502] Synthesis of 9, 12-epoxy staurosporine analogs:
NOTE: 2) 120°, 6) 120°,
Reactantsi 4, Reagents : 7, Catalysts. 2, Solvents 18,
Steps: 9, Stages: 11, Most stages in any one stept 2
[00503] Greater detail is provided in Tetrahedron Letters, 36(46), 8383-6,
1995.
[00504] Alternative synthesis of 9,12-epoxy staurosporine analogs:
NOTEt 1) stereoselective, 5) Raney nickel present,
Reactantsi 5, Reagentsi 6, Catalystsi 4, Solvents! 5,
Stepsi 7, stagest 9, Most stages in any one step: 2 [00505] Greater detail is provided in Organic Letters, 3(11), 1689-1692; 2001.
[00506]
[00507] Synthesis of 9, 13-epoxy staurosporine analogs:
NOTE: 1) STEREOSELECTIVE, 3) (92%/65%/95%/92%) , 4) 100% OVERALL (5.5:1,
ALPHA: BETA), 5) STEREOSELECTIVE, KEY STEP, 8) (97%/91%), 9) PHOTOCHEM.,
12) STEREOSELECTIVE KEY STEP, 14) (92%/81%/82%),
Reactants: 10, Reagents: 20, Catalysts: 5, Solvents: 9,
Steps: 17, Stages: 35, Most stages in any one step: 6
[00508] Whereas, a more thorough description of reagents, reaction conditions, and other pertinent syntheses are described Journal of the American Chemical Society, 117(1), 552-3; 1995. Additionally, syntheses on staurosporine and analogs thereof are described by S.J Danishefsky et al, J.Am.Chem.Soc, 118, 28251996 and J.L.Wood et al, J.Am.Chem.Soc, 118, 106561996.
Table 1 : Staurosporine Analogs
PATENT
https://patents.google.com/patent/WO2020200945A1/enEXAMPLES

.

A2 B2

Example 1Method for obtaining crude midostaurin B1 from crude staurosporine A1


A reactor was loaded with crude staurosporine A1 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C, obtaining crude midostaurin B1 with 95% yield. Example 2Method for obtaining purified midostaurin B2 from crude midostaurin B1 – reduction of 3-hydroxymidostaurin to midostaurin with triethylsilanei. TFA/TESii. NaHC03iii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20


B4 B2

A reactor was loaded with crude midostaurin B1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. At the end of the additions the solution was brought to 20°C. After 3 hours the solution was added with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (30 L) and two changes of solvent at atmospheric pressure were carried out. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield. Example 3Method for obtaining purified staurosporine A2 from crude staurosporine A1 – reduction of 3-hydroxystaurosporine to staurosporine with triethylsilane

A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. After 1 hour from the end of the additions, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 4Method for obtaining purified staurosporine A2 from crude staurosporine A1 – derivatization of 3-hydroxystaurosporine with trifluoroacetic acid and purification by crystallization


A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The mixture was cooled to 0°C and added with TFA (0.50 L), while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 5Method for obtaining purified midostaurin B2 from purified staurosporine A2 i. BzCI/DIPEAii. NH4CI/H2Oiii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20


A2 B2

A reactor was loaded with purified staurosporine A2 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C and subsequently transferred into another reactor. 2-MeTHF (30 L) was added and the suspension was heated under reflux up to complete dissolution. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield.
ClaimsHide Dependent
1) A process for the preparation of midostaurin with high purity, that is with a content of 3-hydroxymidostaurin impurities (III) and (IV) lower than 0.1%, comprising the treatment with strong organic or inorganic acids in a water-immiscible solvent and, optionally, also with reducing silanes.2) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude midostaurin with a reducing silane in the presence of a strong organic or inorganic acid.3) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude staurosporine with a strong organic or inorganic acid, optionally with the concomitant addition of a reducing silane.4) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the water-immiscible solvent is an aprotic polar water-immiscible solvent.5) The process for the preparation of midostaurin according to claim 4 wherein the water-immiscible solvent is dichloromethane, dichloroethane, methyl tetrahydrofuran or methylethylketone, preferably dichloromethane.6) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the strong acid is trifluoroacetic acid.7) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the reducing silane is triethylsilane.8) The process for the preparation of midostaturin according to anyone of the preceding claims, further comprising the benzoylation reaction of staurosporine to midostaurin characterized in that the benzoylation reaction is quenched with an aqueous solution having a slightly acid pH.9) The process for the preparation of midostaurin according to claim 8 wherein the aqueous solution having a slightly acid pH is an aqueous ammonium chloride solution.10) The process for the preparation of midostaurin according to anyone of the preceding claims, comprising the obtainment of purified midostaurin by crystallization from 2-MeTHF and its further isolation by:dissolving the crystallized midostaurin in a water-miscible polar solvent, adding waterisolating purified midostaurin as an amorphous solid obtained by filtering and drying, with a content of organic solvents < 50ppm. 11) The process for the preparation of purified midostaurin according to claim 10, wherein the polar s
Patent
Publication numberPriority datePublication dateAssigneeTitleJPS5247055B21973-12-041977-11-30US5093330A1987-06-151992-03-03Ciba-Geigy CorporationStaurosporine derivatives substituted at methylamino nitrogenEP0575955A11992-06-221993-12-29Kyowa Hakko Kogyo Co., Ltd.Process for producing staurosporine derivativesWO2006048296A12004-11-052006-05-11Novartis AgOrganic compoundsWO2011064355A12009-11-302011-06-03Novartis AgPolymorphous forms iii and iv of n-benzoyl staurosporineWO2018165071A12017-03-062018-09-13Teva Pharmaceutical Works Ltd.Solid state forms of midostaurin
Biological activities
The main biological activity of staurosporine is the inhibition of protein kinases through the prevention of ATP binding to the kinase. This is achieved through the stronger affinity of staurosporine to the ATP-binding site on the kinase. Staurosporine is a prototypical ATP-competitive kinase inhibitor in that it binds to many kinases with high affinity, though with little selectivity.[4] Structural analysis of kinase pockets demonstrated that main chain atoms which are conserved in their relative positions to staurosporine contributes to staurosporine promiscuity.[5] This lack of specificity has precluded its clinical use, but has made it a valuable research tool. In research, staurosporine is used to induce apoptosis. The mechanism of how it mediates this is not well understood. It has been found that one way in which staurosporine induces apoptosis is by activating caspase-3.[6] At lower concentration, depending on the cell type, staurosporine induces specific cell cycle effects arresting cells either in G1 or in G2 phase of the cell cycle.[7]
Chemistry family
Main article: Indolocarbazole
Staurosporine is an indolocarbazole. It belongs to the most frequently isolated group of indolocarbazoles: Indolo(2,3-a)carbazoles. Of these, Staurosporine falls within the most common subgroup, called Indolo(2,3-a)pyrrole(3,4-c)carbazoles. These fall into two classes – halogenated (chlorinated) and non-halogenated. Halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond, while non-halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have both indole nitrogens glycosylated, and a fully reduced C-7 carbon. Staurosporine is in the non-halogenated class.[8]
Staurosporine is the precursor of the novel protein kinase inhibitor midostaurin (PKC412).[9][10] Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
pyrrole(3,4-c)carbazol.svg)
Structure of an Indolo[2,3-a]pyrrole[3,4-c]carbazol

Biosynthesis
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imine by enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450 enzyme to form an aromatic ring system occurs.[8]

This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4’amine by StaMA and N-methylation of the 3′-hydroxy by StaMB leads to the formation of staurosporine.[8]
Research in clinical use
When encapsulated in liposome nanoparticle, staurosporine is shown to suppress tumors in vivo in a mouse model without the toxic side effects which have prohibited its use as an anti-cancer drug with high apoptotic activity. Researchers in UC San Diego Moores Cancer Center develop a platform technology of high drug-loading efficiency by manipulating the pH environment of the cells. When injected into the mouse glioblastoma model, staurosporine is found to accumulate primarily in the tumor via fluorescence confirmation, and the mice did not suffer weight loss compared to the control mice administered with the free compound, an indicator of reduced toxicity.[11][12]
References
- ^ Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchiya H, Takahashi Y, Masuma R (1977). “A new alkaloid AM-2282 of Streptomyces origin taxonomy, fermentation, isolation and preliminary characterization”. J. Antibiot. 30 (4): 275–282. doi:10.7164/antibiotics.30.275. PMID 863788.
- ^ Funato N, Takayanagi H, Konda Y, Toda Y, Harigaya Y, Omura S (1994). “Absolute configuration of staurosporine by X-ray analysis”. Tetrahedron Lett. 35 (8): 1251–1254. doi:10.1016/0040-4039(94)88036-0.
- ^ [1] Rüegg UT, Burgess GM. (1989) Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends in Pharmacological Science 10 (6): 218-220.
- ^ Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP (2008). “A quantitative analysis of kinase inhibitor selectivity”. Nat. Biotechnol. 26 (1): 127–132. doi:10.1038/nbt1358. PMID 18183025. S2CID 205273598.
- ^ Tanramluk D, Schreyer A, Pitt WR, Blundell TL (2009). “On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine”. Chemical Biology & Drug Design. 74 (1): 16–24. doi:10.1111/j.1747-0285.2009.00832.x. PMC 2737611. PMID 19519740.
- ^ Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW, Kim HR (2000). “Molecular mechanism of staurosporine-induced apoptosis in osteoblasts”. Pharmacological Research. 42 (4): 373–381. doi:10.1006/phrs.2000.0700. PMID 10987998.
- ^ Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z (1992). “Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes”. Cancer Res. 52 (2): 470–473. PMID 1728418.
- ^ Jump up to:a b c Ryan KS (2008). “Structural studies of rebeccamycin, staurosporine, and violacein biosynthetic enzymes” (PDF). Ph.D. Thesis. Massachusetts Institute of Technology. Archived from the original (PDF) on 2012-03-14.
- ^ Midostaurin product page, Fermentek
- ^ Wang, Y; Yin, OQ; Graf, P; Kisicki, JC; Schran, H (2008). “Dose- and Time-Dependent Pharmacokinetics of Midostaurin in Patients With Diabetes Mellitus”. J Clin Pharmacol. 48 (6): 763–775. doi:10.1177/0091270008318006. PMID 18508951. S2CID 26657407.
- ^ News Release (21 October 2013). “Study Identifies Safe Delivery System for Tricky Yet Highly Potent Anti-Cancer Compounds”. UC San Diego Health System. Retrieved 27 October 2013.
- ^ Mukthavaram, Rajesh; Jiang, Pengei; Saklecha, Rohit; Simbery, Dmitri; Bharati, Ila; Nomura, Natsuko; Chao, Ying; Pastorino, Sandra (2013). “High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile”. International Journal of Nanomedicine. 8 (1): 3991–4006. doi:10.2147/IJN.S51949. PMC 3808212. PMID 24174874.
| Clinical data | |
|---|---|
| ATC code | none |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62996-74-1 |
| PubChem CID | 44259 |
| IUPHAR/BPS | 346 |
| DrugBank | DB02010 |
| ChemSpider | 40272 |
| UNII | H88EPA0A3N |
| ChEBI | CHEBI:15738 |
| ChEMBL | ChEMBL162 |
| PDB ligand | STU (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID30911019 DTXSID6041131, DTXSID30911019 |
| ECHA InfoCard | 100.109.946 |
| Chemical and physical data | |
| Formula | C28H26N4O3 |
| Molar mass | 466.541 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////STAUROSPORINE, AM-2282, CGP-39360
[H][C@]1(C[C@@]2([H])O[C@](C)(N3C4=CC=CC=C4C4=C5CNC(=O)C5=C5C6=CC=CC=C6N2C5=C34)[C@]1([H])OC)NC

NEW DRUG APPROVALS
ONE TIME
$10.00


{kind=link}
{kind=link}