
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
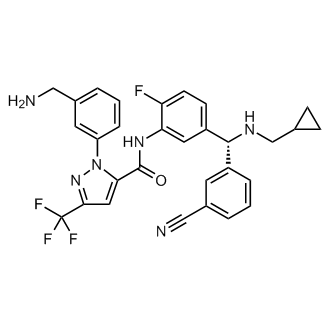

Berotralstat
CAS 1809010-50-1
DIHCl 1809010-52-3
Molecular Formula, C30-H26-F4-N6-O, Molecular Weight, 562.5684
1-(3-(Aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide
1H-Pyrazole-5-carboxamide, 1-(3-(aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-
To treat patients with hereditary angioedema
FDA APPROVED 12/4/2020, Orladeyo, 110MG CAPSULE 0RAL
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
Berotralstat Hydrochloride
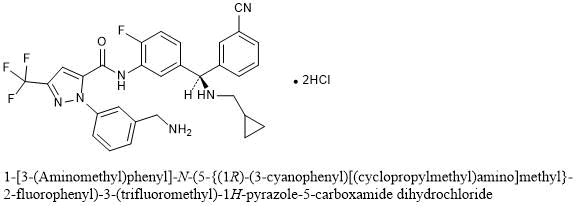
1-[3-(Aminomethyl)phenyl]-N-(5-{(1R)-(3-cyanophenyl)[(cyclopropylmethyl)amino]methyl}-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide dihydrochloride
C30H26F4N6O▪2HCl : 635.48
[1809010-52-3]
Berotralstat, also known as BCX-7353, is a kallikrein inhibitor. BCX7353 is a synthetic, once-daily, small molecule drug that can be taken as an oral capsule to treat HAE attacks and for prophylaxis.
Hereditary angioedema (HAE) is rare disorder caused by a SERPING1 gene mutation that triggers severe swelling of the skin and upper airway. Treatment options for HAE with deficient and dysfunctional C1-inhibitor are expanding to include small-molecule drugs that inhibit protein interactions in the kallikrein-kinin system
Serine proteases make up the largest and most extensively studied group of proteolytic enzymes. Their critical roles in physiological processes extend over such diverse areas as blood coagulation, fibrinolysis, complement activation, reproduction, digestion, and the release of physiologically active peptides. Many of these vital processes begin with cleavage of a single peptide bond or a few peptide bonds in precursor protein or peptides. Sequential limited proteolytic reactions or cascades are involved in blood clotting, fibrinolysis, and complement activation. The biological signals to start these cascades can be controlled and amplified as well. Similarly, controlled proteolysis can shut down or inactivate proteins or peptides through single bond cleavages.
Kallikreins are a subgroup of serine proteases. In humans, plasma kallikrein (KLKB1) has no known homologue, while tissue kallikrein-related peptidases (KLKs) encode a family of fifteen closely related serine proteases. Plasma kallikrein participates in a number of pathways relating to the intrinsic pathway of coagulation, inflammation, and the complement system.
Coagulation is the process by which blood forms clots, for example to stop bleeding. The physiology of coagulation is somewhat complex insofar as it includes two separate initial pathways, which converge into a final common pathway leading to clot formation. In the final common pathway, prothrombin is converted into thrombin, which in turn converts fibrinogen into fibrin, the latter being the principal building block of cross- linked fibrin polymers which form a hemostatic plug. Of the two initial pathways upstream of the final common pathway, one is known as the contact activation or intrinsic pathway, and the other is known as the tissue factor or extrinsic pathway.
The intrinsic pathway begins with formation of a primary complex on collagen by high-molecular- weight kininogen (HMWK), prekallikrein, and FXII (Factor XII; Hageman factor). Prekallikrein is converted to kallikrein, and FXII is activated to become FXIIa. FXIIa then converts Factor XI (FXI) into FXIa, and FXIa in turn activates Factor IX (FIX), which with its co-factor F Villa form the“tenase” complex, which activates Factor X (FX) to FXa. It is FXa which is responsible for the conversion of prothrombin into thrombin within the final common pathway.
Prekallikrein, the inactive precursor of plasma kallikrein, is synthesized in the liver and circulates in the plasma bound to FDVTWK or as a free zymogen. Prekallikrein is cleaved by activated factor XII(FXIIa) to release activated plasma kallikrein (PK). Activated plasma kallikrein displays endopeptidase activity towards peptide bonds after arginine (preferred) and lysine. PK then generates additional FXIIa in a feedback loop which in turn activates factor XI (FXI) to FXIa to connect to the common pathway. Although the initial activation of the intrinsic pathway is through a small amount of FXIIa activating a small amount of PK, it is the subsequent feedback activation of FXII by PK that controls the extent of activation of the intrinsic pathway and hence downstream coagulation. Hathaway, W. E., et al. (1965) Blood 26:521-32.
Activated plasma kallikrein also cleaves HMWK to release the potent vasodilator peptide bradykinin. It is also able to cleave a number of inactive precursor proteins to generate active products, such as plasmin (from plasminogen) and urokinase (from prourokinase). Plasmin, a regulator of coagulation, proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
Patients who have suffered acute myocardial infarction (MI) show clinical evidence of being in a hypercoagulable (clot-promoting) state. This hypercoagulability is
paradoxically additionally aggravated in those receiving fibrinolytic therapy. Increased generation of thrombin, as measured by thrombin-antithrombin III (TAT) levels, is observed in patients undergoing such treatment compared to the already high levels observed in those receiving heparin alone. Hoffmeister, H. M. et al. (1998) Circulation 98:2527-33. The increase in thrombin has been proposed to result from plasmin-mediated activation of the intrinsic pathway by direct activation of FXII by plasmin.
Not only does the fibrinolysis-induced hypercoagulability lead to increased rates of reocclusion, but it is also probably responsible, at least in part, for failure to achieve complete fibrinolysis of the clot (thrombus), a major shortcoming of fibrinolytic therapy (Keeley, E. C. et al. (2003) Lancet 361 : 13-20). Another problem in fibrinolytic therapy is the accompanying elevated risk of intracranial hemorrhage. Menon, V. et al. (2004) (Chest l26:549S-575S; Fibrinolytic Therapy Trialists’ Collaborative Group (1994) Lancet 343 :311-22. Hence, an adjunctive anti -coagulant therapy that does not increase the risk of bleeding, but inhibits the formation of new thrombin, would be greatly beneficial. Plasma kallikrein inhibitors also have therapeutic potential for treating hereditary angioedema (HAE). HAE is is a serious and potentially life-threatening rare genetic illness, caused by mutations in the Cl -esterase inhibitor (C1INH) gene, located on chromosome 1 lq. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by CllNH gene mutations which produce low levels of Cl -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective Cl protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; l00(Suppl2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herein by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that CllNH is the most important inhibitor of kallikrein in human plasma and that CllNH deficiency leads to unopposed activation of the kallikrein- bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack. All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
Therefore, a need exists to develop inhibitors of PK that can tip the balance of fibrinolysis/thrombosis at the occluding thrombus toward dissolution, thereby promoting reperfusion and also attenuating the hypercoagulable state, thus preventing thrombus from reforming and reoccluding the vessel. In particular, the creation of plasma kallikrein inhibitors that are specific and capable of being formulated for in vivo use could lead to a new class of therapeutics. Thus, what is needed are improved compositions and methods for preparing and formulating plasma kallikrein inhibitors.
For example, in patients with angioedema conditions, small polypeptide PK inhibitor DX-88 (ecallantide) alleviates edema in patients with hereditary angioedema (HAE). Williams, A. et al. (2003) Transfus. Apher. Sci. 29:255-8; Schneider, L. et al.
(2007) J Allergy Clin Immunol. 120:416-22; and Levy, J. H. et al. (2006) Expert Opin. Invest. Drugs 15: 1077-90. A bradykinin B2 receptor antagonist, Icatibant, is also effective in treating HAE. Bork, K. et al. (2007) J. Allergy Clin. Immunol. 119:1497-1503. Because plasma kallikrein generates bradykinin, inhibition of plasma kallikrein is expected to inhibit bradykinin production.
For example, in coagulation resulting from fibrinolytic treatment (e.g., treatment with tissue plasminogen activator or streptokinase), higher levels of plasma kallikrein are found in patients undergoing fibrinolysis. Hoffmeister, H. M. et al. (1998) J. Cardiovasc. Pharmacol. 31 :764-72. Plasmin-mediated activation of the intrinsic pathway has been shown to occur in plasma and blood and was markedly attenuated in plasma from individuals deficient in any of the intrinsic pathway components. Ewald, G. A. et al. (1995) Circulation 91 :28-36. Individuals who have had an acute MI were found to have elevated levels of activated plasma kallikrein and thrombin. Hoffmeister, H. M., et al. (1998) Circulation 98:2527-33.
DX-88 reduced brain edema, infarct volume, and neurological deficits in an animal model of ischemic stroke. Storini, C. et al. (2006) J Pharm. Exp. Ther. 318:849-854. Cl- inhibitor reduced infarct size in a mouse model of middle cerebral artery occlusion
(MCAO). De Simoni, M. G. et al. (2004) Am. J. Pathol. 164: 1857-1863; and Akita, N. et al. (2003) Neurosurgery 52:395-400). B2 receptor antagonists were found to reduce the infarct volume, brain swelling, and neutrophil accumulation and were neuroprotective in an MCAO animal model. Zausinger, S. et al. (2003 ) Acta Neurochir. Suppl. 86:205-7;
Lumenta, D. B. et al. (2006) Brain Res. 1069:227-34; Ding-Zhou, L. et al. (2003) Br. J Pharmacol. 139: 1539-47.
Regarding blood loss during cardiopulmonary bypass (CPB), it has been found that the kallikrein-kinin (i.e., contact) system is activated during CABG. Wachtfogel, Y. T. (1989) Blood 73:468. Activation of the contact system during CPB results in up to a 20- fold increase in plasma bradykinin. Cugno, M. et al. (2006) Chest 120:1776-82; and Campbell, D. J. et al. (2001 ) Am. J. Physiol. Reg. Integr. Comp. Physiol. 281 : 1059-70.
Plasma kallikrein inhibitors P8720 and PKSI-527 have also been found to reduce joint swelling in rat models of arthritis. De La Cadena, R. A. et al. (1995) FASEB J. 9:446- 52; Fujimori, Y. (1993) Agents Action 39:42-8. It has also been found that inflammation in animal models of arthritis was accompanied by activation of the contact system. Blais, C. Jr. et al. (1997) Arthritis Rheum. 40: 1327-33.
Additionally, plasma kallikrein inhibitor P8720 has been found to reduce inflammation in an acute and chronic rat model of inflammatory bowel disease (IBD). Stadnicki, A. et al. (1998) FASEB J. 12:325-33; Stadnicki, A. et al. (1996) Dig. Dis. Sci.
41 :9l2-20; and De La Cadena, R. A., et al. (1995) FASEB J. 9:446-52. The contact system is activated during acute and chronic intestinal inflammation. Sartor, R. B. et al. (1996) Gastroenterology 110: 1467-81. It has been found that B2 receptor antagonist, an antibody to high molecular weight kininogen, or reduction in levels of kininogen reduced clinicopathology in animal models of IBD. Ibid !; Arai, Y. et al. (1999) Dig. Dis. Sci.
44:845-51; and Keith, J. C. et al. (2005) Arthritis Res. Therapy 7 :R769-76.
H-D-Pro-Phe-Arg-chloromethylketone (CMK), an inhibitor of PK and FXII and a physiological inhibitor (Cl -inhibitor), has been found to reduce vascular permeability in multiple organs and reduce lesions in lipopolysaccharide (LPS)- or bacterial-induced sepsis in animals. Liu, D. et al. (2005) Blood 105:2350-5; Persson, K. et al. (2000) J. Exp. Med. 192: 1415-24. Clinical improvement was observed in sepsis patients treated with Cl- inhibitor. Zeerleder, S. et al. (2003) Clin. Diagnost. Lab. Immunol. 10:529-35; Caliezi, C., et al. (2002) Crit. Care Med. 30:1722-8; and Marx, G. et al. (1999) Intensive Care Med.
25: 1017-20. Fatal cases of septicemia are found to have a higher degree of contact activation. Martinez-Brotons, F. et al. (1987) Thromb. Haemost. 58:709-713; and Kalter, E. S. et al. (1985) J. Infect. Dis. 151 : 1019-27.
It has also been found that prePK levels are higher in diabetics, especially those with proliferative retinopathy, and correlate with fructosamine levels. Gao, B.-B., et al. (2007) Nature Med. 13: 181-8; and Kedzierska, K. et al. (2005) Archives Med. Res. 36:539- 43. PrePK is also found to be highest in those with a sensorimotor neuropathy. Christie,
M. et al. (1984) Thromb. Haemostas. (Stuttgart) 52:221-3. PrePK levels are elevated in diabetics and are associated with increased blood pressure. PrePK levels independently correlate with the albumin excretion rate and are elevated in diabetics with
macroalbuminuria, suggesting prePK may be a marker for progressive nephropathy. Jaffa, A. A. et al. (2003) Diabetes 52: 1215-21. Bl receptor antagonists have been found to decrease plasma leakage in rats treated with streptozotocin. Lawson, S. R. et al. (2005)
Eur. J. Pharmacol. 514:69-78. Bl receptor antagonists can also prevent streptozotocin- treated mice from developing hyperglycemia and renal dysfunction. Zuccollo, A. et al. (1996) Can. J. Physiol. Pharmacol. 74:586-9.
PATENT
WO 2015134998
https://patents.google.com/patent/WO2015134998A1/en
PATENT
WO 2020092898
https://patents.google.com/patent/WO2020092898A1/en
Example 1 : Synthetic protocol for racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Preparation of 1 -(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide
(54e)
Step-l : Preparation of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b)
To a solution of 3-formylbenzonitrile (54a) (29 g, 217 mmol) in tetrahydrofuran (200 mL) cooled to 0 °C was added freshly prepared Grignard reagent (52c) (245 mL, 221 mmol, ~ 0.9 M in THF) stirred at 0 °C for 1 h, and room temperature for 18 h. The reaction mixture was quenched with 1 N HC1 (aq. 440 mL), stirred for 3 h, neutralized with NaOH (2 N, aq.) to pH = ~ 8. The reaction mixture was extracted with ethyl acetate (600, 300 mL). The combined extracts were washed with brine (120 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column
chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1) to give 3-((3- amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (36.28 g) as a brown gum which was used as such for next step; MS (ES+) 265.3 (M+23).
Step-2: Preparation of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c)
To a solution of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (24.682 g, 102 mmol) in DMF (480 mL) was added l-(3-((tert- butoxycarbonylamino)methyl)phenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxylic acid (lOd) (35.0 g, 91 mmol), N-ethyl-N-isopropylpropan-2-amine (132 mL, 758 mmol), bromotripyrrolidin-l-ylphosphonium hexafluorophosphate(V) (PyBrOP, 42.8 g, 91 mmol) and stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (1000 mL), washed with water (500, 400 mL), brine (400 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1)] to afford tert- butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (4.583 g, 5% for two steps) as a yellow solid; ¾ NMR (300 MHz, DMSO-i¾) d 10.57 (s, 1H), 7.81 (t, J= 1.7 Hz, 1H), 7.73 – 7.66 (m, 2H), 7.64 – 7.19 (m, 10H), 6.25 (d, J= 4.0 Hz, 1H), 5.78 (d, J= 4.0 Hz, 1H), 4.19 (d, J= 6.1 Hz, 2H), 1.37 (s, 9H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -123.09; MS (ES+) 632.3 (M+23).
Step-3: Preparation of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d)
To a solution of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (1.333 g, 2.187 mmol) in dichloromethane (40 mL) at 0°C was added thionyl chloride (0.340 mL, 4.59 mmol) and warmed to room temperature over 2 h. The reaction mixture was quenched with triethyl amine (2.0 mL, 14.35 mmol) stirred at room temperature for 1 h. It was then treated with cyclopropylmethanamine (4.30 mL, 48.0 mmol), concentrated to remove most of dichloromethane followed by addition of acetonitrile (30 mL), stirring at 70 °C for 14 h, and concentration in vacuum to dryness. The residue was treated with chlorofrom (200 mL), washed with water (100 mL), dried over MgS04 followed by filtration and
concentration. The crude product was purified by flash column chromatography [silica gel eluting with hexanes/ethyl acetate (1 :0 to 2: 1)] to afford tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (184 mg, 13%) as colorless gum; ¾ NMR (300 MHz, DMSO-ά) d 10.56 (s, 1H), 7.89 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.70 – 7.30 (m, 10H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 4.19 (d, J= 6.2 Hz, 2H), 2.26 (d, J= 6.6 Hz, 2H), 1.37 (s, 9H), 1.00 – 0.80 (m, 1H), 0.45 – 0.28 (m, 2H), 0.12 – -0.01 (m, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.80 , -123.20; MS (ES+) 663.4 (M+l). Step-4: Preparation of l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e)
To a solution of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (161 mg, 0.243 mmol) in 1,4- Dioxane (18 mL) was added hydrogen chloride (2.60 mL, 10.40 mmol, 4 M in l,4-dioxane) and stirred at room temperature for 16 h. the reaction mixture was treated with hexanes, decanted, washed with hexanes, and decanted again. The insoluble crude product was purified by flash column chromatography [silica gel, eluting with chloroform/CMA80 (1 :0 to 2:1)] to afford l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e). The pure product was dissolved in methanol (10 mL) and added 4 N HC1 (aq. 0.14 mL) followed by concentration in vacuum to dryness to give HC1 salt of l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e) (74 mg, 48%) white solid; ¾ NMR (300 MHz, DMSO- d, D20 ex NMR) d 8.13 (t, J = 1.7 Hz, 1H), 7.98 – 7.84 (m, 3H), 7.73 – 7.64 (m, 3H), 7.63 – 7.48 (m, 4H), 7.44 (dd, J = 10.2, 8.6 Hz, 1H),
5.75 (s, 1H), 4.12 (s, 2H), 2.76 (d, J = 7.2 Hz, 2H), 1.17 – 0.94 (m, 1H), 0.68 – 0.47 (m, 2H), 0.34-0.24 (m, 2H); 19F NMR (282 MHz, DMSO- d) d -60.82, -120.02; MS (ES+): 563.3 (M+l); Analysis calculated for C30H26F4N6O2.O HCT3.0 H2O: C, 52.26; H, 4.97; N, 12.19; Found: C, 52.26; H, 5.00; N, 11.72.
Example 2: Separation of enantiomers of racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Compound I (free base) Separation of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide (Compound I), and (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide ((-
)-enantiomer)
Isomers of Racemic l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lF[- pyrazole-5-carboxamide (54e) (0.4 g) were separated by using preparative SFC method using the following conditions to furnish:
Preparative SFC Method used:
Column 20mm x 25.0 cm ChromegaChiral CCS from
Regis Technologies (Morton Grove, IL)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 1%
Isopropylamine
Isocratic Method 20 % Co-solvent at 80 mL/min
System Pressure 200 bar
Column Temperature 25 °C
Sample Diluent Methanol: Isopropanol
Chiral Purity of peaks was determined by following Analytical SFC Method:
Column 4.6 x 100 mm ChiralPak AS from Chiral
Technologies (West Chester, PA)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 0.1%
Isopropylamine
Isocratic Method 5-65 % Co-solvent Gradient at 4 mL/min System Pressure 100 bar
Column Temperature 25 °C
Sample Diluent Methanol
Peak-l (Compound I) 2.1 min 144 mg >95% ee (UV 254)
98.6 % purity (UV 254)
Peak-2 ((-)-enantiomer) 2.4 min 172 mg 95.5 % ee (UV 254)
96.5 % purity (UV 254) 1. Peak-l assigned as (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (144 mg, >95%ee) free base as white solid; Optical rotation: [O]D = (+) 6.83 [CH3OH, 1.2]; ‘H NMR (300 MHz, DMSO-£¾) d 10.53 (s, 1H, D2O exchangeable), 7.88 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.67 (dt, 7= 7.7, 1.4 Hz, 1H), 7.63 (dd, J= 7.5, 2.1 Hz, 1H), 7.56 (s, 1H), 7.54 – 7.47 (m, 2H), 7.47 – 7.38 (m, 2H), 7.34 (ddt, J= 8.6, 5.9, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.77 (s, 2H), 2.31 – 2.21 (m, 2H), 0.97 – 0.80 (m, 1H), 0.42 – 0.33 (m, 2H), 0.10 – -0.02 (m, 2H); 19F NMR (282 MHz, DMSO-Ts) d -60.73 , -123.20; MS (ES+) 563.3 (M+l), 561.3 (M-l). To a solution of free base mixture of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (120 mg) in methanol (15 mL) was added hydrogen chloride (0.969 mL, 1.938 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (+)-l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (100 mg) hydrochloride salt as white solid; ¾ NMR (300 MHz, DMSO-Ts) d 10.84 (s, 1H, D2O exchangeable), 10.44 (s, 2H, D2O exchangeable), 8.44 (s, 3H, D2O exchangeable), 8.30 (s, 1H, D2O exchangeable), 8.09 (d, J= 7.9 Hz, 1H), 7.99 (d, J = 6.8 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.80 – 7.50 (m, 7H), 7.42 (dd, J= 10.3, 8.6 Hz, 1H), 5.78 (d, J= 6.9 Hz, 1H), 4.13 (d, J= 5.7 Hz, 2H), 2.88 – 2.62 (m, 2H), 1.42 – 0.99 (m, 1H), 0.73 – 0.46 (m, 2H), 0.32 (d, J= 4.4 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -119.99; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-) 561.3 (M-l), 597.3 (M+Cl); Analysis calculated for C30H26F4N6O 2HC1 l.75H20: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97. Peak-2 assigned as (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (172 mg, 95.5 % ee) as free base was repurified by flash column chromatography (silica gel 12 g, eluting 0-30% MeOH in chloroform for 15 min) to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) free base as an off-white solid; Optical rotation: [O]D = (-) 5.44
[CH3OH, 1.25]; ¾ NMR (300 MHz, DMSO-i¾) d 7.88 (t, J= 1.6 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.70 – 7.61 (m, 2H), 7.57 (s, 1H), 7.54 – 7.47 (m, 2H), 7.45 – 7.41 (m,
2H), 7.34 (ddq, J= 8.7, 6.1, 3.5, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.78 (s, 2H), 2.25 (d, J= 6.9 Hz, 2H), 0.90 (ddd, J= 9.8, 8.0, 5.2 Hz, 1H), 0.47 – 0.29 (m, 2H), 0.04 (dd, J= 5.0, 1.5 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.73 , -123.19; MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l). To a solution of free base of (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.124 g, 0.220 mmol) in methanol (15 mL) was added hydrogen chloride (1.102 mL, 2.204 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.121 g) hydrochloride salt as an off-white solid; Ή NMEE ¾ NMR (300 MHz, DMSO-i¾) d 10.82 (s, 1H, D20 exchangeable), 10.36 (s, 2H, D2O exchangeable), 8.38 (s, 3H, D2O exchangeable), 8.27 (s, 1H), 8.06 (d, J= 7.9 Hz, 1H), 7.98 (d, J= 6.7 Hz, 1H), 7.87 (d, J= 7.7 Hz, 1H), 7.78 – 7.49 (m, 7H), 7.48 – 7.37 (m, 1H), 5.78 (s, 1H), 4.13 (d, j= 5.7 Hz, 2H), 2.72 (s, 2H), 1.14 (s, 1H), 0.56 (d, j= 7.7 Hz, 2H), 0.31 (d, J= 5.0 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.82 , -120.03; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l), 597.2 (M+Cl); Analysis calculated for C30H26F4N6O.2HCI. I .75H2O: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97.
Example 3 : Preparation of a Seed Crystal of Compound I*2

A solution of Compound I ( see Example 2) in methyl tert-butyl ether (MTBE) (1 equiv) is added to a solution of HC1 (aq) (2 equiv) in methanol (cold), followed by heating to about 30°C, and keeping it at about 30°C for not longer than 5 hours while stirring at about 115 rpm. Compound I bis(HCl) is collected by filtration and dried. The crystalline material obtained can be used as a seed for the crystallization protocol described in
Example 4. Example 4: Large-Scale Synthetic & Crystallization Protocol for Compound I*2(HC1 )

Compound I (free base) Compound I bis(HCI)
37% Aqueous hydrochloric acid (38.1 kg, 32.3 L, 2.14 equiv.) was charged to a clean and empty crystallization vessel, methanol (228.9 kg, 39.5 equiv.) was added, and the contents were cooled to -7 ± 3°C. A solution of Compound I free base (approx. 101.8 kg; 180.9 moles) in MTBE (approx. 1,300 L) was filtered through a polish filter into the crystallization vessel at temperature -5 ± 5°C. After rinse with MTBE, pre-weighed Compound I»2(HCl) seed crystals (1.39 kg, 0.012 equiv.; Example 3) were charged to the crystallization vessel via the manhole. The vessel content was heated to 30-33°C, and the agitation speed was set to 25-50 rpm. After confirmed crystallization, the slurry was agitated for another three to four hours. The product slurry was transferred to centrifuge and isolated by centrifugation. The product was washed with MTBE (585 L). After dry spinning the wet product, Compound I*2(HC1), it was discharged from the centrifuge, and the product was dried at < 40°C under vacuum in a cone drier. Product Compound I»2(HCl) yield: 100 kg; 157.4 mol; approx. 85%.
‘H NMR (300 MHz, DMSO-c/i,) data is shown in the following table:

19F NMR (282 MHz, DMSO- is) data is shown in the following table:

Compound I has two basic sites. The conjugate acid of the primary amine was calculated to have a pKa value of 8.89, and the conjugate acid of the secondary amine was calculated to have a pKa value of 7.86.
The XRPD pattern of Compound I»2(HCl) is shown in Fig. 1. Compound I»2(HCl) has characteristic peaks in its XRPD pattern at values of two theta (°2Q) of 5.28, 8.96, 14.27, 16.18, 19.79, 21.16, 22.01, 23.31, 24.64, and 30.31. TG-IR analysis indicated two, distinct weight loss regions: the first was completed by 125 °C while the second began at approximately 208 °C. IR analysis of the off gasses from this experiment detected only trace amounts of water at the initial weight loss while HC1 gas was detected at the 208°C event. No other solvents were detected in the sample. Thus, it was determined that Compound I*2(HC1) initially loses water when heated and, when heated to above 200°C, the salt begins to break apart and HC1 gas is evolved. The IR signal for all these events is very weak indicating that they are occurring over a range and not at a specified temperature. An exemplary TG-IR spectrum is shown in Fig. 2.
REFERENCES
1: Sohtome Y, Sodeoka M. Development of Chaetocin and S-Adenosylmethionine Analogues as Tools for Studying Protein Methylation. Chem Rec. 2018 Dec;18(12):1660-1671. doi: 10.1002/tcr.201800118. Epub 2018 Oct 16. Review. PubMed PMID: 30324709.
2: Bensussen A, Torres-Sosa C, Gonzalez RA, Díaz J. Dynamics of the Gene Regulatory Network of HIV-1 and the Role of Viral Non-coding RNAs on Latency Reversion. Front Physiol. 2018 Sep 28;9:1364. doi: 10.3389/fphys.2018.01364. eCollection 2018. PubMed PMID: 30323768; PubMed Central PMCID: PMC6172855.
////////berotralstat, Orladeyo, BIOCRYST, APPROVALS 2020, FDA 2020, ORPHAN DRUG, CX-7353, CX 7353,
NCc1cccc(c1)n2nc(cc2C(=O)Nc3cc(ccc3F)[C@H](NCC4CC4)c5cccc(c5)C#N)C(F)(F)F

