
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
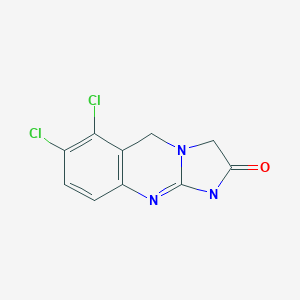

Anagrelide アナグレリド;
QA-0023
BMY-26538-01
GALE-401
KRN-654
SPD-422
| INGREDIENT | UNII | CAS |
|---|---|---|
| Anagrelide Hydrochloride | VNS4435G39 | 58579-51-4 |




EMA
| 2018/2/15 | EMA | APPROVED | Anagrelide | Anagrelide Mylan | Mylan S.A.S. |
Cardiovascular agent
Anagrelide hydrochloride is a cyclic phosphodiesterase III inhibitor that was launched in 1997 in the U.S. by Shire Pharmaceuticals for the treatment of essential thrombocythemia and other myeloproliferative disorders
Anagrelide was assigned orphan drug status by the FDA in 1986, by the Japanese Ministry of Health in 2004 and by the European Commission in January 2001 for the treatment of essential thrombocythemia.
Anagrelide (Agrylin/Xagrid, Shire and Thromboreductin, AOP Orphan Pharmaceuticals AG) is a drug used for the treatment of essential thrombocytosis (ET; essential thrombocythemia), or overproduction of blood platelets. It also has been used in the treatment of chronic myeloid leukemia.[1]
Anagrelide controlled release (GALE-401) is in phase III clinical trials by Galena Biopharma for the treatment of ET.[2]
Medical uses
Anagrelide is used to treat essential thrombocytosis, especially when the current treatment of the patient is insufficient.[3] Essential thrombocytosis patients who are suitable for anagrelide often meet one or more of the following factors:[4][5]
- age over 60 years
- platelet count over 1000×109/L
- a history of thrombosis
According to a 2005 Medical Research Council randomized trial, the combination of hydroxyurea with aspirin is superior to the combination of anagrelide and aspirin for the initial management of ET. The hydroxyurea arm had a lower likelihood of myelofibrosis, arterial thrombosis, and bleeding, but it had a slightly higher rate of venous thrombosis.[3] Anagrelide can be useful in times when hydroxyurea proves ineffective.
Side-effects
Common side effects are headache, diarrhea, unusual weakness/fatigue, hair loss, nausea and dizziness.
The same MRC trial mentioned above also analyzed the effects of anagrelide on bone marrow fibrosis, a common feature in patients with myelofibrosis. The use of anagrelide was associated with a rapid increase in the degree of reticulin deposition (the mechanism by which fibrosis occurs), when compared to those in whom hydroxyurea was used. Patients with myeloproliferative conditions are known to have a very slow and somewhat variable course of marrow fibrosis increase. This trend may be accelerated by anagrelide. Interestingly, this increase in fibrosis appeared to be linked to a drop in hemoglobin as it progressed. Fortunately, stopping the drug (and switching patients to hydroxyurea) appeared to reverse the degree of marrow fibrosis. Thus, patients on anagrelide may need to be monitored on a periodic basis for marrow reticulin scores, especially if anemia develops, or becomes more pronounced if present initially.[6]
Less common side effects include: congestive heart failure, myocardial infarction, cardiomyopathy, cardiomegaly, complete heart block, atrial fibrillation, cerebrovascular accident, pericarditis, pulmonary infiltrates, pulmonary fibrosis, pulmonary hypertension, pancreatitis, gastric/duodenal ulceration, renal impairment/failure and seizure.
Due to these issues, anagrelide should not generally be considered for first line therapy in ET.
Mechanism of action
Anagrelide works by inhibiting the maturation of platelets from megakaryocytes.[7] The exact mechanism of action is unclear, although it is known to be a phosphodiesterase inhibitor.[8] It is a potent (IC50 = 36nM) inhibitor of phosphodiesterase-II.[citation needed] It inhibits PDE-3 and phospholipase A2.[9]
Synthesis
Phosphodiesterase inhibitor with antiplatelet activity.
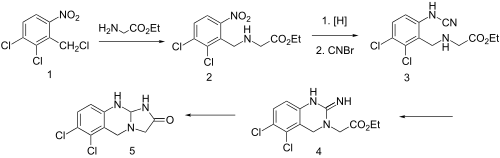

| Synthesis 1[10][11] | Synthesis 2 |
|---|---|
|
|
 |

Condensation of benzyl chloride 1 with ethyl ester of glycine gives alkylated product 2. Reduction of the nitro group leads to the aniline and reaction of this with cyanogen bromidepossibly gives cyanamide 3 as the initial intermediate. Addition of the aliphatic would then lead to formation of the quinazoline ring (4). Amide formation between the newly formed imide and the ester would then serve to form the imidazolone ring, whatever the details of the sequence, there is obtained anagrelide (5).
PATENT
https://patents.google.com/patent/WO2010005480A2/en

PATENT
US20130211083A1

PATENTS
https://patents.google.com/patent/EP2373654A1/en

SYN
CA 1137474, WO 0208228

The nitration of 1,2,3-trichlorobenzene (I) with concentrated HNO3 gives 2,3,4-trichloronitrobenzene (II), which by reaction with cuprous cyanide in hot pyridine is converted to 2,3-dichloro-6-nitrobenzonitrile (III). The reduction of (III) with borane in THF yields 2,3-dichloro-6-nitrobenzylamine (IV), which by reaction with ethyl bromoacetate (V) by means of triethylamine in refluxing dioxane affords ethyl N-(2,3-dichloro-6-nitrobenzyl)glycinate (VI). The reduction of (VI) with SnCl2 in concentrated HCl gives ethyl N-(6-amino-2,3-dichlorobenzyl)glycinate (VII), which is cyclized with cyanogen bromide (VIII) in toluene affording ethyl 5,6-dichloro-3,4-dihydro-2-(1H)-iminoquinazoline-3-acetate (IX). Finally, this compound is submitted to a new cyclization by means of triethylamine in refluxing ethanol.

The reaction of 3-chloroaniline (X) with choral hydrate (XI) and hydroxylamine gives isonitroso-3-chloroacetanilide (XII), which is cyclized by means of H2SO4 to 4-chloroisatin (XIII). Chlorination of (XIII) with SO2Cl2 affords 4,5-dichloroisatin (XIV), which is oxidized with H2O2 yielding 5,6-dichloroanthranilic acid (XV). The reduction of (XV) with borane in THF gives 6-amino-2,3-dichlorobenzyl alcohol (XVI), which by reaction with SOCl2 in benzene is converted to 6-amino-2,3-dichlorobenzyl chloride (XVII). This compound is condensed with ethyl glycinate (XVIII) by means of triethylamine in refluxing methylene chloride to give ethyl N-(6-amino-2,3-dichlorobenzyl)glycinate (VII), which is cyclized with cyanogen bromide (VIII) in toluene affording ethyl 5,6-dichloro-3,4-dihydro-2-(1H)-iminoquinazoline-3-acetate (IX). Finally, this compound is submitted to a new cyclization by means of triethylamine in refluxing ethanol.
SYN
WO 0208228

The nitration of 2,3-dichlorobenzaldehyde (I) with HNO3/H2SO4 gives 2,3-dichloro-6-nitrobenzaldehyde (II), which is reduced with NaBH4 in methanol, yielding 2,3-dichloro-6-nitrobenzyl alcohol (III). The reaction of (III) with SOCl2 and TEA affords the benzyl chloride (IV), which is condensed with glycine ethyl ester (V) by means of TEA to provide the adduct (VI). The reduction of the nitro group of (VI) with SnCl2 in aq. HCl or H2 over PtO2/C in ethanol gives the expected amino derivative (VII), which is cyclized with CN-Br in toluene to yield the iminoquinazoline (VIII). Finally, this compound is further cyclized by means of TEA in water to afford the target imidazoquinazolinone.
| US 3932407 |

The condensation of 2-nitro-6-chlorobenzyl chloride (I) with ethyl glycinate (II) by means of triethylamine in refluxing ethanol gives ethyl N-(2-nitro-6-chlorobenzyl)glycinate (III), which is reduced with H2 over Pd/C in ethanol yielding ethyl N-(2-amino-6-chlorobenzyl)glycinate (IV). The cyclization of (IV) with cyanogen bromide (A) in refluxing ethanol affords 6-chloro-1,2,3,5-tetrahydroimidazo[2,1-b]quinazolin-2-one (V), which is finally chlorinated with Cl2 and FeCl3 in hot nitromethane.
PATENTS
CN 103254197
US 3932407
WO 2002008228
CN 102757434
WO 2012052781
WO 2005080398
PATENT
![]()
| Applicants: | CIPLA LIMITED [IN/IN]; 289 Bellasis Road, Mumbai Central, Mumbai 400 008 (IN) (For All Designated States Except US). PATHI, Srinivas, Laxminarayan [IN/IN]; (IN) (For US Only). KANKAN, Rajendra, Narayanrao [IN/IN]; (IN) (For US Only). RAO, Dharmaraj, Ramachandra [IN/IN]; (IN) (For US Only). CURTIS, Philip, Anthony [GB/GB]; (GB) (MW only) |
| Inventors: | PATHI, Srinivas, Laxminarayan; (IN). KANKAN, Rajendra, Narayanrao; (IN). RAO, Dharmaraj, Ramachandra; (IN) |

Yusuf Hamied
Anagrelide, is a potent reducer of platelet count induced by a variety of aggregating agents and has the following structure

( Formula II)
TJS 4146718 disckre? the process for the preparation ->f ethyl-N-(2,3-dich’oro-6 n:tr^benzyl) glycine hydrochloride from 1,2,3-trichlorobenzene as depicted in Scheme I via 2,3-dichloro-6-nitrobenzonitrile, which involves the use of poisonous reagents, such as cuprous cyanide. Cyanation is carried out at a temperature of 1650C which is highly exothermic, uncontrollable and not scalable. 2, 3-dichloro-6-nitrobenzonitri]e has extreme toxic and skin-irritant properties. Diborane is a flammable gas, used for the reduction of 2, 3-dichloro-6-nitrobenzonitrile. The reduction reaction is exothermic, uncontrollable and not feasible industrially.
Scheme I :

1 ,2,3-Tπchlorobenzene 2,3 ,4-Trichloronitro 2,3-Dichloro-6-nitro
benzene benzonitπle

Ethyl-N-(2,3-dichloro-6-mtrobenzyl) 2,3-Dichloro-6-nitro
glycine hydrochloride benzylamme
US 5801245 discloses process for the preparation of ethyl-N-(2,3-dichloro-6-nitrobenzyl)glycine hydrochloride from 2,3-dichloro toluene as depicted in Scheme II.

2,3-dichloro-toluene 2,3-dichloro-6-nιtrotoluene
+ H2NCH2COOEt HCI HCI 

2,3-dιchloro-6-nitro Glycine ethyl ester ethyl-N-(2,3-dιchloro-6-nιtro benzyl bromide hydrochloride benzyl)glycιne HCI
The reaction involves a radical halogenation of the toluene group. The material is purified by column chromatography at each stage which makes the process more tedious and it is not viable industrially. The use of a chromatographic solvent, such as chloroform (which is a known carcinogen), is disadvantageous with respect to industrial application.
US 2003/0060630 discloses a method for making ethyl-N-(2, 3-dichloro-6-nitro benzyl)glycine hydrochloride form 2,3-dichloro benzaldehyde as depicted in Scheme III.
Scheme III :

2,3-Dichloro benzaldehyde 2,3-Dichloro-6-mtro 2,3-Dichloro-6-nitro
benzaldehyde benzylalcohol
Step c Thionyl chloride

Ethyl-N-(2,3-dichloro-6-nitrobenzyl) 2,3 -Dichloro-6-nitro
glycine hydrochloride benzyl chloride
In step (b), the reduction reaction is earned out in high boiling solvents like toluene. The reduction in step (b) and the chlorination in step (c) are sluggish. Also, the chlorination reaction is exothermic and uncontrollable, which leads to formation of more impurities and thereby resulting in low yield (page 4, column 2, and page 5, column 1 : 65 %) . Hence, this prior art process is not viable for industrial scale up.
Because of the difficulties encountered in the processes disclosed in the prior art, there is a need to develop more efficient and economical synthetic route for the preparation of ethyl-N- (2,3-dichloro-6-nitrobenzyl)glycine hydrochloride, which is suitable for industrial scale up. The present invention relates to a new process for the synthesis of Ethyl-N-(2, 3-dichloro-6-nitrobenzyl)glycine hydrochloride.
Scheme IV :

2,3-Dichloro-6-nitro 2, 3-Dichloro-6-nitro
benzaldehyde benzylalcohol
( III ) ( IV ) ( V )
Acetonitπle
H2NCH9COOEt
HCl(g) in DPA / Ethyl acetate

Ethyl-N-(2,3-dichloro-6-nitroberizyl)
glycine hydrochloride ( I )
EXAMPLES
Example 1
Preparation of 2, 3-dichloro-6-nitro benzyl methane sulphonate, a compound of formula
(V):
Methylene chloride (2000 ml) and sodium borohydride (120 g) were charged to a clean and dry flask and chilled to 0-50C. Methanol (100 ml) was added slowly over a period of 20 minutes followed by 2,3-dichloro-6-nitro benzaldehyde solution (500 g in 2000 ml of methylene chloride) over a period of 2 hours maintaining the temperature at 0-50C and the contents were stirred at 0-50C for 1 hour. After completion of reaction, water (3000 ml) was added and stirred for 10 minutes. The organic layer was separated, dried over sodium sulphate and was filtered to get a clear filtrate.
To the clear filtrate triethylamine (460 ml), was slowly added over a period of 1 hour at 10- 5 150C, then methane sulphonyl chloride (325 ml) was added drop wise over a period of 2 hours maintaining temperature of 10-150C and the reaction mass was allowed to attain room temperature. Further the reaction mass was stirred at room temperature for 5 hours and after completion of reaction, the organic layer was washed with water (1000 ml) twice, followed by IN HCl solution (1000 ml) twice, 5% Sodium bicarbonate solution (1000 ml) twice, water 0 (1000 ml) twice and was dried over sodium sulfate. The clear organic layer was concentrated under vacuum below 4O0C to give the title compound which was used in the next step.
Example 2
Preparation of ethyl N-(2,3-dichIoro-6-nitrobenzyl)gIycine hydrochloride, a compound of formula (I) :
2,3-dichloro-6-nitro benzyl methane sulphonate ( Examplel ) was dissolved in acetonitrile (2400 ml). To this reaction mass were charged anhydrous Potassium carbonate (480 g), dimethyl amino pyridine (480 mg) and glycine ethyl ester (240 g) at room temperature. The contents were stirred at 37-4O0C for 24 hours. After completion of reaction, the insolubles were filtered, washed with acetonitrile (120 ml). The clear filtrate was concentrated and stripped off usin” ethyl acetate (240 ml).
Further ethyl acetate (1200 ml) was added, chilled the contents to 5-100C, adjusted the pH to 2.0 using IP A-HCl at 5-1O0C. The contents were stirred at 5-100C for 1 hour. The solids were filtered, washed with chilled ethyl acetate (120 ml) and dried under vacuum at room temperature for 4 hours to give the title compound (595 g, 76 % yield, 98.5% HPLC purity).
Example 3
Preparation of Anagrelide , a compound of formula (II)
a) Preparation of Ethyl-5,6-dichloro-3,4-dihydro-2[lH]-imino quinazolin-3-acetate hydrobromide A solution of stannous chloride dihydrate (1850 gms) in concentrated HCl (6.7 liters ) was added slowly to a cooled solution of ethyl-N-(2,3-dichloro-6-nitrobenzyl)glycine hydrochloride (595gms) in concentrated HCl (5.15 liters) maintaining temperature 15-200C over a period of 2 hours. The contents were heated slowly to 40-450C and stirred for 1 hour at 40-450C. After completion of reaction, the contents were cooled to 15-2O0C, maintained for 15 minutes and filtered.
The solids thus obtained were suspended in water (2.9 liters), adjusted the pH of the reaction mass to 8.0-9.0 using potassium carbonate solution (prepared by dissolving 376 gms of potassium carbonate in 4.25 liters of water) at 0-50C, extracted into toluene (3.0 liters><3), dried over sodium sulphate and clarified.
To the clear toluene layer, added Cyanogen bromide solution (prepared by dissolving 222 gms of cyanogen bromide in 655 ml of toluene) in 30 minutes maintaining temperature 15-200C and stirred at 25-300C for 2 hours. The contents were heated slowly to 105-1100C and maintained for 16 hours at 105-1100C. After completion of reaction, the mass was cooled to 15-2O0C and stirred for 45 minutes. Filtered the material, washed with chilled toluene (1.3 liters). The material was slurried in toluene (470 ml) at 15-200C for 1 hour, filtered, washed with cold toluene (160 ml) and dried under vacuum at 50-600C for 8 hours to give the title compound (445 gms ).
b) Preparation of 6,7-Dichloro-l,5-dihydroimidazo[2,l-b]quinazolin-2(3H)-one [Anagrelide]
A mixture of ethyl-5,6-dichloro-3,4-dihydro-2(lH)-iminoquinazolin-3-acetate hydrobromide (445 gms), isopropyl alcohol (4.45 liters) and triethylamine (246 ml) was refluxed for 2 hours. After completion of reaction, the mixture was cooled to 20-250C, filtered, washed with chilled isopropyl alcohol (1.0 liters) and dried under vacuum at 50-550C for 6 hours to give the title compound (285 gms).
REF
- Jump up^ Voglová J, Maisnar V, Beránek M, Chrobák L (2006). “[Combination of imatinib and anagrelide in treatment of chronic myeloid leukemia in blastic phase]”. Vnitr̆ní lékar̆ství (in Czech). 52 (9): 819–22. PMID 17091608.
- Jump up^ https://globenewswire.com/news-release/2016/12/28/901925/0/en/Galena-Biopharma-Confirms-Regulatory-Pathway-for-GALE-401-Anagrelide-Controlled-Release.html
- ^ Jump up to:a b Harrison CN, Campbell PJ, Buck G, et al. (July 2005). “Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia”. N. Engl. J. Med. 353 (1): 33–45. doi:10.1056/NEJMoa043800. PMID 16000354.
- Jump up^ Reilly, John T. (1 February 2009). “Anagrelide for the treatment of essential thrombocythemia: a survey among European hematologists/oncologists”. Hematology. 14(1): 1–10. doi:10.1179/102453309X385115. PMID 19154658.
- Jump up^ Brière, Jean B (1 January 2007). “Essential thrombocythemia”. Orphanet Journal of Rare Diseases. 2 (1): 3. doi:10.1186/1750-1172-2-3. PMC 1781427
 . PMID 17210076.
. PMID 17210076. - Jump up^ Campbell PJ, Bareford D, Erber WN, et al. (June 2009). “Reticulin accumulation in essential thrombocythemia: prognostic significance and relationship to therapy”. J. Clin. Oncol. 27 (18): 2991–9. doi:10.1200/JCO.2008.20.3174. PMC 3398138 . PMID 19364963.
- Jump up^ Petrides PE (2006). “Anagrelide: what was new in 2004 and 2005?”. Semin. Thromb. Hemost. 32 (4 Pt 2): 399–408. doi:10.1055/s-2006-942760. PMID 16810615.
- Jump up^ Jones GH, Venuti MC, Alvarez R, Bruno JJ, Berks AH, Prince A (February 1987). “Inhibitors of cyclic AMP phosphodiesterase. 1. Analogues of cilostamide and anagrelide”. J. Med. Chem. 30 (2): 295–303. doi:10.1021/jm00385a011. PMID 3027338.
- Jump up^ Harrison CN, Bareford D, Butt N, et al. (May 2010). “Guideline for investigation and management of adults and children presenting with a thrombocytosis”. Br. J. Haematol. 149(3): 352–75. doi:10.1111/j.1365-2141.2010.08122.x. PMID 20331456.
- Jump up^ W. N. Beverung, A. Partyka, U.S. Patent 3,932,407; USRE 31617; T. A. Jenks et al., U.S. Patent 4,146,718 (1976, 1984, 1979 all to Bristol-Myers).
- Jump up^ Yamaguchi, Hitoshi; Ishikawa, Fumiyoshi (1981). “Synthesis and reactions of 2-chloro-3,4-dihydrothienopyrimidines and -quinazolines”. Journal of Heterocyclic Chemistry. 18: 67. doi:10.1002/jhet.5570180114.
External links
- Shire Pharmaceuticals Agrylin/Xagrid website for international visitors
- Shire Pharmaceuticals Xagrid website for UK visitors
- AOP Orphan Pharmceuticals website

 |
|
| Clinical data | |
|---|---|
| Trade names | Agrylin |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601020 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Metabolism | Hepatic, partially through CYP1A2 |
| Biological half-life | 1.3 hours |
| Excretion | Urine (<1%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C10H7Cl2N3O |
| Molar mass | 256.088 g/mol |
| 3D model (JSmol) | |
/////////Anagrelide, アナグレリド , EU 2018, EMA 2018, SHIRE, FDA 1997. orphan drug status

